Embed Size (px)
Citation preview

Exon skipping of FceRIβ eliminates expression of thehigh-affinity IgE receptor in mast cells with therapeuticpotential for allergyGlenn Crusea,b,1, Yuzhi Yinb, Tomoki Fukuyamaa, Avanti Desaib, Greer K. Arthura, Wolfgang Bäumera,Michael A. Beavenc, and Dean D. Metcalfeb
aDepartment of Molecular Biomedical Sciences, College of Veterinary Medicine, North Carolina State University, Raleigh, NC 27607; bLaboratory of AllergicDiseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892; and cLaboratory of MolecularImmunology, National Heart, Lung and Blood Institute, National Institutes of Health, Bethesda, MD 20892
Edited by K. Frank Austen, Brigham and Women’s Hospital, Boston, MA, and approved October 26, 2016 (received for review May 27, 2016)
Allergic diseases are driven by activation of mast cells and release ofmediators in response to IgE-directed antigens. However, there are nodrugs currently available that can specifically down-regulate mast cellfunction in vivo when chronically administered. Here, we describe aninnovative approach for targeting mast cells in vitro and in vivo usingantisense oligonucleotide-mediated exon skipping of the β-subunit ofthe high-affinity IgE receptor (FceRIβ) to eliminate surface high-affinityIgE receptor (FceRI) expression and function, rendering mast cellsunresponsive to IgE-mediated activation. As FceRIβ expression is re-stricted to mast cells and basophils, this approach would selectivelytarget these cell types. Given the success of exon skipping in clinicaltrials to treat genetic diseases such as Duchenne muscular dystrophy,we propose that exon skipping of FceRIβ is a potential approach formast cell-specific treatment of allergic diseases.
mast cell | allergy | IgE receptor | oligonucleotides | dermatitis
Asthma and related allergic diseases affect up to 1 in 10people in developed countries, and about 10% of patients
with asthma cannot be controlled with current therapeutic ap-proaches. The most widely used therapies for asthma rely upondampening the inflammatory response with either oral or inhaledglucocorticosteroids and relaxing the constricted airway smoothmuscle cells with β-adrenoreceptor agonists. However, high doses ofsteroids when needed for severe asthma are associated with un-desirable side effects and inhaled β-adrenoreceptor agonists can in-crease the risk of death from asthma if not used in combination withglucocorticosteroids. It is suggested that β-adrenoreceptor agonistsmay promote underlying inflammation that could contribute to theairway wall remodeling seen in asthma (1). Other clinical approachesthat aim for more long-term alleviation of symptoms, including de-sensitization with incremental increases in dose of allergen or hypo-sensitization to induce immune tolerance, have proven beneficial forsome, but not all, patients, and serious adverse effects can occur (2).In addition to asthma, other allergic diseases have similar treatment
strategies and also have an unmet clinical need. An example is atopicdermatitis (AD), which like asthma, is also a multifactorial diseasewith complex pathophysiology, remains incurable, and affects 10–20%of children in the United States (3). The two major symptoms of ADare inflammatory lesions and severe pruritus. Both the prevalence andthe economic burden of AD are increasing worldwide. Symptomatictreatment with topical and/or systemic glucocorticosteroids or calci-neurin inhibitors are still considered the gold standard. However, dueto the known adverse effects of these drugs, prescription rates are lowand patient compliance is often poor (3). Thus, there is still a need fornew therapeutic approaches with a better safety profile.An approach for intervention in allergic inflammation that we
have pursued is based on the finding that the gene loci 11q12-q13are strongly linked to allergy and asthma susceptibility and that themembrane spanning 4A (MS4A) gene family is clustered around11q12-q13 (4–7). Moreover, the genes MS4A1 (encoding the pro-tein CD20) andMS4A2 [encoding the protein FceRIβ (β-subunit ofthe high-affinity IgE receptor)] are associated with the activation
