Embed Size (px)
Citation preview

Ž .Brain Research 826 1999 104–111
Research report
Facilitation by arachidonic acid of acetylcholine release from the rathippocampus
Teresa Almeida a, Rodrigo A. Cunha a,b,) , J.A. Ribeiro a
a Laboratory of Neurosciences, Faculty of Medicine, UniÕersity of Lisbon, AÕ. Prof. Egas Moniz, 1649-035, Lisbon, Portugalb Department of Chemistry and Biochemistry, Faculty of Sciences, UniÕersity of Lisbon, Lisbon, Portugal
Accepted 16 February 1999
Abstract
Ž . w3 x Žw3 x .We investigated the effect of arachidonic acid AA on the release of H acetylcholine H ACh from the rat hippocampus. AAŽ . w3 x3–30 mM increased the basal tritium outflow and the field-electrically evoked release of H ACh from hippocampal slices in a
Ž .concentration-dependent manner. AA 30 mM produced a 69"7% facilitation of the evoked and a 36"3% facilitation of basal tritiumŽ . Ž .outflow. The effect of AA 30 mM on the evoked tritium release was prevented by bovine serum albumin BSA, 1% , which quenches
Ž .AA, and was unaffected by the cyclooxygenase inhibitor, indomethacin 100 mM , and the lipooxygenase inhibitor, nordihydroguaiareticŽ . Ž .acid 50 mM . Phospholipase A PLA , 2 Urml , an enzyme that releases AA from the sn-2 position of phospholipids, mimicked the2 2
Ž . Ž .facilitatory effect of AA on the evoked tritium release 86"14% facilitation , an effect prevented by BSA 1% . The PLA activator,2Ž .melittin 1 mM , enhanced the evoked tritium release by 98"11%, an effect prevented by the PLA inhibitor, arachidonyl2
Ž . Ž . Ž . Ž . Ž .trifluromethylketone AACOCF , 20 mM , and by BSA 1% . AA 30 mM , but not arachidic acid 30 mM , also facilitated 72"9%3Ž . w3 x Ž . Žthe veratridine 10 mM -evoked H ACh release from superfused hippocampal synaptosomes, whereas PLA 2 Urml and melittin 12
. Ž .mM caused a lower facilitation 46"1% and 38"5%, respectively . The present results show that both exogenously added andw3 xendogenously produced AA increase the evoked release of H ACh from rat hippocampal nerve terminals. Since muscarinic activation
triggers AA production and we now observed that AA enhances ACh release, it is proposed that AA may act as a facilitatory retrogrademessenger in hippocampal cholinergic muscarinic transmission as it has been proposed to act in glutamatergic transmission. q 1999Elsevier Science B.V. All rights reserved.
Keywords: Acetylcholine; Arachidonic acid; Hippocampus; Neuromodulation
1. Introduction
Ž .Arachidonic acid AA is a 20-carbon unsaturated fattyŽ .acid 20:4 , which is mostly found sterified in the sn-2
w xposition of membrane phospholipids 23 . AA is an intra-w xcellular second messenger 23 and also acts as a neuro-
modulator that is released in situations of increased neu-ronal activity, such as synaptic plasticity, epilepsy or hy-
w xpoxia 4,25 . AA is proposed to act as a facilitatoryw xretrograde neuromodulator in glutamatergic synapses 17 ,
since it is released upon activation of glutamate receptorw x w x10,20 and also increases glutamate release 11,17 .
The recent observation that AA metabolism in the CNSis particularly enhanced upon muscarinic stimulationw x13,29,32 and that muscarinic activation causes AA re-
) Corresponding author. Fax: q 351-1-7936787; E-mail:[email protected]
w xlease 14,32 might lead to the hypothesis that AA mayalso act as a retrograde messenger in cholinergic mus-carinic transmission, if AA would be able to modulateacetylcholine release. Thus, we now tested whether AA
w3 xmight modify H acetylcholine release from the rat hip-pocampus.
2. Materials and methods
2.1. Chemicals
Ž .Arachidonic acid AA , arachidic acid, bovine serumŽ . Žalbumin BSA, fatty acid free , phospholipase A EC2
.3.1.1.4, PLA from bee venom, melittin, indomethacin,2
nordihydroguaiaretic acid, hemicholinium-3, neostigmineŽ .bromide, choline kinase EC 2.7.1.32 , veratridine and
tetraphenylboron sodium salt were from Sigma. Arachi-Ž .donyl trifluromethylketone AACOCF was from Cal-3
w3 x Žbiochem. Methyl- H choline chloride specific activity
0006-8993r99r$ - see front matter q 1999 Elsevier Science B.V. All rights reserved.Ž .PII: S0006-8993 99 01267-6

( )T. Almeida et al.rBrain Research 826 1999 104–111 105
.76–86.3 Cirmmol was obtained from Amersham. Allother reagents were of the highest purity available.
AA and arachidic acid were made up into a 30 mMstock solution and AACOCF was made up into a 10 mM3
stock solution in ethanol, both being aliquoted and storedunder nitrogen atmosphere at y208C. Indomethacin wasmade up into a 20 mM stock in methanol and nordihydro-guaiaretic acid was made up into a 20 mM stock inethanol. Aqueous dilutions of these stock solutions weremade daily. The maximal concentration of ethanol ormethanol used was devoid of effects on both basal and
Ž .evoked tritium release data not shown .
