Embed Size (px)
Citation preview

MECHANISMS OF EXOCYTOSIS
Fluorescent Cargo Proteins in PeptidergicEndocrine Cells
Cell Type Determines Secretion Kineticsat Exocytosis
Darren J. Michael,a Sompol Tapechum,a,b Joyce G. Rohan,a
Joseph M. Johnson,a and Robert H. Chowa
aDepartment of Physiology and Biophysics, Keck School of Medicine, Zilkha NeurogeneticInstitute, University of Southern California, Los Angeles, California, USA
bDepartment of Physiology, Mahidol University, Bangkok, Thailand
Fluorescent fusion proteins are an important tool for the study of vesicle trafficking andexocytosis, especially when combined with newer types of microscopy. We previouslyreported that the design of a vesicle-targeted fluorescent fusion construct strongly in-fluences the kinetics of fluorescence change at exocytosis. In the present study wedemonstrate that the cell in which a construct is expressed also affects the kineticsof fluorescence change at exocytosis. We fused enhanced green fluorescent protein tothe carboxy terminus of the vesicular cargo protein rodent islet amyloid polypeptide.The two proteins were separated by a “linker” sequence of 18 amino acids. We thencompared kinetics of fluorescence change at exocytosis for this fluorescent cargo pro-tein expressed in three different types of peptidergic endocrine cell: pancreatic alphacell, pancreatic beta cell, and adrenal chromaffin cell. In resting cells of all three types,fluorescent spots of similar size and membrane-proximal density appeared near theplasma membrane as expected if the probe is stored in large dense-core secretory vesi-cles. Upon stimulation, the fluorescent spots displayed sudden changes in fluorescenceintensity that were consistent with exocytosis. In beta and alpha cells the fluorescentspots consistently brightened and persisted, whereas in chromaffin cells the fluores-cent spots always dispersed rapidly. Thus, for fluorescent cargo proteins in peptidergicendocrine cells, cell type influences the kinetics of fluorescence change at exocytosis.Together with our previous findings, this observation strongly highlights the fact thatthe behavior of vesicle-targeted fluorescent cargo may be unrelated to that of nativecargo, and it emphasizes the need for caution in interpreting fluorescence kinetics interms of an exocytosis mechanism.
Key words: peptide hormones; islets of langerhans; insulin; insulin-secreting cells;glucagon-secreting cells; green fluorescent proteins; chromaffin granules; secretoryvesicles; membrane fusion
Introduction
Peptidergic endocrine cells are specializedfor the synthesis, sorting, trafficking, storage,and secretion of peptide hormones.1,2 In gen-
Address for correspondence: Robert H. Chow, 1501 San Pablo St.,ZNI325, Los Angeles, CA 90033. Voice: +323-442-2901; fax: +323-442-4466. [email protected]
eral, peptide hormones are synthesized as pre-cursor polypeptides that exit the Golgi body inlarge dense-core vesicles (LDCVs). Precursorpolypeptides are then converted to mature pep-tide hormones inside LDCVs, while the vesi-cles are transported to the plasma membraneand made ready for secretion.3 In addition topeptide hormones, LDCVs also contain a richmixture of ions, small molecules, and otherproteins.4,5 The sizes and chemical properties
Mechanisms of Exocytosis: Ann. N.Y. Acad. Sci. 1152: 7–17 (2009).doi: 10.1111/j.1749-6632.2008.04006.x C© 2009 New York Academy of Sciences.
7

8 Annals of the New York Academy of Sciences
of these contents vary widely. Moreover, as LD-CVs mature, this cargo partitions between asolution phase and a condensed core that con-tains most of the peptide hormone. Because thecomponents of the stored cargo are of differentsizes and different physical states within a sin-gle vesicle, it is unclear whether all cargo leavesLDCVs at the same rate or to the same extent.6
At the plasma membrane, secretion-competent LDCVs await the elevation of cy-toplasmic calcium to trigger secretion of theircargo; but before vesicles can release theircargo, the vesicle membrane must fuse withthe plasma membrane. Both proteins and lipidscontribute to this process, which begins withthe formation of a narrow aqueous canal—or“fusion pore”—connecting the vesicle lumenwith the extracellular space and having proper-ties similar to those of an ion channel.7 Recentstudies have clearly shown that this fusion poreis dynamic, sometimes opening rapidly and ir-reversibly, at other times opening repeatedlyand briefly and then closing. In the latter case,both the lifetime and the size of the fusion porevary widely. From experiments combining elec-trophysiological8 and electrochemical9,10 mea-surements of single vesicle exocytosis, it is nowclear that some small molecules and ions can es-cape during transient opening of fusion pores.11
Yet the scarcity of other methods for measur-ing secretion of peptide hormones from singleLDCVs has left unanswered the questions ofhow much and how quickly peptide cargo is re-leased from individual LDCVs during a singleround of exocytosis.
