Embed Size (px)
Citation preview

Fa 06 CSE182
CSE182-L6
Protein sequence analysis

Fa 06 CSE182
Possible domain queries
• Case 1: – You have a collection of sequences that belong to a
family (contain a functional domain).– Given an ‘orphan’ sequence, does it belong to the
family?– There are different solutions depending upon the
representation of the domain (patterns/alignments/HMM/profiles)
• Case 2: – You have an orphan sequence from an
uncharacterized family. Can you identify other members of the family, and create a representation of them (Harder problem).

Fa 06 CSE182
EX: Innexins
• The Macagno lab is studying Gap junction proteins, Innexins (invertebrate analogs of connexins) in Hirudo
• Innexins have been found in C. elegans, and Drosophila.
• In C. elegans, 25 members of this family have been found, and partially categorized.
QuickTime™ and aTIFF (LZW) decompressor
are needed to see this picture.

Fa 06 CSE182
Innexins in Hirudo
• When certain Innexins are knocked out, they cause serious defects in cells in the ganglia.
• The EST database (partial gene sequences) contains a number of putative Innexins, discovered via BLAST.
• Project:• Q: Can you confirm that these are Innexins. Can you
find more members? (this lecture)• Q: Can you characterize them w.r.t known innexins in C.
elegans, and Drosophila?• Q: Use your method for other families of interest.
Netrins, and their receptors.

Fa 06 CSE182
Not all features(residues) are important
Skin patternsFacial Features

Fa 06 CSE182
Protein sequence motifs
• Premise: • The sequence of a protein sequence gives clues about its
structure and function.• Not all residues are equally important in determining
function.• Suppose we knew the key residues of a family. If our query
matches in those residues, it is a member. Otherwise, it is not.
• The key residues can be identified if we had structural information, or through conserved residues in an alignment of the family.

Fa 06 CSE182
Representation of domains/families.
• We will consider a number of representations that describe key residues, characteristic of a family– Patterns (regular expressions)– Alignments– Profiles– HMMs
• Start with the following:– A collection of sequences with the same function.– Region/residues known to be significant for maintaining structure and
function. • Develop a pattern of conserved residues around the
residues of interest• Iterate for appropriate sensitivity and specificity
Fa 06 CSE182
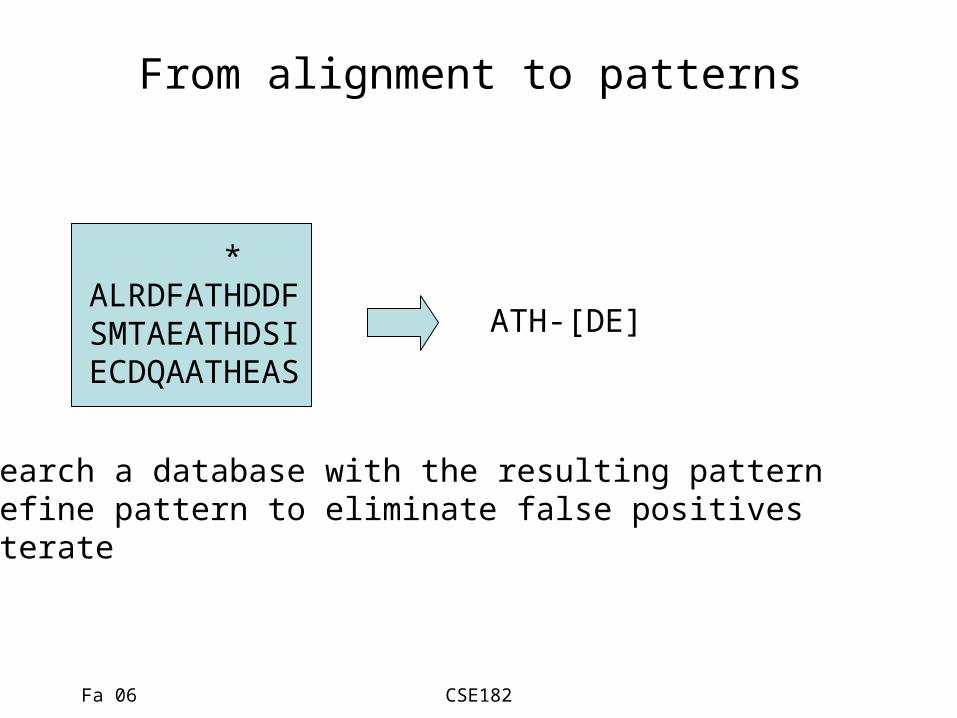
From alignment to patterns
* ALRDFATHDDF SMTAEATHDSI ECDQAATHEAS
ATH-[DE]
• Search a database with the resulting pattern• Refine pattern to eliminate false positives• Iterate