[3 ]2.2. H acetylcholine release experiments from hip-pocampal slices
Experiments performed on hippocampal slices takenŽ .from young adult male Wistar rats 6 weeks old , were
w xcarried as previously described 7 . The animals wereanaesthetised with halothane, decapitated, and both hip-pocampi lobes dissected free within an ice-cold Krebssolution, gassed with 95% O and 5% CO mixture, of the2 2
Ž .following composition mM : NaCl 124, KCl 3, KH PO2 4
1.25, MgSO 1, CaCl 2, NaHCO 26, glucose 10, pH4 2 3Ž .7.4. Slices 400 mm thick were cut perpendicularly to the
long axis of the hippocampus and kept in gassed KrebsŽ .solution at room temperature 22–258C . After 1 h recov-
ery period, four slices were incubated at 378C with 2 ml ofw3 x Žgassed Krebs solution containing H choline 10 mCirml,
.0.125 mM . After a 30-min loading period, the 4 sliceswere washed twice with 2 ml of gassed Krebs solutioncontaining 10 mM of the choline uptake blocker, hemi-cholinium-3, placed in 100 ml perspex chambers andsuperfused with gassed Krebs with 10 mM hemicholinium-3 at 378C with a flow rate of 0.6 mlrmin. From this time
Ž .onward, hemicholinium-3 10 mM was present in theperfusion solutions. After 45 min superfusion, the effluentwas collected in 3 min fractions and 500 ml used forscintillation counting analysis. Six minutes after startingsample collection, the slices were field-electrically stimu-
Ž .lated S with monopolar square-wave pulses with a1
duration of 3 ms and an amplitude of 40 V, delivered withŽ .a frequency of 5 Hz for a period of 2 min 600 pulses ,
through platinum electrodes. The electrical pulses werecontinuously monitored with an oscilloscope in parallelwith the perfusion chambers and the pulses remainedconstant throughout the stimulation period. Thirty-six min-utes after starting sample collection the slices were again
Ž .electrically stimulated S as in S . Under these condi-2 1
tions, the field-electrically evoked tritium release is Ca2q-dependent, tedrodotoxin-sensitive and is mainly due tow3 x Žw3 x . w xH acetylcholine H ACh release 7 . Thus, in most
w3 xexperiments we measured H ACh release as tritium re-Žlease. At the end of the release experiments 57 min after
.starting sample collection , the slices were removed fromthe chamber and homogenised by sonication in 0.5 ml of 3
Ž .M perchloric acid and 2% vrv Triton X-100, and a 100ml aliquot analysed by scintillation counting for determina-tion of the tritium retained in the preparations.
[3 ]2.3. H acetylcholine release experiments from hip-pocampal slices
w3 xThe release of H ACh from a rat hippocampal synap-tosomal fraction was performed as previously describedw x8 . Briefly, the synaptosomes were equilibrated at 378Cfor 10 min in Krebs solution. From this time onwards, allsolutions applied to the synaptosomes were kept at 378Cand gassed with 95% O and 5% CO . After the equilibra-2 2
w3 xtion period, the synaptosomes were loaded with H cholineŽ .10 mCirml, 0.125 mM for 10 min, centrifuged, andwashed twice with 1 ml of Krebs solution containing
Ž .hemicholinium-3 10 mM , which was present up to theend of the experiment to prevent the reuptake of choline.The synaptosomes were then resuspended in 10 ml ofKrebs solution and layered over Whatman GFrC filters
Žinto 8 parallel 90 ml superfusion chambers adapted from.Swinny filter holders, Millipore with the aid of a roller
Žpump flow rate: 0.6 mlrmin, which was kept constant.through the experiment . The chamber volume plus dead
volume was approximately 0.6 ml. A series of 8 parallelsuperfusion chambers was used to enable both control andtest conditions to be performed in duplicate from the samebatch of synaptosomes. After a 15-min washout period, theeffluent was collected in 2-min fractions for scintillation
w xcounting 8 . The synaptosomes were stimulated with vera-Ž .tridine 10 mM for 2 min at 6 and 20 min after starting
Ž .sample collection S and S . At the end of the experi-1 2
ments, the filters were removed from the superfusionchambers and analysed by scintillation counting for deter-mination of tritium retained by the synaptosomes.
At least 12 h after addition of 5 ml of scintillationŽ .cocktail Scintran T, BDH to the aliquots of effluent
samples and either homogenised slices or synaptosomesretained in the filters, scintillation counting was performedwith a 55–60% efficiency during 2 min to ensure acounting error -5%. Radioactivity was expressed in termsof disintegrations per min per mg of protein in each
Ž .chamber dpmrmg . The amount of protein in the ho-mogenised slices or in the synaptosomal aliquots was
w xmeasured according to Peterson 22 . The fractional releasewas expressed in terms of percentage of total radioactivitypresent in the tissue at the time of sample collection. Therelease of tritium evoked by each period of electricalstimulation or by each veratridine pulse, i.e., the evoked
Ž .release expressed as fractional release was calculated byintegration of the area of the peak upon subtraction of theestimated basal tritium outflow from the total outflow oftritium due to stimulation.
When the effect of any drug on the release of ACh wasinvestigated, the tested drug was added to the perfusion
Ž . Ž .medium 21 min slices or 8 min synaptosomes before

( )T. Almeida et al.rBrain Research 826 1999 104–111106
S , i.e., 16–18 min after starting sample collection, and2
remained in the bath up to the end of the experiment. Theeffect of drugs on basal tritium outflow was quantified bythe percent variation of baseline before and after superfu-sion of the preparations with a drug. The effect of drugs onthe evoked release of ACh was expressed by alterations ofthe ratio between the evoked release due to second stimu-lation period and the evoked release due to the first
Ž .stimulation period S rS ratio . When we evaluated the2 1
modifications of the effect of a test drug by another drug,this drug was applied 15 min before starting sample collec-tion and hence was present during S and S . When1 2
present during S and S , none of the drugs significantly1 2Ž .altered P)0.05 the S rS as compared to the S rS2 1 2 1
ratio obtained in the absence of drugs.