For more than 50 years, there has beenkeen interest to understand how peptidergic en-docrine cells regulate the secretion of LDCVsand their peptide hormone cargo. This interestarises for at least two different reasons. First,peptidergic endocrine cells secrete LDCVs,using much of the same protein machinerythat neurons use to secrete synaptic vesicles.3
However, secretion from the larger LDCVs(diameters 200–500 nm) is much easier to studythan it is from synaptic vesicles (diameters40–50 nm). Second, and more important, re-
search on secretion from peptidergic endocrinecells has been driven by the increasing preva-lence of diseases characterized by impaired se-cretion of peptide hormones, such as type 2diabetes mellitus.12 Much of the recent work atthe level of the single cell and the single vesiclehas relied on electrophysiological and electro-chemical measures of secretion, which have suf-ficient temporal resolution to monitor the stepsof membrane fusion and cargo secretion, re-spectively.13,14 But neither of these techniquesis able to follow the behavior of vesicle cargobefore, during, and after a single round of ex-ocytosis. Such studies are now possible usingfluorescent probes and specialized forms of op-tical microscopy.
During the last decade, major advances inoptical microscopy, molecular biology, virology,and protein engineering have made it possi-ble to use fluorescent probes to study regu-lated secretion in living peptidergic endocrinecells. Particularly important has been the de-velopment of fluorescent proteins,15 which canserve to label LDCVs specifically using ge-netically encoded probes.16 In general, thesefluorescent protein constructs consist of threedistinct components: a vesicle protein, a fluo-rescent protein, and a short sequence of aminoacids, or “linker,” that links the vesicle pro-tein and the fluorescent protein.17 These flu-orescent constructs have been made eitherwith cargo proteins that are stored in thevesicle lumen and secreted during exocyto-sis or with membrane proteins that are pre-sumably retained by the vesicle membraneand reused in multiple rounds of exocytosis.The list of fluorescently tagged vesicle pro-teins used to date includes luminal proteins,such as chromogranin B,16 neuropeptide Y,18–21
islet amyloid polypeptide,17,22 insulin,23 insulinC-peptide,17,24,25 atrial naturetic protein,26,27
tissue plasminogen activator,19,20,28 and syn-collin,17,29 as well as membrane proteins,such as phogrin,23,30,31 syntaxin,20,32 VAMP(vesicle-associated membrane protein),21,33 andSNAP-25 (synaptosome-associated protein of25 dKa).32,34

Michael et al.: Fluorescent Cargo Proteins in Peptidergic Endocrine Cells 9
Despite the increasing use of fluorescentcargo proteins in studies of regulated exocy-tosis, few studies have directly compared se-cretion kinetics at the level of the single vesiclefor the same fluorescent cargo protein being ex-pressed in different endocrine cell types. Such acomparison is especially important because thebehavior of a fluorescent cargo protein is notalways directly related to the behavior of nativecargo found in LDCVs.24 In fact, we have pre-viously shown that small changes in the designof a fluorescent cargo protein can significantlychange its behavior during exocytosis.17 Here,we compare the behavior of the same fluores-cent cargo protein in three types of endocrinecell, all of which use LDCVs to transport, store,and secrete peptide hormones. Specifically, weinvestigate single vesicle secretion in living pri-mary cultures of pancreatic alpha cells, pancre-atic beta cells, and adrenal chromaffin cells.
Materials and Methods
Tissue Collection and Cell Culture:Pancreatic Alpha and Beta Cells
Adult (8- to 12-week-old) male rats (Sprague-Dawley, Charles River, Wilmington, MA) wereanesthetized (50 mg/kg pentobarbital, in-traperitoneal) and sacrificed according to pro-cedures approved by the University of SouthernCalifornia Institutional Animal Care and UseCommittee. The pancreas was inflated via thepancreatic duct with 10 mL of cold digestionsolution: Hank’s balanced salt solution (HBSS)containing 0.233 mg/mL collagenase (LiberaseRI, Roche, Indianapolis, IN) and 0.1 mg/mLDNase I (Roche). The excised pancreas wastransferred to a glass vial (20 mL capacity) con-taining 5 mL of cold digestion solution andimmediately incubated (static, 33 min) in a wa-ter bath at 37◦C. After incubation, the vialwas shaken vigorously and the contents werewashed at least four times with cold HBSS.The tissue was centrifuged (200 g, 1 min), andthe resulting supernatant was discarded. Islets
were enriched by centrifugation (15 min, 200 g)in a discontinuous bovine serum albumin (BSA)gradient (35%, 31%, 27%, and HBSS). Isletswere washed once more, picked by hand us-ing a pipette tip (200-μL capacity), and col-lected in a microcentrifuge vial (approximately300 islets/vial). For dispersion, islets were incu-bated at 37◦C for 5 min in Ca2+-free solution(0.5 mL, HBSS without Mg2+ or Ca2+ contain-ing 1 mmol/L EGTA) and thereafter for 5 minin HBSS (0.5 mL) containing 0.03 units/mLneutral protease (Sigma, St. Louis, MO). Uponremoval of the enzyme solution, islets were re-suspended in culture medium [approximately1.33 μL/islet RPMI 1640 without fetal bovineserum (FBS), 8 mmol/L glucose] and dispersedinto small clusters of cells using gentle trit-uration with a pipette tip (1.0 mL capacity).Cells were plated (40 μL/dish) either on bareglass cover slips or on cover slips coated with alaminin-rich matrix35 and allowed to attach forapproximately 30 min. Thereafter, cells werecultured (RPMI 1640, 10% FBS, 8 mmol/Lglucose) at 37◦C with 5% CO2 and used within5 days.