Fa 06 CSE182
Regular Expression Patterns
• Zinc Finger motif– C-x(2,4)-C-x(3)-[LIVMFYWC]-x(8)-H-x(3,5)-H – 2 conserved C, and 2 conserved H
• How can we search a database using these motifs?– The motif is described using a regular expression.
What is a regular expression?

Fa 06 CSE182
Regular Expressions
• Concise representation of a set of strings over alphabet .
• Described by a string over• R is a r.e. if and only if
€
Σ,⋅,∗,+{ }
€
R = {ε} Base caseR = {σ },σ ∈ ΣR = R1 + R2 Union of stringsR = R1 ⋅R2 ConcatenationR = R
1
* 0 or more repetitions

Fa 06 CSE182
Regular Expression
• Q: Let ={A,C,E}– Is (A+C)*EEC* a regular expression?– Is *(A+C) regular?
• Q: When is a string s in a regular expression?– R =(A+C)*EEC*– Is CEEC in R?– AEC?– ACEE?

Fa 06 CSE182
Regular Expression & Automata
Every R.E can be expressed by an automaton (a directed graph) with the following properties:– The automaton has a start and end node– Each edge is labeled with a symbol from , or
Suppose R is described by automaton AS R if and only if there is a path from start to end in A, labeled with s.

Fa 06 CSE182
Examples: Regular Expression & Automata
• (A+C)*EEC*
CA
C
start endE E
–Is CEEC in R?–AEC?–ACEE?–ACE?

Fa 06 CSE182
Constructing automata from R.E
• R = {}• R = {}, • R = R1 + R2
• R = R1 · R2
• R = R1*

