Embed Size (px)
Citation preview

Expanded Access Programsfor Drugs and Biologics
_________________________________________________________
Richard Klein
Office of Health and Constituent Affairs
Food and Drug Administration February 27, 2014

2
What is Expanded Access?
• Use of an investigational drug or biologic to treat a patient with a serious disease or condition who does not have comparable or satisfactory alternative therapies to treat the disease or condition.– Intent is clearly treatment- not primarily intended to obtain information about the safety
or effectiveness of a drug
• Contrast with investigational drug in a clinical trial where the primary intent is research– systematic collection of data with the intent to analyze it to
learn about the drug
also called Treatment Use, Compassionate Use

3
What is Expanded Access?
• Patient is not considered part of clinical trial
• Minimal data collecting:– Serious/unexpected adverse event– Outcome summary
• Not part of study data from trials

4
Expanded Access Programs (EAP) Should Be Option of Last Resort
• Approved Drugs: – Studied and characterized– labeled – broadest availability– 3rd party reimbursement
• Clinical Trials: – Provide necessary data to determine safety &
effectiveness
• EAP: – Represent opportunity when other options exhausted

5
FDA Published Regulations in 2009
• Subpart I consolidates treatment use into a separate subpart of the IND regulations containing all necessary information• Describes three distinct categories of access
– Individual– Intermediate-Size– Treatment IND/protocol
• Describes the general criteria applicable to all categories of access, and additional criteria that must be met for each access category
• Describes requirements for submission • Describes the safeguards applicable to EAPs (e.g.,
informed consent, IRB review, reporting requirements)

6
Requirements for all EAPs21 CFR 312.305
• Serious or immediately life threatening illness or condition
• No comparable or satisfactory alternative therapy
• Potential benefit justifies the potential risks of the treatment, and those risks are not unreasonable in the context of the disease or condition being treated
• Providing drug will not interfere with or compromise development for the expanded access use

7
Human Subject Protections Apply to All EAPs
Drugs in EAPs are investigational drugs, and they are subject to the following requirements from 21 CFR:
– Part 50- Protection of Human Subjects (informed consent)
– Part 56- Institutional Review Board
– Part 312 - including Clinical Holds based on safety and reporting requirements (adverse event reports, annual reports)

Requirements for Individual Patient EAPs
21 CFR 312.310
8
• Physician must determine probable risk from drug does not exceed that from disease
• FDA must determine that the patient cannot obtain access under another type of IND
• Procedures for emergency use (where there is not time to make a written IND submission) – FDA may authorize starting access without submission, with very quick turn-around (F/U written submission required within 15 working days of authorization)

9
Requirements for Individual Patient EAPs
– Treatment generally limited to one course (though FDA may ok ongoing therapy)
– FDA requires written summary report, and may require special monitoring
Physician often takes role of sponsor/investigator

Intermediate Size Population 21 CFR 312.315
• Intended for situations where multiple patients with the same condition might benefit from a particular investigational product
• No set numerical requirements – meant to be practical and flexible– more than a few, and less than a lot
10

11
Requirements for Intermediate Size Population
21 CFR 312.315
• Can be used when a drug is
– Being developed (e.g., patients not eligible for trial)
– Not being developed (e.g., rare disease, cannot recruit for a trial)
– Approved (e.g., drug withdrawn, drug shortage situation- e.g., foreign version of a U.S. approved drug)

12
Requirements for Intermediate Size Population
• Intended for patient populations smaller than intended for Treatment IND (generally up to 100 patients)
• FDA can request consolidation when a number of individual requests are received for the same use
• Sufficient evidence drug is safe at proposed dose and duration to justify size of exposed population
• Preliminary evidence (clinical or plausible pharmacological) of effect
• Annual review to determine whether treatment use should be continued and whether a T-IND would be a more appropriate mechanism

13
Requirements for Treatment IND or Protocol
21 CFR 321.320
• Drug is being investigated in clinical trial designed to support marketing, or trials are complete
• Company is actively pursuing marketing approval
• Sufficient evidence of safety and effectiveness– Serious disease: evidence from phase 3 or
compelling data from phase 2 clinical trials– Immediately life-threatening disease: evidence
from phase 3 or phase 2 studies, but could be based on more preliminary clinical evidence

14
FDA Recognizes a Need for Balance• Treatment access must be balanced against the
systematic collection of clinical data to characterize safety and effectiveness
• Patient autonomy must be balanced against exposure to unreasonable risks and the potential for health fraud, potential exploitation of desperate patients
• Individual needs must be balanced against societal needs – Clinical trials are the best mechanism to provide
evidence of safety and effectiveness for potential new treatments
– FDA approval for marketing is the most efficient means to make safe and effective treatments available to the greatest number of patients.