Tissue Collection and Cell Culture:Bovine Adrenal Chromaffin Cells
Adrenal glands were obtained from steers(1–2-years old) killed at a local abattoir. Glandswere immediately perfused with cold Locke’ssalt solution and returned to the laboratory onice. In the laboratory, fat was trimmed from thegland before the gland was perfused with warmenzyme solution [type 2 collagenase (Worthing-ton Biochemical Corporation, Lakewood, NJ),final concentration of 500 units/mL; DNase I(Roche), final concentration of 0.02 mg/mL].The inflated gland was incubated for 5 min at37◦C. The previous two steps—inflation andincubation—were repeated three times. A su-perficial equatorial incision was made alongthe perimeter of the gland, and forceps wereused to pull the gland apart, thereby expos-ing the central adrenal medulla. Tissue in themedulla was identified by its pale yellow color

10 Annals of the New York Academy of Sciences
and transferred to a small sterile flask that con-tained a spin bar and 20 mL of enzyme solu-tion. Tissue was incubated for 20 min by plac-ing the bottom of the flask in a water bath at37◦C and using a magnetic stir bar/plate toprovide gentle mixing throughout the incuba-tion period. Digested tissue was then passedthrough a filter (100-μm nylon mesh) and cen-trifuged (60 g, 5 min). Cells were washed oncewith Locke’s salt solution and then resuspendedwith culture medium [Dulbecco’s modified Ea-gle’s medium (DMEM)/F-12 with 1% insulin-transferrin-selenium-X (Invitrogen, Carlsbad,CA) and 0.1% penicillin-streptomycin] at adensity of 100–200 cells/mm3. Cells were cul-tured on collagen-coated glass cover slips andused within 5 days.
Recombinant Adenovirusand DNA Delivery
Construction of plasmid and adenoviruscontaining the rodent isoform of islet amy-loid polypeptide (rIAPP)-enhanced green flu-orescent protein (EGFP) has been reported.17
Following overnight incubation after plating,cells were transduced with adenovirus (5 × 107
plaque-forming units/mL in RPMI 1640 with10% FBS and 8 mmol/L glucose) contain-ing the gene for rIAPP-EGFP. Following in-cubation (4 h), the medium was removedand replaced with RPMI 1640 (10% FBS,8 mmol/L glucose, pancreatic alpha and betacells) or DMEM/F-12 (1% insulin-transferrin-selenium-X, adrenal chromaffin cells). Cellswere imaged between 16 and 48 h followingtransduction.
Solutions and Chemicals
Extracellular solution used in imaging ex-periments was composed of the following[mmol/L]: 136 NaCl, 4.2 KCl, 2.4 CaCl2, 1.2MgSO4, 1.2 KH2PO4, 1 L-glutamine, 4 glu-cose, and 10 HEPES (pH 7.4). In the stimulat-ing solution, an equal quantity of KCl replacedNaCl to yield a final potassium concentration of
50 mmol/L. All solutions were prepared withwater from a commercial purification system(Nanopure Infinity, Barnstead Thermolyne,Dubuque, IA). The osmolalities of extracellularand stimulating solutions were matched and fellbetween 280 and 320 mOsm. All salts for so-lutions were obtained from Sigma-Aldrich (St.Louis, MO) with the highest purity available.
Total Internal ReflectionFluorescence Imaging
We have previously published the detailsfor our total internal reflection fluorescence(TIRF) microscope.17 For two color TIRF mi-croscopy, we added the following components:1) helium-neon laser (543 nm; JDS Uniphase,San Jose, CA), 2) dichroic mirror (530dclp;Chroma, Rockingham, VT), 3) dichroic mir-ror (XF2046; Omega, Brattleboro, VT), and4) dual emission pathway [DualView; OpticalInsights, Santa Fe, NM equipped with dichroicmirror (560dcxr, Chroma) and emission filters(HQ515/30 and D605/55, Chroma)]. Beforeand after each two-color TIRF experiment, weimaged a field of stationary beads labeled withmultiple fluorescent dyes (100-nm diameter;TetraSpeck, Invitrogen). For most beads, redand green fluorescence profiles agreed within1 pixel in both the x- and y-axis. If an adjust-ment was necessary to align red and green flu-orescence profiles for beads at a given location,a similar adjustment was applied to the sameregions for all images when we analyzed datafrom the corresponding experiments.
Immunocytochemical Labelingof Pancreatic Alpha Cells
Living cultures of pancreatic islet cells werefirst imaged by TIRF microscopy and thenstimulated. If a cell responded, bright-fieldimages were taken in the region surround-ing the cell of interest. These images werealigned and used as a map for finding the cellfollowing fixation and immunocytochemicallabeling.

Michael et al.: Fluorescent Cargo Proteins in Peptidergic Endocrine Cells 11
For immunocytochemical labeling, cells werefixed [2% paraformaldehyde in phosphatebuffered saline (PBS), 15 min], washed (PBS),and permeabilized (0.2% Triton-X 100 inPBS). Thereafter, nonspecific antigens wereblocked [1% BSA, twice for 5 min followedby 5% normal rabbit serum (NRS), 30 min].Next, samples were treated with primary [1:50,rabbit antiglucagon antibody (Zymed Labora-tories, South San Francisco, CA), 1 h] and sec-ondary antibody (1:1000, AlexaFluor 555 goatantirabbit antibody, 30 min). Both antibody so-lutions were prepared in PBS with 5% NRS.Finally, cells were washed three times with PBS(5 min each wash).
