Embed Size (px)
Citation preview

1
Expression cloning identifies Transgelin (SM22) as a novel repressor of 92 kDa type IV collagenase (MMP-9) expression
Rajesh R. Nair1, Julian Solway2 and Douglas D. Boyd1,3
1Department of Cancer Biology, MD Anderson Cancer Center, Houston, Texas, and 2Department of Medicine, University of Chicago, Illinois
Running Title: Transgelin represses MMP-9 expression
3 To whom correspondence should be addressed: Department of Cancer Biology, Box 173 M.D. Anderson Cancer Center 1515 Holcombe Blvd. Houston, Texas 77030 USA. Tel 713 563 4918 Fax: 713 563 5489 Email: [email protected]
http://www.jbc.org/cgi/doi/10.1074/jbc.M602703200The latest version is at JBC Papers in Press. Published on July 10, 2006 as Manuscript M602703200
Copyright 2006 by The American Society for Biochemistry and Molecular Biology, Inc.
by guest on July 8, 2019http://w
ww
.jbc.org/D
ownloaded from

2
SUMMARY The 92 kDa gelatinase (MMP-9) expres-sion is prerequisite for tissue remodeling in physiology and cancer. However, there are few known regulators of MMP-9 expression. Using an expression cloning strategy, we identified transgelin (SM22), a 22-25 kDa actin-binding protein localized to the cell membrane and cyto-plasm, as a novel regulator of MMP-9 expres-sion. Overexpression of a SM22 cDNA in HT1080 cells decreased MMP-9 mRNA/protein levels and diminished in vitro invasion the latter rescued with exogenous MMP-9. Conversely, siRNA-mediated knockdown of SM22 elevated MMP-9 synthesis and uterus from SM22-null mice showed strong MMP-9 immunoreactivity compared with wild type animals. The ability of SM22 to repress MMP-9 expression required an intact amino-terminus calponin homology do-
main. MMP-9 expression is driven by ERK signal-ing and SM22 targeted this pathway as evidenced by (a) the transience in MAPK activation and (b) blunted stimulation of the MMP-9 promoter by a constitutively active MEK expression vector. Pro-gressive deletion analysis located the SM22-responsive region of the MMP-9 promoter to the proximal 90 bp a region harboring an AP-1 motif subsequently implicated by site-directed mutagene-sis. Further, nuclear extract from the SM22 transfec-tants showed diminished c-Fos binding to this motif and SM22 expression reduced the activity of an AP-1-driven reporter by 75 %. Thus, SM22 adds to a short list of repressors of MMP-9 expression, achieving this by reducing AP-1-dependent trans-activation of the gene by way of compromised ERK activation. Diminished transgelin expression in sev-eral cancers, may thus partly account for the ele-vated MMP-9 expression evident in these tumors.
by guest on July 8, 2019http://w
ww
.jbc.org/D
ownloaded from

3
INTRODUCTION The 92 kDa type IV collagenase (MMP-9) contributes to tissue remodeling both in physiology and pathology. In pregnancy, expres-sion of this metalloproteinase by invading tro-phoblasts is prerequisite for implantation into the maternal decidua (1) while smooth muscle cell replication and migration into the neointima after denuding injury requires MMP-9 expres-sion (2;3). Similarly, in bone development, mi-gration of osteoclasts into cartilage matrix is dependent on their expression of this metallo-proteinase and defective endochondral bone formation is evident in mice null for this metal-loproteinase (4). MMP-9 also plays a key role in angiogenesis with knockout mice manifesting abnormal skeletal growth plate vascularization (5). In cancer, MMP-9 enhances tumor pro-gression in some, but not all, malignancies. In an earlier study, expression of an anti-MMP-9 ri-bozyme effectively blocked metastasis of rat sarcoma cells (6) while, more recently, in an elegant series of experiments primary tumor growth, angiogenesis and lung metastasis were diminished in animals null for this collagenase (7). Recent studies also indicate a function of MMP-9 in cell transformation (8) suggesting a role in an early malignant event. Surprisingly, MMP-9 can also suppress tumor progression depending on the tumor type and/or stage with siRNA-targeting this metalloproteinase yielding increased intravasation and metastasis in the chorioallantoic membrane assay (9). The original proposal that MMP-9 solely functions in the cleavage of extracellular matrix components including elastin, type III, IV and V collagen (10) has had to be broadened in light of recent findings. Indeed, it is becoming increasingly evident that MMP-9 is a multi-functional protein that also regulates angiogene-sis (11;12) by generating both pro- (VEGF and TGF-β) and anti-angiogenic (angiostatin and tumstatin) proteins as well as a cryptic pro-migratory epitope (for endothelial cells) from type IV collagen (11-14). Further, transforma-tion of immortalized human mammary epithelial cells by a constitutively active Stat-3-C requires MMP-9 and indeed the amounts of the transcrip-tion factor and metalloproteinase correlate in primary breast cancer specimens (8).
MMP-9 expression is largely controlled by transcription of the gene encoded on chromosome 20 (15) although mRNA stability (16-18) and transla-tional efficiency (19) also play a role in regulating the amounts of the protein product. Regulation of transcription is achieved via 2.2 kb of upstream regulatory sequence containing binding sites for AP-1, NF-kB, Sp1, PEA3/Ets (20-23) and is also de-pendent on the state of chromatin condensation (24;25) dictated by recruitment of histone deacety-lase 2 and the Brg-1 subunit of the SWI/SNF chro-matin-remodeling motor to the promoter via c-Fos/JunD complexes. Translation of the 2.5 kb mRNA yields a latent 92 kDa protein subsequently activated by the enzymatic removal of 73 amino ac-ids at the amino-terminus (26;27). The activity of MMP-9, in turn, is titrated by the endogenous tissue inhibitor of metalloproteinases -TIMPs (1-3) (28;29). To date, a mere handful of inducers of MMP-9 expression including KGF, HGF, TGF-β, TNFα (30-33) and the proto-oncogenes c-Src and H-Ras have been identified (20;23). The Snail tran-scription factor, stimulatory for an invasive pheno-type, also up-regulates MMP-9 transcription (34). Additionally, MMP-9 expression is a target of the Wnt pathway as evidenced by the ability of Frizzled-related protein 3, a secreted Wnt antagonist, to sup-press MMP-9 expression and PC-3 prostate cancer cell invasiveness (35). Conversely, the metastases suppressor Kiss-1 is one of the few negative regula-tors of MMP-9 expression identified to date achiev-ing this repression by down-regulating NF-kB trans-location (36).
However, so far, there have been no ge-nome-wide studies to identify novel regulators of MMP-9 expression in a systematic manner. Consid-ering the important role of MMP-9 in physiology and pathology we have employed expression cloning to identify hitherto unknown regulators of expres-sion of this collagenase. We report herein SM22 (transgelin) as a novel regulator of MMP-9 expres-sion repressing its expression by interfering with ERK activation and AP-1 signaling. EXPERIMENTAL PROCEDURES Cell Culture HT-1080 cells were maintained in McCoys 5A medium supplemented with 10 % fetal bovine
by guest on July 8, 2019http://w
ww
.jbc.org/D
ownloaded from