and proliferation of B cells (8) andmast cells (9–11), respectively, andare therefore considered potential candidates for the linkage of thesegenetic regions with allergy. In particular, FceRIβ contributes to IgE-dependent mast cell signaling by trafficking the FceRI receptorcomplex to the cell surface and amplifying FceRI-induced signaling(12, 13). The first transmembrane domain of FceRIβ is required fortrafficking the receptor complex (14), whereas the C-terminalimmunoreceptor tyrosine-based activation motif (ITAM) amplifiessignaling (15). Thus, a report that polymorphisms in MS4A2 wereassociated with development of asthma gained interest (16), butstudies into the functional consequence of mutations in MS4A2 didnot appear to affect the function of FceRIβ (17). However, we haveidentified the expression of a truncated isoform of FceRIβ(t-FceRIβ) that lacks exon 3 of MS4A2, which encodes the firsttwo transmembrane domains of FceRIβ (10). This truncated isoformdoes not traffic to the plasma membrane (9, 10) and appears tofunction in intracellular rather than plasma transmembrane signaling(9). Therefore, our hypothesis is that perturbation of MS4A2 splicingcould lead to disproportionate expression of the t-FceRIβ isoform atthe expense of full-length (FL) FceRIβ isoform and thus perturbtrafficking of the FceRI receptor complex to the plasma membrane aswell as mast cell responses to IgE-directed antigens.Here, we have examined whether manipulation ofMS4A2 splicing
favors t-FceRIβ formation, disrupts FceRI expression and signaling,and has functional consequences. We identified that forced expres-sion of t-FceRIβ using antisense oligonucleotide (AON)-mediatedexon skipping of MS4A2 exon 3 eliminated expression of FceRI in
Significance
We identified an innovative use for the technique of antisenseoligonucleotide-mediated exon skipping to specifically targetand down-regulate IgE receptor expression in mast cells. Exonskipping is typically used as part of personalized medicine,where a mutant exon is skipped after sequencing the patients’affected genes. Our approach, however, targets a nonmutatedgene and an exon that is critical for surface IgE receptor ex-pression. It does not require a personalized approach with geneticsequencing or multiple iterations of oligonucleotides that wouldrequire clinical trials. Furthermore, the diseases to be treated withthis technology are ideal for local delivery of the oligonucleotidesby aerosols or topical cream formulations.
Author contributions: G.C. conceived research; G.C., W.B., M.A.B., and D.D.M. designedresearch; G.C., Y.Y., T.F., A.D., G.K.A., W.B., and M.A.B. performed research; G.C., Y.Y.,T.F., G.K.A., and W.B. analyzed data; and G.C., M.A.B., and D.D.M. wrote the paper.
Conflict of interest statement: Carrying out this study resulted in the filing of US Pro-visional Patent Application No. 62/289,447 by G.C. and D.D.M.
This article is a PNAS Direct Submission.
Freely available online through the PNAS open access option.1To whom correspondence should be addressed. Email: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1608520113/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1608520113 PNAS Early Edition | 1 of 6
IMMUNOLO
GYAND
INFLAMMATION

mast cells and resulted in mast cells that were functionally un-responsive to IgE-mediated antigen challenge. Given the recentpromising results of using AONs to alter splicing in diseases (forreviews, see refs. 18–20), and their success in clinical trials forDuchenne muscular dystrophy (21, 22), we propose that our re-sults warrant further study to develop this approach as a potentialmast cell-specific treatment for allergic diseases.
ResultsLoss of FceRI with FceRIβ Exon Skipping. We first tested whetherAONs could be efficiently transfected into mast cells using a control25-mer FITC-conjugated morpholino AON in primary mouse bonemarrow-derived mast cells (BMMCs). We achieved >95% effi-ciency in mouse BMMCs at 24 h (Fig. 1 A and B) with no evidenceof cytotoxicity as determined by propidium iodide staining (Fig. 1A).The naturally occurring truncation of MS4A2 exon 3 leads to
loss of the first two transmembrane domains of FceRIβ resultingin the expression of t-FceRIβ that does not traffic to the plasmamembrane nor associate with FceRI (9, 10). Therefore, we pre-dicted that skipping exon 3 of MS4A2 following FceRIβ AONtreatment would result in preferential production of t-FceRIβinstead of FL FceRIβ as well as loss of expression of surfaceFceRI, which is dependent on FL FceRIβ (9, 12–14). Weattempted to induce exon skipping with AONs designed to targetMs4a2 exon 3 at the intron–exon boundary and identified thatFceRIβ AONs dose-dependently induced exon skipping of FceRIβmRNA as indicated by RT-PCR compared with cells transfectedwith an equivalent 25-mer standard control AON (Fig. 1C).To test whether exon skipping leads to loss of surface expression
of FceRI, we used flow cytometry to measure surface FceRI ex-pression in mouse BMMCs and found that it was reduced after10 μM FceRIβ AON treatment by 95.6 ± 0.4% (n = 5; P < 0.001)(Fig. 1D), thus virtually eliminating FceRI expression (efficiencyof transfection was ∼95%). In agreement with the exon-skippingdata (Fig. 1C), the effects of FceRIβ AONs on surface FceRIexpression were also dose dependent (Fig. 1E).
