Embed Size (px)
Citation preview

83041Federal Register / Vol. 65, No. 251 / Friday, December 29, 2000 / Notices
evaluating the quality and reliability ofinformation and data for use indeveloping the VADS system contents;(3) apply the principles ofpharmacology in constructingtherapeutic regimens for use whenapproved antimicrobial products are noteffective as labeled; (4) design arelational database allowing a user toefficiently search the VADS system forlabel and extralabel regimens based ontherapeutic applications, and to thenreview regulatory and food safetyinformation applicable to theseregimens; and (5) subject the VADSsystem content to review prior to releaseand then constantly upgrade the contenton the basis of new information andreview by users.
II. Eligible ApplicantsAssistance may only be provided to
Iowa State University because of thefollowing:
1. Iowa State University is the onlyorganization that submitted anunsolicited application for the purposestated above.
2. The project proposed by theapplicant is unique and innovative inthat pharmacokinetic,pharmacodynamic, clinical trial, andpathogen susceptibility information willbe interpreted by clinicalpharmacologists and reviewed by otherexperts in the appropriate fields prior toinclusion in the system. Users mayeither use the information as providedor examine the transparent developmentprocess used in constructing the system.In addition, by compiling availableinformation to support prudentantimicrobial use, the VADS system willemphasize what information is notavailable, thereby aiding researchers intargeting research goals.
3. The team assembled to carry outthe proposed work is uniquely qualifiedto achieve the goals of this application.Their combined experienceencompasses practice in academic,general, and specialized productionmedicine settings as well asdemonstrated competence in theapplication of clinical pharmacologyand informatics in veterinary medicine.Support for the research team and theVADS system project has already beenexpressed in the form of start upfunding provided by veterinary andproducer organizations.
III. FundingWe anticipate that approximately
$250,000 may be made available infiscal year (FY) 2001 to support thisproject. If funded the award will beginsometime in FY 2001 and will be madefor a 12-month budget period within a
project period of up to 5 years. Fundingestimates may change. Continuationawards within an approved projectperiod will be made on the basis ofsatisfactory progress as evidenced byrequired reports and the availability offunds.
Dated: December 22, 2000.Margaret M. Dotzel,Associate Commissioner for Policy.[FR Doc. 00–33372 Filed 12–28–01; 8:45 am]BILLING CODE: 4160–01–S
DEPARTMENT OF HEALTH ANDHUMAN SERVICES
Food and Drug Administration
[Docket No. 97D–0448]
International Conference onHarmonisation; Guidance on Q6ASpecifications: Test Procedures andAcceptance Criteria for New DrugSubstances and New Drug Products:Chemical Substances
AGENCY: Food and Drug Administration,HHS.ACTION: Notice.
SUMMARY: The Food and DrugAdministration (FDA) is publishing aguidance entitled ‘‘Q6A Specifications:Test Procedures and Acceptance Criteriafor New Drug Substances and New DrugProducts: Chemical Substances.’’ Theguidance was prepared under theauspices of the International Conferenceon Harmonisation of TechnicalRequirements for Registration ofPharmaceuticals for Human Use (ICH).The guidance describes or providesrecommendations concerning theselection of test procedures and thesetting and justification of acceptancecriteria for new chemical drugsubstances and new drug productsproduced from them. The guidance isintended to assist in the establishmentof a single set of global specifications fornew drug substances and new drugproducts.DATES: Submit written comments byMarch 29, 2001.ADDRESSES: Submit written commentson the guidance to the DocketsManagement Branch (HFA–305), Foodand Drug Administration, 5630 FishersLane, rm. 1061, Rockville, MD 20852.Copies of the guidance are availablefrom the Drug Information Branch(HFD–210), Center for Drug Evaluationand Research, Food and DrugAdministration, 5600 Fishers Lane,Rockville, MD 20857, 301–827–4573.FOR FURTHER INFORMATION CONTACT:
Regarding the guidance: Eric B.
Sheinin, Center for Drug Evaluationand Research (HFD–003), Food andDrug Administration, 5600 FishersLane, Rockville, MD 20857, 301–594–2847, or Neil D. Goldman,Center for Biologics Evaluation andResearch (HFM–20), Food and DrugAdministration, 1401 RockvillePike, Rockville, MD 20852, 301–827–0377.
Regarding the ICH: Janet J. Showalter,Office of Health Affairs (HFY–20),Food and Drug Administration,5600 Fishers Lane, Rockville, MD20857, 301–827–0864.
SUPPLEMENTARY INFORMATION: In recentyears, many important initiatives havebeen undertaken by regulatoryauthorities and industry associations topromote international harmonization ofregulatory requirements. FDA hasparticipated in many meetings designedto enhance harmonization and iscommitted to seeking scientificallybased harmonized technical proceduresfor pharmaceutical development. One ofthe goals of harmonization is to identifyand then reduce differences in technicalrequirements for drug developmentamong regulatory agencies.
ICH was organized to provide anopportunity for tripartite harmonizationinitiatives to be developed with inputfrom both regulatory and industryrepresentatives. FDA also seeks inputfrom consumer representatives andothers. ICH is concerned withharmonization of technicalrequirements for the registration ofpharmaceutical products among threeregions: The European Union, Japan,and the United States. The six ICHsponsors are the European Commission,the European Federation ofPharmaceutical Industries Associations,the Japanese Ministry of Health andWelfare, the Japanese PharmaceuticalManufacturers Association, the Centersfor Drug Evaluation and Research andBiologics Evaluation and Research,FDA, and the Pharmaceutical Researchand Manufacturers of America. The ICHSecretariat, which coordinates thepreparation of documentation, isprovided by the InternationalFederation of PharmaceuticalManufacturers Associations (IFPMA).
The ICH Steering Committee includesrepresentatives from each of the ICHsponsors and the IFPMA, as well asobservers from the World HealthOrganization, the Canadian HealthProtection Branch, and the EuropeanFree Trade Area.
In the Federal Register of November25, 1997 (62 FR 62890), FDA publisheda draft tripartite guidance entitled ‘‘Q6ASpecifications: Test Procedures and
VerDate 11<MAY>2000 19:54 Dec 28, 2000 Jkt 194001 PO 00000 Frm 00068 Fmt 4703 Sfmt 4703 E:\FR\FM\29DEN1.SGM pfrm01 PsN: 29DEN1

83042 Federal Register / Vol. 65, No. 251 / Friday, December 29, 2000 / Notices
1 This guidance represents the Food and DrugAdministration’s current thinking on this topic. Itdoes not create or confer any rights for or on anyperson and does not operate to bind FDA or thepublic. An alternative approach may be used if suchapproach satisfies the requirements of theapplicable statutes and regulations.
Acceptance Criteria for New DrugSubstances and New Drug Products:Chemical Substances.’’ The notice gaveinterested persons an opportunity tosubmit comments by January 26, 1998.
After consideration of the commentsreceived and revisions to the guidance,a final draft of the guidance wassubmitted to the ICH SteeringCommittee and endorsed by the threeparticipating regulatory agencies onOctober 6, 1999.
In accordance with FDA’s goodguidance practices regulation (65 FR56468, September 19, 2000), thisdocument has been designated aguidance, rather than a guideline.
The guidance providesrecommendations on the selection oftest procedures and the setting andjustification of acceptance criteria fornew drug substances of syntheticchemical origin, and new drug productsproduced from them, that have not beenregistered previously in the UnitedStates, the European Union, or Japan.This guidance is intended to assist inthe establishment of a single set ofglobal specifications for new drugsubstances and new drug products.
This guidance represents the agency’scurrent thinking on the selection of testsprocedures and the setting andjustification of acceptance criteria fornew chemical drug substances and newdrug products. It does not create orconfer any rights for or on any personand does not operate to bind FDA or thepublic. An alternative approach may beused if such approach satisfies therequirements of the applicable statutesand regulations.
Interested persons may submit to theDockets Management Branch (addressabove) written comments on theguidance at any time. Two copies of anycomments are to be submitted, exceptthat individuals may submit one copy.Comments are to be identified with thedocket number found in brackets in theheading of this document. The guidanceand received comments may be seen inthe Dockets Management Branchbetween 9 a.m. and 4 p.m., Mondaythrough Friday. An electronic version ofthis guidance is available on the Internetat http://www.fda.gov/cder/guidance/index.htm or at http://www.fda.gov/cber/publications.htm.
The text of the guidance follows:
Q6A Specifications: Test Proceduresand Acceptance Criteria for New DrugSubstances and New Drug Products:Chemical Substances 1
Table of Contents1. Introduction
1.1 Objective of the Guidance1.2 Background1.3 Scope of the Guidance
2. General Concepts2.1 Periodic or Skip Testing2.2 Release vs. Shelf-Life Acceptance
Criteria2.3 In-Process Tests2.4 Design and Development
Considerations2.5 Limited Data Available at Filing2.6 Parametric Release2.7 Alternative Procedures2.8 Pharmacopeial Tests and Acceptance
Criteria2.9 Evolving Technologies2.10 Impact of Drug Substance on Drug
Product Specifications2.11 Reference Standard
3. Guidance3.1 Specifications: Definition and
Justification3.1.1 Definition of Specifications3.1.2 Justification of Specifications3.2 Universal Tests/Criteria3.2.1 New Drug Substances3.2.2 New Drug Products3.3 Specific Tests/Criteria3.3.1 New Drug Substances3.3.2 New Drug Products
4. Glossary5. References6. Attachments: Decision Trees #1 Through
#8
1. Introduction
1.1 Objective of the GuidanceThis guidance is intended to assist, to
the extent possible, in the establishmentof a single set of global specifications fornew drug substances and new drugproducts. It provides guidance on thesetting and justification of acceptancecriteria and the selection of testprocedures for new drug substances ofsynthetic chemical origin, and new drugproducts produced from them, that havenot been registered previously in theUnited States, the European Union, orJapan.
1.2 BackgroundA specification is defined as a list of
tests, references to analyticalprocedures, and appropriate acceptancecriteria that are numerical limits, ranges,or other criteria for the tests described.It establishes the set of criteria to which
a drug substance or drug product shouldconform to be considered acceptable forits intended use. ‘‘Conformance tospecifications’’ means that the drugsubstance and/or drug product, whentested according to the listed analyticalprocedures, will meet the listedacceptance criteria. Specifications arecritical quality standards that areproposed and justified by themanufacturer and approved byregulatory authorities as conditions ofapproval.
Specifications are one part of a totalcontrol strategy for the drug substanceand drug product designed to ensureproduct quality and consistency. Otherparts of this strategy include thoroughproduct characterization duringdevelopment, upon whichspecifications are based, and adherenceto good manufacturing practices(GMP’s), e.g., suitable facilities, avalidated manufacturing process,validated test procedures, raw materialstesting, in-process testing, stabilitytesting.
Specifications are chosen to confirmthe quality of the drug substance anddrug product rather than to establishfull characterization, and should focuson those characteristics found to beuseful in ensuring the safety andefficacy of the drug substance and drugproduct.
1.3 Scope of the GuidanceThe quality of drug substances and
drug products is determined by theirdesign, development, in-processcontrols, GMP controls, processvalidation, and by specificationsapplied to them throughoutdevelopment and manufacture. Thisguidance addresses specifications, i.e.,those tests, procedures, and acceptancecriteria that play a major role in assuringthe quality of the new drug substanceand new drug product at release andduring shelf life. Specifications are animportant component of qualityassurance, but are not its onlycomponent. All of the factors listedabove are considered necessary toensure consistent production of drugsubstances and drug products of highquality.
This guidance addresses only themarketing approval of new drugproducts (including combinationproducts) and, where applicable, newdrug substances; it does not addressdrug substances or drug products duringthe clinical research stages of drugdevelopment. This guidance may beapplicable to synthetic andsemisynthetic antibiotics and syntheticpeptides of low molecular weight;however, it is not sufficient to
VerDate 11<MAY>2000 19:54 Dec 28, 2000 Jkt 194001 PO 00000 Frm 00069 Fmt 4703 Sfmt 4703 E:\FR\FM\29DEN1.SGM pfrm01 PsN: 29DEN1