4
serum. WI-38 and VA-13 cells were maintained in DMEM medium supplemented with 10 % FBS. For stable transfections, cells were trans-fected at ~80% confluence using Lipofectamine 2000 as described by the manufacturer (Invitro-gen). Briefly, the cells were transfected with 8 µg of DNA and 24 µg of lipofectamine 2000 for 24 h, after which the transfection mixture was replaced with fresh medium. Cells were in-cubated for another 48 h and then selected with 1 mg/ml of G418. Clones were isolated, expanded, and screened for SM22 expression. Constructs The pQE-30 bacterial expression vector harboring the SM22 cDNA fragments (1-201; 1-186; 1-166 and 1-151) was used as template to PCR-amplify (with Pfu Turbo) and subclone these fragments into the Hind III/Kpn I-digested pEGFP-C1 expression vector. Similarly, the SM22 cDNA fragments encoding amino acids 51-201, 76-201 and 101-201 were generated by PCR and subcloned into the Hind III/Kpn I-digested pEGFP-N1 expression vector. Expressing Cloning Strategy These assays were performed as de-scribed previously (37). Briefly, an arrayed hu-man colon cDNA library (#LCO-1001, Origene Technologies, Rockville, MD) in which cDNAs were constructed in the pCMV6-XL4 expression vector was employed. DH10B E. Coli bacteria were transformed with DNA from the library and seeded on ampicillin LB-agar plates to ob-tain approximately 100 colonies per plate and cDNA pooled from each plate. In the primary screen cells in a 24 well format were co-transfected using Lipofectamine 2000 (Invitrogen) with a DNA mixture com-prised of the cDNA pool (700 ng), the MMP-9 promoter (2.2 kb) -luciferase reporter (100 ng), and 1 ng of an pRL-SV40 internal control. After 24 h, cells were lysed and assayed for luciferase activity using the Dual-Luciferase Reporter As-say System (Promega). In secondary screens, positive pools were further subdivided with 10 colonies per plate. cDNA sub-pools were prepared and as-sayed as described above. Finally, in a tertiary screen, single colonies from positive sub-pools (selected in the secondary screen) were identi-
fied as described above. Immunostaining: Uterus was fixed in 10 % formalin and em-bedded in paraffin. Sections were then de-paraffinized, treated with 3 % H2O2 in methanol and then blocked with 5 % normal horse serum and 1 % normal goat serum. Sections were incubated over-night with a 1:500 rabbit polyclonal anti-mouse MMP-9 antibody (Santa Cruz Biotechnology, CA) or an equivalent amount of an anti-Rabbit IgG and subsequently with an anti-rabbit peroxidase-conjugated F(ab)2 fragment. The diaminobenzidine (DAB) substrate was used to visualize immunoreac-tivity after counterstaining with hematoxylin. Mobility Shift Assays Nuclear extracts and EMSA were carried out as described by us elsewhere (36). EMSA was per-formed using 10 μg nuclear extract, 0.6 µg of poly dI/dC and (2 x 104 cpm) of a [γ32P] ATP T4 polynucleotide kinase-labeled oligonucleotide. Invasion Assays These were as described by this lab previ-ously (36). Cells (25,000) in 10 % albumin (serum-free) were seeded on Matrigel-coated porous filter (8 μm) using 10 % FBS as chemoattractant. After 24 h, cells on the upper aspect of the membrane were re-moved by scrubbing and the cells on the lower as-pect counted. Reporter Assays These were carried out as previously re-ported (36). Cells were co-transfected (using Li-pofectamine 2000) 24 h post-seeding with a pro-moter-driven luciferase reporter (0.1 μg) and, where indicated, an expression vector (pEGFP-C1) encod-ing the full length or truncated SM22 fragments. A SV40 promoter (unless specified otherwise)-driven Renilla luciferase (4 ng) was included as internal control. Cells were washed 24 h later, lysed and as-sayed for luciferase activity. Semi-quantitation of MMP-9 mRNA by RT-PCR Total RNA was isolated from cultured cells or mouse uterus using the TRIZOL reagent (Invitro-gen) according to the manufacturer's instructions. RNA (2 µg) and oligo dt primer (1µg) were heated at 70 °C for 5 min, and chilled. To this was added
by guest on July 8, 2019http://w
ww
.jbc.org/D
ownloaded from

5
1× RT buffer (Promega), 0.5 mM each of four deoxynucleosides, 1 unit/µl RNasein (Promega), 4 mM of sodium pyrophosphate (Sigma) and 30 units of avian myeloblastosis virus reverse transcriptase (Promega). The RNA was reverse transcribed at 42 °C, and 2 µl of the products were used as the template for multiplex PCR using MMP-9 and β-actin primers. The primers (5'-GAGGTTCGACGTGAAGGCGCAGATG-3' and 5'-CATAGGT-CACGTAGCCCAC-TTGGTC-3') used to amplify the human MMP-9
(product size, 120 bp), and -actin (product size, 621 bp), 5'-ACACTGTGCCCATCTACG-AGG-3' and 5'-AGGGGCCGGACTCGTCATACT-3'. For mouse tissues, primers used were (5’-ATTGGTGAACAGCCTGTATCCT-3’ and 5’-CTCCACGGTAGT-TTCCATCG-3’) to amplify a 251-bp product for the SM22 transcript. The primers for mouse GAPDH (5’-ACCCAGAAGACTGTGGATGG-3’ and 5’-CACATTGGGGGTAGGAACAC-3’) amplify a 171-bp product. siRNA targeting of SM22 Transfection efficiency using lipofec-tamine 2000 was optimized using siTOX (Dharmacon RNA technologies, cat #. D-001500-01-05). Cells were transfected with ei-ther siGenome SMARTpool reagent targeting SM22 (Dharmacon RNA Technologies, cat #. M-003714-01-0005) or siCONTROL non-targeting siRNA (Dharmacon RNA Technolo-gies, cat # D-001210-01-05) at a concentration of 100 nM. The cells were incubated for 48 h post transfection after which MMP-9 and SM22 expression was determined. Western Blotting For ERK, MEK, p38 and JNK-1 im-munoblotting, cells were serum starved over-night and then treated with 100 nM PMA for various times. The cells were lysed in a buffer (10 mM Tris-HCl, pH 7.4, 0.5% Nonidet P-40, 1% Triton X-100, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM Na3VO4, 1 mM PMSF, and 1 µg/ml aprotinin, leupeptin, and pepstatin) on ice for 30 min. Cell lysates were cleared by cen-trifugation, proteins (15-50 µg) resolved by polyacrylamide gel electrophoresis and then transferred to a nitrocellulose membrane. After blocking with 5% milk solution, blots were in-
cubated with primary antibodies to phosphorylated JNK1, and p38 (Santa Cruz sc6254 and sc-7973 re-spectively), total ERKs (Cell Signaling cat # 4696) and phosphorylated ERKs (Cell Signaling cat #9106) and total and phosphorylated MEKs 1 & 2 (Cell Signaling cat # 9122 and 9121 respectively) at 4 °C overnight. Blots were washed with TBS (10 mM Tris-HCl, pH 8.0, 150 mM NaCl) buffer, and then incubated with a horseradish peroxidase-conjugated secondary antibody. Proteins were visu-alized with ECL reagents. MMP-9 and SM22 protein were quantified essentially as described above but using polyclonal anti-MMP-9 (Biomol International, LP (Cat No: SA-106) and anti-SM22 antibodies the latter described elsewhere (38). Zymography Zymography was carried out as described previously (36), using aliquots of conditioned me-dium corrected for any differences in cell number. Quantitation Band intensity was quantified by densitome-try using Quantity One Software (v4.1) (Bio-Rad, Hercules, CA). Statistical Analysis Differences were tested for statistical sig-nificance using an Unpaired t test and GraphPad, Prism software (v3.03).
by guest on July 8, 2019http://w
ww
.jbc.org/D
ownloaded from