Cells were then imaged using an invertedmicroscope with both low- (Olympus, 20×,0.5 NA, phase contrast) and high-magnification(Zeiss, 100×, 1.4 NA) objectives. When EGFPis viewed through the filters described above,red intensity is approximately 10% green inten-sity. To assess objectively the overlap of EGFPand AlexaFluor 555-labeled antibody, we com-puted correlation coefficients for each plane inrepresentative z-stacks. Additionally, we veri-fied that regions of overlap shared similar mor-phological structure by comparing red andgreen fluorescence intensity along lines drawnthrough a single plane (see Fig. 2).
Deconvolution Wide-fieldFluorescence Microscopy
Z -stacks were collected with 100-nm spac-ing by using commercially available software(Metamorph, Molecular Devices, Downing-town, PA) to control a piezoelectric posi-tioner (PiFoc, Physik Instrumente, Walbronn,Germany) placed under the microscope ob-jective. For deconvolution, we used commer-cially available software (Metamorph) that usesa measured point spread function. To measurethe point spread function of our microscope,we acquired z-stacks with 100-nm spacing for100-nm beads labeled with multiple fluorescentdyes (TetraSpeck, Invitrogen).
Results and Discussion
A Simple Method to Enrich Alpha Cellsin Primary Cultures of Rat Pancreatic
Endocrine Cells
The Islets of Langerhans contain multipletypes of endocrine cells, including glucagon-secreting alpha cells, insulin-secreting betacells, somatostatin-secreting delta cells, andpancreatic-polypeptide-secreting PP cells. Theproportion of each endocrine cell type withinan islet varies according to species. In rodents,beta cells account for roughly 70–80% of thecells,36,37 and they are located primarily at thecenter of the islet, while the other cell typessegregate mostly to the edges of the islet. Thus,when rodent pancreatic islets are extractedfrom tissue, they often lose a large fraction of theendocrine cell types found at the edges while re-taining most of the centrally located beta cells.For these reasons, primary cultures of rodentislet endocrine cells typically are enriched inpancreatic beta cells, making it challenging tostudy nonbeta islet cells.
The morphological and physiological prop-erties of pancreatic alpha and beta cells differ,and these properties have been used to distin-guish the types of cells in culture.37 For exam-ple, pancreatic beta cells tend to be larger thanpancreatic alpha cells. These two cell types alsohave different ionic currents, and it is possi-ble to determine a cell’s type by characterizingits sodium currents; under typical conditions,mouse pancreatic beta cells lack the rapidly in-activating sodium current that is found in bothpancreatic alpha and delta cells. There are im-portant limitations, however, to using cell sizeand sodium current electrophysiology to distin-guish islet cells. First, the difference in sodiumcurrents for alpha and beta cells may be lim-ited to mice. In addition, the characterization ofsodium currents is a relatively slow process thatis not possible in laboratories lacking electro-physiology equipment or expertise to evaluateionic currents.
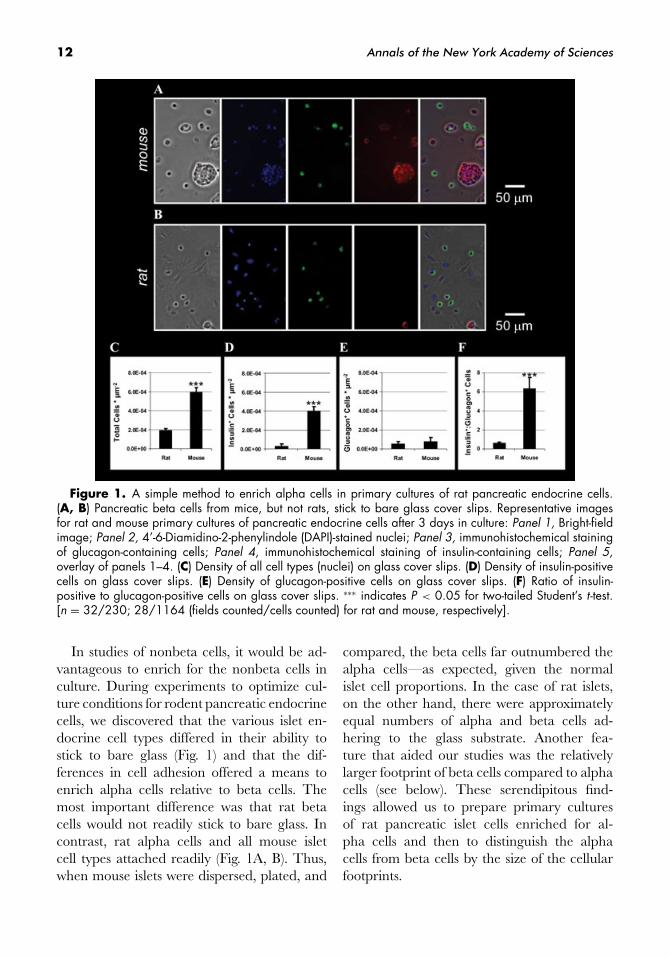
12 Annals of the New York Academy of Sciences
Figure 1. A simple method to enrich alpha cells in primary cultures of rat pancreatic endocrine cells.(A, B) Pancreatic beta cells from mice, but not rats, stick to bare glass cover slips. Representative imagesfor rat and mouse primary cultures of pancreatic endocrine cells after 3 days in culture: Panel 1, Bright-fieldimage; Panel 2, 4’-6-Diamidino-2-phenylindole (DAPI)-stained nuclei; Panel 3, immunohistochemical stainingof glucagon-containing cells; Panel 4, immunohistochemical staining of insulin-containing cells; Panel 5,overlay of panels 1–4. (C) Density of all cell types (nuclei) on glass cover slips. (D) Density of insulin-positivecells on glass cover slips. (E) Density of glucagon-positive cells on glass cover slips. (F) Ratio of insulin-positive to glucagon-positive cells on glass cover slips. ∗∗∗ indicates P < 0.05 for two-tailed Student’s t -test.[n = 32/230; 28/1164 (fields counted/cells counted) for rat and mouse, respectively].