83043Federal Register / Vol. 65, No. 251 / Friday, December 29, 2000 / Notices
adequately describe specifications ofhigher molecular weight peptides andpolypeptides, and biotechnological/biological products. The ICH guidanceon ‘‘Q6B Specifications: Test Proceduresand Acceptance Criteria forBiotechnological/Biological Products’’addresses guidance specifications, tests,and procedures for biotechnological/biological products.Radiopharmaceuticals, products offermentation, oligonucleotides, herbalproducts, and crude products of animalor plant origin are similarly not covered.
Guidance is provided with regard toacceptance criteria that should beestablished for all new drug substancesand new drug products, i.e., universalacceptance criteria, and those that areconsidered specific to individual drugsubstances and/or dosage forms. Thisguidance should not be considered allencompassing. New analyticaltechnologies, and modifications toexisting technology, are continuallybeing developed. Such technologiesshould be used when justified.
Dosage forms addressed in thisguidance include solid oral dosageforms, liquid oral dosage forms, andparenterals (small and large volume).This is not meant to be an all-inclusivelist, or to limit the number of dosageforms to which this guidance applies.The dosage forms presented serve asmodels that may be applicable to otherdosage forms that have not beendiscussed. The extended application ofthe concepts in this guidance to otherdosage forms, e.g., to inhalation dosageforms (powders, solutions, etc.), totopical formulations (creams, ointments,gels), and to transdermal systems, isencouraged.
2. General ConceptsThe following concepts are important
in the development and setting ofharmonized specifications. They are notuniversally applicable, but each shouldbe considered in particularcircumstances. This guidance presents abrief definition of each concept and anindication of the circumstances underwhich it may be applicable. Generally,proposals to implement these conceptsshould be justified by the applicant andapproved by the appropriate regulatoryauthority before being put into effect.
2.1 Periodic or Skip TestingPeriodic or skip testing is the
performance of specified tests at releaseon preselected batches and/or atpredetermined intervals, rather than ona batch-by-batch basis, with theunderstanding that those batches notbeing tested still meet all acceptancecriteria established for that product.
This represents a less than full scheduleof testing and should therefore bejustified and presented to and approvedby the regulatory authority prior toimplementation. This concept may beapplicable to, for example, residualsolvents and microbiological testing forsolid oral dosage forms. It is recognizedthat only limited data may be availableat the time of submission of anapplication (see section 2.5). Thisconcept should therefore generally beimplemented postapproval. Whentested, any failure to meet acceptancecriteria established for the periodic testshould be handled by propernotification of the appropriateregulatory authority(ies). If these datademonstrate a need to restore routinetesting, then batch-by-batch releasetesting should be reinstated.
2.2 Release vs. Shelf-Life AcceptanceCriteria
The concept of different acceptancecriteria for release vs. shelf-lifespecifications applies to drug productsonly; it pertains to the establishment ofmore restrictive criteria for the release ofa drug product than are applied to theshelf life. Examples where this may beapplicable include assay and impurity(degradation product) levels. In Japanand the United States, this concept mayonly be applicable to in-house criteria,and not to the regulatory release criteria.Thus, in these regions, the regulatoryacceptance criteria are the same fromrelease throughout shelf life; however,an applicant may choose to have tighterin-house limits at the time of release toprovide increased assurance to theapplicant that the product will remainwithin the regulatory acceptance criteriathroughout its shelf life. In the EuropeanUnion there is a regulatory requirementfor distinct specifications for release andfor shelf life where different.
2.3 In-Process TestsIn-process tests, as presented in this
guidance, are tests that may beperformed during the manufacture ofeither the drug substance or drugproduct, rather than as part of theformal battery of tests that areconducted prior to release.
In-process tests that are only used forthe purpose of adjusting processparameters within an operating range,e.g., hardness and friability of tabletcores that will be coated and individualtablet weights, are not included in thespecification.
Certain tests conducted during themanufacturing process, where theacceptance criterion is identical to ortighter than the release requirement,(e.g., pH (hydrogen-ion concentration)
of a solution) may be sufficient to satisfyspecification requirements when the testis included in the specification.However, this approach should bevalidated to show that test results orproduct performance characteristics donot change from the in-process stage tofinished product.
2.4 Design and DevelopmentConsiderations
The experience and data accumulatedduring the development of a new drugsubstance or product should form thebasis for the setting of specifications. Itmay be possible to propose excluding orreplacing certain tests on this basis.Some examples are:
• Microbiological testing for drugsubstances and solid dosage forms thathave been shown during developmentnot to support microbial viability orgrowth (see Decision Trees #6 and #8).
• Extractables from productcontainers where it has beenreproducibly shown that either noextractables are found in the drugproduct or the levels meet acceptedstandards for safety.
• Particle size testing may fall intothis category, may be performed as anin-process test, or may be performed asa release test, depending on itsrelevance to product performance.
• Dissolution testing for immediaterelease solid oral drug products madefrom highly water soluble drugsubstances may be replaced bydisintegration testing, if these productshave been demonstrated duringdevelopment to have consistently rapiddrug release characteristics (seeDecision Trees #7(1) through #7(2)).
2.5 Limited Data Available at Filing
It is recognized that only a limitedamount of data may be available at thetime of filing, which can influence theprocess of setting acceptance criteria. Asa result, it may be necessary to proposerevised acceptance criteria as additionalexperience is gained with themanufacture of a particular drugsubstance or drug product (example:acceptance limits for a specificimpurity). The basis for the acceptancecriteria at the time of filing shouldnecessarily focus on safety and efficacy.
When only limited data are available,the initially approved tests andacceptance criteria should be reviewedas more information is collected, with aview towards possible modification.This could involve loosening, as well astightening, acceptance criteria, asappropriate.
VerDate 11<MAY>2000 19:54 Dec 28, 2000 Jkt 194001 PO 00000 Frm 00070 Fmt 4703 Sfmt 4703 E:\FR\FM\29DEN1.SGM pfrm01 PsN: 29DEN1

83044 Federal Register / Vol. 65, No. 251 / Friday, December 29, 2000 / Notices
2.6 Parametric Release
Parametric release can be used as anoperational alternative to routine releasetesting for the drug product in certaincases, when approved by the regulatoryauthority. Sterility testing for terminallysterilized drug products is one example.In this case, the release of each batch isbased on satisfactory results frommonitoring specific parameters, e.g.,temperature, pressure, and time duringthe terminal sterilization phase(s) ofdrug product manufacturing. Theseparameters can generally be moreaccurately controlled and measured, sothey are more reliable in predictingsterility assurance than is end-productsterility testing. Appropriate laboratorytests (e.g., chemical or physicalindicator) may be included in theparametric release program. It isimportant to note that the sterilizationprocess should be adequately validatedbefore parametric release is proposed,and maintenance of a validated stateshould be demonstrated by revalidationat established intervals. Whenparametric release is performed, theattribute that is indirectly controlled(e.g., sterility), together with a referenceto the associated test procedure, stillshould be included in thespecifications.
2.7 Alternative Procedures
Alternative procedures are those thatmay be used to measure an attributewhen such procedures control thequality of the drug substance or drugproduct to an extent that is comparableor superior to the official procedure.Example: For tablets that have beenshown not to degrade duringmanufacture, it may be permissible touse a spectrophotometric procedure forrelease as opposed to the officialprocedure, which is chromatographic.However, the chromatographicprocedure should still be used todemonstrate compliance with theacceptance criteria during the shelf lifeof the product.
2.8 Pharmacopeial Tests andAcceptance Criteria
References to certain procedures arefound in pharmacopeias in each region.Wherever they are appropriate,pharmacopeial procedures should beused. Whereas differences inpharmacopeial procedures and/oracceptance criteria have existed amongthe regions, a harmonized specificationis possible only if the procedures andacceptance criteria defined areacceptable to regulatory authorities inall regions.
The full utility of this guidance isdependent on the successful completionof harmonization of pharmacopeialprocedures for several attributescommonly considered in thespecification for new drug substances ornew drug products. The PharmacopoeialDiscussion Group (PDG) of theEuropean Pharmacopeia, the JapanesePharmacopoeia (JP), and the UnitedStates Pharmacopeia has expressed acommitment to achievingharmonization of the procedures in atimely fashion.
Where harmonization has beenachieved, an appropriate reference tothe harmonized procedure andacceptance criteria is consideredacceptable for a specification in all threeregions. For example, afterharmonization, sterility data generatedusing the JP procedure, as well as the JPprocedure itself and its acceptancecriteria, will be considered acceptablefor registration in all three regions. Tosignify the harmonized status of theseprocedures, the pharmacopeias haveagreed to include a statement in theirrespective texts that indicates that theprocedures and acceptance criteria fromall three pharmacopeias are consideredequivalent and are, therefore,interchangeable.
Since the overall value of thisguidance is linked to the extent ofharmonization of the analyticalprocedures and acceptance criteria ofthe pharmacopeias, it is agreed by themembers of the Q6A expert workinggroup that none of the threepharmacopeias should change aharmonized monograph unilaterally.According to the PDG procedure for therevision of harmonized monographs andchapters, ‘‘no pharmacopoeia shallrevise unilaterally any monograph orchapter after sign-off or afterpublication.’’
2.9 Evolving TechnologiesNew analytical technologies, and
modifications to existing technology, arecontinually being developed. Suchtechnologies should be used when theyare considered to offer additionalassurance of quality, or are otherwisejustified.
2.10 Impact of Drug Substance on DrugProduct Specifications
In general, it should not be necessaryto test the drug product for qualityattributes uniquely associated with thedrug substance. Example: It is normallynot considered necessary to test thedrug product for synthesis impuritiesthat are controlled in the drug substanceand are not degradation products. Referto the ICH guidance on ‘‘Q3B Impurities
in New Drug Products’’ for detailedinformation.
2.11 Reference StandardA reference standard, or reference
material, is a substance prepared for useas the standard in an assay,identification, or purity test. It shouldhave a quality appropriate to its use. Itis often characterized and evaluated forits intended purpose by additionalprocedures other than those used inroutine testing. For new drug substancereference standards intended for use inassays, the impurities should beadequately identified and/or controlled,and purity should be measured by aquantitative procedure.
3. Guidance
3.1 Specifications: Definition andJustification
3.1.1 Definition of SpecificationsA specification is defined as a list of
tests, references to analyticalprocedures, and appropriate acceptancecriteria that are numerical limits, ranges,or other criteria for the tests described.It establishes the set of criteria to whicha new drug substance or new drugproduct should conform to beconsidered acceptable for its intendeduse. ‘‘Conformance to specifications’’means that the drug substance and/ordrug product, when tested according tothe listed analytical procedures, willmeet the listed acceptance criteria.Specifications are critical qualitystandards that are proposed andjustified by the manufacturer andapproved by regulatory authorities asconditions of approval.
It is possible that, in addition torelease tests, a specification may list in-process tests as defined in section 2.3,periodic or skip tests, and other teststhat are not always conducted on abatch-by-batch basis. In such cases theapplicant should specify which tests areroutinely conducted batch by batch, andwhich tests are not, with an indicationand justification of the actual testingfrequency. In this situation, the drugsubstance and/or drug product shouldmeet the acceptance criteria if tested.
It should be noted that changes in thespecification after approval of theapplication may need prior approval bythe regulatory authority.
3.1.2 Justification of SpecificationsWhen a specification is first proposed,
justification should be presented foreach procedure and each acceptancecriterion included. The justificationshould refer to relevant developmentdata, pharmacopeial standards, test datafor drug substances and drug products
VerDate 11<MAY>2000 19:54 Dec 28, 2000 Jkt 194001 PO 00000 Frm 00071 Fmt 4703 Sfmt 4703 E:\FR\FM\29DEN1.SGM pfrm01 PsN: 29DEN1