6
RESULTS There has been no genome-wide search
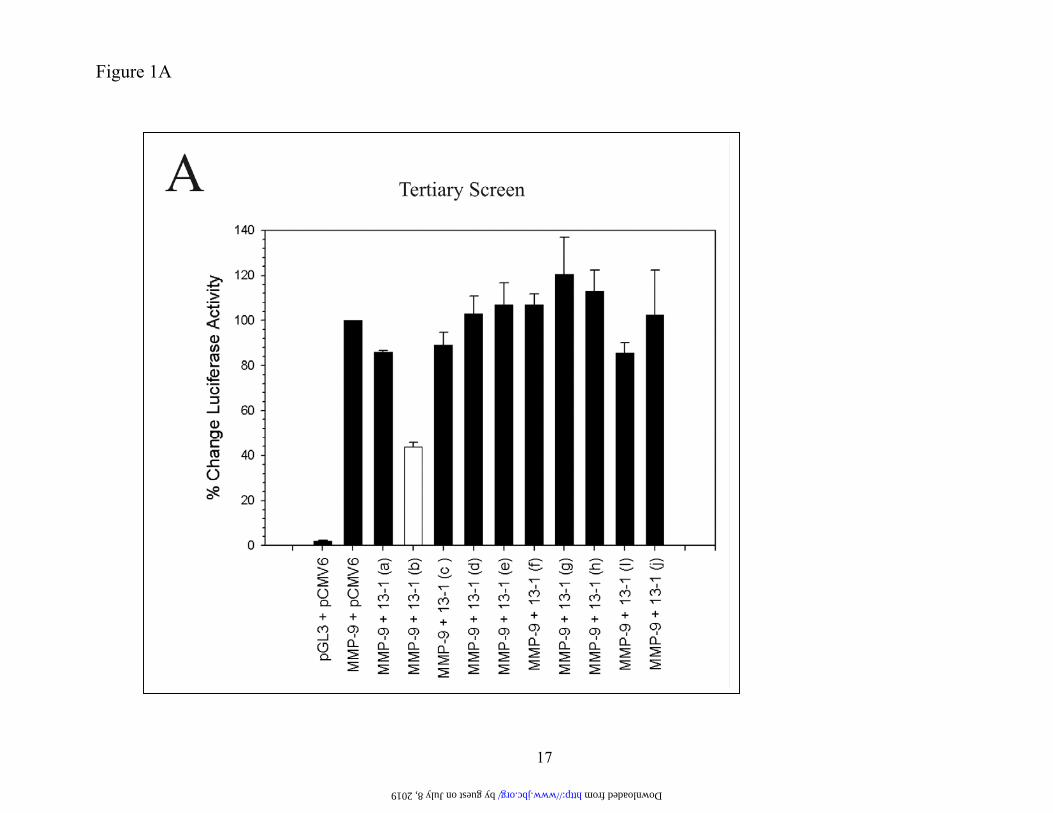
for regulators of MMP-9 expression and, to date, only a few regulators have been identified by empirical “guesswork.” Therefore to identify novel regulators of MMP-9 expression we em-ployed a cDNA library comprised of 500,000 cDNAs. In this approach, the library was subdi-vided with primary screenings undertaken with pools of 100 cDNAs. For screening, we used HT1080 cells since it constitutively expresses moderate MMP-9 amounts allowing a search for both activators and repressors of MMP-9 ex-pression. Identification of SM22 as a putative regulator of MMP-9 expression. HT1080 cells were co-transfected with cDNA pools and a luciferase reporter regulated by 2.2 kb of MMP-9 upstream sequence and subsequently assayed for luciferase activity. This promoter sequence includes all the regula-tory elements necessary for appropriate MMP-9 expression (39). Primary and secondary screens led to a sub-pool (13-1) of 10 cDNAs that di-minished MMP-9 promoter activity (data not shown). This sub-pool was divided into individ-ual clones and in a tertiary screen (Figure 1A) clone 13-1 (b) (open bar) repressed MMP-9 promoter activity. DNA sequencing revealed that clone 13-1 (b) corresponded to an open reading frame showing 98 % homology with the full length human transgelin (also known as SM22) cDNA coding sequence. MMP-9 pro-moter activity was decreased in a dose-dependent manner (Figure 1B) with a 5:1 ratio of expression vector to the MMP-9 reporter re-ducing luciferase activity by over 60 %.
While several putative regulators of MMP-9 expression were identified, we pursued SM22 as a candidate regulator for three reasons. First, there are few known repressors of MMP-9 expression. Second, data mining of expression profiling studies (Oncomine- http://www.oncomine.org/main/index.jsp) re-vealed that SM22 expression is attenuated in some metastatic cancers (e.g. lung and prostate) characterized by their elevated MMP-9 mRNA/protein expression. (40-42;42;43). Third, little is known as to SM22 function. The transge-lin gene (TAGLN) is located on chromosome
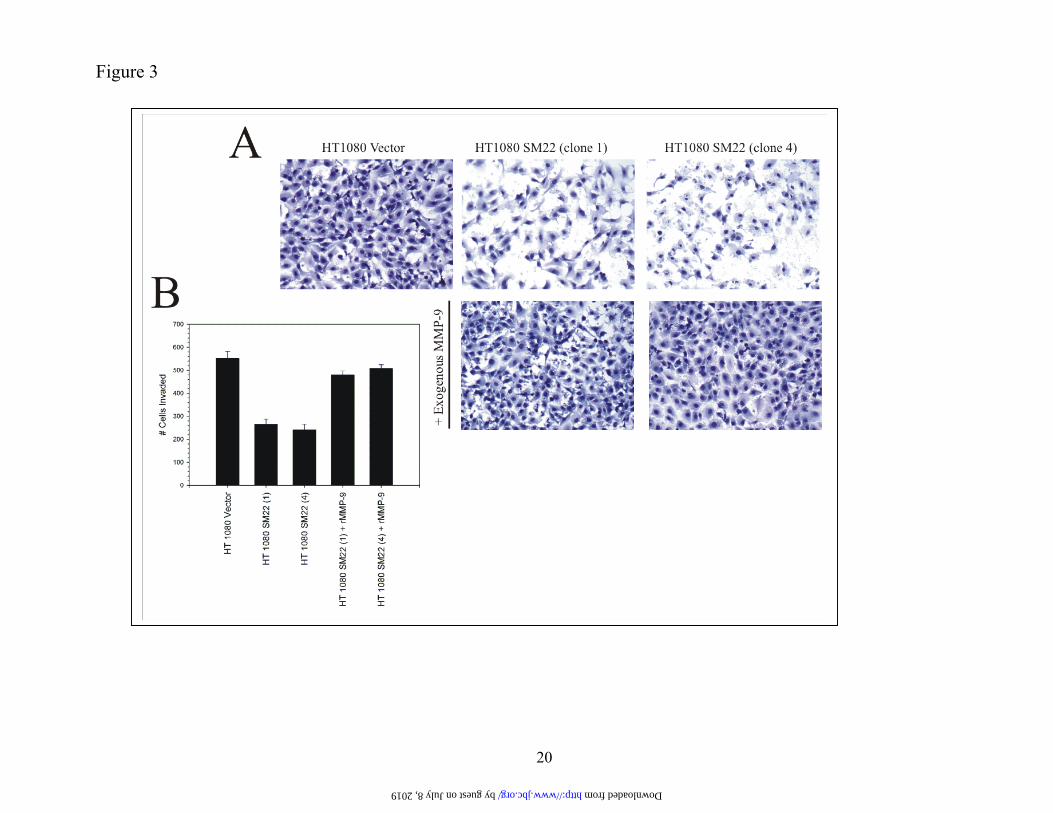
11q23.2 and generates a 1.3 kb mRNA. TAGLN expression is repressed by cell transformation (41). The encoded 25 and 22 kDa protein products local-ize (44) to the cytoplasm and cell membrane (45). SM22 binds actin via its carboxy-terminus residues (38) and has a putative role in actin gelation. Stable SM22 expression represses MMP-9 ex-pression. To validate the reporter assays, we deter-mined the effect of SM22 on endogenous MMP-9 expression. HT1080 cells were stably transfected with an expression construct bearing the SM22 cod-ing sequence, G418-resistant clones expanded and analyzed for SM22 protein by Western blotting (Figure 2A). Three HT1080 clones (1, 4, 6) were positive for SM22 protein whereas the parental cells and an empty vector control were negative for this protein. The various clones were then analyzed for MMP-9 activity by zymography (Figure 2B). A ge-latinolytic band identical in size (92 kDa) to MMP-9 was evident (Figure 2B) in conditioned medium from both parental HT1080 and cells harboring the empty vector whereas the intensity of the band was substantially diminished in the three SM22-expressing clones. In contrast, the 72 kDa gelati-nolytic band, representing the product of the MMP-2 gene (46) was unchanged by SM22 expression. To corroborate these data, MMP-9 mRNA levels were semi-quantitifed by multiplex RT-PCR. Again, while an amplified product of the predicted size (120 base pairs) was easily detected with the parental and empty vector-transfected HT1080 cells (Figure 2C), the intensity of the signal was clearly attenuated in the three separate SM22-expressing HT1080 clones (1, 4, 6). To further confirm its role as a suppressor of MMP-9 expression, HT1080 cells bearing the empty vector or clones expressing SM22 were ana-lyzed for in vitro invasion. Expectedly (47), HT1080 cells expressing the empty vector were highly inva-sive through an extracellular matrix-coated porous filter (Figure 3A) whereas two independent clones expressing the SM22 cDNA showed about a 50 % reduction in this assay. Diminished invasiveness was effectively rescued by the addition of exogenous MMP-9 protein suggesting that the attenuated be-havior was largely due to repressed MMP-9 expres-sion. The reduced invasiveness of the SM22-expressing clones was not a consequence of dimin-ished proliferation (data not shown). Thus, HT1080 cells stably over-expressing SM22 show attenuated
by guest on July 8, 2019http://w
ww
.jbc.org/D
ownloaded from