2.4. Biochemical analysis
w3 xFor the measurement of the amounts of H ACh in thetotal tritium outflow, the evoked tritium outflow experi-
Žments were performed in the presence of neostigmine 20. w3 xmM , H ACh was separated using a cation exchanger,
w3 xtetraphenylboron, after phosphorylation of H choline, asŽ w x.previously described see Ref. 7 . For the determination
Ž . Žof the energy charge, defined as ATPq1r2 ADP r ATP.qADPqAMP , the slices were incubated without or with
AA for 30 min, homogenised and purines were extractedŽ w x.and analysed by HPLC see Ref. 7 . To evaluate cellular
integrity, either the slices or the synaptosomes were placedin the superfusion chambers and superfused for 30 minwith gassed Krebs without or with AA in a close-circuitmanner. Cellular disruption was determined by comparingthe lactate dehydrogenase activity in the superfusionmedium with that found in the slices or the synaptosomes
Ž .upon their solubilization, by sonication, with 2% vrvw xTriton X-100 6 .
2.5. Statistics
The values are presented as mean"S.E.M. To test thesignificance of the effect of a drug vs. control, a pairedStudent’s t-test was used. When making comparisons froma different set of experiments with control, one way analy-
Ž .sis of variance ANOVA was used, followed by Dunnett’stest. P-0.05 was considered to represent a significantdifference.
3. Results
3.1. Effect of exogenously added arachidonic acid on[3 ]H ACh release from hippocampal slices
Under control conditions, the electrically evoked tritiumrelease from hippocampal slices during the two episodes of
Ž .electrical stimulation S and S was similar with an1 2Ž .S rS ratio of 1.12"0.03 ns10 . As shown in Fig. 1A,2 1
Ž .superfusion of slices with arachidonic acid AA, 30 mM ,15 min before S , increased both the basal outflow and the2
evoked release of tritium. To check whether the facilitatoryeffect of AA on tritium release corresponded to a facilita-
w3 xtion of H ACh release, we compared the release ofw3 xtritium and of H ACh in the absence and in the presence
Ž . w3 xof AA 30 mM . It was found that H ACh accounted forŽ .79"5% ns4 of the evoked tritium release in the
Ž .absence and for 83"5% ns4 in the presence of AAŽ . w3 x30 mM . Concerning the basal outflow of tritium, H ACh
Ž .accounted for 48"6% ns4 of the evoked tritiumŽ .release in the absence and for 43"4% ns4 in the
Ž .presence of AA 30 mM .Fig. 1B shows the concentration-response curve for the
Ž .facilitatory effect of AA 3–100 mM on the evokedŽ .release of tritium from hippocampal slices. AA 30 mM
Ž .produced a maximal facilitation of 69"7% ns6 of theevoked tritium release. Increasing concentrations of AAŽ .100 mM caused a lower facilitation of evoked tritium
Ž .release 38"1%, ns3 . In contrast, as shown in Fig. 1B,there was a monotonous increase in the basal tritium
Ž .outflow with increasing concentrations of AA 3–100 mM .To investigate whether the effect of AA on tritium releasewas influenced by detergent-like effects or uncoupling ofoxidative phosphorylation caused by AA, we tested theeffect of AA on the energy charge and the release oflactate dehydrogenase, an intracellular marker, from super-
Žfused rat hippocampal slices. As shown in Table 1, AA 30.mM did not significantly change either the energy charge
or the release of lactate dehydrogenase. In contrast, AAŽ .100 mM decreased the energy charge by 1.84"0.01%Ž .ns4, P-0.05 and caused the release of 14"2%Ž .ns4, P-0.05 of total lactate dehydrogenase during the
Ž .45 min superfusion period Table 1 .Incubation of the slices with bovine serum albumin
Ž .BSA, 1% from 15 min before starting sample collectionŽ .onward hence present during S and S , prevented the1 2
Ž .facilitatory effect of AA 30 mM on the evoked tritiumŽ . Ž .release Fig. 1C and attenuated by 81"6% ns4 the
Ž .AA 30 mM -induced increase in basal tritium outflow.Incubation of the slices in the simultaneous presence of
Ž .the cyclooxygenase inhibitor, indomethacin 100 mM , andthe lipooxygenase inhibitor, nordihydroguaiaretic acidŽ .NDGA, 50 mM , from 15 min before starting sample
Ž .collection onward hence present during S and S , did1 2Ž .not change the effect of AA 30 mM on the evoked
Ž . Ž .tritium release Fig. 1C and tended to attenuate P)0.05Ž .the effect of AA 30 mM on the basal tritium outflow
Ž43"4% facilitation in the absence and 32"7% facilita-tion in the presence of nordihydroguaiaretic acid and indo-
.methacin, ns4 .