83045Federal Register / Vol. 65, No. 251 / Friday, December 29, 2000 / Notices
used in toxicology and clinical studies,and results from accelerated and long-term stability studies, as appropriate.Additionally, a reasonable range ofexpected analytical and manufacturingvariability should be considered. It isimportant to consider all of thisinformation.
Approaches other than those set forthin this guidance may be applicable andacceptable. The applicant should justifyalternative approaches. Suchjustification should be based on dataderived from the new drug substancesynthesis and/or the new drug productmanufacturing process. Thisjustification may consider theoreticaltolerances for a given procedure oracceptance criterion, but the actualresults obtained should form theprimary basis for whatever approach istaken.
Test results from stability andscaleup/validation batches, withemphasis on the primary stabilitybatches, should be considered in settingand justifying specifications. If multiplemanufacturing sites are planned, it maybe valuable to consider data from thesesites in establishing the initial tests andacceptance criteria. This is particularlytrue when there is limited initialexperience with the manufacture of thedrug substance or drug product at anyparticular site. If data from a singlerepresentative manufacturing site areused in setting tests and acceptancecriteria, product manufactured at allsites should still comply with thesecriteria.
Presentation of test results in graphicformat may be helpful in justifyingindividual acceptance criteria,particularly for assay values andimpurity levels. Data from developmentwork should be included in such apresentation, along with stability dataavailable for new drug substance or newdrug product batches manufactured bythe proposed commercial processes.Justification for proposing exclusion ofa test from the specification should bebased on development data and onprocess validation data (whereappropriate).
3.2 Universal Tests/CriteriaImplementation of the
recommendations in the followingsection should take into account the ICHguidances ‘‘Q2A Text on Validation ofAnalytical Procedures’’ and ‘‘Q2BValidation of Analytical Procedures:Methodology.’’
3.2.1 New Drug SubstancesThe following tests and acceptance
criteria are considered generallyapplicable to all new drug substances.
(a) Description: A qualitativestatement about the state (e.g., solid,liquid) and color of the new drugsubstance. If any of these characteristicschange during storage, this changeshould be investigated and appropriateaction taken.
(b) Identification: Identificationtesting should optimally be able todiscriminate between compounds ofclosely related structure that are likelyto be present. Identification tests shouldbe specific for the new drug substance,e.g., infrared spectroscopy (IR).Identification solely by a singlechromatographic retention time, forexample, is not regarded as beingspecific. However, the use of twochromatographic procedures, where theseparation is based on differentprinciples or a combination of tests intoa single procedure, such as HPLC (high-pressure liquid chromatography)/UV(ultraviolet) diode array, HPLC/MS(mass spectroscopy), or GC (gaschromatography)/MS is generallyacceptable. If the new drug substance isa salt, identification testing should bespecific for the individual ions. Anidentification test that is specific for thesalt itself should suffice.
New drug substances that areoptically active may also need specificidentification testing or performance ofa chiral assay. Please refer to section3.3.1(d) in this guidance for furtherdiscussion of this topic.
(c) Assay: A specific, stability-indicating procedure should beincluded to determine the content of thenew drug substance. In many cases it ispossible to employ the same procedure(e.g., HPLC) for both assay of the newdrug substance and quantitation ofimpurities.
In cases where use of a nonspecificassay is justified, other supportinganalytical procedures should be used toachieve overall specificity. For example,where titration is adopted to assay thedrug substance, the combination of theassay and a suitable test for impuritiesshould be used.
(d) Impurities: Organic and inorganicimpurities and residual solvents areincluded in this category. Refer to theICH guidances on ‘‘Q3A Impurities inNew Drug Substances’’ and ‘‘Q3CImpurities: Residual Solvents’’ fordetailed information.
Decision Tree #1 addresses theextrapolation of meaningful limits onimpurities from the body of datagenerated during development. At thetime of filing it is unlikely thatsufficient data will be available to assessprocess consistency. Therefore it isconsidered inappropriate to establishacceptance criteria that tightly
encompass the batch data at the time offiling (see section 2.5).
3.2.2 New Drug ProductsThe following tests and acceptance
criteria are considered generallyapplicable to all new drug products:
(a) Description: A qualitativedescription of the dosage form shouldbe provided (e.g., size, shape, andcolor). If any of these characteristicschange during manufacture or storage,this change should be investigated andappropriate action taken. Theacceptance criteria should include thefinal acceptable appearance. If colorchanges during storage, a quantitativeprocedure may be appropriate.
(b) Identification: Identificationtesting should establish the identity ofthe new drug substance(s) in the newdrug product and should be able todiscriminate between compounds ofclosely related structure that are likelyto be present. Identity tests should bespecific for the new drug substance, e.g.,infrared spectroscopy. Identificationsolely by a single chromatographicretention time, for example, is notregarded as being specific. However, theuse of two chromatographic procedures,where the separation is based ondifferent principles, or a combination oftests into a single procedure, such asHPLC/UV diode array, HPLC/MS, orGC/MS, is generally acceptable.
(c) Assay: A specific, stability-indicating assay to determine strength(content) should be included for all newdrug products. In many cases it ispossible to employ the same procedure(e.g., HPLC) for both assay of the newdrug substance and quantitation ofimpurities. Results of contentuniformity testing for new drugproducts can be used for quantitation ofdrug product strength, if the methodsused for content uniformity are alsoappropriate as assays.
In cases where use of a nonspecificassay is justified, other supportinganalytical procedures should be used toachieve overall specificity. For example,where titration is adopted to assay thedrug substance for release, thecombination of the assay and a suitabletest for impurities can be used. Aspecific procedure should be used whenthere is evidence of excipientinterference with the nonspecific assay.
(d) Impurities: Organic and inorganicimpurities (degradation products) andresidual solvents are included in thiscategory. Refer to the ICH guidances on‘‘Q3B Impurities in New Drug Products’’and ‘‘Q3C Impurities: ResidualSolvents’’ for detailed information.
Organic impurities arising fromdegradation of the new drug substance
VerDate 11<MAY>2000 19:54 Dec 28, 2000 Jkt 194001 PO 00000 Frm 00072 Fmt 4703 Sfmt 4703 E:\FR\FM\29DEN1.SGM pfrm01 PsN: 29DEN1