7
MMP-9 expression. Suppression of SM22 expression up-regulates MMP-9 activity. To accrue further evidence implicating SM22 as a regulator of MMP-9 expression we determined if its knockdown elevates synthesis of this metalloproteinase. Towards this end, we used normal lung fibroblasts (WI-38) which ex-press SM22 (41). The cells were transfected with a pool of 4 siRNAs targeting SM22 or, as a control, a non-targeting siRNA. SM22 protein levels were clearly reduced in the WI-38 fibro-blasts transfected with the siRNA pool targeting SM22 (Figure 4A). More importantly, zymogra-phy and Western blotting indicated up to a 3 fold increase in MMP-9 protein/activity in lysates and conditioned medium from cells treated with the siRNA targeting SM22 (Figure 4B, C).
Since cell transformation by SV40 abol-ishes SM22 expression (41) MMP-9 induction should also be apparent in the transformed cells. Consequently, we compared MMP-9 expression in the normal human lung fibroblasts (WI-38) and their SV40-transformed counterparts (VA-13). As expected, SM22 protein levels were ex-tinguished in the transformed cells (VA-13) (Figure 5A) and this observation was paralleled by a 2 fold increase in 92 kDa gelatinase activity (Figure 5B). The elevated MMP-9 activity in the VA-13 cells, extinguished for SM22 expression, reflected a parallel increase in MMP-9 mRNA (Figure 5C) as revealed by RT-PCR.
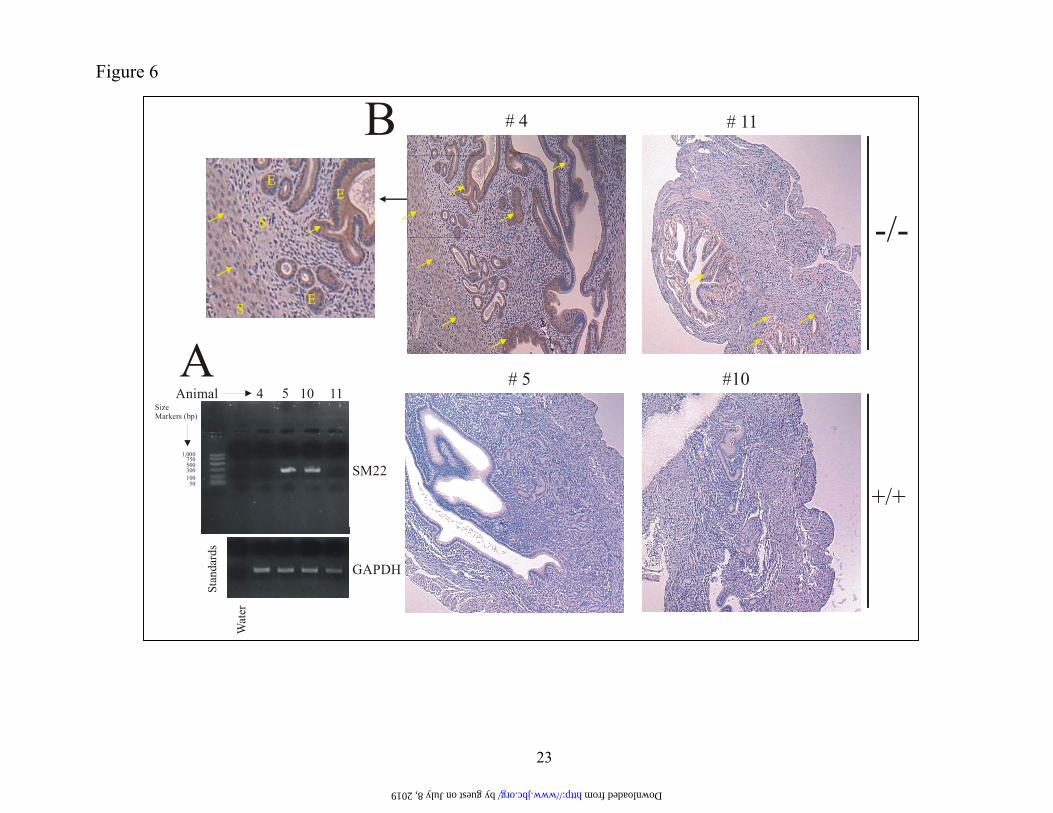
We then determined if MMP-9 expres-sion was up-regulated in SM22 null mice (48). Uterine tissue, which constitutively expresses transgelin (45), was obtained from mice wild type or null for SM22 (Figure 6A) and stained for MMP-9 protein. Mice were synchronized in their estrus cycle by housing in the same cage. Tissue from 2 independent SM22 knockout mice (-/-) showed clear MMP-9 immunoreactivity both in the stromal and epithelial compartments (arrows) whereas little immunoreactivity was evident with tissues derived from the wild type (+/+) animals (Figure 6B). Thus, taken together, these studies indicate SM22 as a bona fide regu-lator of MMP-9 expression. The actin-binding region of SM22 is dispen-sable with respect to MMP-9 repression.
How does SM22 regulate MMP-9 expres-sion? SM22 contains a single amino-terminal-located calponin-homology domain (49) and cal-ponin-like-repeats at the carboxy-terminus (http://smart.embl-heidelberg.de/). Since its binding to actin is mediated through the SM22 carboxy-terminus (38), we asked whether this region is re-quired for MMP-9 repression and towards this end, we employed expression constructs encoding the full length (1-201 amino acids) SM22 or truncations thereof (Figure 7A Left Panel). HT1080 cells were co-transfected with these various SM22 expression constructs and a luciferase reporter driven by the MMP-9 promoter. Luciferase assays (Figure 7B) indicated a strong repression of MMP-9 promoter activity by the full length SM22 (wt) whereas an unrelated promoter (thymidine kinase) was unaf-fected (data not shown). The carboxy-terminus-deleted construct (SM22 (1-151)), unable to bind actin, as determined by actin-co-sedimentation (38), was equiactive with the full length SM22 in repress-ing MMP-9 promoter activity. In contrast, amino-terminal deletions gener-ating constructs 51-201, 76-201 and 101-201 (Figure 7A Right Panel), thus lacking an intact calponin ho-mology domain (amino acids 25-132), yielded SM22 fragments far less effective (Figure 7B) in repressing MMP-9 promoter activity when compared with the full length (1-201) protein (SM22 wt). These data would suggest that the region harboring the calponin homology domain, but not the actin-binding region, is required for optimal MMP-9 repression by SM22. Transient ERK activation in SM22 transfectants. The cytoplasmic localization of SM22 (45) coupled with the absence of a nuclear localization signal suggested that SM22 represses MMP-9 ex-pression indirectly. Further, we noted that the ability of SM22 to repress MMP-9 promoter activity de-pended on the presence of an intact amino-terminal type 3 calponin homology domain (see above). Since the amino-terminal type 3 calponin homology do-main physically interacts with ERKs 1 and 2 (49), SM22 may interfere with signal transduction path-ways impinging on MMP-9 expression (see Discus-sion also). Indeed, the regulation of MMP-9 expres-sion by the ERK signaling module has been previ-ously reported by several laboratories (50-52). To determine if SM22 targets the ERK pathway, acti-vated (phosphorylated) forms of ERKs, were meas-ured in the HT1080 SM22 transfectants and the vec-
by guest on July 8, 2019http://w
ww
.jbc.org/D
ownloaded from