3.2. Effect of endogenously produced arachidonic acid on[3 ]H ACh release from hippocampal slices
Ž .When phospholipase A PLA , 5 Urml , an enzyme2 2
that releases fatty acids from the sn-2 position of phospho-

( )T. Almeida et al.rBrain Research 826 1999 104–111 107
lipids where AA is mostly bound, was applied to the slices15 min before S , it enhanced the evoked release of tritium2
Ž . Ž .by 86"14% ns4 . PLA 5 Urml also enhanced the2Ž .basal outflow of tritium by 29"3% ns4 . Incubation of
Ž .the slices with BSA 1% from 15 min before startingŽsample collection onward hence present during S and1
. Ž .S , prevented the facilitatory effect of PLA 5 Urml on2 2Ž .the evoked tritium release Fig. 2A and attenuated by
Ž .68"8% ns4 the PLA -induced increase in basal tri-2
tium outflow.Ž .When melittin 1 mM , an activator of endogenous
PLA , was applied to the slices 15 min before S , it2 2Ženhanced the evoked release of tritium by 98"11% ns
. Ž .4 . Melittin 1 mM also enhanced the basal outflow ofŽ .tritium by 30"5% ns4 . Incubation of the slices with
Ž .arachidonyl trifluromethylketone AACOCF , 20 mM , a3
PLA inhibitor, from 15 min before starting sample collec-2
Table 1Ž . Ž .Lack of effect of AA 30 mM and modification by AA 100 mM of the
energy charge and the release of lactate dehydrogenase, an intracellularmarker, from superfused rat hippocampal slices
Ž . Ž .Control AA 30 mM AA 100 mMUEnergy charge 0.811"0.003 0.809"0.004 0.796"0.003
U% total lactate 6"2 5"2 14"2dehydrogenasereleased
The values are mean"S.E.M. of 4 experiments performed in duplicate.U P -0.05 vs. control.
Ž .tion onward hence present during S and S , prevented1 2Ž .the facilitatory effect of melittin 1 mM on the evoked
Ž . Ž .tritium release Fig. 2B and attenuated by 71"4% ns4the melittin-induced increase in basal tritium outflow. In-
Ž .cubation of the slices with BSA 1% from 15 min beforeŽstarting sample collection onward hence present during S1
. Žand S , prevented the facilitatory effect of melittin 12. Ž .mM on evoked tritium release Fig. 2B and attenuated by
Ž .54"6% ns4 the melittin-induced increase in basaltritium outflow.
To test whether endogenous AA was tonically modulat-ing acetylcholine release from hippocampal slices underthe experimental conditions used, we tested the effect of
Ž . Ž .BSA 1% and AACOCF 20 mM on basal outflow and3Ž .evoked release of tritium. When BSA 1% was superfused
15 min before S , it slightly decreased the evoked release2Ž .of tritium by 9"3% ns4 and decreased the basal
Fig. 1. Facilitation by exogenously added arachidonic acid ofw3 xH acetylcholine release from rat hippocampal slices. Rat hippocampal
w3 xslices were loaded with H choline, superfused and effluent samplesŽ .analysed by scintillation counting. A Time course of tritium release in a
Žtypical experiment. The slices were field-electrically stimulated 40 V, 3. Ž . Ž .ms, 5 Hz for 2 min 6 min S and 36 min S after starting sample1 2
Ž .collection, as indicated by the bars above the abscissa. AA 30 mM wasapplied through the superfusate to two of the four parallel superfusionchambers containing the slices 15 min before S , as indicated by the bar2
above the abscissa. The open symbols represent the averaged tritiumŽ .release from the two control slices to which no AA was added and the
Žfilled symbols represent the averaged tritium release of the test slices to. Ž .which AA was added 15 min before S onwards . B The2
Ž .concentration-dependent effect of AA 3–100 mM on basal tritiumŽ . Ž .outflow B and on evoked tritium release v . The effect of each
concentration of AA on evoked release was calculated as the percentagevariation of the amount of tritium released in S ramount of tritium2
released in S in the presence of AA during S vs. the S rS ratio in1 2 2 1
control conditions in the same experiment. The results are mean"S.E.M.of 3–4 experiments. U P -0.05 vs. 0%. In C is shown the ability of
Ž .bovine serum albumin BSA, 1% , which quenches AA, to prevent theŽ .facilitatory effect of AA 30 mM on the evoked tritium release, and the
inability of the lipooxygenase inhibitor, nordihydroguaiaretic acidŽ . ŽNDGA, 50 mM , and the cyclooxigenase inhibitor, indomethacin 100
.mM , to modify this AA-induced facilitation. The substances were pre-sent in S or in S and S as indicated under the bars. The results are2 1 2
mean"S.E.M. of 3–4 experiments. U P -0.05 vs. 0%. UU P -0.05 whenŽ .compared with the effect of AA 30 mM .