Degranulation and Calcium Influx. In view of the loss of surfaceFceRI expression with FceRIβ AON transfection, we tested whetherthere were corresponding reductions in responses to antigen in
BMMCs. There was a dose-dependent decrease in degranulation inresponse to DNP with increasing concentrations of FceRIβ AON,and degranulation was eliminated with 10 μM FceRIβ AON (Fig.2A). One micromolar FceRIβ AON resulted in 80% reduction insurface FceRI expression (Fig. 1C) whereas the reduction in de-granulation was lower (∼25%), although significant (Fig. 2A). Apossible explanation for this finding is that the number of FceRIreceptors and signaling capacity appears to exceed the requirementsfor degranulation, an explanation in agreement with studies reportedin RBL-2H3 mast cells (23). Although FceRI numbers vary duringthe cell cycle (24) and among different mast cell types, with estimatesfrom 130,000 in human lung mast cells (25) to ∼290,000 per cell inRBL-2H3 cells (23) and 120,000–380,000 in human cord blood-derived mast cell/basophil cultures (26), it is likely that mast cells ingeneral harbor surplus FceRI as significant degranulation is ob-served with aggregation of a few hundred receptors (23).The specificity of FceRIβ AON treatment was next determined by
its effect on thapsigargin-induced degranulation. Although FceRI-dependent degranulation was eliminated in BMMCs, thapsigargin-induced degranulation was unaffected by FceRIβ exon skipping (Fig.2B). In addition, we found the same pattern for calciummobilizationwith robust inhibition of the calcium signal in response to FceRIaggregation whereas the response to thapsigargin was unaffected(Fig. 2C). As with thapsigargin, IgE-mediated calcium influx is de-pendent upon store-operated calcium entry (27). Therefore, FceRIβexon skipping appears to selectively target IgE-dependent activationwithout disrupting cell responses to other stimuli.
Signaling, Cytokine Release, and Migration. We next examined theeffects of FceRIβ exon skipping in BMMCs on cell signaling eventsthat regulate both degranulation and de novo cytokine synthesisand in particular whether residual weak signals that fail to stimulatedegranulation were still sufficient to induce synthesis of cytokines.As expected from the diminished calcium signal, we found nosignificant FceRI-mediated phosphorylation of PLCγ1 with FceRIβAON transfection, and unlike FceRI-mediated activation,thapsigargin does not induce phosphorylation and acts in-dependently of PLCγ (Fig. 3 A and B). Phosphorylation of AKTand ERK are more distal signals than PLCγ1 phosphorylation,but phosphorylation of both of AKT and ERK were also markedly
Fig. 1. Transfection of AONs induce exon skippingand loss of surface FceRI expression. (A) Density dotplots of FITC positivity (x axes) versus propidium iodidepositivity (y axes) of mock-treated BMMCs (Top) andFITC morpholino AON-transfected BMMCs (Bottom).(B) Transfection efficiency data displayed as histogramwith mock-treated (red line) versus FITC AON-treated(blue line) BMMCs. (C) Transfection of Ms4a2 exon3-targeted AONs (FceRIβ AON) induces exon skippingin mouse BMMCs. Qualitative RT-PCR bands corre-spond to full-length (FL) FceRIβ and t-FceRIβ (exon 3truncation), shown with arrows as determined byDNA size markers. (D) Flow-cytometric analysis ofsurface FceRIα expression in BMMCs 48 h aftertransfection with either the standard control AON(Std Con AON, blue lines) or FceRIβ AONs (red lines).Black line is the isotype control. Data are represen-tative of five experiments. (E) Loss of surface FceRIαexpression with FceRIβ AON transfection is dose de-pendent. Combined data from five experiments.Data are the mean ± SEM. ****P < 0.0001.
2 of 6 | www.pnas.org/cgi/doi/10.1073/pnas.1608520113 Cruse et al.
