Embed Size (px)
Citation preview

Flexible, accessible & reproducible workflows for tandem proteogenomic and metaproteomic analysis using the GalaxyP platform.
Pratik Jagtap1; Brian Sandri2; Julie Yang2; Kevin Murray2; Joel Kooren2; James Johnson3; Getiria Onsongo3; Joel Rudney2; Christine Wendt2 and Tim Griffin11,2 1. Center for Mass Spectrometry and Proteomics, UMN, St. Paul, MN; 2. University of Minnesota, Minneapolis, MN; 3. Minnesota Supercomputing Institute, Minneapolis, MN.
• Salivary proteome was analyzed using search algorithms for high mass accuracy datasets to generate a confident list of proteins
and peptides.
FUTURE PLAN
• We plan to search the entire salivary proteome using ProteinPilot so as to analyze and compare it with MaxQuant results at
protein
ACKNOWLEDGEMENTS
Funding Sources : NIH/NIDCR R01 DE 017734 for Dr. Griffin and Dr. Bandhakavi. Multiple funding resources enlisted on
http://www.cbs.umn.edu/msp/about.shtml for Thomas McGowan.
REFERENCES
Salivary Proteome : Bandhakavi et al J. Proteome Res. 8, 5590 (2009)).
CONCLUSIONS
INTRODUCTION
GALAXYP
GalaxyP Workflow GalaxyP Tools
GalaxyP has multiple software tools
- some proteomics-specific - and
others from the genomics Galaxy
framework.
• Proteogenomics (for identifying unannotated proteoforms) and
metaproteomics (for characterizing non-host/multi-organism proteomes) are
research areas that extend discoveries beyond the reference proteome.
• For biomedically-relevant proteomics studies, tandem proteogenomic and
metaproteomic analysis offers great promise for new discoveries.
• We describe effective and accessible bioinformatic analytical workflows,
amenable to creative customization and sharing to foster collaborative
research efforts.
Tools can be used in a sequential manner to generate
workflows that can be reused, shared and creatively
modified for multiple studies.
Benefits of Galaxy / GalaxyP:
• Software accessibility and usability.
• Share-ability of tools, workflows and histories.
• Reproducibility and ability to test and compare
results after using multiple parameters.
METHODS & DATASETS
• Analytical transparency
• Scalability of data
RAW files from multiple datasets (see below) were generated from Orbitrap Velos instrument.
The processed peak lists were searched using ProteinPilot ™ version 4.5 (AB Sciex) within GalaxyP (usegalaxyp.org). the datasets were searched against 3-frame translated cDNA database
and the human oral microbial database by using two-step method (Jagtap et al 2013). After
optimization & testing, multiple workflows were used in a sequential manner to generate inputs
for the subsequent workflow. Microbial peptides were identified after using metaproteomic
workflows & novel proteoforms were identified after using proteogenomic workflows.
UNLABELED SAMPLE:
• Oral pre-malignant lesion (OPML) dataset was collected as oral exudate using PerioPaper
strip method (Kooren et al 2011) from an individual with pre-malignant lesion & a matched
control sample from adjacent oral cavity.
4-plex iTRAQ LABELED SAMPLE:
• Brush biopsies were collected from patients diagnosed with OPML and from patients with
Oral Squamous Cell Carcinoma (OSCC). For each patient, brush biopsies from the lesion &
the healthy mucosa of corresponding contralateral area were collected (Yang et al 2014).
8-plex iTRAQ LABELED SAMPLE:
• Chronic Obstructive Pulmonary Disease (COPD) – linked lung cancer tissue samples were
collected & subjected to iTRAQ labeling and 2D LC-MS. Ten replicates of this dataset were
searched against the 3-frame translated cDNA database & human oral microbiome database
(HOMD) using the two-step method.
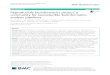
WORKFLOWS FOR TANDEM PROTEOGENOMIC AND METAPROTEOMIC ANALYSIS.