( )T. Almeida et al.rBrain Research 826 1999 104–111108
Ž .Fig. 2. Facilitation by phospholipase A PLA , which releases AA2 2
from the sn-2 position of phospholipids, and by melittin, which stimu-w3 xlates endogenous PLA , of H acetylcholine release from rat hippocam-2
w3 xpal slices. Rat hippocampal slices were loaded with H choline, super-fused and effluent samples analysed by scintillation counting. The slices
Ž . Ž .were field-electrically stimulated 40 V, 3 ms, 5 Hz for 2 min 6 min S1Ž .and 36 min S after starting sample collection. PLA or melittin were2 2
Ž .added 15 min before S , whereas bovine serum albumin BSA or2Ž .arachidonyl trifluromethylketone AACOCF were added 15 min before3
starting sample collection and thus were present during S and S . The1 2Ž .effect of drugs was calculated by modification of the S rS ratio. A2 1
Ž .The facilitatory effect of PLA 2 Urml on evoked tritium release and2Ž .the ability of BSA 1% , which quenches AA, to prevent this effect ofŽ . Ž .PLA . The absence y or presence q of each drug during S or2 2
during S and S is indicated below each bar. The results are mean"1 2
S.E.M. of 4 experiments. U P -0.05 vs. 0%. UU P -0.05 when comparedŽ . Ž .with the effect of PLA 2 Urml . B The facilitatory effect of melittin2
Ž . Ž .1 mM on evoked tritium release and the ability of AACOCF 20 mM ,3Ž .an inhibitor of PLA , and of BSA 1% to prevent this effect of melittin.2
Ž . Ž .The absence y or presence q of each drug during S or during S2 1
and S is indicated below each bar. The results are mean"S.E.M. of 3–42
experiments. U P -0.05 vs. 0%. UU P -0.05 when compared with theŽ .effect of melittin 1 mM .
Ž .outflow of tritium by 9"3% ns4 . When AACOCF3Ž .20 mM was superfused 15 min before S , it slightly2
Ž .decreased the evoked release of tritium by 7"2% ns4and decreased the basal outflow of tritium by 6"1%Ž .ns4 .
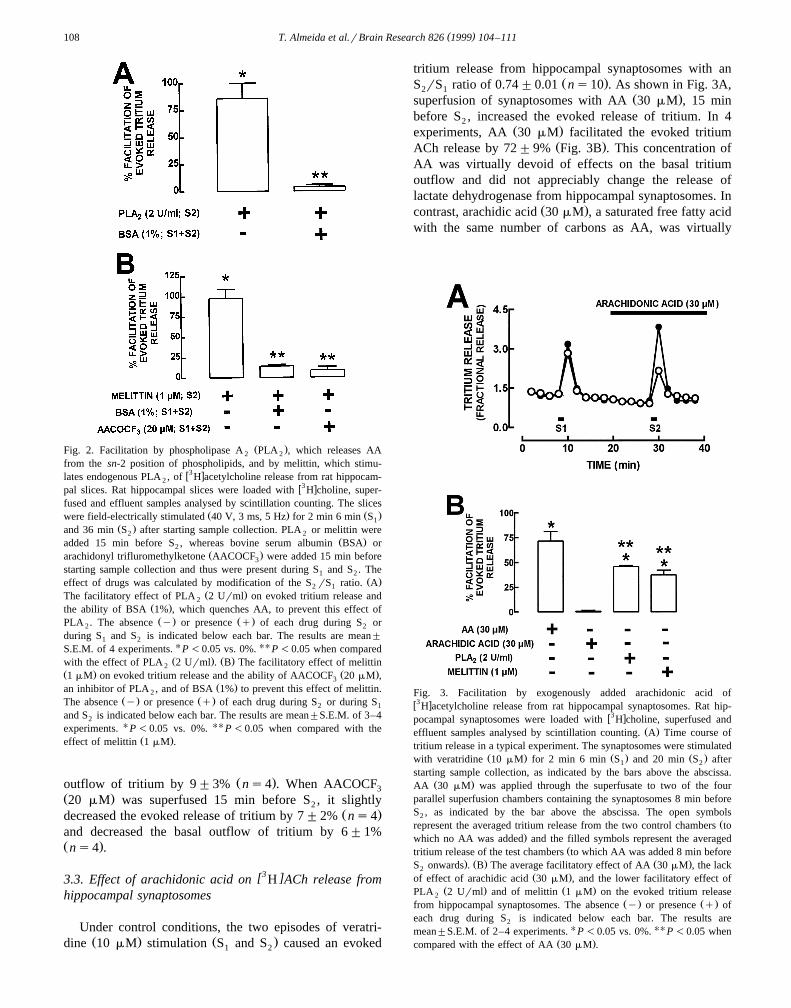
[3 ]3.3. Effect of arachidonic acid on H ACh release fromhippocampal synaptosomes
Under control conditions, the two episodes of veratri-Ž . Ž .dine 10 mM stimulation S and S caused an evoked1 2
tritium release from hippocampal synaptosomes with anŽ .S rS ratio of 0.74"0.01 ns10 . As shown in Fig. 3A,2 1
Ž .superfusion of synaptosomes with AA 30 mM , 15 minbefore S , increased the evoked release of tritium. In 42
Ž .experiments, AA 30 mM facilitated the evoked tritiumŽ .ACh release by 72"9% Fig. 3B . This concentration of
AA was virtually devoid of effects on the basal tritiumoutflow and did not appreciably change the release oflactate dehydrogenase from hippocampal synaptosomes. In
Ž .contrast, arachidic acid 30 mM , a saturated free fatty acidwith the same number of carbons as AA, was virtually
Fig. 3. Facilitation by exogenously added arachidonic acid ofw3 xH acetylcholine release from rat hippocampal synaptosomes. Rat hip-
w3 xpocampal synaptosomes were loaded with H choline, superfused andŽ .effluent samples analysed by scintillation counting. A Time course of
tritium release in a typical experiment. The synaptosomes were stimulatedŽ . Ž . Ž .with veratridine 10 mM for 2 min 6 min S and 20 min S after1 2
starting sample collection, as indicated by the bars above the abscissa.Ž .AA 30 mM was applied through the superfusate to two of the four
parallel superfusion chambers containing the synaptosomes 8 min beforeS , as indicated by the bar above the abscissa. The open symbols2
Žrepresent the averaged tritium release from the two control chambers to.which no AA was added and the filled symbols represent the averaged
Žtritium release of the test chambers to which AA was added 8 min before. Ž . Ž .S onwards . B The average facilitatory effect of AA 30 mM , the lack2
Ž .of effect of arachidic acid 30 mM , and the lower facilitatory effect ofŽ . Ž .PLA 2 Urml and of melittin 1 mM on the evoked tritium release2
Ž . Ž .from hippocampal synaptosomes. The absence y or presence q ofeach drug during S is indicated below each bar. The results are2
mean"S.E.M. of 2–4 experiments. U P -0.05 vs. 0%. UU P -0.05 whenŽ .compared with the effect of AA 30 mM .