In studies of nonbeta cells, it would be ad-vantageous to enrich for the nonbeta cells inculture. During experiments to optimize cul-ture conditions for rodent pancreatic endocrinecells, we discovered that the various islet en-docrine cell types differed in their ability tostick to bare glass (Fig. 1) and that the dif-ferences in cell adhesion offered a means toenrich alpha cells relative to beta cells. Themost important difference was that rat betacells would not readily stick to bare glass. Incontrast, rat alpha cells and all mouse isletcell types attached readily (Fig. 1A, B). Thus,when mouse islets were dispersed, plated, and
compared, the beta cells far outnumbered thealpha cells—as expected, given the normalislet cell proportions. In the case of rat islets,on the other hand, there were approximatelyequal numbers of alpha and beta cells ad-hering to the glass substrate. Another fea-ture that aided our studies was the relativelylarger footprint of beta cells compared to alphacells (see below). These serendipitous find-ings allowed us to prepare primary culturesof rat pancreatic islet cells enriched for al-pha cells and then to distinguish the alphacells from beta cells by the size of the cellularfootprints.

Michael et al.: Fluorescent Cargo Proteins in Peptidergic Endocrine Cells 13
A Fluorescent Cargo Protein: RodentIslet Amyloid Polypeptide-enhanced GFP
IAPP is a small cargo protein found in insulinvesicles in many species.38 The human isoform(hIAPP) of this protein is the self-associatingprecursor polypeptide of amyloid plaques com-monly seen in type 2 diabetes mellitus.39 Selfassociation of hIAPP is believed to be mediatedby the so-called GAILS sequence, a consensussequence of amino acids that is shared by IAPPfrom nearly all species whose IAPP is self aggre-gating. The rodent isoform of IAPP lacks theGAILS sequence; therefore, rodent pancreasdoes not develop spontaneous amyloid plaques,even when animals are maintained under con-ditions that would lead to metabolic diseases inhumans similar to type 2 diabetes mellitus.
The inability of rIAPP to self aggregate ledus, in a previous study,17 to select it as thetargeting moiety for a fluorescent cargo pro-tein designed to label individual insulin secre-tory vesicles. We attached rIAPP to enhancedgreen fluorescent protein (EGFP), which is ap-proximately three times brighter than wild-typeGFP and sensitive to the proton concentrationof its surroundings.15 The fluorescence inten-sity of EGFP is quenched (pKa approximately6.0) in the normally acidic lumen of LDCVsand rapidly brightens when the vesicles fusewith the plasma membrane and the luminalcontents quickly attain neutral pH.40 Whetherthis brightening of EGFP is detected duringexocytosis depends upon how the rate of neu-tralization for the dense core cargo comparesto the rate of escape for the same cargo aswell as the rate at which the camera collectsimages.17
Endocrine Cell Type DeterminesSecretion Kinetics for Fluorescent
Cargo Proteins
Using previously described protocols, weprepared primary cultures of pancreatic alphaand beta cells from rats and primary cultures ofadrenal chromaffin cells from steers. In experi-
ments conducted in parallel, we used recombi-nant adenovirus to deliver the gene for rIAPP-EGFP to living primary cultures of all three celltypes. We used TIRF microscopy41 to imageindividual vesicles near the plasma membraneat the base of single cells. Within 48 h, cellsinfected with adenovirus displayed large num-bers of membrane-proximal vesicles containingfluorescent cargo. The size and membrane-proximal density of these fluorescent vesicleswere similar for all three cell types. In rest-ing cells, most membrane-proximal vesicles dis-played little mobility, only jiggling around acentral point.42–44
Most adrenal chromaffin cells and most pan-creatic beta cells flattened when maintained inprimary cultures. In contrast, most alpha cellsdid not flatten when maintained in culture onbare glass cover slips, giving the cells a roundedappearance with a smaller footprint relative tothe other two types of endocrine cell (Fig. 1).This morphological feature along with thesmaller overall size of alpha cells compared tobeta cells allowed us tentatively to identify cellslikely to be pancreatic alpha cells, even thoughthese cells are much less prevalent than betacells within rat islets. After live-cell imaging,we confirmed the identity of alpha cells in allexperiments by postexperiment immunocyto-chemistry (Fig. 2).