8
tor control by Western blotting (Figure 8A). In-terestingly, while phorbol ester activated ERKs 1 & 2 in both the vector and SM22-expressing HT1080 cells, induction in the former was sus-tained (up to 6 h) while returning to baseline by 2 h in the latter cells. These findings are note-worthy since sustained, but not transient, ERK activation drives MMP-9 expression (51). The absence of activated ERKs at time 0 probably reflects prior serum-starvation. Interestingly, the upstream activator of the ERKs, namely, MEKs 1 and 2 showed little difference in their activa-tion in the SM22 transfectants suggesting that the transient ERK activation was not reflective of regulation at this higher level signaling kinase. We also determined if the JNK or p38 signaling modules were targeted by SM22. However, little change in the levels of these dual-activity kinases was evident between the SM22 and vector-expressing HT1080 cells (Fig-ure 8A). To accrue further evidence that the ERK pathway was blunted by SM22, HT1080 cells bearing either the empty vector or stably ex-pressing transgelin, were transiently co-transfected with the MMP-9 promoter-driven luciferase reporter and an expression vector en-coding a constitutively activated MEK (MEK ΔN3S218E-S222D) 60-400 X more active in phosphorylating ERKs than the wild type MEK used as control (53;54). The mutation-activated MEK stimulated a robust increase (~ 300 %) in MMP-9 promoter activity in the HT1080 cells bearing the empty vector (Figure 8B). In con-trast, the % increase in MMP-9 promoter activ-ity achieved with MEK ΔN3S218E-S222D was significantly (<0.0001) less with the HT1080 cells stably expressing SM22. These data further support the notion that SM22 interferes with ERK signaling impinging on MMP-9 expres-sion. SM22 reduces AP-1 trans activation of the MMP-9 promoter. To determine the transcriptional ele-ments mediating MMP-9 repression by SM22, we transiently transfected HT1080 cells with 5’ deleted MMP-9 promoter fragments fused to a luciferase reporter. SM22 reduced transcription by 50 % (Figure 9) from the longest (2.2 kb) MMP-9 promoter fragment. However, this re-
pression was unimpaired by progressive 5’ deletions with the shortest (90 bp) and longest (2.2 kb) frag-ments showing equal sensitivity. This minimal pro-moter region contains an AP-1 binding motif, a GC box, as well as the KRE-M9 element recognizing the differentiation-repressing factor 1 (55).
Considering the transient ERK signal evi-dent in the SM22 transfectants together with the well-established role of the AP-1 motif in regulating expression of this metalloproteinase downstream of this signaling module (50), we performed two ex-periments to determine the role of this motif in the SM22-dependent MMP-9 repression. First, the effect of mutating the proximal (-79) and distal (-533) (20;56) AP-1 motifs on MMP-9 repression by SM22 was determined. Interestingly, only the simultaneous mutation of both proximal and distal AP-1 motifs in context of the 670 bp MMP-9 promoter impaired the repressive effect of SM22 (data not shown) arguing that these motifs are redundant with respect to the suppressive effect of transgelin. Second, we deter-mined if transcription factors binding to the proxi-mal AP-1 site was altered in the SM22 transfectants. Nuclear extract from the parental and vector-transfected HT1080 cells gave a shifted band (paren-thesis) with an oligonucleotide bearing the proximal AP-1 motif (Figure 10A-Lanes 3, 4). This shifted band represented specific binding since it was abol-ished with an excess of non-radioactive probe (Fig-ure 10A- Lane 2). More importantly, the intensity of this shifted band was reduced with the three inde-pendent SM22 clones (Lanes 6, 7, 8). In contrast, binding to the MMP-9 promoter-derived KRE-M9 element, located immediately downstream of this AP-1 motif, and which recognizes the differentiation repressing factor-1 (DRF)) (55) was unaffected (Figure 10B) with nuclear extract from the SM22 transfectants.
Since transient ERK activation (as evident with the SM22 transfectants) is known to decrease c-Fos protein amount via de-stabilization (57) we per-formed super-shifting experiments (Figure 10C) us-ing an antibody directed at this AP-1-binding pro-tein. A super shift was evident with nuclear extract derived from the HT1080 cells bearing the vector only (Figure 10C-Lane 4, arrow) at the expense of the retarded band (parenthesis) clearly indicating the presence of this DNA-binding protein in the com-plex. In contrast, the intensity of this supershifted band was greatly diminished with nuclear extract generated from two independent HT1080 clones ex-
by guest on July 8, 2019http://w
ww
.jbc.org/D
ownloaded from

9
pressing the exogenous SM22 cDNA (Figure 10C-lanes 7 and 10-arrow). To accrue further evidence that SM22 was targeting AP-1-dependent regulation of MMP-9 expression, HT1080 cells were co-transfected with a luciferase reporter regulated by 7X tandem AP-1 repeats (Stratagene # 219074) and an expression vector encoding nothing (pEGFP-C1) or the SM22 coding sequence (pEGFP-SM22). Ex-pression of the SM22 cDNA yielded ~ 75 % reduction in promoter activity (Figure 10D) when compared with that achieved with the empty expression vector a difference that was statistically significant (P<0.0001). Taken to-gether, these data suggest that SM22 represses MMP-9 expression at least in part via reduced trans-activation of the promoter through its proximal AP-1 motif. DISCUSSION
Using an expression cloning strategy, we have identified transgelin (SM22) as a novel regulator of MMP-9 expression. SM22 adds to a short list of proteins, which includes the metas-tases suppressor Kiss-1 (36) and the yet unchar-acterized differentiating-repressing factor-1 (55) that attenuate MMP-9 expression. SM22 dimin-ishes MMP-9 expression by blunting ERK acti-vation leading to suppressed trans-activation of the promoter through the proximal AP-1 motifs.
Currently, little is known regarding SM22 function. Certainly, transgelin binds actin (38) and this association may contribute to actin gelation although such a role has been ques-tioned since it does not occur at physiological pH (58). Nevertheless, a truncated SM22 frag-ment, incapable of binding actin, was equi-active with the full length protein in repressing MMP-9 promoter activity making it unlikely that diminished MMP-9 expression reflects its capacity to associate with this cytoskeletal pro-tein. More plausible is that transgelin regulates MMP-9 expression via modulation of ERK acti-vation. Indeed, this signaling module impinges on MMP-9 expression in MDCK epithelial cells (34), keratinocytes (51) and oral squamous cell carcinoma (50). While sustained (up to 6 h) ERK activation was evident in HT1080 vector controls, phosphorylated ERK levels in the SM22 transfectants were transient, returning to base-line within 2 h. Sustained, but not transient,
ERK activation is critical for elevated MMP-9 ex-pression as demonstrated previously with EGF and hepatocyte growth factor-stimulated keratinocytes (51). Thus, SM22-dependent MMP-9 repression likely reflects blunted ERK activation.
How then does SM22 interfere with ERK activation? SM22 contains a type 3 calponin homol-ogy domain (CH3) located within its amino-terminal 132 amino acids (http://smart.embl-heidelberg.de/) and the calponin protein, bearing this domain, physi-cally interacts with ERKs 1 and 2 (59). Indeed, cDNAs encoding truncated SM22 proteins lacking an intact CH3 domain were poor repressors of MMP-9 expression when expressed in HT1080 cells raising the possibility that binding of transgelin to these MAPKs somehow blunts their activation. However, our co-immunoprecipitation attempts to show interaction between these proteins failed. An alternate possibility is that transgelin increases the activity and/or amount of MKP-1, or another dual activity phosphatase, yielding the transient ERK ac-tivation evident in the SM22 transfectants. Irrespec-tive of the ERK-regulatory mechanism, the transient activation of this MAPK subset was clearly evident in the SM22 transfectants and probably contributes to MMP-9 repression (51). On the other hand, unlike oral keratinocytes (52), it does not appear that p38 (60) signaling contributes to the SM22-dependent MMP-9 repression insofar as the activated form was unaffected by transgelin expression. Likewise, JNK activation prerequisite for MMP-9 induction by the Ras oncogene in ovarian cancer cells (50), was also unchanged by transgelin. Presumably, these two pathways are non-redundant with the ERK signaling module and therefore unable to compensate for the deficient signaling through the latter. Our studies indicated that repressed MMP-9 expression by SM22 was at least partly due to di-minished transcription from the MMP-9 promoter. However, we cannot presently exclude the possibil-ity that reduced expression also reflects, in part, a post-transcriptional component. While our reporter assay indicated a 50-60 % reduction in promoter activity, enzyme (zymography) and mRNA determi-nations indicated a more pronounced effect invoking the possibility of post-transcriptional control. Post-transcriptional modulation of MMP-9 expression has been reported previously with TGF-β and LPS both mediating MMP-9 induction via stabilization of the transcript (18;61). In keratinocytes, ERK activation by a mutation-activated Ras, yields MMP-9 mRNA
by guest on July 8, 2019http://w
ww
.jbc.org/D
ownloaded from