( )T. Almeida et al.rBrain Research 826 1999 104–111 109
devoid of effects on the evoked tritium release fromŽ . Ž . Žhippocampal synaptosomes ns2 Fig. 3B . PLA 22
. Ž .Urml and melittin 1 mM also facilitated by 46"1%Ž . Ž .ns4 and 38"5% ns3 the veratridine-evoked tri-
Ž .tium release from hippocampal synaptosomes Fig. 3B .
4. Discussion
Ž .The present results show that arachidonic acid AAw3 xstimulated the evoked release of H acetylcholine
Žw3 x .H ACh from the rat hippocampus. Low micromolarconcentrations of AA caused a concentration-dependent
w3 xenhancement of the evoked H ACh release from hip-pocampal slices with a maximal effect recorded at 30 mM.
Ž .AA 3–100 mM also enhanced the basal tritium release,Ž .but the effect of exogenously added AA 3–30 mM was
w3 xmore marked on evoked H ACh release than on basaltritium release. These concentrations of AA are within therange of concentrations estimated to be reached by AA in
w xthe extracellular medium upon neuronal activity 4,11 . AtŽ .higher concentrations of AA 100 mM , which are esti-
mated to be reached during pathological conditions such asw xupon ischemic insults 1,25 , there was a decreased facilita-
tion of evoked tritium release, but an higher facilitation ofbasal tritium release. These differential effects of 100 mMof exogenously added AA on basal tritium outflow and
w3 xevoked H ACh release may be due to disturbance ofmembrane integrity andror uncoupling of oxidative phos-
w xphorylation 2,27 , since 100 mM AA, in contrast with thelower concentrations of AA tested, caused a reduction inthe energy charge of the slice and an increase in therelease of lactate dehydrogenase, an intracellular marker.Furthermore, the lack of effect of arachidic acid on the
w3 xevoked H ACh release reinforces the idea that changes inmembrane integrity are unlikely to be the underlying causeof the facilitatory effect of low micromolar concentrations
w3 xof AA on H ACh release.AA, in concentrations similar to those used in the
present work, also increases the release of other neuro-w x w xtransmitters such as glutamate 11 , GABA 2,3 , or
w xdopamine 16 . These facilitatory effects are observed bothw xin slice preparations 3 as well as in synaptosomal prepa-
w xrations 3,11 , suggesting that the facilitatory effect of lowmicromolar concentrations of AA may be a direct effect onneurotransmitter release from nerve terminals. The ob-served increase of ACh release from hippocampal synapto-somes by AA suggests that AA causes a direct increase ofACh release from cholinergic nerve terminals. Thus, theuse of superfused hippocampal synaptosomes makes itunlikely that the effect of AA may be an indirect effectmediated by modification of the extracellular levels of
w xmodulators of acetylcholine release 24 , e.g., adenosinew x9 . In this respect, it is elucidating that the muscarinic
w xautoinhibition, which is evident in hippocampal slices 30 ,is not observed in superfused hippocampal synaptosomes
w x18 , although muscarinic receptor agonists depress theevoked ACh release in both hippocampal slices and
w xsynaptosomes 18,30 .In several preparations, some metabolites of AA also
w xmodulate the release of neurotransmitters 23 . However,the observation that inhibition of cyclooxygenase, by usingindomethacin, and lipooxygenase, by using NDGA, failedto modify the effect of exogenously added AA indicatesthat the facilitatory effect of AA on acetylcholine releaseresults from the action of AA itself rather from that of itsmetabolites.
The present results also suggest that endogenously pro-w3 xduced AA is able to enhance evoked H ACh release.
Superfusion of the slices with PLA , which hydrolyses the2
sn-2 position of phospholipids where AA is normallysterified, and melittin, a peptide that activates endogenous
Ž w x.PLA e.g., Ref. 5 , mimicked the effect of exogenously2
added AA in low micromolar concentrations. However, theobservation that the amplitude of the facilitatory effect ofboth PLA and melittin at facilitating the evoked release2
w3 x Žof H ACh from hippocampal slices was greater P-.0.05 than that of exogenously added AA, suggests that the
generation of endogenous AA was more effective thanexogenously added AA at raising free arachidonic acid
w xlevels 11,28 . The effect of both PLA and melittin was2
prevented by albumin, a water soluble protein that bindswith high affinity free, but not lipid-sterified, fatty acidsw x15 , supporting the requirement of AA production forPLA and melittin to exert their facilitatory effects on2w3 xH ACh release. The observation that the effect of both
w3 xPLA and melittin on the evoked release of H ACh was2
almost abolished by albumin, which binds AA with signifi-w xcantly higher affinity than its metabolites 21 , also sug-
gests that the facilitation of acetylcholine release is causedby endogenous AA itself rather than by its cyclooxigenaseor lipooxygenase metabolites. The prevention of the effectsof PLA and melittin by albumin also suggests that the2
endogenously produced AA has to be available to bequenched in the extracellular space. The observation that,in contrast to what was observed in slices, the facilitatoryeffect of PLA and melittin on the evoked release of2w3 x ŽH ACh from hippocampal synaptosomes was lower P
.-0.05 than the effect of exogenously added AA, sug-gests that AA is generated in structures other than cholin-ergic nerve terminals, e.g., from post-synaptic componentsw x w x17 or from glial cells 19 rather than being generated incholinergic nerve terminals and rapidly equilibratingthrough the plasma membrane.