Fa 06 CSE182
Regular Expression Matching
• Given a database D, and a regular expression R, is a substring of D in R?
• Is there a string D[l..c] that is accepted by the automaton of R?
• Simpler Q: Is D[1..c] accepted by the automaton of R?
Fa 06 CSE182
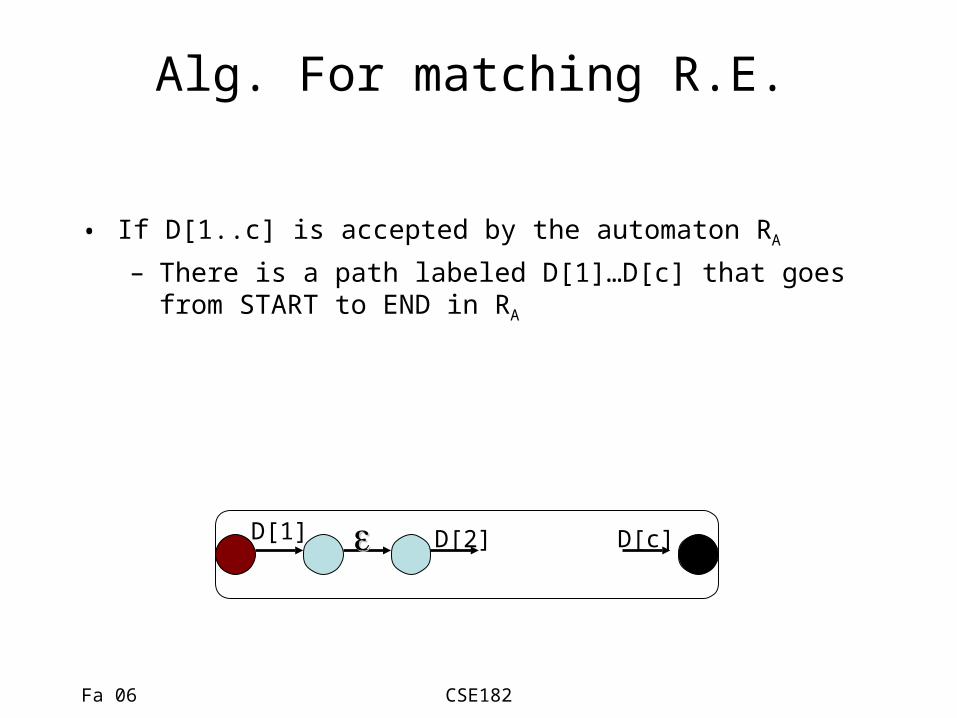
Alg. For matching R.E.
• If D[1..c] is accepted by the automaton RA
– There is a path labeled D[1]…D[c] that goes from START to END in RA
D[1] D[2] D[c]
Fa 06 CSE182
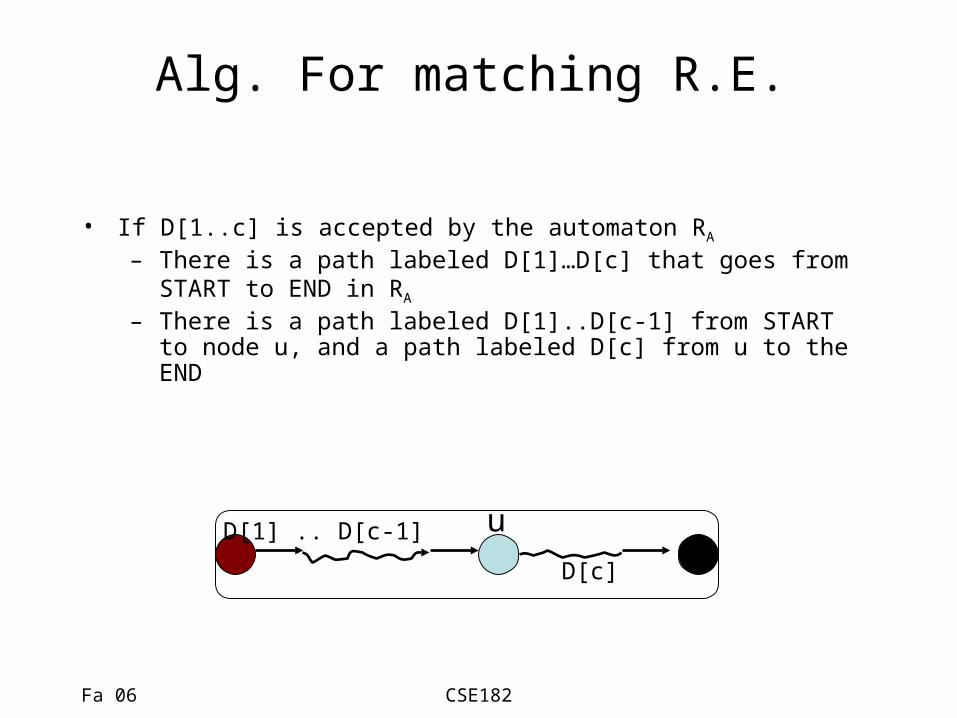
Alg. For matching R.E.
• If D[1..c] is accepted by the automaton RA
– There is a path labeled D[1]…D[c] that goes from START to END in RA
– There is a path labeled D[1]..D[c-1] from START to node u, and a path labeled D[c] from u to the END
D[1] .. D[c-1]
D[c]
u

Fa 06 CSE182
D.P. to match regular expression
• Define:– A[u,] = Automaton node
reached from u after reading
– Eps(u): set of all nodes reachable from node u using epsilon transitions.
– N[c] = subset of nodes reachable from START node after reading D[1..c]
– Q: when is v N[c]
uu vv
uu Eps(u)Eps(u)

Fa 06 CSE182
• Q: when is v N[c]?• A: If for some u N[c-1], w = A[u,D[c]],
• v {w}+ Eps(w)
D.P. to match regular expression

Fa 06 CSE182
Algorithm

Fa 06 CSE182
The final step
• We have answered the question:– Is D[1..c] accepted by R?– Yes, if END N[c]
• We need to answer – Is D[l..c] (for some l, and some c) accepted by R
€
D[l..c]∈ R⇔ D[1..c]∈ Σ∗R

Fa 06 CSE182
Representation 2: Profiles
• Profiles versus regular expressions – Regular expressions are intolerant to an occasional
mis-match.– The Union operation (I+V+L) does not quantify the
relative importance of I,V,L. It could be that V occurs in 80% of the family members.
– Profiles capture some of these ideas.