83046 Federal Register / Vol. 65, No. 251 / Friday, December 29, 2000 / Notices
and impurities that arise during themanufacturing process for the drugproduct should be monitored in the newdrug product. Acceptance limits shouldbe stated for individual specifieddegradation products, which mayinclude both identified and unidentifieddegradation products, as appropriate,and total degradation products. Processimpurities from the new drug substancesynthesis are normally controlledduring drug substance testing, andtherefore are not included in the totalimpurities limit. However, when asynthesis impurity is also a degradationproduct, its level should be monitoredand included in the total degradationproduct limit. When it has beenconclusively demonstrated viaappropriate analytical methodology thatthe drug substance does not degrade inthe specific formulation, and under thespecific storage conditions proposed inthe new drug application, degradationproduct testing may be reduced oreliminated upon approval by theregulatory authorities.
Decision Tree #2 addresses theextrapolation of meaningful limits ondegradation products from the body ofdata generated during development. Atthe time of filing it is unlikely thatsufficient data will be available to assessprocess consistency. Therefore it isconsidered inappropriate to establishacceptance criteria that tightlyencompass the batch data at the time offiling (see section 2.5).
3.3 Specific Tests/CriteriaIn addition to the universal tests
listed above, the following tests may beconsidered on a case-by-case basis fordrug substances and/or drug products.Individual tests/criteria should beincluded in the specification when thetests have an impact on the quality ofthe drug substance and drug product forbatch control. Tests other than thoselisted below may be needed inparticular situations or as newinformation becomes available.
3.3.1 New Drug Substances(a) Physicochemical properties: These
are properties such as pH of an aqueoussolution, melting point/range, andrefractive index. The procedures usedfor the measurement of these propertiesare usually unique and do not needmuch elaboration, e.g., capillary meltingpoint, Abbe refractometry. The testsperformed in this category should bedetermined by the physical nature of thenew drug substance and by its intendeduse.
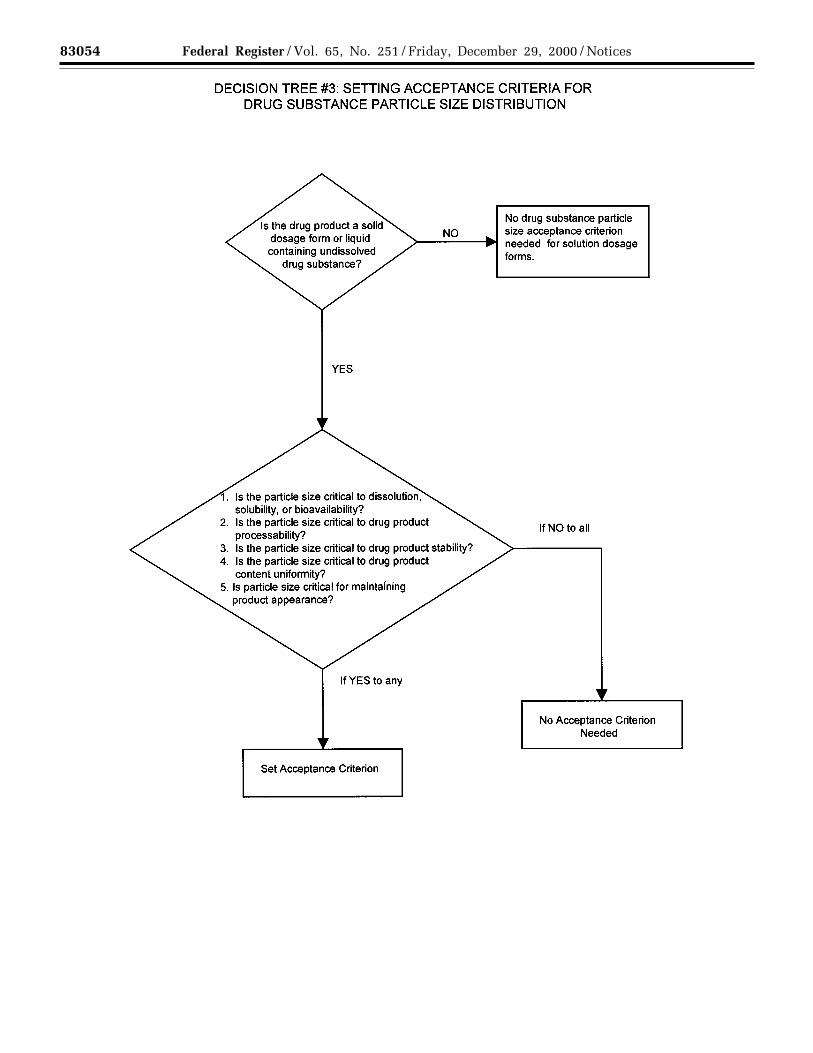
(b) Particle size: For some new drugsubstances intended for use in solid orsuspension drug products, particle size
can have a significant effect ondissolution rates, bioavailability, and/orstability. In such instances, testing forparticle size distribution should becarried out using an appropriateprocedure, and acceptance criteriashould be provided.
Decision Tree #3 provides additionalguidance on when particle size testingshould be considered.
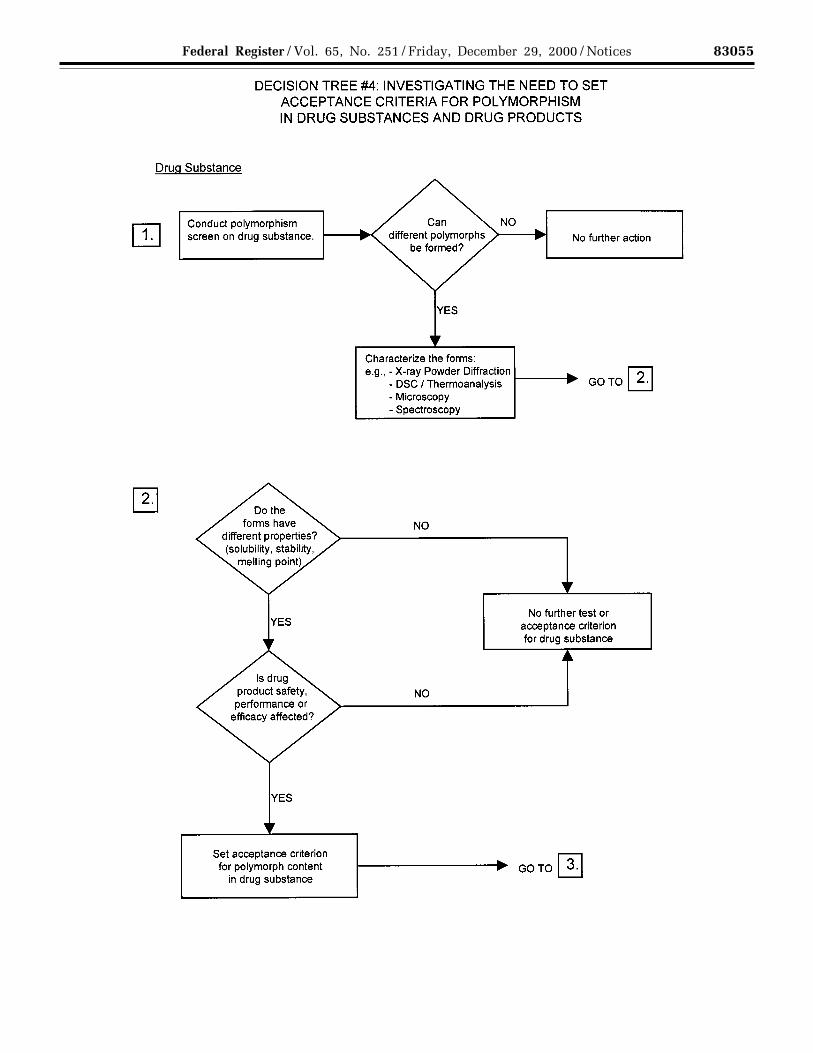
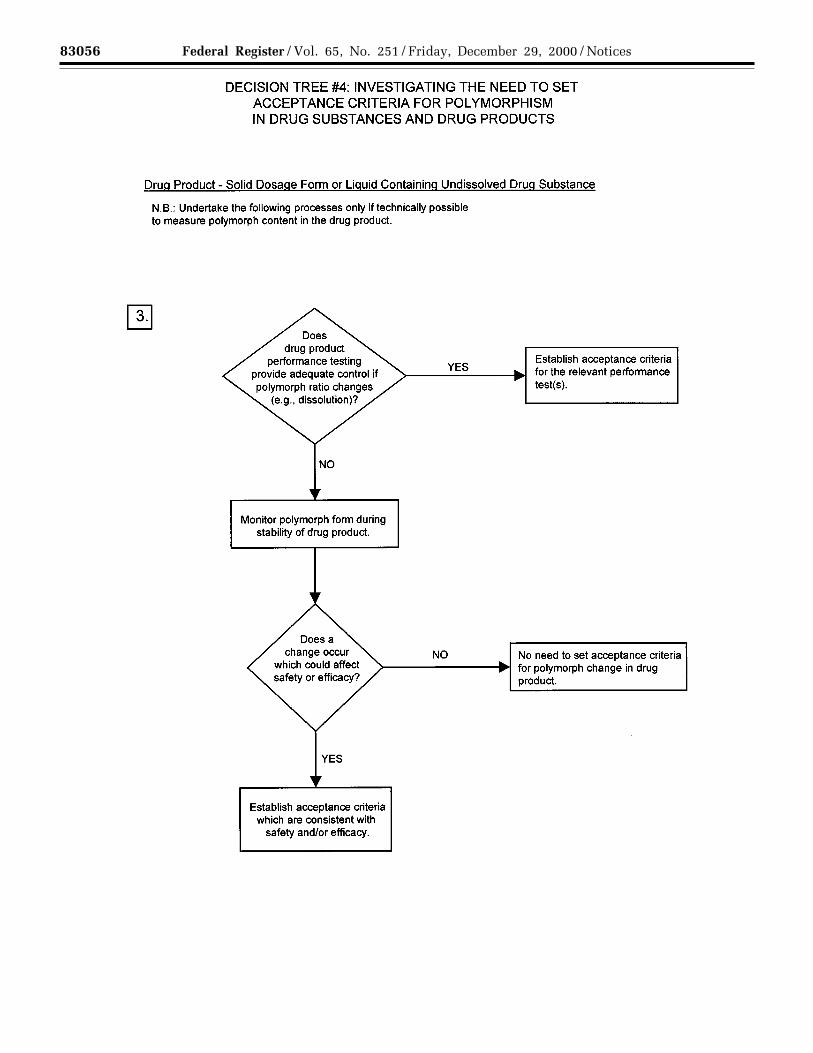
(c) Polymorphic forms: Some newdrug substances exist in differentcrystalline forms that differ in theirphysical properties. Polymorphism mayalso include solvation or hydrationproducts (also known aspseudopolymorphs) and amorphousforms. Differences in these forms could,in some cases, affect the quality orperformance of the new drug products.In cases where differences exist thathave been shown to affect drug productperformance, bioavailability, orstability, then the appropriate solid stateshould be specified.
Physicochemical measurements andtechniques are commonly used todetermine whether multiple forms exist.Examples of these procedures are:Melting point (including hot-stagemicroscopy), solid state IR, X-raypowder diffraction, thermal analysisprocedures (like DSC (differentialscanning calorimetry), TGA(thermogravimetric analysis) and DTA(differential thermal analysis)), Ramanspectroscopy, optical microscopy, andsolid state NMR (nuclear magneticresonance) spectroscopy.
Decision Trees #4(1) through #4(3)provide additional guidance on when,and how, polymorphic forms should bemonitored and controlled.
Note: These decision trees should befollowed sequentially. Trees #4(1) and#4(2) consider whether polymorphismis exhibited by the drug substance, andwhether the different polymorphicforms can affect performance of the drugproduct. Tree #4(3) should only beapplied when polymorphism has beendemonstrated for the drug substance,and shown to affect these properties.Tree #4(3) considers the potential forchange in polymorphic forms in thedrug product and whether such achange has any effect on productperformance.
It is generally technically verydifficult to measure polymorphicchanges in drug products. A surrogatetest (e.g., dissolution) (see Decision Tree#4(3)) can generally be used to monitorproduct performance, and polymorphcontent should only be used as a testand acceptance criterion of last resort.
(d) Tests for chiral new drugsubstances: Where a new drugsubstance is predominantly one
enantiomer, the opposite enantiomer isexcluded from the qualification andidentification thresholds given in theICH guidances on ‘‘Q3A Impurities inNew Drug Substances’’ and ‘‘Q3BImpurities in New Drug Products’’because of practical difficulties inquantifying it at those levels. However,that impurity in the chiral new drugsubstance and the resulting new drugproduct(s) should otherwise be treatedaccording to the principles establishedin those guidances.
Decision Tree #5 summarizes whenand if chiral identity tests, impuritytests, and assays may be needed for bothnew drug substances and new drugproducts, according to the followingconcepts:
Drug Substance: Impurities. For chiraldrug substances that are developed as asingle enantiomer, control of the otherenantiomer should be considered in thesame manner as for other impurities.However, technical limitations maypreclude the same limits ofquantification or qualification frombeing applied. Assurance of control alsocould be given by appropriate testing ofa starting material or intermediate, withsuitable justification.
Assay. An enantioselectivedetermination of the drug substanceshould be part of the specification. It isconsidered acceptable for this to beachieved either through use of a chiralassay procedure or by the combinationof an achiral assay together withappropriate methods of controlling theenantiomeric impurity.
Identity. For a drug substancedeveloped as a single enantiomer, theidentity test(s) should be capable ofdistinguishing both enantiomers and theracemic mixture. For a racemic drugsubstance, there are generally twosituations where a stereospecificidentity test is appropriate for release/acceptance testing: (1) Where there is asignificant possibility that theenantiomer might be substituted for theracemate, or (2) when there is evidencethat preferential crystallization may leadto unintentional production of anonracemic mixture.
Drug Product: Degradation products.Control of the other enantiomer in adrug product is considered necessaryunless racemization has been shown tobe insignificant during manufacture ofthe dosage form and on storage.
Assay. An achiral assay may besufficient where racemization has beenshown to be insignificant duringmanufacture of the dosage form and onstorage. Otherwise a chiral assay shouldbe used. Alternatively, the combinationof an achiral assay plus a validated
VerDate 11<MAY>2000 19:54 Dec 28, 2000 Jkt 194001 PO 00000 Frm 00073 Fmt 4703 Sfmt 4703 E:\FR\FM\29DEN1.SGM pfrm01 PsN: 29DEN1