The mechanism by which AA facilitates evokedw3 xH ACh release remains to be established. We have ex-
w3 xcluded the possibility that any effects of AA on H AChrelease may be caused by inhibition of choline uptakew x1,27 , since the experiments were performed in the pres-ence of supra-maximal concentrations of the choline up-take blocker, hemicholinium-3. Furthermore, we have ob-served that the AA-facilitation of electrically evoked tri-

( )T. Almeida et al.rBrain Research 826 1999 104–111110
w3 xtium outflow is mostly due to increased H ACh releasew3 xrather than extracellular H choline accumulation.
These results show that AA facilitates both basal andevoked ACh release in the hippocampus, as has previouslybeen reported to occur at the myenteric plexus of the
w xguinea pig ileum 26,31 . The observation that AAmetabolism in the CNS is particularly intense upon mus-
w xcarinic stimulation 13,29,32 and that muscarinic activa-w xtion triggers the release of arachidonic acid 14,32 to-
gether with the present observation that AA directly facili-tates acetylcholine release from cholinergic nerve termi-nals, raises the hypothesis that AA may act as a facilitatoryretrograde messenger in cholinergic muscarinic transmis-sion, as it has been proposed to act in glutamatergic
w xtransmission 17 . In glutamatergic synapses, endogenousAA only fulfils a tonic modulatory role during intense
w xglutamatergic transmission 17 . The inability to detect anyeffect of endogenous AA as a tonic modulator of AChrelease might also suggest that intense cholinergic mus-carinic transmission might be required to recruit the AAneuromodulatory pathway. However, this possible role ofAA as a retrograde messenger in cholinergic muscarinictransmission still remains to be directly demonstrated, andits physiological role has to be explored. Never the less,this hypothesis presents a new facilitatory control mecha-nism in cholinergic pathways that may be explored tocorrect deficits of cholinergic activity underlying different
Ž w x.neurodegenerative diseases e.g., Ref. 12 .
Acknowledgements
The technical assistance of Mrs. M.D. Constantino isgreatly acknowledged. This work was supported by
Ž .PRAXIS XXI SAUr44r96 .
References
w x1 P. Boksa, S. Mykita, B. Collier, Arachidonic acid inhibits cholineuptake and depletes acetylcholine content in rat cerebral cortical
Ž .synaptosomes, J. Neurochem. 50 1988 1309–1318.w x2 A.I.M. Breukel, E. Besselsen, F.H. Lopes da Silva, W.E.J.M. Ghi-
jsen, Arachidonic acid inhibits uptake of amino acids and potentiatesPKC effects on glutamate, but not GABA, exocytosis in isolated
Ž .hippocampal nerve terminals, Brain Res. 773 1997 90–97.w x3 A. Cheramy, F. Artaud, G. Godeheu, M. L’hirondel, J. Glowinski,´
Stimulatory effect of arachidonic acid on the release of GABA inŽ .matrix-enriched areas from the rat striatum, Brain Res. 742 1996
185–194.w x4 M.P. Clements, T.V.P. Bliss, M.A. Lynch, Increase in arachidonic
acid concentration in a postsynaptic membrane fraction followingthe induction of long-term potentiation in the dentate gyrus, Neuro-
Ž .science 45 1991 379–389.w x5 K.M. Conricode, R.S. Ochs, Mechanism for the inhibitory and
stimulatory actions of proteins on the activity of phospholipase A ,2Ž .Biochim. Biophys. Acta 1003 1989 36–43.
w x X6 R.A. Cunha, A.M. Sebastiao, J.A. Ribeiro, Ecto-5 -nucleotidase is˜associated with cholinergic nerve terminals in the hippocampus but
Ž .not in the cerebral cortex of the rat, J. Neurochem. 59 1992657–666.
w x7 R.A. Cunha, E. Milusheva, E.S. Vizi, J.A. Ribeiro, A.M. Sebastiao,˜Excitatory and inhibitory effects of A and A adenosine receptor1 2A
w3 xactivation on the electrically evoked H acetylcholine release fromŽ .different areas of the rat hippocampus, J. Neurochem. 63 1994
207–214.w x8 R.A. Cunha, J.A. Ribeiro, A.M. Sebastiao, Purinergic modulation of˜
w3 xthe evoked release of H acetylcholine from the hippocampus andcerebral cortex of the rat: role of the ectonucleotidases, Eur. J.