Overall, in alpha cells there was only amoderate correlation between the intensities ofEGFP and fluorescently tagged antiglucagonantibody. This moderate correlation was notsurprising because LDCVs in alpha cells havelifetimes on the order of several days to weeks,so only the newly synthesized vesicles shouldcontain rIAPP-EGFP when imaged 48 h afterviral transduction. In contrast, older LDCVsshould contain glucagon and should be stainedby fluorescent antibody yet should not containany EGFP.
Primary cultures of peptidergic endocrinecells expressing rIAPP-EGFP were stimulatedby a solution containing an elevated concen-tration of potassium ions. Upon stimulation,a fraction of the labeled vesicles in all cell

14 Annals of the New York Academy of Sciences
Figure 2. The rodent isoform of islet amyloid polypeptide (rIAPP)-enhanced green fluorescent protein(EGFP) labels glucagon vesicles in pancreatic alpha cells. (A-C) These images were deconvolved as describedin the methods section. For this cell, the correlation coefficient for red and green fluorescence intensity wascalculated for every plane in the z-stack. Before calculating the correlation coefficient for each plane, afilter and a threshold were applied to flatten the background fluorescence intensity and to eliminate positivecorrelation from any low-intensity systematic noise that might be present. For this data set, the averagecorrelation coefficient was 0.41 ± 0.01 (range, 0.56 to 0.26). The specificity of the correlation was confirmedby scrambling the pixel intensities in each plane for data in the red channel and then calculating the correlationcoefficient for each plane (after scrambling, average correlation coefficient was 0.00 ± 0.04, range: 0.03to −0.03). (D) Line scan along dotted line in C, showing fluorescence intensity. A.U. = arbitrary units.
Figure 3. Fluorescent cargo proteins in peptidergic endocrine cells; cell type determinessecretion kinetics during exocytosis. Labels at top identify the cell type for the data appearingbelow. (A-C) Cell micrographs show a representative cell expressing rIAPP-EGFP; montagesshow consecutive images for a single representative vesicle undergoing exocytosis; line traceswere prepared as follows: For a group of representative vesicles, individual fluorescence in-tensity traces were first corrected for local background, then normalized and aligned, andfinally averaged to produce the trace shown (n = [103,29]∗, [25,8], [27,6], [vesicles, cells];∗data from pancreatic beta cells are reproduced with permission from a previous publica-tion17). For each point, the vertical bars represent SE. Fluorescence intensity was averaged ina 1-μm × 1-μm box centered on the vesicle.
types displayed sudden changes in fluorescenceintensity. The exact pattern of fluorescencechange varied according to the cell type butwas consistent for every vesicle for a givencell type. As previously reported,17 in pan-creatic beta cells all vesicles undergoing ex-
ocytosis were seen as fluorescent spots thatbrightened and persisted (Fig. 3A). In alphacells, all secreted glucagon vesicles labeled withrIAPP-EGFP behaved similarly (Fig. 3B). Incontrast, in chromaffin cells, every secretedcatecholamine-containing vesicle labeled with

Michael et al.: Fluorescent Cargo Proteins in Peptidergic Endocrine Cells 15
rIAPP-EGFP disappeared in a rapidly dispers-ing cloud of fluorescence (Fig. 3C). Of note,others have also reported that small fluorescentcargo proteins leave chromaffin vesicles rapidlyand completely during exocytosis.26
In a previous report,17 we demonstratedin pancreatic beta cells that the behavior ofrIAPP-EGFP at exocytosis changes dramati-cally when the linker sequence is removed.Here, we observed the same behavior for “link-erless” rIAPP-EGFP in pancreatic alpha cells(data not shown). Thus, as previously reportedfor fluorescent cargo proteins in pancreatic betacells,17 the behavior of fluorescent cargo pro-teins in pancreatic alpha cells is determined bythe design of a fluorescent cargo protein.
In conclusion, we have compared the be-havior of the same fluorescent cargo protein inthree different endocrine cell types at the levelof the single vesicle. In all three cell types, weobserved fluorescent spots whose fluorescenceintensity changed suddenly in response to stim-ulation. The pattern of fluorescence change dif-fered according to the type of endocrine cell,but it was consistent in a given cell type for allvesicles undergoing exocytosis.
In a previous publication, we demonstratedthat the design of rIAPP-EGFP fluorescent con-structs determines fluorescence kinetics at exo-cytosis. When that previous observation is com-bined with our current observation, it is clearthat the use of a single fluorescent marker tomonitor exocytosis must be carried out with ex-treme caution. Clearly, the behavior of foreignfluorescent proteins introduced into the reg-ulated secretory pathway may be completelyunrelated to the behavior of native cargo. Dif-ferences in behavior between native cargo andforeign proteins could arise for many reasons,including differences in the way they are pro-cessed or differences in the way they bind orotherwise interact with other vesicle compo-nents, which themselves may display diversebehavior. In the extreme case, foreign proteinsintroduced into the regulated secretory path-way could conceivably lead to the productionof a novel class of vesicles whose behavior is
unrelated to that of native vesicles. Fluores-cent marker behavior should be interpreted interms of exocytosis mechanisms only when themarker has been thoroughly characterized incontrol experiments, and, ideally, studies shouldbe corroborated with other experimental meth-ods conducted in parallel.