83047Federal Register / Vol. 65, No. 251 / Friday, December 29, 2000 / Notices
procedure to control the presence of theopposite enantiomer may be used.
Identity. A stereospecific identity testis not generally needed in the drugproduct release specification. Whenracemization is insignificant duringmanufacture of the dosage form and onstorage, stereospecific identity testing ismore appropriately addressed as part ofthe drug substance specification. Whenracemization in the dosage form is aconcern, chiral assay or enantiomericimpurity testing of the drug product willserve to verify identity.
(e) Water content: This test isimportant in cases where the new drugsubstance is known to be hygroscopic ordegraded by moisture or when the drugsubstance is known to be astoichiometric hydrate. The acceptancecriteria may be justified with data on theeffects of hydration or moistureabsorption. In some cases, a loss ondrying procedure may be consideredadequate; however, a detectionprocedure that is specific for water (e.g.,Karl Fischer titration) is preferred.
(f) Inorganic impurities: The need forinclusion of tests and acceptancecriteria for inorganic impurities (e.g.,catalysts) should be studied duringdevelopment and based on knowledgeof the manufacturing process.Procedures and acceptance criteria forsulfated ash/residue on ignition shouldfollow pharmacopeial precedents; otherinorganic impurities may be determinedby other appropriate procedures, e.g.,atomic absorption spectroscopy.
(g) Microbial limits: There may be aneed to specify the total count of aerobicmicroorganisms, the total count ofyeasts and molds, and the absence ofspecific objectionable bacteria (e.g.,Staphylococcus aureus, Escherichiacoli, Salmonella, Pseudomonasaeruginosa). These should be suitablydetermined using pharmacopeialprocedures. The type of microbial test(s)and acceptance criteria should be basedon the nature of the drug substance,method of manufacture, and theintended use of the drug product. Forexample, sterility testing may beappropriate for drug substancesmanufactured as sterile, and endotoxintesting may be appropriate for drugsubstances used to formulate aninjectable drug product.
Decision Tree #6 provides additionalguidance on when microbial limitsshould be included.
3.3.2 New Drug ProductsAdditional tests and acceptance
criteria generally should be included forparticular new drug products. Thefollowing selection presents arepresentative sample of both the drug
products and the types of tests andacceptance criteria that may beappropriate. The specific dosage formsaddressed include solid oral drugproducts, liquid oral drug products, andparenterals (small and large volume).Application of the concepts in thisguidance to other dosage forms isencouraged. Note that issues related tooptically active drug substances and tosolid state considerations for drugproducts are discussed in section 3.3.1of this guidance.
3.3.2.1 The following tests areapplicable to tablets (coated anduncoated) and hard capsules. One ormore of these tests may also beapplicable to soft capsules and granules.
(a) Dissolution: The specification forsolid oral dosage forms normallyincludes a test to measure release ofdrug substance from the drug product.Single-point measurements are normallyconsidered to be suitable for immediate-release dosage forms. For modified-release dosage forms, appropriate testconditions and sampling proceduresshould be established. For example,multiple time-point sampling should beperformed for extended-release dosageforms, and two-stage testing (usingdifferent media in succession or inparallel, as appropriate) may beappropriate for delayed-release dosageforms. In these cases it is important toconsider the populations of individualswho will be taking the drug product(e.g., achlorhydric elderly) whendesigning the tests and acceptancecriteria. In some cases (see section3.3.2.1(b) Disintegration) dissolutiontesting may be replaced bydisintegration testing (see Decision Tree#7(1)).
For immediate-release drug productswhere changes in dissolution rate havebeen demonstrated to significantly affectbioavailability, it is desirable to developtest conditions that can distinguishbatches with unacceptablebioavailability. If changes informulation or process variablessignificantly affect dissolution, and suchchanges are not controlled by anotheraspect of the specification, it may alsobe appropriate to adopt dissolution testconditions that can distinguish thesechanges (see Decision Tree #7(2)).
Where dissolution significantly affectsbioavailability, the acceptance criteriashould be set to reject batches withunacceptable bioavailability. Otherwise,test conditions and acceptance criteriashould be established that passclinically acceptable batches (seeDecision Tree #7(2)).
For extended-release drug products,in vitro/in vivo correlation may be used
to establish acceptance criteria whenhuman bioavailability data are availablefor formulations exhibiting differentrelease rates. Where such data are notavailable, and drug release cannot beshown to be independent of in vitro testconditions, then acceptance criteriashould be established on the basis ofavailable batch data. Normally, thepermitted variability in mean releaserate at any given time point should notexceed a total numerical difference of±10 percent of the labeled content ofdrug substance (i.e., a total variability of20 percent: a requirement of 50±10percent thus means an acceptable rangefrom 40 percent to 60 percent), unlessa wider range is supported by abioequivalency study (see Decision Tree#7(3)).
(b) Disintegration: For rapidlydissolving (dissolution >80 percent in15 minutes at pH 1.2, 4.0, and 6.8)products containing drugs that arehighly soluble throughout thephysiological range (dose/solubilityvolume ≤ 250 milliliters (mL) from pH1.2 to 6.8), disintegration may besubstituted for dissolution.Disintegration testing is consideredmost appropriate when a relationship todissolution has been established orwhen disintegration is shown to bemore discriminating than dissolution. Insuch cases dissolution testing may notbe necessary. It is expected thatdevelopment information will beprovided to support the robustness ofthe formulation and manufacturingprocess with respect to the selection ofdissolution versus disintegration testing(see Decision Tree #7(1)).
(c) Hardness/friability: It is normallyappropriate to perform hardness and/orfriability testing as an in-process control(see section 2.3). Under thesecircumstances, it is normally notnecessary to include these attributes inthe specification. If the characteristics ofhardness and friability have a criticalimpact on drug product quality (e.g.,chewable tablets), acceptance criteriashould be included in the specification.
(d) Uniformity of dosage units: Thisterm includes both the mass of thedosage form and the content of theactive substance in the dosage form; apharmacopeial procedure should beused. In general, the specificationshould include one or the other, but notboth. If appropriate, these tests may beperformed in-process; the acceptancecriteria should be included in thespecification. When weight variation isapplied to new drug products exceedingthe threshold value to allow testinguniformity by weight variation,applicants should verify during drug
VerDate 11<MAY>2000 19:54 Dec 28, 2000 Jkt 194001 PO 00000 Frm 00074 Fmt 4703 Sfmt 4703 E:\FR\FM\29DEN1.SGM pfrm01 PsN: 29DEN1

83048 Federal Register / Vol. 65, No. 251 / Friday, December 29, 2000 / Notices
development that the homogeneity ofthe product is adequate.
(e) Water content: A test for watercontent should be included whenappropriate. The acceptance criteriamay be justified with data on the effectsof hydration or water absorption on thedrug product. In some cases, a loss ondrying procedure may be consideredadequate; however, a detectionprocedure that is specific for water (e.g.,Karl Fischer titration) is preferred.
(f) Microbial limits: Microbial limittesting is seen as an attribute of GMP,as well as of quality assurance. Ingeneral, it is advisable to test the drugproduct unless its components aretested before manufacture and themanufacturing process is known,through validation studies, not to carrya significant risk of microbialcontamination or proliferation. It shouldbe noted that, whereas this guidancedoes not directly address excipients, theprinciples discussed here may beapplicable to excipients as well as tonew drug products. Skip testing may bean appropriate approach in both cases,where permissible (see Decision Tree #6for microbial testing of excipients).
Acceptance criteria should be set forthe total count of aerobicmicroorganisms, the total count ofyeasts and molds, and the absence ofspecific objectionable bacteria (e.g.,Staphylococcus aureus, Escherichiacoli, Salmonella, Pseudomonasaeruginosa). These should bedetermined by suitable procedures,using pharmacopeial procedures, and ata sampling frequency or time point inmanufacture that is justified by data andexperience. The type of microbial test(s)and acceptance criteria should be basedon the nature of the drug substance,method of manufacture, and theintended use of the drug product. Withacceptable scientific justification, itshould be possible to propose nomicrobial limit testing for solid oraldosage forms.
Decision Tree #8 provides additionalguidance on the use of microbial limitstesting.
3.3.2.2 Oral liquids: One or more of thefollowing specific tests will normally beapplicable to oral liquids and topowders intended for reconstitution asoral liquids.
(a) Uniformity of dosage units: Thisterm includes both the mass of thedosage form and the content of theactive drug substance in the dosageform; a pharmacopeial procedureshould be used. In general, thespecification should include one or theother, but not both. When weightvariation is applied to new drug
products exceeding the threshold valueto allow testing uniformity by weightvariation, applicants should verifyduring drug development that thehomogeneity of the product is adequate.
If appropriate, tests may be performedin-process; however, the acceptancecriteria should be included in thespecification. This concept may beapplied to both single-dose andmultiple-dose packages.
The dosage unit is considered to bethe typical dose taken by the patient. Ifthe actual unit dose, as taken by thepatient, is controlled, it may either bemeasured directly or calculated, basedon the total measured weight or volumeof drug divided by the total number ofdoses expected. If dispensing equipment(such as medicine droppers or droppertips for bottles) is an integral part of thepackaging, this equipment should beused to measure the dose. Otherwise, astandard volume measure should beused. The dispensing equipment to beused is normally determined duringdevelopment. For powders forreconstitution, uniformity of masstesting is generally consideredacceptable.
(b) pH: Acceptance criteria for pHshould be provided where applicableand the proposed range justified.
(c) Microbial limits: Microbial limittesting is seen as an attribute of GMP,as well as of quality assurance. Ingeneral, it is advisable to test the drugproduct unless its components aretested before manufacture and themanufacturing process is known,through validation studies, not to carrya significant risk of microbialcontamination or proliferation. It shouldbe noted that, whereas this guidancedoes not directly address excipients, theprinciples discussed here may beapplicable to excipients as well as tonew drug products. Skip testing may bean appropriate approach in both cases,where permissible. With acceptablescientific justification, it may bepossible to propose no microbial limittesting for powders intended forreconstitution as oral liquids.
Acceptance criteria should be set forthe total count of aerobicmicroorganisms, total count of yeastsand molds, and the absence of specificobjectionable bacteria (e.g.,Staphylococcus aureus, Escherichiacoli, Salmonella, Pseudomonasaeruginosa). These should bedetermined by suitable procedures,using pharmacopeial procedures, and ata sampling frequency or time point inmanufacture that is justified by data andexperience.
Decision Tree #8 provides additionalguidance on the use of microbial limitstesting.
(d) Antimicrobial preservativecontent: For oral liquids needing anantimicrobial preservative, acceptancecriteria for preservative content shouldbe established. Acceptance criteria forpreservative content should be basedupon the levels of antimicrobialpreservative necessary to maintainmicrobiological quality of the product atall stages throughout its proposed usageand shelf life. The lowest specifiedconcentration of antimicrobialpreservative should be demonstrated tobe effective in controllingmicroorganisms by using apharmacopeial antimicrobialpreservative effectiveness test.
Testing for antimicrobial preservativecontent should normally be performedat release. Under certain circumstances,in-process testing may suffice in lieu ofrelease testing. When antimicrobialpreservative content testing isperformed as an in-process test, theacceptance criteria should remain partof the specification.
Antimicrobial preservativeeffectiveness should be demonstratedduring development, during scaleup,and throughout the shelf life (e.g., instability testing: see the ICH guidance‘‘Q1A Stability Testing of New DrugSubstances and Products’’), althoughchemical testing for preservative contentis the attribute normally included in thespecification.
(e) Antioxidant preservative content:Release testing for antioxidant contentshould normally be performed. Undercertain circumstances where justified bydevelopmental and stability data, shelf-life testing may be unnecessary, and in-process testing may suffice in lieu ofrelease testing where permitted. Whenantioxidant content testing is performedas an in-process test, the acceptancecriteria should remain part of thespecification. If only release testing isperformed, this decision should bereinvestigated whenever either themanufacturing procedure or thecontainer/closure system changes.
(f) Extractables: Generally, wheredevelopment and stability data showevidence that extractables from thecontainer/closure systems areconsistently below levels that aredemonstrated to be acceptable and safe,elimination of this test can normally beaccepted. This should be reinvestigatedif the container/closure system orformulation changes.
Where data demonstrate the need,tests and acceptance criteria forextractables from the container/closuresystem components (e.g., rubber
VerDate 11<MAY>2000 19:54 Dec 28, 2000 Jkt 194001 PO 00000 Frm 00075 Fmt 4703 Sfmt 4703 E:\FR\FM\29DEN1.SGM pfrm01 PsN: 29DEN1