Ž .Neurosci. 6 1994 33–42.w x9 R.A. Cunha, M.D. Constantino, J.A. Ribeiro, Modification of adeno-
sine modulation by arachidonic acid in the rat hippocampus, Phar-Ž .macol. Toxicol. 81 1997 41, Suppl. 1.
w x10 A. Dumuis, J.P. Pin, K. Oomagari, M. Sebben, J. Bockaert, Arachi-donic acid released from striatal neurons by joint stimulation of
Ž .ionotropic and metabotropic quisqualate receptors, Nature 347 1990182–184.
w x11 E.J. Freeman, D.M. Terrian, R.V. Dorman, Presynaptic facilitationof glutamate release from isolated hippocampal mossy fiber nerve
Ž .endings by arachidonic acid, Neurochem. Res. 15 1990 743–750.w x12 M.E. Hasselmo, J.M. Bower, Acetylcholine and memory, TINS 16
Ž .1993 218–222.w x13 C.R. Jones, T. Arai, J.M. Bell, S.I. Rapoport, Preferential in vivo
w3 xincorporation of H arachidonic acid from blood into rat brainsynaptosomal fractions before and after cholinergic stimulation, J.
Ž .Neurochem. 67 1996 822–829.w x14 R.Y. Kanterman, A.L. Ma, E.M. Briley, J. Axelrod, C.C. Felder,
Muscarinic receptors mediate the release of arachidonic acid fromspinal cord and hippocampal neurons in primary culture, Neurosci.
Ž .Lett. 118 1990 235–237.w x15 U. Kragh-Hansen, Molecular aspects of ligand binding to serum
Ž .albumin, Pharmacol. Rev. 33 1981 17–53.w x16 M. L’hirondel, A. Cheramy, G. Godeheu, J. Glowinski, Effects of´
arachidonic acid on dopamine synthesis, spontaneous release, anduptake in striatal synaptosomes from the rat, J. Neurochem. 64Ž .1995 1406–1409.
w x17 M.A. Lynch, M.P. Clements, K.L. Voss, C.R. Bramham, T.V.P.Bliss, Is arachidonic acid a retrograde messenger in long-term
Ž .potentiation?, Biochem. Soc. Trans. 19 1991 391–396.w x18 M. Marchi, P. Paudice, M. Raiteri, Autoregulation of acetylcholine
release in isolated hippocampal nerve endings, Eur. J. Pharmacol. 73Ž .1981 75–79.
w x19 S.A. Moore, E. Yoder, S. Murphy, G.R. Dutton, A.A. Spector,Ž .Astrocytes, not neurons, produce docosahexaenoic acid 22:6v y3
Ž . Ž .and arachidonic acid 20:4v y6 , J. Neurochem. 56 1981 518–524.w x20 L. Pellerin, L.S. Wolfe, Release of arachidonic acid by NMDA-re-
Ž .ceptor activation in the rat hippocampus, Neurochem. Res. 9 1991983–989.
w x21 T. Peters, Jr., Serum albumin, in: C.B. Anfisen, J.T. Edsall, F.M.Ž .Richards Eds. , Advances in Protein Chemistry, Vol. 37, Academic
Press, Orlando, 1985, pp. 161–245.w x22 G.L. Peterson, A simplification of the protein assay method of
Lowry et al. which is more generally applicable, Anal. Biochem. 83Ž .1977 346–356.
w x23 D. Piomelli, Eicosanoids in synaptic transmission, Crit. Rev. Neuro-Ž .biol. 8 1994 65–83.
w x24 M. Raiteri, F. Angelini, G. Levi, A simple apparatus for studying therelease of neurotransmitters from synaptosomes, Eur. J. Pharmacol.
Ž .25 1974 411–414.w x25 S. Rehncrona, E. Westerberg, B. Akesson, B.K. Siejo, Brain cortical¨
fatty acid phospholipids during and following complete and severeŽ .incomplete ischemia, J. Neurochem. 38 1982 84–93.
w x26 N. Saitoh, R. Fujimoto, T. Ishii, H. Nishio, T. Takeuchi, F. Hata,Muscarinic autoinhibition and modulatory role of protein kinase C inacetylcholine release from the myenteric plexus of guinea pig ileum,
Ž .Jpn. J. Pharmacol. 74 1997 155–163.

( )T. Almeida et al.rBrain Research 826 1999 104–111 111
w x27 M.D. Saltarelli, K. Yamada, J.T. Coyle, Phospholipase A and2w3 xH -hemicholinium-3 binding sites in rat brain: a potential second-messenger role for fatty acids in the regulation of high-affinity
Ž .choline uptake, J. Neurosci. 10 1990 62–72.w x28 D.R. Samples, E.A. Sprague, M.J.K. Harper, J.T. Herlihy, In vitro
adsorption losses of arachidonic acid and calcium ionophore A23187,Ž .Am. J. Physiol. 257 1989 C1166–C1170.
w x29 J. Strosznajder, M. Samochocki, M. Duran, Aging diminishes sero-tonin-stimulated arachidonic acid uptake and cholinergic receptor-activated arachidonic acid release in rat brain cortex membrane, J.
Ž .Neurochem. 62 1994 1048–1054.
w x w3 x30 J.C. Szerb, P. Hadhazy, J.D. Dudar, Release of H acetylcholine´from rat hippocampal slices: effect of septal lesion and of gradedconcentrations of muscarinic agonists and antagonists, Brain Res.
Ž .128 1977 285–291.w x31 T. Takeuchi, O. Yagasaki, Modulation of acetylcholine release from
guinea-pig ileum myenteric plexus by arachidonic acid cascadeŽ .inhibitors, Jpn. J. Pharmacol. 45 1987 434–437.
w x32 M. Tence, J. Cordier, J. Premont, J. Glowinski, Muscarinic choliner-´gic agonists stimulate arachidonic acid release from mouse striatal
Ž .neurons in primary culture, J. Pharmacol. Exp. Ther. 269 1994646–653.





