Acknowledgments
We thank members of the Chow labora-tory for informative discussions and for com-ments on this manuscript. We are also gratefulfor the contributions from two students fromBravo Medical Magnet High School, Los An-geles, CA: Junia Song and James Yu. This workwas funded by a generous gift from the familyof Betty Smith Rose and grants from the USNational Institutes of Health: DK10181 (DJM)and DK60623 (RHC).
Conflicts of Interest
The authors declare no conflicts of interest.
References
1. Arvan, P. & D. Castle. 1998. Sorting and storage dur-ing secretory granule biogenesis: looking backwardand looking forward. Biochem. J. 332: 593–610.
2. Arvan P. & P.A. Halban. 2004. Sorting ourselvesout: seeking consensus on trafficking in the beta-cell.Traffic 5: 53–61.
3. Jahn, R., T. Lang & T.C. Sudhof. 2003. Membranefusion. Cell 112: 519–533.
4. Njus, D., P.M. Kelley & G.J. Harnadek. 1986. Bioen-ergetics of secretory vesicles. Biochim. Biophys. Acta.
853: 237–265.5. Hutton, J.C., E.J. Penn & M. Peshavaria. 1983. Low-
molecular-weight constituents of isolated insulin-secretory granules. Bivalent cations, adenine nu-cleotides and inorganic phosphate. Biochem. J. 210:297–305.
6. Michael, D.J., H. Cai, W. Xiong, et al. 2006. Mecha-nisms of peptide hormone secretion. Trends Endocrinol.
Metab. 17: 408–415.7. Jackson, M.B. & E.R. Chapman. 2006. Fusion pores
and fusion machines in Ca2+-triggered exocytosis.Annu. Rev. Biophys. Biomol. Struct. 35: 135–160.

16 Annals of the New York Academy of Sciences
8. Neher, E. & A. Marty. 1982. Discrete changes of cellmembrane capacitance observed under conditionsof enhanced secretion in bovine adrenal chromaffincells. Proc. Natl. Acad. Sci. USA 79: 6712–6716.
9. Chow, R.H., L. Von Ruden & E. Neher. 1992. Delayin vesicle fusion revealed by electrochemical monitor-ing of single secretory events in adrenal chromaffincells. Nature 356: 60–63.
10. Leszczyszyn, D.J., J.A. Jankowski, O.H. Viveros, et al.
1990. Nicotinic receptor-mediated catecholamine se-cretion from individual chromaffin cells. Chemicalevidence for exocytosis. J. Biol. Chem. 265: 14736–14737.
11. G.A. de Toledo, R. Fernandez-chacon & J.M.Fernandez. 1993. Release of secretory products dur-ing transient vesicle fusion. Nature 363: 554–558.
12. Weir, G.C. & S. Bonner-Weir. 2004. Five stages ofevolving beta-cell dysfunction during progression todiabetes. Diabetes 53: S16–S21.
13. Travis, E.R. & R.M. Wightman. 1998. Spatio-temporal resolution of exocytosis from individualcells. Annu. Rev. Biophys. Biomol. Struct. 27: 77–103.
14. Angleson, J.K. & W.J. Betz. 1997. Monitoring se-cretion in real time: capacitance, amperometry andfluorescence compared. Trends Neurosci. 20: 281–287.
15. R.Y. Tsien. 1998. The green fluorescent protein.Annu. Rev. Biochem. 67: 509–544.
16. Kaether, C., T. Salm, M. Glombik, et al. 1997. Tar-geting of green fluorescent protein to neuroendocrinesecretory granules: a new tool for real time studies ofregulated protein secretion. Eur. J. Cell. Biol. 74: 133–142.
17. Michael, D.J., X. Geng, N.X. Cawley, et al. 2004. Flu-orescent cargo proteins in pancreatic beta-cells: de-sign determines secretion kinetics at exocytosis. Bio-
phys. J. 87: L3-L5.18. Lang, T., I. Wacker, J. Steyer, et al. 1997. Ca2+-
triggered peptide secretion in single cells imaged withgreen fluorescent protein and evanescent-wave mi-croscopy. Neuron 18: 857–863.
19. Perrais, D., I.C. Kleppe, J.W. Taraska & W. Almers.2004. Recapture after exocytosis causes differentialretention of protein in granules of bovine chromaffincells. J. Physiol. (Oxford) 560: 413–428.
20. Taraska, J.W., D. Perrais, M. Ohara-Imaizumi, et al.
2003. Secretory granules are recaptured largely in-tact after stimulated exocytosis in cultured endocrinecells. Proc. Natl. Acad. Sci. USA 100: 2070–2075.
21. Tsuboi, T. & G.A. Rutter. 2003. Multiple formsof “kiss-and-run” exocytosis revealed by evanescentwave microscopy. Curr. Biol. 13: 563–567.
22. Barg, S., C.S. Olofsson, J. Schriever-Abeln, et al.
2002. Delay between fusion pore opening and pep-tide release from large dense-core vesicles in neuroen-docrine cells. Neuron 33: 287–299.