Fa 06 CSE182
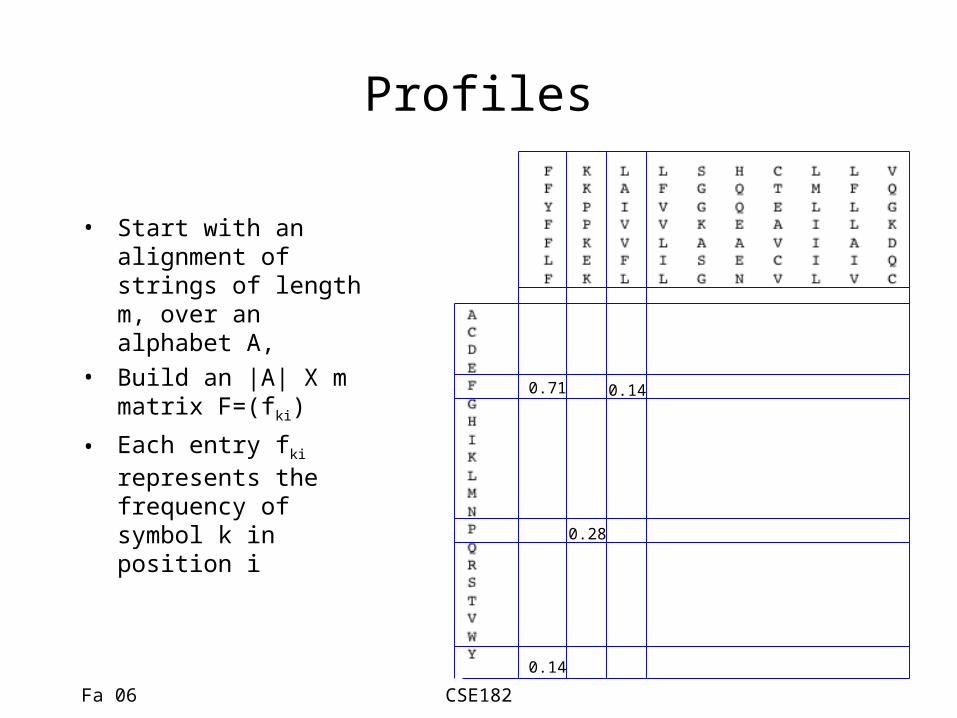
Profiles
• Start with an alignment of strings of length m, over an alphabet A,
• Build an |A| X m matrix F=(fki)
• Each entry fki represents the frequency of symbol k in position i
0.71
0.14
0.14
0.28
Fa 06 CSE182
Profiles
• Start with an alignment of strings of length m, over an alphabet A,
• Build an |A| X m matrix F=(fki)
• Each entry fki represents the frequency of symbol k in position i
0.71
0.14
0.14
0.28