83049Federal Register / Vol. 65, No. 251 / Friday, December 29, 2000 / Notices
stopper, cap liner, plastic bottle, etc.)are considered appropriate for oralsolutions packaged in nonglass systems,or in glass containers with nonglassclosures. The container/closurecomponents should be listed, and datacollected for these components as earlyin the development process as possible.
(g) Alcohol content: Where it isdeclared quantitatively on the label inaccordance with pertinent regulations,the alcohol content should be specified.It may be assayed or calculated.
(h) Dissolution: In addition to theattributes recommended immediatelyabove, it may be appropriate (e.g.,insoluble drug substance) to includedissolution testing and acceptancecriteria for oral suspensions and drypowder products for resuspension.Dissolution testing should be performedat release. This test may be performedas an in-process test when justified byproduct development data. The testingapparatus, media, and conditionsshould be pharmacopeial, if possible, orotherwise justified. Dissolutionprocedures using either apharmacopeial or nonpharmacopeialapparatus and conditions should bevalidated.
Single-point measurements arenormally considered suitable forimmediate-release dosage forms.Multiple-point sampling, at appropriateintervals, should be performed formodified-release dosage forms.Acceptance criteria should be set basedon the observed range of variation, andshould take into account the dissolutionprofiles of the batches that showedacceptable performance in vivo.Developmental data should beconsidered when determining the needfor either a dissolution procedure or aparticle size distribution procedure.
(i) Particle size distribution:Quantitative acceptance criteria and aprocedure for determination of particlesize distribution may be appropriate fororal suspensions. Developmental datashould be considered when determiningthe need for either a dissolutionprocedure or a particle size distributionprocedure for these formulations.
Particle size distribution testingshould be performed at release. It maybe performed as an in-process test whenjustified by product development data.If these products have beendemonstrated during development tohave consistently rapid drug releasecharacteristics, exclusion of a particlesize distribution test from thespecification may be proposed.
Particle size distribution testing mayalso be proposed in place of dissolutiontesting; justification should be provided.The acceptance criteria should include
acceptable particle size distribution interms of the percent of total particles ingiven size ranges. The mean, upper,and/or lower particle size limits shouldbe well defined.
Acceptance criteria should be setbased on the observed range ofvariation, and should take into accountthe dissolution profiles of the batchesthat showed acceptable performance invivo, as well as the intended use of theproduct. The potential for particlegrowth should be investigated duringproduct development; the acceptancecriteria should take the results of thesestudies into account.
(j) Redispersibility: For oralsuspensions that settle on storage(produce sediment), acceptance criteriafor redispersibility may be appropriate.Shaking may be an appropriateprocedure.
The procedure (mechanical ormanual) should be indicated. Timerequired to achieve resuspension by theindicated procedure should be clearlydefined. Data generated during productdevelopment may be sufficient to justifyperiodic or skip testing, or eliminationof this attribute from the specificationmay be proposed.
(k) Rheological properties: Forrelatively viscous solutions orsuspensions, it may be appropriate toinclude rheological properties(viscosity/specific gravity) in thespecification. The test and acceptancecriteria should be stated. Data generatedduring product development may besufficient to justify periodic or skiptesting, or elimination of this attributefrom the specification may be proposed.
(l) Reconstitution time: Acceptancecriteria for reconstitution time should beprovided for dry powder products thatrequire reconstitution. The choice ofdiluent should be justified. Datagenerated during product developmentmay be sufficient to justify periodic orskip testing, or elimination of thisattribute from the specification may beproposed.
(m) Water content: For oral productsrequiring reconstitution, a test andacceptance criterion for water contentshould be proposed when appropriate.Loss on drying is generally consideredsufficient if the effect of absorbedmoisture versus water of hydration hasbeen adequately characterized duringthe development of the product. Incertain cases a more specific procedure(e.g., Karl Fischer titration) may bepreferable.
3.3.2.3 Parenteral Drug Products: Thefollowing tests may be applicable toparenteral drug products.
(a) Uniformity of dosage units: Thisterm includes both the mass of thedosage form and the content of theactive drug substance in the dosageform; a pharmacopeial procedureshould be used. In general, thespecification should be one or the other,but not both, and is applicable topowders for reconstitution. Whenweight variation is applied to new drugproducts exceeding the threshold valueto allow testing uniformity by weightvariation, applicants should verifyduring drug development that thehomogeneity of the product is adequate.
If appropriate (see section 2.3), thesetests may be performed in-process; theacceptance criteria should be includedin the specification. This test may beapplied to both single-dose andmultiple-dose packages.
For powders for reconstitution,uniformity of mass testing is generallyconsidered acceptable.
(b) pH: Acceptance criteria for pHshould be provided where applicable,and the proposed range justified.
(c) Sterility: All parenteral productsshould have a test procedure andacceptance criterion for evaluation ofsterility. Where data generated duringdevelopment and validation justifyparametric release, this approach maybe proposed for terminally sterilizeddrug products (see section 2.6).
(d) Endotoxins/Pyrogens: A testprocedure and acceptance criterion forendotoxins, using a procedure such asthe limulus amoebocyte lysate test,should be included in the specification.Pyrogenicity testing may be proposed asan alternative to endotoxin testingwhere justified.
(e) Particulate matter: Parenteralproducts should have appropriateacceptance criteria for particulatematter. This will normally includeacceptance criteria for visibleparticulates and/or clarity of solution, aswell as for subvisible particulates, asappropriate.
(f) Water content: For nonaqueousparenterals, and for parenteral productsfor reconstitution, a test procedure andacceptance criterion for water contentshould be proposed when appropriate.Loss on drying is generally consideredsufficient for parenteral products, if theeffect of absorbed moisture versus waterof hydration has been adequatelycharacterized during development. Incertain cases a more specific procedure(e.g., Karl Fischer titration) may bepreferred.
(g) Antimicrobial preservativecontent: For parenteral products
VerDate 11<MAY>2000 19:54 Dec 28, 2000 Jkt 194001 PO 00000 Frm 00076 Fmt 4703 Sfmt 4703 E:\FR\FM\29DEN1.SGM pfrm01 PsN: 29DEN1

83050 Federal Register / Vol. 65, No. 251 / Friday, December 29, 2000 / Notices
needing an antimicrobial preservative,acceptance criteria for preservativecontent should be established.Acceptance criteria for preservativecontent should be based upon the levelsof antimicrobial preservative necessaryto maintain microbiological quality ofthe product at all stages throughout itsproposed usage and shelf life. Thelowest specified concentration ofantimicrobial preservative should bedemonstrated to be effective incontrolling microorganisms by using apharmacopeial antimicrobialpreservative effectiveness test.
Testing for antimicrobial preservativecontent should normally be performedat release. Under certain circumstances,in-process testing may suffice in lieu ofrelease testing, where permitted. Whenantimicrobial preservative contenttesting is performed as an in-processtest, the acceptance criteria shouldremain part of the specification.
Antimicrobial preservativeeffectiveness should be demonstratedduring development, during scaleup,and throughout the shelf life (e.g., instability testing: see the ICH guidance‘‘Q1A Stability Testing of New DrugSubstances and Products’’), althoughchemical testing for preservative contentis the attribute normally included in thespecification.
(h) Antioxidant preservative content:Release testing for antioxidant contentshould normally be performed. Undercertain circumstances, where justifiedby developmental and stability data,shelf-life testing may be unnecessaryand in-process testing may suffice inlieu of release testing. When antioxidantcontent testing is performed as an in-process test, the acceptance criteriashould remain part of the specification.If only release testing is performed, thisdecision should be reinvestigatedwhenever either the manufacturingprocedure or the container/closuresystem changes.
(i) Extractables: Control ofextractables from container/closuresystems is considered significantly moreimportant for parenteral products thanfor oral liquids. However, wheredevelopment and stability data showevidence that extractables areconsistently below the levels that aredemonstrated to be acceptable and safe,elimination of this test can normally beaccepted. This should be reinvestigatedif the container/closure system orformulation changes.
Where data demonstrate the need,acceptance criteria for extractables fromthe container/closure components areconsidered appropriate for parenteralproducts packaged in nonglass systemsor in glass containers with elastomeric
closures. This testing may be performedat release only, where justified by dataobtained during development. Thecontainer/closure system components(e.g., rubber stopper, etc.) should belisted, and data collected for thesecomponents as early in the developmentprocess as possible.
(j) Functionality testing of deliverysystems: Parenteral formulationspackaged in prefilled syringes,autoinjector cartridges, or the equivalentshould have test procedures andacceptance criteria related to thefunctionality of the delivery system.These may include control ofsyringeability, pressure, and sealintegrity (leakage), and/or parameterssuch as tip cap removal force, pistonrelease force, piston travel force, andpower injector function force. Undercertain circumstances these tests may beperformed in-process. Data generatedduring product development may besufficient to justify skip lot testing orelimination of some or all attributesfrom the specification.
(k) Osmolarity: When the tonicity of aproduct is declared in its labeling,appropriate control of its osmolarityshould be performed. Data generatedduring development and validation maybe sufficient to justify performance ofthis procedure as an in-process control,skip lot testing, or direct calculation ofthis attribute.
(l) Particle size distribution:Quantitative acceptance criteria and aprocedure for determination of particlesize distribution may be appropriate forinjectable suspensions. Developmentaldata should be considered whendetermining the need for either adissolution procedure or a particle sizedistribution procedure.
Particle size distribution testingshould be performed at release. It maybe performed as an in-process test whenjustified by product development data.If the product has been demonstratedduring development to haveconsistently rapid drug releasecharacteristics, exclusion of particle sizecontrols from the specification may beproposed.
Particle size distribution testing mayalso be proposed in place of dissolutiontesting when development studiesdemonstrate that particle size is theprimary factor influencing dissolution;justification should be provided. Theacceptance criteria should includeacceptable particle size distribution interms of the percent of total particles ingiven size ranges. The mean, upper,and/or lower particle size limits shouldbe well defined.
Acceptance criteria should be setbased on the observed range of
variation, and should take into accountthe dissolution profiles of the batchesthat showed acceptable performance invivo and the intended use of theproduct. The potential for particlegrowth should be investigated duringproduct development; the acceptancecriteria should take the results of thesestudies into account.
(m) Redispersibility: For injectablesuspensions that settle on storage(produce sediment), acceptance criteriafor redispersibility may be appropriate.Shaking may be an appropriateprocedure. The procedure (mechanicalor manual) should be indicated. Timerequired to achieve resuspension by theindicated procedure should be clearlydefined. Data generated during productdevelopment may be sufficient to justifyskip lot testing, or elimination of thisattribute from the specification may beproposed.
(n) Reconstitution time: Acceptancecriteria for reconstitution time should beprovided for all parenteral products thatrequire reconstitution. The choice ofdiluent should be justified. Datagenerated during product developmentand process validation may be sufficientto justify skip lot testing or eliminationof this attribute from the specificationfor rapidly dissolving products.
4. Glossary (the following definitionsare presented for the purpose of thisguidance)
Acceptance criteria: Numerical limits,ranges, or other suitable measures foracceptance of the results of analyticalprocedures.
Chiral: Not superimposable with itsmirror image, as applied to molecules,conformations, and macroscopic objects,such as crystals. The term has beenextended to samples of substanceswhose molecules are chiral, even if themacroscopic assembly of suchmolecules is racemic.
Combination product: A drug productthat contains more than one drugsubstance.
Degradation product: A moleculeresulting from a chemical change in thedrug molecule brought about over timeand/or by the action of light,temperature, pH, water, or by reactionwith an excipient and/or the immediatecontainer/closure system. Also calleddecomposition product.
Delayed release: Release of a drug (ordrugs) at a time other than immediatelyfollowing oral administration.
Enantiomers: Compounds with thesame molecular formula as the drugsubstance, which differ in the spatialarrangement of atoms within themolecule and are nonsuperimposablemirror images.
VerDate 11<MAY>2000 19:54 Dec 28, 2000 Jkt 194001 PO 00000 Frm 00077 Fmt 4703 Sfmt 4703 E:\FR\FM\29DEN1.SGM pfrm01 PsN: 29DEN1