> ENST00000 cdna:
TACGGCCGTCGTGCCC
> ENST000000 cdna:
TCGTGCCGCTTACGGC
Two-step Database
Search method
translation
Peptide Summary from
Second-Step Search with
novel proteoform PSMs and
microbial peptides
Transcriptomic (cDNA or
RNASeq) or Genomic Database
Database
C
PTNTIALNEWPRTEFRM
PEPTIDESINFSTAFRMT
MICRPEPTIDES
ARCHAEALPEPTIDES
Identifying peptides from
microbial db
Data
Processing
Microbial
Peptides D
BLAST-P Analysis E
> ENST|Potential new Microbe1
PTNTIALNEWPRTEFRM
> ENST|Potential new Microbe2
PEPTIDESINFSTAFRMT
Peptides in
FASTA format
Submit for
MEGAN5 Anaysis
Submit to UniPept
for analysis
Peak list (MGF or mzml)
msconvert
MGF
Formatter
Mass spectra
Peak processing
A
> ENST00000 cdna:
TACGGCCGTCGTGCCC
> ENST000000 cdna:
TCGTGCCGCTTACGGC
> ENST00000 protein
ITSAPRTEINDATASET
> ENST000000 protein
INANTHERFRAMETHGH
> sp|Acc No 1|Human
MANPRTEINS
> sp|Acc No 2|Human
MANYHMANPRTEINS
> ENST00000 protein
ITSAPRTEINDATASET
> ENST000000 protein
INANTHERFRAMETHGH
> sp|Acc No 1|Human
MANPRTEINS
> sp|Acc No 2|Human
MANYHMANPRTEINS
Database generation B
translation
Merge
FASTA
Target
database
Host Protein
Database
Metagenomic
Database
Microbial
protein db
> ENST00000 protein
ITSAPRTEINDATASET
> ENST000000 protein
INANTHERFRAMETHGH
> sp|Acc No 1|Human
MANPRTEINS
> sp|Acc No 2|Human
MANYHMANPRTEINS
Translated
genomic db
Host Protein
Database
> ENST00000 protein
ITSAPRTEINDATASET
> ENST000000 protein
INANTHERFRAMETHGH
> sp|Acc No 1|Human
MANPRTEINS
> sp|Acc No 2|Human
MANYHMANPRTEINS
Merge
FASTA
Target
database
Peptide Summary
Peptide Summary
Data
Processing
Data
Processing
> ENST00000 protein
ITSAPRTEINDATASET
> ENST000000 protein
INANTHERFRAMETHGH
> sp|Acc No 1|Human
MANPRTEINS
> sp|Acc No 2|Human
MANYHMANPRTEINS
> ENST00000 protein (decoy)
TESATADNIIETRPASTI
> ENST000000 protein (decoy)
HGHTEMARFREHTNANI
> sp|Acc No 1|Human (decoy)
SNIETRPNAM
> sp|Acc No 2|Human (decoy)
SNIETRPNAMHYNAM
Target-Decoy database
from accession numbers
from first step search
Screenshot of a novel proteoform peptide within Integrated Genomic Viewer. Novel proteoform peptides
identified at 5% local FDR were used to generate GTF file so that the genomic localization of these peptides
can be viewed in genomic context. Proteoform corresponding to HNRNPA2B1 was identified to have an
alternative start site and was identified in 4 replicates.
RESULTS SUMMARY
Short peptides
(<30 aas) Long peptides
(<30 aas)
Spectral
Visualization
Filtering of Peptide Spectral
Matching Metrics
Identifying peptides from
translated genomic db
Data
Processing
F
PTNTIALNEWPRTEFRM
PEPTIDESINFSTAFRMT
MICRPEPTIDES
ARCHAEALPEPTIDES
Potential Novel
Proteoform
Peptides
> ENST|Potential new Microbe1
PTNTIALNEWPRTEFRM
> ENST|Potential new Microbe2
PEPTIDESINFSTAFRMT
Peptides in
FASTA format Short peptides
(<30 aas)
Long peptides
(<30 aas)
BLAST-P Analysis
Filter peptides
with mismatches
to human NCBI
database.
Peak list
(mzml)
Peptide Summary of new proteoform peptides.