Fa 06 CSE182
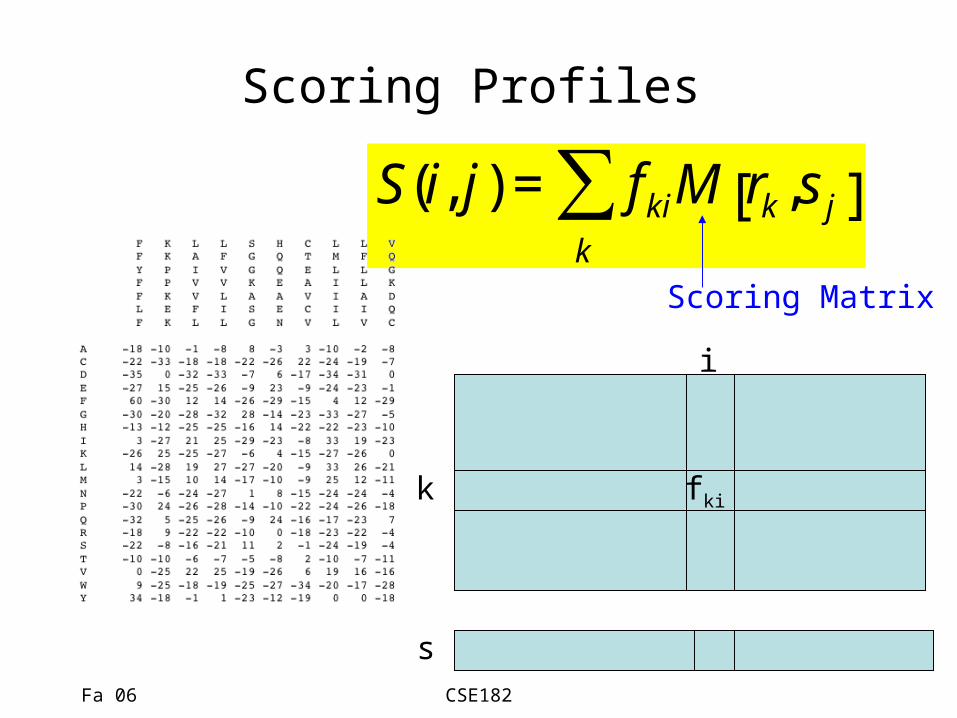
Scoring matrices
• Given a sequence s, does it belong to the family described by a profile?
• We align the sequence to the profile, and score it
• Let S(i,j) be the score of aligning position i of the profile to residue sj
• The score of an alignment is the sum of column scores.
s
sj
i
Fa 06 CSE182
Scoring Profiles
€
S(i, j) = fkik
∑ M rk,s j[ ]
k
i
s
fki
Scoring Matrix

Fa 06 CSE182
Domain analysis via profiles
• Given a database of profiles of known domains/families, we can query our sequence against each of them, and choose the high scoring ones to functionally characterize our sequences.
• What if the sequence matches some other sequences weakly (using BLAST), but does not match any Profile?
Fa 06 CSE182
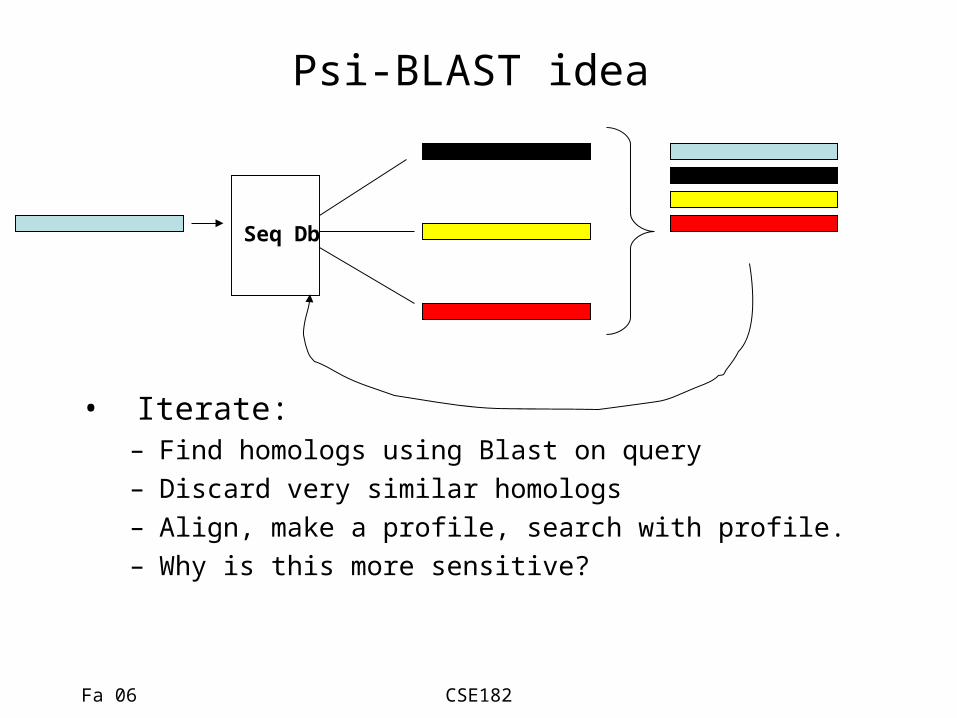
Psi-BLAST idea
• Iterate:– Find homologs using Blast on query– Discard very similar homologs– Align, make a profile, search with profile.– Why is this more sensitive?
Seq Db