10
stabilization (16) a finding pertinent to our study which implicated transiency in ERK activation in the SM22-dependent MMP-9 repression. Thus, it is possible that SM22 represses MMP-9 expression not only by targeting the transcrip-tion machinery but also by regulating mRNA stability or possibly even translational efficiency as evident in myc/ras-transformed murine uro-genital sinus cells (19). The MMP-9 promoter contains multiple cis elements regulatory for its expression includ-ing Ets, NF-κB and Sp1 binding sites residing between –600 and –533 relative to the transcrip-tion start site (20). However, our 5’ deletion analysis of the promoter revealed that only the proximal 90 base pairs of the MMP-9 promoter was required for the SM22-dependent MMP-9 repression thus arguing against the contribution of these upstream cis elements (20). These find-ings distinguish the MMP-9 repression by SM22 from that achieved by Kiss-1. This metastases suppressor blocks p65/p50 nuclear translocation and hence trans -activation of the MMP-9 pro-moter through its NF-κB site at –600 (36). The 90 base pairs of 5’ flanking MMP-9 sequence contains a well-characterized AP-1 motif previ-ously shown to mediate MMP-9 induction by diverse stimuli including integrin linked kinase (ILK), oncogenic Ras, Src, myc, and phorbol ester (20;21;23;25;62). In fact, our observations of a marked reduction in transcription factor (in-cluding c-Fos) binding to this element in the SM22 transfectants reflecting either ERK-dependent decreased c-Fos protein (57), nuclear-cytoplasmic shuttling (63) or altered expression of a dimerizing partner (64), strongly argue that this site mediates, to a large extent, MMP-9 re-pression. Nevertheless, the responsive promoter region (-90) also harbors a GC-box located
proximal of this AP-1 site that could feasibly also contribute to the SM22-dependent regulation of MMP-9 expression. Indeed this element mediates, at least in part, MMP-9 induction by the Snail tran-scription factor (34) and v-src (23). Additionally, the proximal MMP-9 sequence also harbors a KRE-M9 element but since no change in transcription factor binding to this motif was evident with nuclear ex-tract from the SM22 transfectants it is unlikely that transgelin represses MMP-9 expression via this binding site unless transcriptional activity of the cor-responding DNA-binding protein (DRF-1) is tar-geted.
SM22 was originally identified as a trans-formation-sensitive (diminished expression) protein (41;44) and mRNA levels are down-regulated in re-sected breast and colon cancers when compared with adjacent non-malignant mucosa (65). Interestingly, these finding parallel a robust expression of MMP-9 in these two malignancies (66-69). Similarly, ex-pression profiling studies revealed 85 % lower levels of SM22 in metastatic prostate cancer when com-pared with the primary tumor (70) and, like breast and colon cancer, MMP-9 expression is elevated in advanced disease (42). These findings raise the pos-sibility that diminished SM22 expression account, at least in part, for the higher levels of this metallopro-teinase in these cancers. In conclusion, using an unbiased expression cloning strategy, we have identified a novel regula-tor (SM22/transgelin) of MMP-9 expression. SM22 represses MMP-9 promoter activity in a manner in-dependent of its actin-binding arguing for a hitherto unknown function for this protein previously recog-nized only for its actin-binding capacity. The loss of transgelin expression, evident in cancers of diverse origin (40;70), may contribute to the well-oft obser-vation of elevated levels of MMP-9 in these malig-nancies.
REFERENCES
1. Folgueras, A. R., Pendas, A. M., Sanchez, L. M., and Lopez-Otin, C. (2004) International Jour-nal of Developmental Biology 48, 411-424
2. Cho, A. and Reidy, M. A. (2002) Circulation Res. 91, 845-851
3. Galis, Z. S., Johnson, C., Godin, D., Magid, R., Shipley, J. M., Senior, R. M., and Ivan, E. (2002) Circulation Res. 91, 852-859
by guest on July 8, 2019http://w
ww
.jbc.org/D
ownloaded from

11
4. Vu, T. H. and Werb, Z. (2000) Genes and Development 14, 2123-2133
5. Vu, T. H., Shipley, J. M., Bergers, G., Berger, J. E., Helms, J. A., Hanahan, D., Shapiro, S. D., Senior, R. M., and Werb, Z. (1998) Cell 93, 411-422
6. Hua, J. and Muschel, R. J. (1996) Cancer Res. 56, 5279-5284
7. Hiratsuka, S., Nakamura, K., Iwai, S., Murakami, M., Itoh, T., Kijima, H., Shipley, J. M., Senior, R. M., and Shibuya, M. (2002) Cancer Cell 2, 289-300
8. Dechow, T. N., Pedranzinin, L., Leithch, A., Leslie, K., Gerald, W. L., Linkov, I., and Bromberg, J. F. (2004) Proc.Natl.Acad.Sci USA 101, 10602-10607
9. Deryugina, E. I., Zijlstra, A., Partridge, J. J., Kupriyanova, T. A., Madsen, M. A., Papagianna-kopoulos, T., and Quigley, J. P. (2005) Cancer Res. 65, 10959-10969
10. Murphy, G., Cockett, M. I., Ward, R. V., and Docherty, A. J. P. (1991) Biochem.J. 277, 277-279
11. Handsley, M. M. and Edwards, D. R. (2005) Int.J.Cancer 115, 849-860
12. Jodele, S., Chantrain, C. F., Blavier, L., Lutzko, C., Crooks, G. M., Shimada, H., Coussens, L., and DeClerck, Y. A. (2005) Cancer Res. 65, 3200-3208
13. Yu, Q. and Stamenkovic, I. (2000) Genes and Development 14, 163-176
14. Bergers, G., Brekken, R., McMahon, G., Vu, T. H., Itoh, T., Tamaki, K., Tanzawa, K., Thorpe, P., Itohara, S., Werb, Z., and Hanahan, D. (2000) Nature Cell Biology 2, 737-744
15. Linn, R., DuPont, B. R., Knight, C. B., Plaetke, R., and Leach, R. J. (1996) Cytogenetics and Cell Genetics 72, 159-161
16. Iyer, V., Pumiglia, K., and DiPersio, C. M. (2005) J.Cell Science 118, 1185-1195
17. Eberhardt, W., Akool, E.-S., Rebhan, J., Frank, S., Beck, K.-F., Franzen, R., Hamada, F. M. A., and Pfeilschifter, J. (2002) J.Biol.Chem. 277, 33518-33528
18. Sehgal, I. and Thompson, T. C. (1999) Molecular Biology of the Cell 10, 407-416
19. Jiang, Y. and Muschel, R. J. (2002) Cancer Res. 62, 1910-1914
20. Gum, R., Lengyel, E., Juarez, J., Chen, J.-H., Sato, H., Seiki, M., and Boyd, D. (1996) J.Biol.Chem. 271, 10672-10682
21. Himelstein, B. P., Lee, E. J., Sato, H., Seiki, M., and Muschel, R. J. (1997) Oncogene 14, 1995-1998
22. Mohan, R., Sivak, J., Ashton, P., Russo, L. A., Pham, B. Q., Kasahara, N., Raizman, M. B., and Fini, M. E. (2000) J.Biol.Chem. 275, 10405-10412
23. Sato, H., Kita, M., and Seiki, M. (1993) J.Biol.Chem. 268, 23460-23468
24. Yan, C., Wang, H., Toh, Y., and Boyd, D. D. (2003) J.Biol.Chem. 278, 2309-2316
by guest on July 8, 2019http://w
ww
.jbc.org/D
ownloaded from

12
25. Ma, Z., Shah, R. C., Chang, M. J., and Benveniste, E. N. (2004) Mol.Cell.Biol. 24, 5496-5509
26. Wilhelm, S. M., Collier, I. E., Marmer, B. L., Eisen, A. Z., Grant, G., and Goldberg, G. (1989) J.Biol.Chem. 264, 17213-17221
27. Shapiro, S. D., Fliszar, C. J., Broekelmann, T. J., Mecham, R. P., Senior, R. M., and Welgus, H. G. (1995) J.Biol.Chem. 270, 6351-6356
28. Olson, M. W., Gervasi, D. C., Mobashery, S., and Fridman, R. (1997) J.Biol.Chem. 272, 29975-29983
29. Testa, J. E. (1992) Cancer Res. 52, 5597-5603
30. Hurwitz, A., Dushnik, M., Solomon, H., Ben-Chetrit, A., Finci-Yeheskel, Z., Milwidsky, A., Mayer, M., Adashi, E. Y., and Yagel, S. (1993) Endocrinology 132, 2709-2714
31. Putnins, E. E., Firth, J. D., and Uitto, V.-J. (1995) J.Invest.Dermatology. 104, 989-994
32. Hanzawa, M., Shindoh, M., Higashino, F., Yasuda, M., Inoue, N., Hida, K., One, M., Kohga, T., Nakamura, M., Notani, K., Fukuda, H., Totsuka, Y., Yoshida, K., and Fujinaga, K. (2000) Car-cinogenesis 21, 1079-1085
33. Stuelten, C. H., DaCosta Byfield, S., Arany, P. R., Karpova, T. S., Stetler-Stevenson, W. G., and Roberts, A. B. (2005) J.Cell Science 118, 2143-2153
34. Jorda, M., Olmeda, D., Vinyals, A., Valero, E., Cubillo, E., Llorens, A., Cano, A., and Fabra, A. (2005) J.Cell Science 118, 3371-3385
35. Zi, X., Guo, Y., Simoneau, A. R., Hope, C., Xie, J., Holcombe, R. F., and Hoang, B. H. (2005) Cancer Res. 65, 9762-9770
36. Yan, C., Wang, H., and Boyd, D. D. (2001) J.Biol.Chem. 276, 1164-1172
37. Wang, H., Yang, L., Jamaluddin, M. d. S., and Boyd, D. D. (2004) J.Biol.Chem. 279, 22674-22683
38. Fu, Y., Liu, H. W., Forsythe, S. M., Kogut, P., McConville, J. F., Halayko, A. J., Camoretti-Mercado, B., and Solway, J. (2000) Journal of Applied Physiology 89, 1985-1990
39. Yan, C., Wang, H., Aggarwal, B. B., and Boyd, D. D. (2004) FASEB J. 18, 540-541
40. Garber, M. E., Troyanskaya, O. G., Schluens, K., Petersen, S., Thaesler, Z., Pacyna-Gengelbach, M., van de Rijn, M., Rosen, G. D., Perou, C. M., Whyte, R. I., Altman, R. B., Brown, P. O., Bot-stein, D., and Petersen, I. (2001) Proc.Natl.Acad.Sci.USA 98, 13784-13789
41. Schenker, T. and Trueb, B. (1998) Experimental Cell Res. 239, 161-168
42. Wood, M., Fudge, K., Mohler, J. L., Frost, A. R., Garcia, F., Wang, M., and Stearns, M. E. (1997) Clin.Exp.Metastasis 15, 246-258
43. Moses, M. A., Wiederschain, D., Loughlin, K. R., Zurakowski, D., Lamb, C. C., and Freeman, M. R. (1998) Cancer Res. 58, 1395-1399
by guest on July 8, 2019http://w
ww
.jbc.org/D
ownloaded from