Peptide Spectral
Match Evaluation
Dataset
RAW
Files
Distinct peptides
of microbial
origin
Number of
unique
peptides
(Species
Identified )
Novel proteoform
peptides
OPML Control
(unlabeled) 7 637 136 (6) 50
OPML Lesion
(non-labeled) 7 688 136 (3) 47
Brush Biopsy OSCC
(4-plex iTRAQ) 15 1118 166 (6) 6
COPD
(10 replicates)
(8-plex iTRAQ) 150 87 9 (9) 7
RESULTS
G
H
Peptide to GTF
conversion
Peptide Summary of new proteoform peptides with
quality peptide spectral matching characteristics.
General Transfer Format
file for Human genome
Peptide to
GTF
I
Visualization in
genomic context
In COPD dataset, five lung-infecting organisms were identified. Actinomyces viscosus – a bacterium that causes
Actinomycosis (granulomatous infection with the formation of abscesses) in the lungs was identified in five
replicates.
EFFECT OF SEARCH DATABASES ON MICROBIAL &
NOVEL PROTEOFORM IDENTIFICATIONS.
598 30 39
627 33 61
22 6 28
30 11 17
OPML dataset
(CONTROL)
OPML dataset
(LESION)
MICROBIAL PEPTIDES PROTEOFORM PEPTIDES
Search against microbial db
Search against microbial and
translated cDNA db
Search against microbial and
translated cDNA db
Search against translated cDNA db
METAPROTEOMICS
PROTEOGENOMICS
0
1
2
3
4
5
CONTROL
LESION
M1 M2
Microbial peptides identified at 5% local FDR were analyzed using UniPept and MEGAN5.
Fig M1: Organisms were identified at species level only when assigned 3 unique peptides or more.
Fig M2: Functional groups were assigned using SEED program within MEGAN5.
# o
f Id
enti
fied
Pep
tid
es
# o
f Id
enti
fied
Pep
tid
es
Workflow Link:
z.umn.edu/peaklistconversion
Workflow Link: z.umn.edu/dbgenmp
Workflow Link:
z.umn.edu/mn2stp
Workflow Link: z.umn.edu/dbgen
Workflow Link:
z.umn.edu/pepfrommicrobialdb
Workflow Link: z.umn.edu/blastp Workflow Link: z.umn.edu/pep2gtf
Workflow Link: z.umn.edu/blastp
Workflow Link:
z.umn.edu/peptidesfromcdnadb
Workflow Link:
z.umn.edu/psme
Using correct search databases that contribute to the proteome under study (both metaproteomic &
proteogenomic databases in tandem) help in confident identification of microbial peptides & novel proteoforms.
PSM EVALUATION & GENOME VISUALIZATION PSME Input Parameters
HTML
Output
Spectral
Visualization
Tabular Output
PSME (Peptide-Spectral-Match Evaluator)
uses spectral summary as an input to parse
mzml data and generates a tabular format
output with customized spectral features.
PSME also generates HTML links that are
used to visualize spectral assignments.
Interactive boxes can be used to change ion
assignments and other parameters.
GENOME VISUALIZATION
• We demonstrate the use of a complete platform for tandem
metaproteomic / proteogenomic analysis. Workflow for each
module/step have been shared for use within Galaxy environment.
• Using both metaproteomic & proteogenomic databases in tandem help
in confident identification of microbial peptides & novel proteoforms.
• Using this platform, we have identified microbial peptides & novel
proteoforms from both labeled & unlabeled datasets. For example for
COPD datasets, the number of identifications from both
proteogenomic & metaproteomic databases is limited yet consistent
across replicates..
• For metaproteomics studies, identified microbial peptides were used
for taxonomic classification (Unipept and MEGAN5) for functional
classification (MEGAN5).
• For proteogenomic studies, identified novel proteoforms were
validated using PSM evaluation tool and visualizing peptides against
the genome.
ACKNOWLEDGEMENTS: GalaxyP is supported through the National Science Foundation Grant
1147079. Many thanks to John Chilton (PennState) for GalaxyP development. Also thanks to LeeAnn
Higgins, Todd Markowski (CMSP, UMN), Bart Gottschalk and Anne Lamblin (MSI), Katie Vermillion
(UMD-Duluth) and Gloria Shenykman (UW-Madison) for helpful discussions.