Fa 06 CSE182
Psi-BLAST speed
• Two time consuming steps.1. Multiple alignment of homologs2. Searching with Profiles.
1. Does the keyword search idea work?
• Multiple alignment:– Use ungapped multiple
alignments only
• Pigeonhole principle again: – If profile of length m must score >= T– Then, a sub-profile of length l must
score >= lT|/m– Generate all l-mers that score at least
lT|/M– Search using an automaton

Fa 06 CSE182
Representation 3: HMMs
• Question:• your ‘friend’ likes to gamble. • He tosses a coin: HEADS, he gives you a dollar.
TAILS, you give him a dollar.• Usually, he uses a fair coin, but ‘once in a
while’, he uses a loaded coin. • Can you say what fraction of the times he
loads the coin?

Fa 06 CSE182
Representation 3: HMMs
• Building good profiles relies upon good alignments.– Difficult if there are gaps in the
alignment.– Psi-BLAST/BLOCKS etc. work
with gapless alignments.
• An HMM representation of Profiles helps put the alignment construction/membership query in a uniform framework.
V

Fa 06 CSE182
The generative model
• Think of each column in the alignment as generating a distribution.
• For each column, build a node that outputs a residue with the appropriate distribution
0.71
0.14
Pr[F]=0.71Pr[Y]=0.14

Fa 06 CSE182
A simple Profile HMM
• Connect nodes for each column into a chain. Thie chain generates random sequences.
• What is the probability of generating FKVVGQVILD?• In this representation
– Prob [New sequence S belongs to a family]= Prob[HMM generates sequence S]
• What is the difference with Profiles?
Fa 06 CSE182
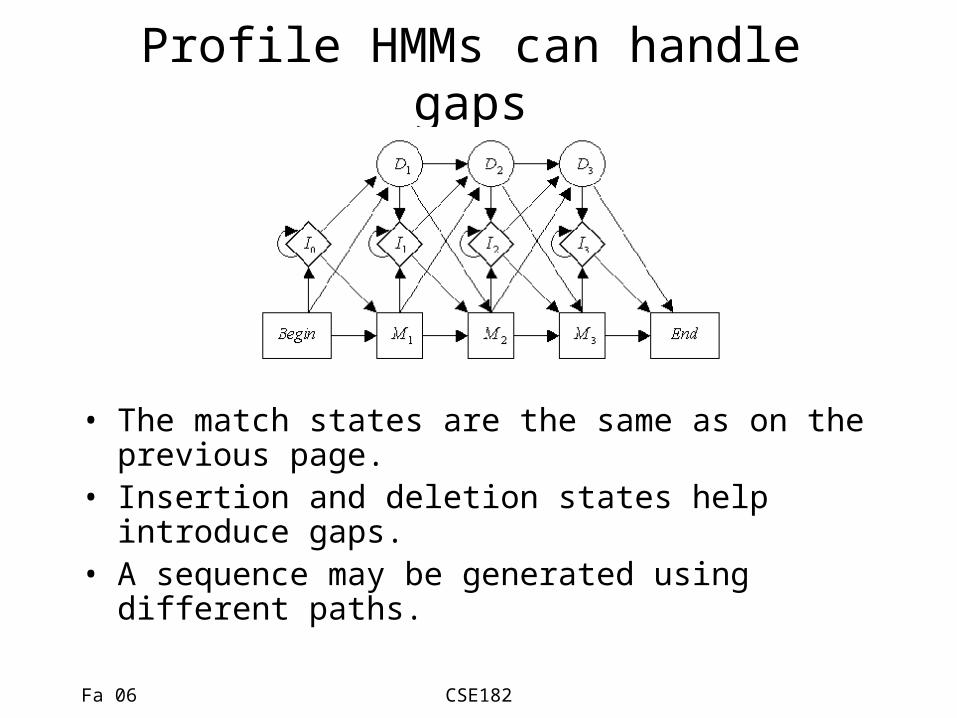
Profile HMMs can handle gaps
• The match states are the same as on the previous page.
• Insertion and deletion states help introduce gaps.
• A sequence may be generated using different paths.