13
44. Shapland, C., Hsuan, J. J., Totty, N. F., and Lawson, D. (1993) J.Cell Biol. 121, 1065-1073
45. Camoretti-Mercado, B., Forsythe, S. M., LeBeau, M. M., Espinosa, R., Vieira, J. E., Halayko, A. J., Willadsen, S., Kurtz, B., Ober, C., Evans, G. A., Thweatt, R., Shapiro, S., Niu, Q., Qin, Y., Padrid, P. A., and Solway, J. (1998) Genomics 49, 452-457
46. Collier, I. E., Bruns, G. A. P., Goldberg, G. I., and Gerhard, D. S. (1991) Genomics 9, 429-434
47. Sato, T., Koike, L., Miyata, Y., Hirata, M., Mimaki, Y., Sashida, Y., Yano, M., and Ito, A. (2002) cancer res 62, 1025-1029
48. Zhang, J. C., Kim, S., Helmke, B. P., Yu, W. W., Du, K. L., Lu, M. M., Strobeck, M. W., Yu, Q., and Parmacek, M. S. (2001) Mol.Cell.Biol. 21, 1336-1344
49. Gimona, M., Djinovic-Carugo, K., Kranewitter, W. J., and Winder, S. J. (2002) FEBS Letters 513, 98-106
50. Gum, R., Wang, H., Lengyel, E., Juarez, J., and Boyd, D. (1997) Oncogene 14, 1481-1493
51. McCawley, L. J., Li, S., Wattenberg, E. V., and Hudson, L. G. (1999) J.Biol.Chem. 274, 4347-4353
52. Mukhopadhyay, S., Munishi, H. G., Kambhampati, S., Sassano, A., Platanias, L. C., and Stack, M. S. (2004) J.Biol.Chem. 279, 33139-33146
53. Lengyel, E., Wang, H., Gum, R., Simon, C., Wang, Y., and Boyd, D. (1997) Oncogene 14, 2563-2573
54. Mansour, S. J., Matten, W. T., Hermann, A. S., Candia, J. M., Rong, S., Fukusawa, K., Vande Woude, G. F., and Ahn, N. G. (1994) Science 265, 966-970
55. Kobayashi, T., Kishiimoto, J., Hattori, S., Wachi, H., Shinkai, H., and Burgeson, R. E. (2004) J.Invest.Dermatology. 122, 278-285
56. Sato, H. and Seiki, M. (1993) Oncogene 8, 395-405
57. Murphy, L. O., Smith, S., Chen, R.-H., Fingar, D. C., and Blenis, J. (2002) Nature Cell Biology 4, 556-564
58. Goodman, A., Goode, B. L., Matsudaira, P., and Fink, G. R. (2003) Molecular Biology of the Cell 14, 2617-2629
59. Menice, C. B., Hulvershorn, J., Adam, L. P., Wang, C.-L. A., and Morgan, K. G. (1997) J.Biol.Chem. 272, 25157-25161
60. Simon, C., Goepfert, H., and Boyd, D. (1998) Cancer Res. 58, 1135-1139
61. Yao, P. M., Buhler, J.-M., d'Ortho, M. P., Lebargy, F., Delclaux, C., Harf, A., and Lafuma, C. (1996) J.Biol.Chem. 271, 15580-15589
62. Troussard, A. A., Costello, P., Yoganathan, T. N., Kumagai, S., Roskelley, C. D., and Dedhar, S. (2000) Oncogene 19, 5444-5452
by guest on July 8, 2019http://w
ww
.jbc.org/D
ownloaded from

14
63. Higashi, N., Kunimoto, H., Kaneko, S., Sasaki, T., Ishii, M., Kojima, H., and Nakajima, K. (2004) Genes to Cells 9, 233-242
64. Eferl, R. and Wagner, E. F. (2003) Nature Reviews 3, 895-868
65. Shields, J. M., Rogers-Graham, K., and Der, C. (2002) J.Biol.Chem. 277, 9790-9799
66. Soini, Y., Hurskainen, T., Hoyhtya, M., Oikarinen, A., and Autio-Harmainen, H. (1994) The Journal of Histochemistry and Cytochemistry 42, 945-951
67. Zucker, S., Lysik, R. M., Zarrabi, M. H., and Moll, U. (1993) Cancer Res. 53, 140-146
68. Pyke, C., Ralfkiaer, E., Tryggvason, K., and Dano, K. (1993) American Journal of Pathology 142, 359-365
69. Nielsen, B. S., Timshel, S., Kjeldsen, L., Sehested, M., Pyke, C., Borregaard, N., and Dano, K. (1996) Int.J.Cancer 65, 57-62
70. LaTulippe, E., Satagopan, J., Smith, A., Scher, H., Scardino, P., Reuter, V., and Gerald, W. L. (2002) Cancer Res. 62, 4499-4506
Acknowledgments This work was supported by NIH grants R01DE10845 and CA58311 to DB. We are grateful to Dr. Mi-chael Parmacek (University of Pennsylvania) for providing the SM22 null mice.
by guest on July 8, 2019http://w
ww
.jbc.org/D
ownloaded from

15
FIGURE LEGENDS Figure 1. Expression cloning identifies a putative regulator of MMP-9 expression. PANEL A. HT1080 cells were co-transfected with 100 ng of a 2.2 kb MMP-9 promoter-luciferase (or pGL3) plasmid, a CMV6-driven expression vector encoding nothing or individual cDNAs (identified from primary and secondary screens) (700 ng) and pRL-SV40 (1 ng). After 24 h, cells were lysed and luciferase activity determined. Data, normalized for differences in transfection efficiency, are shown as % change (+ S.D. values of 3 independent experiments) with the value derived from co-transfection with the MMP-9 promoter and the empty expression construct set at 100 %. PANEL B. Same as Panel A except that the 1:1 MMP-9-Luc: clone 13-1(b)-ratio indicates 100 ng each of the reporter and expression constructs. Figure 2. Stable SM22 expression in HT1080 cells reduces MMP-9 expression. PANEL A. HT1080 cells were transfected with an SM22 expression construct or the empty vec-tor (pEGFP-C1), G418-resistant clones expanded and analyzed for SM22 protein by Western blotting us-ing a polyclonal anti-SM22 antibody. PANEL B. Conditioned medium (in serum free medium) normal-ized for cell number differences was analyzed by gelatin zymography. PANEL C. Total RNA from the indicated clones was subjected to multiplex RT-PCR using primers generating the MMP-9 (120 bp) and actin (621 bp) amplicons. +ve control- PCR of a MMP-9 coding sequence plasmid. The data are typical of duplicate experiments. Figure 3. Reduced in vitro invasion of SM22-transfected HT1080 cells. The indicated clones (25,000) were seeded on Matrigel-coated porous filters with, or without ex-ogenous (33 ng/ml) MMP-9 protein. After 18, h, cells were removed from the top aspect of the filter. The filter was stained with Diff-Quik and cells on the lower aspect of the filter enumerated. The data in Panel B represent average + S.D of quadruplicate assays. Figure 4. Knockdown of SM22 with siRNA up-regulates MMP-9 synthesis. WI-38 cells were transfected with a pool of 4 siRNA (100 nM final concentration) targeting SM22 or a control mismatched siRNA (Cntrl) using lipofectamine 2000. PANEL A and B. After 72 h, the cells were analyzed by Western blotting using polyclonal antibodies directed at SM22 or MMP-9. PANEL C. One day post-transfection, cells were changed to serum-free medium for a 24 h period, condi-tioned medium harvested, adjusted for differences in cell numbers and subjected to zymography using 1 % gelatin. Band intensity was quantified by densitometry. The experiment was performed twice. Figure 5. Extinguishing SM22 expression in lung fibroblasts is accompanied by induced MMP-9 expression. PANEL A. Equal protein from WI-38 fibroblasts, or their SV40-transformed counterparts (VA-13), was analyzed for SM22 protein by Western blotting. PANEL B. Conditioned medium from the indi-cated cells was normalized for cell number differences and analyzed for MMP-9 activity by gelatin zy-mography. PANEL C. Total RNA was extracted from the indicated cells, and subjected to multiplex PCR using primers specific for the actin and MMP-9 transcripts. The arrow points to the MMP-9 amplicon and the * indicates primers. The MMP-9 (+ve Cntrl) represents amplified product from a plasmid bearing the MMP-9 coding sequence. For Panels B and C, the intensity of the 92 kDa gelatinolytic band and the am-plified cDNA corresponding to the MMP-9 transcript was quantified by densitometry. Data are typical of duplicate experiments. Figure 6. Increased MMP-9 protein levels in uterus derived from SM22 null mice. PANEL A. Total RNA from mouse uterus was analyzed for SM22 and GAPDH transcripts by RT-PCR. PANEL B. Sections of the uterus derived from the indicated mice were fixed and subjected to immunohistochemistry with a rabbit anti mouse-MMP-9 antibody followed by an HRP-conjugated anti
by guest on July 8, 2019http://w
ww
.jbc.org/D
ownloaded from