23. M. Ohara-Imaizumi, Y. Nakamichi, T. Tanaka, et al.
2002. Monitoring of exocytosis and endocytosis ofinsulin secretory granules in the pancreatic beta-cell line MIN6 using pH-sensitive green fluorescentprotein (pHluorin) and confocal laser microscopy.Biochem. J. 363: 73–80.
24. Michael, D.J., L. Haataja, R.A. Ritzel & R.H. Chow.2006. Pancreatic beta-cells secrete insulin in fast- andslow-release forms. Diabetes 55: 600–607.
25. Watkins, S., X. Geng, L. Li, et al. 2002. Imagingsecretory vesicles by fluorescent protein insertionin propeptide rather than mature secreted peptide.Traffic 3: 461–471.
26. Duncan, R.R., J. Greaves, U.K. Wiegand, et al. 2003.Functional and spatial segregation of secretory vesi-cle pools according to vesicle age. Nature 422: 176–180.
27. Han, W.P., D.Q. Li, A.K. Stout, et al. 1999. Ca2+-induced deprotonation of peptide hormones insidesecretory vesicles in preparation for release. J. Neu-
rosci. 19: 900–905.28. Lochner, J.E., M. Kingma, S. Kuhn, et al. 1998. Real-
time imaging of the axonal transport of granules con-taining a tissue plasminogen activator/green fluores-cent protein hybrid. Mol. Biol. Cell. 9: 2463–2476.
29. Ma, L., V.P. Bindokas, A. Kuznetsov, et al. 2004. Di-rect imaging shows that insulin granule exocytosisoccurs by complete vesicle fusion. Proc. Natl. Acad. Sci.
USA 101: 9266–9271.30. Tsuboi, T., C. Zhao, S. Terakawa & G.A. Rutter.
2000. Simultaneous evanescent wave imaging of in-sulin vesicle membrane and cargo during a singleexocytotic event. Curr. Biol. 10: 1307–1310.
31. Taraska, J.W. & W. Almers. 2004. Bilayers merge evenwhen exocytosis is transient. Proc. Natl. Acad. Sci. USA
101: 8780–8785.32. An, S.J. & W. Almers. 2004. Tracking SNARE com-
plex formation in live endocrine cells. Science 306:1042–1046.
33. Allersma, M., L. Wang, D. Axelrod & R. Holz. 2004.Visualization of regulated exocytosis with a granule-membrane probe using total internal reflection mi-croscopy. Mol. Biol. Cell 15: 4658–4668.
34. Takahashi, N., H. Hatakeyama, H. Okado, et al.
2004. Sequential exocytosis of insulin granules is as-sociated with redistribution of SNAP25. J. Cell. Biol.
165: 255–262.35. Beattie, G.M., V. Cirulli, A.D. Lopez & A. Hayek.
1997. Ex vivo expansion of human pancreatic en-docrine cells. J. Clin. Endocrinol. Metab. 82: 1852–1856.
36. Gopel, S.O., T. Kanno, S. Barg & P. Rorsman.2000. Patch-clamp characterisation of somatostatin-secreting -cells in intact mouse pancreatic islets. J.
Physiol. (London) 528: 497–507.

Michael et al.: Fluorescent Cargo Proteins in Peptidergic Endocrine Cells 17
37. Gopel, S., Q. Zhang, L. Eliasson, et al. 2004. Ca-pacitance measurements of exocytosis in mouse pan-creatic alpha-, beta- and delta-cells within intactislets of Langerhans. J. Physiol. (London) 556: 711–726.
38. Nishi, M., T. Sanke, S. Nagamatsu, et al. 1990. Isletamyloid polypeptide. A new beta cell secretory prod-uct related to islet amyloid deposits. J. Biol. Chem.
265: 4173–4176.39. Westermark, P., C. Wernstedt, E. Wilander & K. Slet-
ten. 1986. A novel peptide in the calcitonin generelated peptide family as an amyloid fibril protein inthe endocrine pancreas. Biochem. Biophys. Res. Commun.
140: 827–831.40. Breckenridge, L.J. & W. Almers. 1987. Currents
through the fusion pore that forms during exocytosisof a secretory vesicle. Nature 328: 814–817.
41. D. Axelrod. 2001. Selective imaging of surface fluo-rescence with very high aperture microscope objec-tives. J. Biomed. Opt. 6: 6–13.
42. Ivarsson, R., S. Obermuller, G.A. Rutter, et al. 2004.Temperature-sensitive random insulin granule diffu-sion is a prerequisite for recruiting granules for re-lease. Traffic 5: 750–762.
43. Oheim, M., D. Loerke, W. Stuhmer & R.H. Chow.1998. The last few milliseconds in the life ofa secretory granule. Docking, dynamics and fu-sion visualized by total internal reflection fluores-cence microscopy (TIRFM). Eur. Biophys. J. 27: 83–98.
44. Steyer, J.A. & W. Almers. 1999. Tracking single secre-tory granules in live chromaffin cells by evanescent-field fluorescence microscopy. Biophys J. 76: 2262–2271.





