83051Federal Register / Vol. 65, No. 251 / Friday, December 29, 2000 / Notices
Extended release: Products that areformulated to make the drug availableover an extended period afteradministration.
Highly water soluble drugs: Drugswith a dose/solubility volume of lessthan or equal to 250 mL over a pH rangeof 1.2 to 6.8. (Example: Compound Ahas as its lowest solubility at 37±0.5 °C,1.0 milligram (mg)/milliliter (mL) at pH6.8, and is available in 100 mg, 200 mg,and 400 mg strengths. This drug wouldbe considered a low solubility drug, asits dose/solubility volume is greaterthan 250 mL (400 mg/1.0 mg/mL = 400mL)).
Immediate release: Allows the drug todissolve in the gastrointestinal contents,with no intention of delaying orprolonging the dissolution or absorptionof the drug.
Impurity: (1) Any component of thenew drug substance that is not thechemical entity defined as the new drugsubstance. (2) Any component of thedrug product that is not the chemicalentity defined as the drug substance oran excipient in the drug product.
Identified impurity: An impurity forwhich a structural characterization hasbeen achieved.
In-process tests: Tests that may beperformed during the manufacture ofeither the drug substance or drugproduct, rather than as part of theformal battery of tests that areconducted prior to release.
Modified release: Dosage forms whosedrug release characteristics of timecourse and/or location are chosen toaccomplish therapeutic or convenienceobjectives not offered by conventionaldosage forms, such as a solution or animmediate-release dosage form.Modified-release solid oral dosage formsinclude both delayed- and extended-release drug products.
New drug product: A pharmaceuticalproduct type, e.g., tablet, capsule,solution, cream, etc., that has notpreviously been registered in a region orMember State, and that contains a drugingredient generally, but notnecessarily, in association withexcipients.
New drug substance: The designatedtherapeutic moiety that has not
previously been registered in a region orMember State (also referred to as a newmolecular entity or new chemicalentity). It may be a complex, simpleester, or salt of a previously approveddrug substance.
Polymorphism: The occurrence ofdifferent crystalline forms of the samedrug substance. This may includesolvation or hydration products (alsoknown as pseudopolymorphs) andamorphous forms.
Quality: The suitability of either adrug substance or drug product for itsintended use. This term includes suchattributes as the identity, strength, andpurity.
Racemate: A composite (solid, liquid,gaseous, or in solution) of equimolarquantities of two enantiomeric species.It is devoid of optical activity.
Rapidly dissolving products: Animmediate release solid oral drugproduct is considered rapidly dissolvingwhen not less than 80 percent of thelabel amount of the drug substancedissolves within 15 minutes in each ofthe following media: (1) pH 1.2, (2) pH4.0, and (3) pH 6.8.
Reagent: A substance, other than astarting material or solvent, that is usedin the manufacture of a new drugsubstance.
Solvent: An inorganic or an organicliquid used as a vehicle for thepreparation of solutions or suspensionsin the synthesis of a new drug substanceor the manufacture of a new drugproduct.
Specification: A list of tests,references to analytical procedures, andappropriate acceptance criteria that arenumerical limits, ranges, or othercriteria for the tests described. Itestablishes the set of criteria to which adrug substance or drug product shouldconform to be considered acceptable forits intended use. ‘‘Conformance tospecifications’’ means that the drugsubstance and/or drug product, whentested according to the listed analyticalprocedures, will meet the listedacceptance criteria. Specifications arecritical quality standards that areproposed and justified by themanufacturer and approved byregulatory authorities as conditions ofapproval.
Specific test: A test that is consideredto be applicable to particular new drugsubstances or particular new drugproducts, depending on their specificproperties and/or intended use.
Specified impurity: An identified orunidentified impurity that is selectedfor inclusion in the new drug substanceor new drug product specification andis individually listed and limited toensure the quality of the new drugsubstance or new drug product.
Unidentified impurity: An impuritythat is defined solely by qualitativeanalytical properties (e.g.,chromatographic retention time).
Universal test: A test that isconsidered potentially applicable to allnew drug substances, or all new drugproducts; e.g., appearance,identification, assay, and impurity tests.
5. References
International Conference onHarmonisation, ‘‘Q3A Impurities inNew Drug Substances,’’ 1996.
International Conference onHarmonisation, ‘‘Q3B Impurities in NewDrug Products,’’ 1997.
International Conference onHarmonisation, ‘‘Q1A Stability Testingof New Drug Substances and Products,’’1994.
International Conference onHarmonisation, ‘‘Q2A Text onValidation of Analytical Procedures,’’1995.
International Conference onHarmonisation, ‘‘Q2B Validation ofAnalytical Procedures: Methodology,’’1996.
International Conference onHarmonisation, ‘‘Q3C Impurities:Residual Solvents,’’ 1997.
International Conference onHarmonisation, ‘‘Q6B Specifications:Test Procedures and Acceptance Criteriafor Biotechnological/BiologicalProducts,’’ 1999.
6. Attachments: Decision Trees #1through #8
For the decision trees referenced inthis guidance, see the following pages.
BILLING CODE 4160–01–F
VerDate 11<MAY>2000 19:54 Dec 28, 2000 Jkt 194001 PO 00000 Frm 00078 Fmt 4703 Sfmt 4703 E:\FR\FM\29DEN1.SGM pfrm01 PsN: 29DEN1

83052 Federal Register / Vol. 65, No. 251 / Friday, December 29, 2000 / Notices
VerDate 11<MAY>2000 19:54 Dec 28, 2000 Jkt 194001 PO 00000 Frm 00079 Fmt 4703 Sfmt 4725 E:\FR\FM\29DEN1.SGM pfrm01 PsN: 29DEN1

83053Federal Register / Vol. 65, No. 251 / Friday, December 29, 2000 / Notices
VerDate 11<MAY>2000 19:54 Dec 28, 2000 Jkt 194001 PO 00000 Frm 00080 Fmt 4703 Sfmt 4725 E:\FR\FM\29DEN1.SGM pfrm01 PsN: 29DEN1
83054 Federal Register / Vol. 65, No. 251 / Friday, December 29, 2000 / Notices
VerDate 11<MAY>2000 19:54 Dec 28, 2000 Jkt 194001 PO 00000 Frm 00081 Fmt 4703 Sfmt 4725 E:\FR\FM\29DEN1.SGM pfrm01 PsN: 29DEN1
83055Federal Register / Vol. 65, No. 251 / Friday, December 29, 2000 / Notices
VerDate 11<MAY>2000 19:54 Dec 28, 2000 Jkt 194001 PO 00000 Frm 00082 Fmt 4703 Sfmt 4725 E:\FR\FM\29DEN1.SGM pfrm01 PsN: 29DEN1
83056 Federal Register / Vol. 65, No. 251 / Friday, December 29, 2000 / Notices
VerDate 11<MAY>2000 19:54 Dec 28, 2000 Jkt 194001 PO 00000 Frm 00083 Fmt 4703 Sfmt 4725 E:\FR\FM\29DEN1.SGM pfrm01 PsN: 29DEN1

83057Federal Register / Vol. 65, No. 251 / Friday, December 29, 2000 / Notices
VerDate 11<MAY>2000 19:54 Dec 28, 2000 Jkt 194001 PO 00000 Frm 00084 Fmt 4703 Sfmt 4725 E:\FR\FM\29DEN1.SGM pfrm01 PsN: 29DEN1