16
rabbit antibody. MMP-9 immunoreactivity (arrows) was visualized with DAB. A zoomed image of the indicated boxed area is shown depicting epithelial (E) and stromal (S) compartments. Figure 7. The actin-binding domain of SM22 is dispensable with respect to repression of MMP-9 promoter activity. PANEL A. SM22 cDNA fragments cloned into the pEGF-C1/N1 expression constructs were di-gested with HindIII/KpnI and the products resolved in an agarose gel. The faster migrating bands in each lane represent the SM22 coding sequence. Actin-binding affinity was as published (38) with -, +, ++, and +++ indicating undetectable, undetectable-10; 10-20 and >20 % SM22 fragment co-sedimenting with ac-tin respectively. PANEL B; The indicated SM22 expression constructs (700 ng) were co-transfected with a luciferase reporter regulated by 2.2 kb of the MMP-9 regulatory sequence (100 ng). After 24 h, cells were lysed and, after normalization for differences in transfection efficiency, assayed for luciferase activ-ity. The data are typical of 4 independent experiments. Figure 8. Transient activation of signaling kinases in SM22-transfectants. PANEL A. The indicated cells were serum-starved for 16 h and then treated with PMA (100 nM). At the stated times thereafter, cells were extracted and analyzed for phosphorylated (activated) or total amounts of the indicated kinase by Western blotting. The experiment was performed twice. PANEL B. The indicated cells were cotransfected with 100 ng of a MMP-9 promoter fragment (2.2 kb)-firefly luciferase reporter and 700 ng of either the wt MEK 1 or a constitutively active MEK 1 expression con-struct (MEK ΔN3S218E-S222D). A β-actin promoter-driven renilla reporter (4 ng) was included as in-ternal control. Cells were lysed 24 hr post transfection and luciferase activity determined. The activity of the MEKwt was set to 100 % and data represent average ± S.D. % values of 4 independent experiments. Figure 9. Identification of the SM22-responsive region of the MMP-9 promoter. PANEL A. HT1080 cells were co-transfected with the indicated MMP-9 promoter fragment-luciferase reporter (100 ng) and the SM22 expression construct or the empty vector pEGFP-C1 (700 ng) as described in Figure 1. After 24 h, cell lysates were assayed for luciferase activity. Data are expressed as % change (+ S.D of triplicate experiments) with 100 % representing the value achieved by co-transfection with the 2.2 kb MMP-9 promoter fragment and the empty expression vector. Figure 10. Nuclear extract from the SM22-transfectants shows diminished binding to the MMP-9-derived AP-1 consensus sequence. PANEL A, B. Nuclear extract (10 μg protein) was subjected to EMSA using an oligonucleotide (2 X 104 cpm) spanning the proximal AP-1 motif (-79/-73) (Panel A) or the KRE-M9 cis element (-66/-57) (Panel B) in the MMP-9 promoter. The nucleotide sequence of the corresponding oligonucleotide is indicated with the consensus sequence bolded. PANEL C. EMSA was as in Panel A with the exception that the anti-c-Fos antibody or IgG (1 μg) was added to the nuclear extract. The experiment was per-formed twice. PANEL D. HT1080 cells were cotransfected with 100 ng of an AP-1-regulated firefly luciferase reporter and 700 ng of either pEGFP or this vector encoding SM22. All transfections included a β-actin promoter-driven Renilla reporter (4 ng). Cells were lysed 24 h later and luciferase activity de-termined. Data are expressed as average ± S.D. values of 4 independent experiments.
by guest on July 8, 2019http://w
ww
.jbc.org/D
ownloaded from

19
Figure 2 .
HT
1080
HT1
080
Vect
or
HT1
080
SM22
(1)
HT1
080
SM22
(4)
HT1
080
SM22
(6)
HT
1080
HT1
080
Vect
or
HT1
080
SM22
(1)
HT1
080
SM22
(4)
HT1
080
SM22
(6)
pCM
V-M
MP-
9 (+
ve C
ontro
l)
Wat
er
CB
92 kDa72 kDa
621 bp
120 bp
HT
1080
HT1
080
Vect
or
HT1
080
SM22
(1)
HT1
080
SM22
(4)
HT1
080
SM22
(6)
SM22
Actin
A
50
100
300
500
7501000
SizeMarkers (bp)
by guest on July 8, 2019 http://www.jbc.org/ Downloaded from

21
Figure 4
SM22
Actin
92 kDa
B
A Sm22
siR
NA
Cnt
rl Si
RN
A
Sm22
siR
NA
Cnt
rl Si
RN
A
48 h Time post-transfectionC
MMP-9
ActinDensitometric
Value26,163 46,862
DensitometricValue47 150
by guest on July 8, 2019 http://www.jbc.org/ Downloaded from

22
Figure 5
92 kDa
WI-
38
VA-1
3
Wat
er
WI-
38
VA-1
3
MM
P-9
(+ve
Cnt
rl)
SM22
Actin
WI-
38
VA-1
3
B
A
C
*
72 kDa
560
1315
DensitometricValue135 275
DensitometricValue
100200300400500700
Size Markers (bp)
1,5001,000
by guest on July 8, 2019 http://www.jbc.org/ Downloaded from
23
Figure 6
-/-
+/+
# 4
#10
# 11
# 54 5 10 11Animal
Stan
dard
s
Wat
er
SM22
GAPDH
B
A
100300500750
Size Markers (bp)
1,000
50
by guest on July 8, 2019 http://www.jbc.org/ Downloaded from

25
Figure 8A
Time with PMA (h)0 1 2 3 60.5 0 1 2 3 60.5HT1080 Vector HT1080 SM22 (clone 1)
Phosphorylated ERK 1/2
Total ERK 1/2
Phosphorylated MEK 1/2
Total MEK 1/2
Phosphorylated JNK
Phosphorylated p38
Time with PMA (h)0 1 2 3 6 0 1 2 3 6
A
by guest on July 8, 2019 http://www.jbc.org/ Downloaded from

28
Figure 10A,B
Nuclear Extract
100 X oligonucleotide competitor
HT
1080
HT
1080
SM
22 (6
)
CTGACCCC GCACTTTGAGTCA
- -- - - - -+
HT
1080
Non
e
HT
1080
HT
1080
Vec
tor
HT1
080
SM22
(1)
HT
1080
SM
22 (4
)
HT
1080
SM
22 (6
)
HT
1080
Non
e
- -- - - - -+
Nuclear Extract
AGCACTT GAGCCTGTCAAG
HT
1080
Vec
tor
HT1
080
SM22
(1)
HT
1080
SM
22 (4
)
Lane 1 2 3 4 5 6 7 8
by guest on July 8, 2019 http://www.jbc.org/ Downloaded from

Rajesh Nair, Julian Solway and Douglas D. Boydcollagenase (MMP-9) expression
Expression cloning identifies transgelin (SM22 ) as a novel repressor of 92 kDa type IV
published online July 10, 2006J. Biol. Chem.
10.1074/jbc.M602703200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on July 8, 2019http://w
ww
.jbc.org/D
ownloaded from