15
Concern about Trial Enrollment
• Early access to investigational therapies could make clinical trials more difficult to perform– E.g., AZT for HIV, High Dose Chemotherapy +
bone marrow transplant for stage IV breast cancer
• Clinical trial enrollment and conduct is a factor in consideration of treatment access to experimental drugs
• Manufacturing capacity is often limitation in early phases – supply of drug for expanded access could limit supply for trials

16
EAP-Implementing the process
• A community responsibility– the patient– the doctor– the sponsor– FDA– IRB

17
EAP-Implementing the process
• The patient– Facing difficult medical circumstances and confusing decisions– Need to discover options when others are exhausted– Need to understand and accepts potential risks– Patients may face costs that are not reimbursed by health insurers– Navigating uncharted waters that differ significantly from standard
health care, e.g., IRB involvement, informed consent
– Patients (and their advising physicians) may have limited information about a drug
• confidential commercial information that FDA has access to• may not have access to developing efficacy and/or safety information

18
EAP-Implementing the process
• The doctor– Helps initiate the process for the patient– requires commitment to contacting company and filing
paperwork• may represent unfamiliar processes for many treating
physicians– responsible for ongoing support and monitoring of patient– responsible for adverse event and outcome reporting– Physicians costs of providing access may not be fully
compensated– liability issues?

19
EAP-Implementing the process
• The sponsor– must be able and willing to provide the product– work with doctor to provide product and monitor
product – develop mid-size and large scale program
protocols and support program infrastructure• administration • monitoring and reporting responsibilities• IRB review and continuing review

20
EAP-Implementing the process
• FDA• IND paperwork• medical records review• quick turn-around time• requires resources
– assessment of existing data for safety and evidence of effectiveness
– assurance of patient protections (IRB review, informed consent)

21
EAP-Implementing the process
• Institutional Review Board (IRB)– outside physician looking for review for their patient– not all IRBs are familiar with expanded access protocols
slant of review (intent is treatment, not clinical research)– may miscalculate risk patient is willing to accept– workload and scheduling issues for IRB can delay review– requires entire committee to review (no expedited review
procedures)– cost concerns and reimbursement for services

22
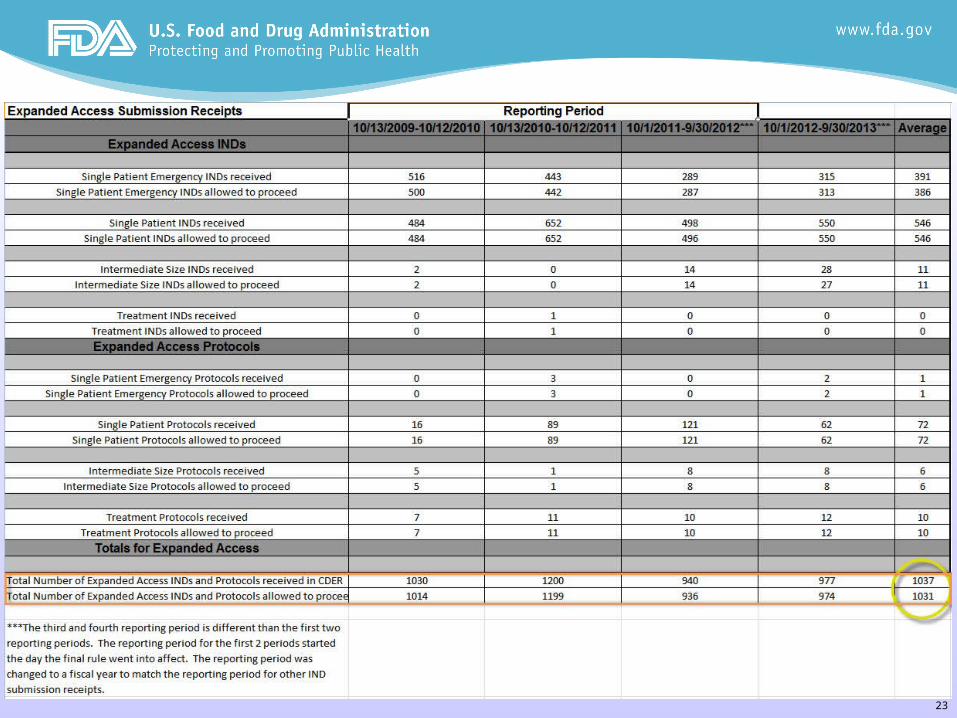
23

24
How do patients find access programs?
– Through their healthcare provider– Internet
• ClinicalTrials.gov• Patient organizations• Patient forums
– Other patients

Take-Away Messages
• Patient, physician and sponsor all play integral parts
• FDA provides the pathway/mechanism
• Sponsor must be able/willing to provide product
• Investigational product with more unknowns
• Investigator status, IRB, and informed consent
25

26
Web Resources
Access to Investigational Drugs
• http://www.patientnetwork.fda.gov/expanded-access
• http://www.fda.gov/ForConsumers/ByAudience/ForPatientAdvocates/AccesstoInvestigationalDrugs/default.htm
Physician Request for an Individual Patient IND under Expanded Access for Non-emergency or Emergency Use
• http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/InvestigationalNewDrugINDApplication/ucm107434.htm
www.fda.gov, www.fda.gov, search “expanded access”search “expanded access”

27
For Further Information
Richard Klein
Office of Health and Constituent Affairs
(301) 796.8460
www.fda.gov, search “expanded access”





