83058 Federal Register / Vol. 65, No. 251 / Friday, December 29, 2000 / Notices
VerDate 11<MAY>2000 19:54 Dec 28, 2000 Jkt 194001 PO 00000 Frm 00085 Fmt 4703 Sfmt 4725 E:\FR\FM\29DEN1.SGM pfrm01 PsN: 29DEN1
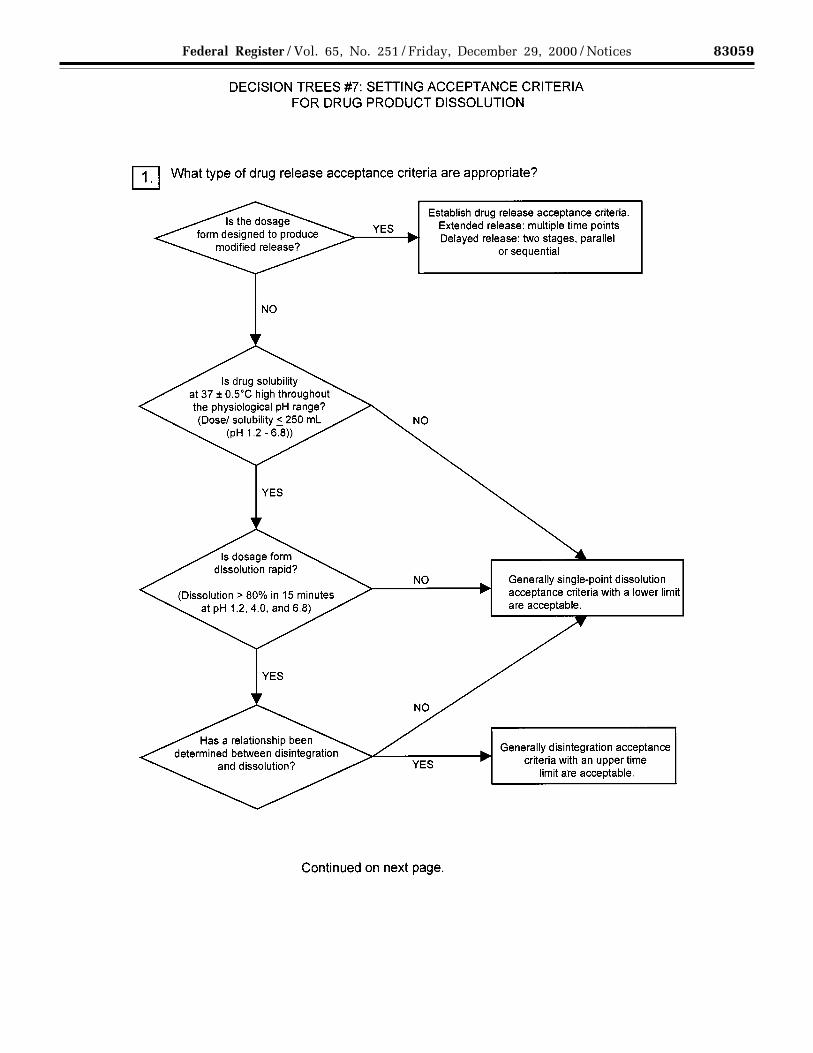
83059Federal Register / Vol. 65, No. 251 / Friday, December 29, 2000 / Notices
VerDate 11<MAY>2000 19:54 Dec 28, 2000 Jkt 194001 PO 00000 Frm 00086 Fmt 4703 Sfmt 4725 E:\FR\FM\29DEN1.SGM pfrm01 PsN: 29DEN1
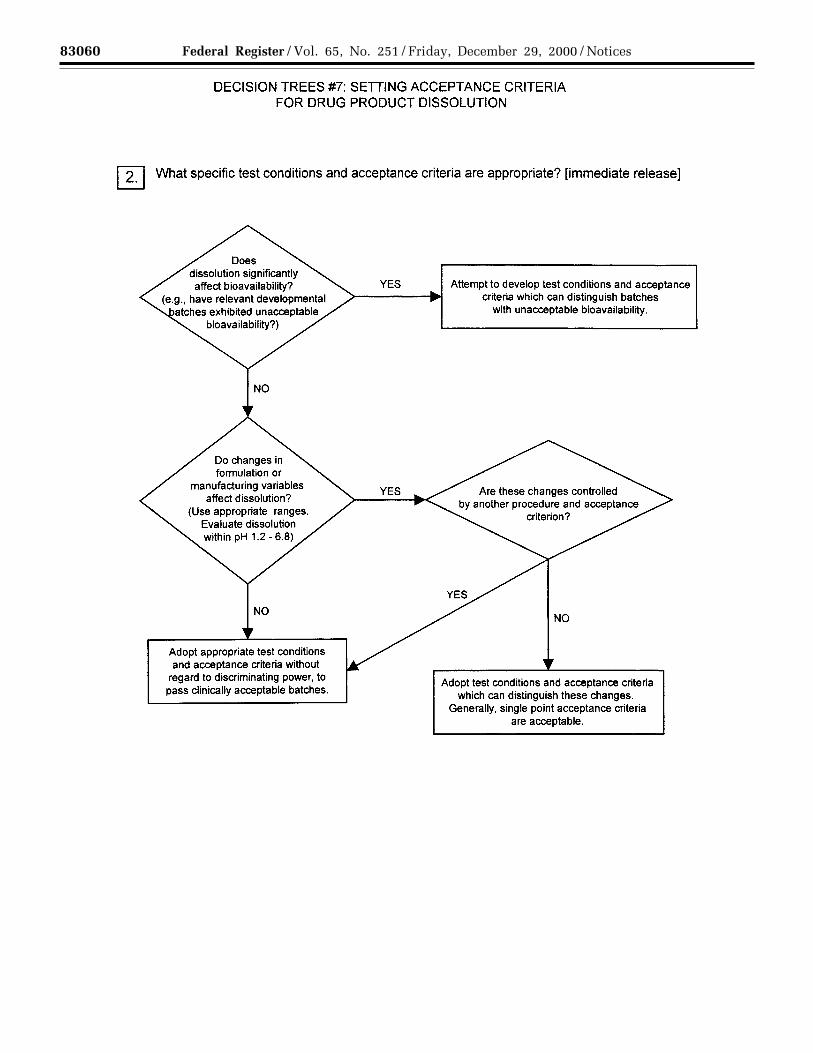
83060 Federal Register / Vol. 65, No. 251 / Friday, December 29, 2000 / Notices
VerDate 11<MAY>2000 19:54 Dec 28, 2000 Jkt 194001 PO 00000 Frm 00087 Fmt 4703 Sfmt 4725 E:\FR\FM\29DEN1.SGM pfrm01 PsN: 29DEN1

83061Federal Register / Vol. 65, No. 251 / Friday, December 29, 2000 / Notices
VerDate 11<MAY>2000 19:54 Dec 28, 2000 Jkt 194001 PO 00000 Frm 00088 Fmt 4703 Sfmt 4725 E:\FR\FM\29DEN1.SGM pfrm01 PsN: 29DEN1
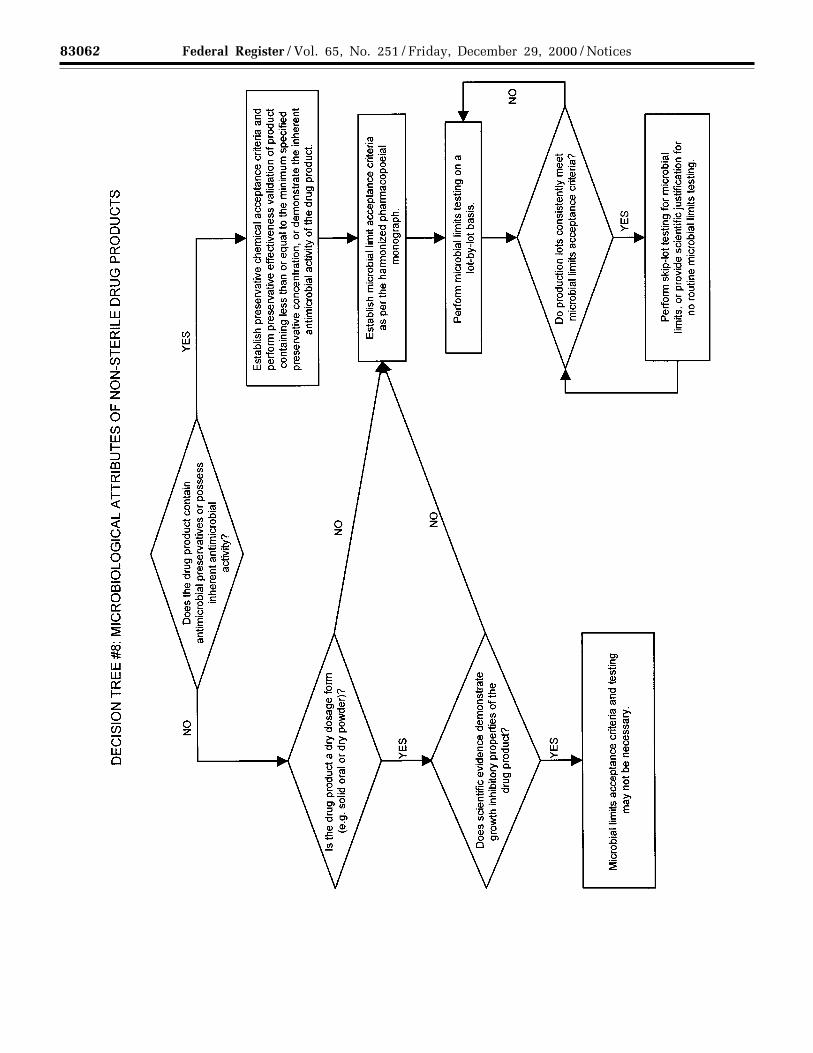
83062 Federal Register / Vol. 65, No. 251 / Friday, December 29, 2000 / Notices
VerDate 11<MAY>2000 19:54 Dec 28, 2000 Jkt 194001 PO 00000 Frm 00089 Fmt 4703 Sfmt 4725 E:\FR\FM\29DEN1.SGM pfrm01 PsN: 29DEN1

83063Federal Register / Vol. 65, No. 251 / Friday, December 29, 2000 / Notices
Dated: December 20, 2000.Margaret M. Dotzel,Associate Commissioner for Policy.[FR Doc. 00–33369 Filed 12–28–00; 8:45 am]BILLING CODE 4160–01–C
DEPARTMENT OF HEALTH ANDHUMAN SERVICES
Food and Drug Administration
Cooperative Arrangement Between theUnited States Food and DrugAdministration and Therapeutic GoodsAdministration, Republic of AustraliaRegarding the Exchange ofInformation on Current GoodManufacturing Practice Inspections ofHuman Pharmaceutical Facilities
AGENCY: Food and Drug Administration,HHS.
ACTION: Notice.
SUMMARY: The Food and DrugAdministration (FDA) is providingnotice of cooperative arrangementbetween the Food and DrugAdministration, Department of Healthand Human Services, United States ofAmerica and the Therapeutic GoodsAdministration, Department of Healthand Aged Care, Commonwealth ofAustralia. The purpose of thearrangement is to enable eachadministration to obtain informationthat will enable it to make its ownindependent facility and/or productregulatory decisions in the assessmentof current good manufacturing practicescompliance, public health protection,and approval of new drugs. It also willfacilitate more efficient use of resourcesfor each organization in meeting theirstatutory requirements withoutreduction of public safety or regulatoryresponsibilities.DATES: The arrangement becameeffective October 11, 2000.
FOR FURTHER INFORMATION CONTACT:Merton V. Smith, Office of InternationalPrograms, International AgreementsStaff (HFG–1), Food and DrugAdministration, 5600 Fishers Lane,Rockville, MD 20857, 301–827–0910.
SUPPLEMENTARY INFORMATION: Thiscooperative arrangement is subject toFDA’s regulations in 21 CFR 20.108 forcooperative agreements. Therefore, inaccordance with 21 CFR 20.108(c),which states that all written agreementsand memoranda of understandingbetween FDA and others shall bepublished in the Federal Register, theagency is publishing notice of thiswritten arrangement.
Dated: December 20, 2000.Margaret M. Dotzel,Associate Commissioner for Policy.
The arrangement is set forth in itsentirety as follows:BILLING CODE 4160–01–F
VerDate 11<MAY>2000 19:54 Dec 28, 2000 Jkt 194001 PO 00000 Frm 00090 Fmt 4703 Sfmt 4703 E:\FR\FM\29DEN1.SGM pfrm01 PsN: 29DEN1





