Fa 06 CSE182
Example
• Probability [ALIL] is part of the family?• Note that multiple paths can generate this sequence.
– M1I1M2M3
– M1M2I2M3
• In order to compute the probabilities, we must assign probabilities of transition between states
A L - LA I V LA I - L

Fa 06 CSE182
Profile HMMs
• Directed Automaton M with nodes and edges. – Nodes emit symbols according to ‘emission
probabilities’– Transition from node to node is guided by ‘transition
probabilities’
• Joint probability of seeing a sequence S, and path P– Pr[S,P|M] = Pr[S|P,M] Pr[P|M]– Pr[ALIL AND M1I1M2M3]
= Pr[ALIL| M1I1M2M3,M] Pr[M1I1M2M3| M]
• Pr[ALIL | M] = ?

Fa 06 CSE182
Protein structure basics

Fa 06 CSE182
Side chains determine amino-acid type
QuickTime™ and aTIFF (Uncompressed) decompressorare needed to see this picture.QuickTime™ and aTIFF (Uncompressed) decompressorare needed to see this picture.
• The residues may have different properties.• Aspartic acid (D), and Glutamic Acid (E) are acidic
residues

Fa 06 CSE182
Bond angles form structural constraints

Fa 06 CSE182
Various constraints determine 3d structure
• Constraints– Structural constraints due to physiochemical
properties– Constraints due to bond angles– H-bond formation
• Surprisingly, a few conformations are seen over and over again.

Fa 06 CSE182
Alpha-helix
• 3.6 residues per turn• H-bonds between 1st
and 4th residue stabilize the structure.
• First discovered by Linus Pauling

Fa 06 CSE182
Beta-sheet
• Each strand by itself has 2 residues per turn, and is not stable.• Adjacent strands hydrogen-bond to form stable beta-sheets, parallel or anti-parallel.• Beta sheets have long range interactions that stabilize the structure, while alpha-helices have local
interactions.

Fa 06 CSE182
Domains
• The basic structures (helix, strand, loop) combine to form complex 3D structures.
• Certain combinations are popular. Many sequences, but only a few folds

Fa 06 CSE182
3D structure
• Predicting tertiary structure is an important problem in Bioinformatics.
• Premise: Clues to structure can be found in the sequence.• While de novo tertiary structure prediction is hard, there are
many intermediate, and tractable goals.• The PDB database is a compendium of structures
PDB

Fa 06 CSE182
Searching structure databases
• Threading, and other 3d Alignments can be used to align structures.
• Database filtering is possible through geometric hashing.

Fa 06 CSE182
Trivia Quiz
• What research won the Nobel prize in Chemistry in 2004?
• In 2002?

Fa 06 CSE182
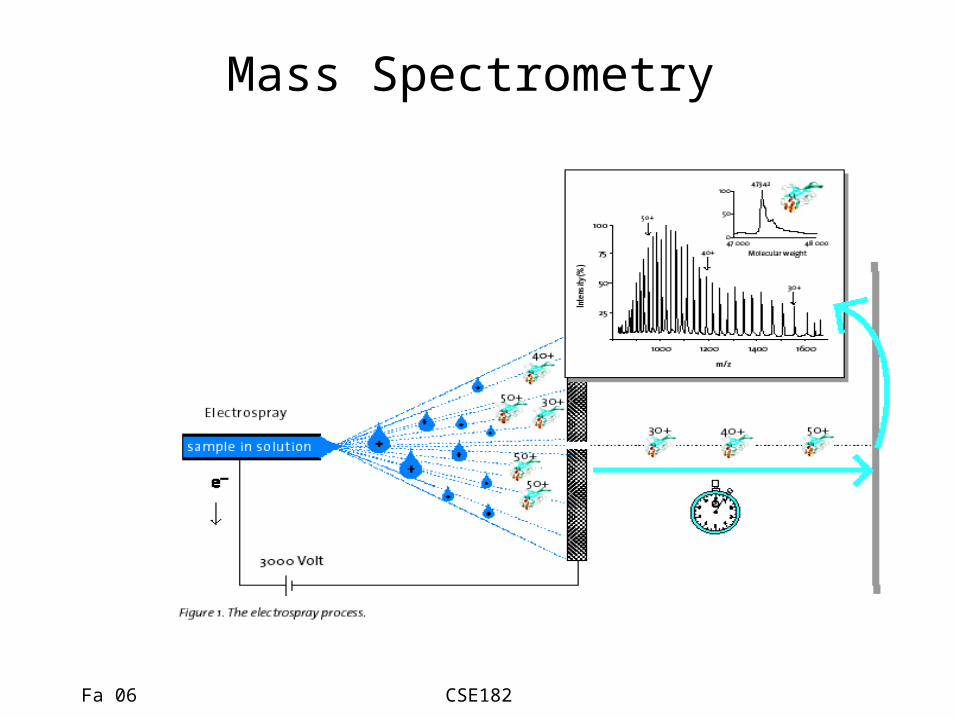
How are Proteins Sequenced? Mass Spec 101:

Fa 06 CSE182
Nobel Citation 2002

Fa 06 CSE182
Nobel Citation, 2002
Fa 06 CSE182
Mass Spectrometry
Fa 06 CSE182
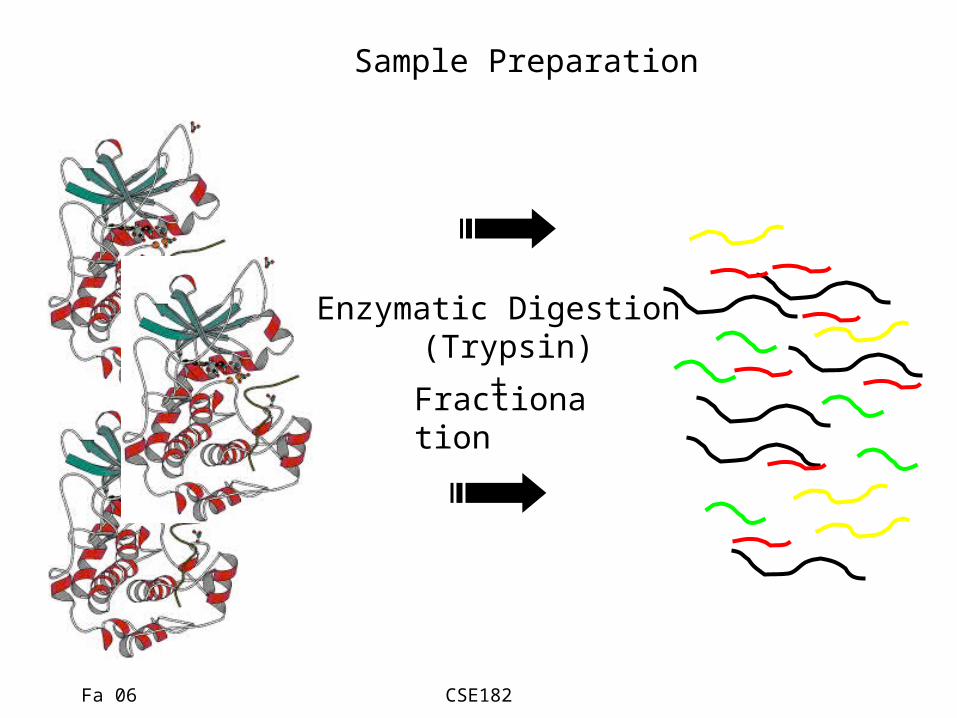
Sample Preparation
Enzymatic Digestion (Trypsin)
+Fractionation

Fa 06 CSE182
Single Stage MS
MassSpectrometry
LC-MS: 1 MS spectrum / second

Fa 06 CSE182
Tandem MS
Secondary Fragmentation
Ionized parent peptide


![[fa] Validity date from کشور [fa] Viet Nam 00269 [FA ... · 5 / 33 [fa] List in force شماره تایید نام شهر [fa] Regions [fa] Activities [fa] Remark [fa] Date of](https://img.dokumen.tips/doc/110x75/5e0e403e2c91e71788574ed3/fa-validity-date-from-fa-viet-nam-00269-fa-5-33-fa-list-in.jpg)

















![fa-ellipsis-v fa-eye []](https://img.dokumen.tips/doc/110x75/5697bf731a28abf838c7eca9/-fa-ellipsis-v-fa-eye-.jpg)