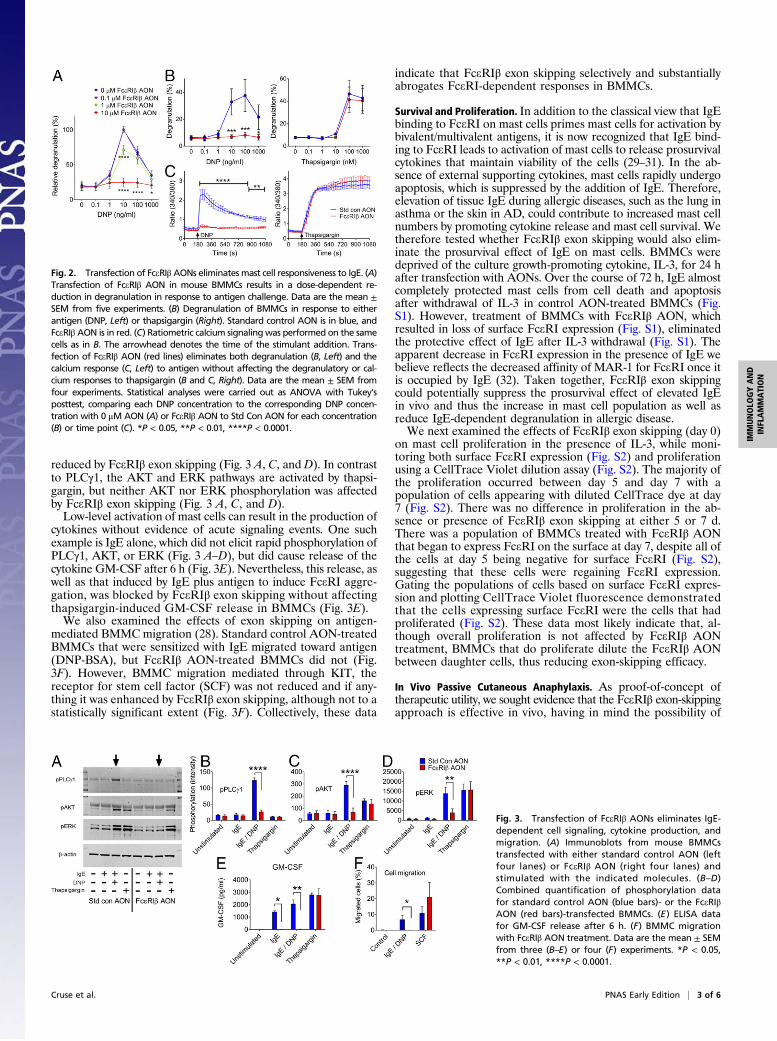
reduced by FceRIβ exon skipping (Fig. 3 A, C, and D). In contrastto PLCγ1, the AKT and ERK pathways are activated by thapsi-gargin, but neither AKT nor ERK phosphorylation was affectedby FceRIβ exon skipping (Fig. 3 A, C, and D).Low-level activation of mast cells can result in the production of
cytokines without evidence of acute signaling events. One suchexample is IgE alone, which did not elicit rapid phosphorylation ofPLCγ1, AKT, or ERK (Fig. 3 A–D), but did cause release of thecytokine GM-CSF after 6 h (Fig. 3E). Nevertheless, this release, aswell as that induced by IgE plus antigen to induce FceRI aggre-gation, was blocked by FceRIβ exon skipping without affectingthapsigargin-induced GM-CSF release in BMMCs (Fig. 3E).We also examined the effects of exon skipping on antigen-
mediated BMMC migration (28). Standard control AON-treatedBMMCs that were sensitized with IgE migrated toward antigen(DNP-BSA), but FceRIβ AON-treated BMMCs did not (Fig.3F). However, BMMC migration mediated through KIT, thereceptor for stem cell factor (SCF) was not reduced and if any-thing it was enhanced by FceRIβ exon skipping, although not to astatistically significant extent (Fig. 3F). Collectively, these data
indicate that FceRIβ exon skipping selectively and substantiallyabrogates FceRI-dependent responses in BMMCs.
Survival and Proliferation. In addition to the classical view that IgEbinding to FceRI on mast cells primes mast cells for activation bybivalent/multivalent antigens, it is now recognized that IgE bind-ing to FceRI leads to activation of mast cells to release prosurvivalcytokines that maintain viability of the cells (29–31). In the ab-sence of external supporting cytokines, mast cells rapidly undergoapoptosis, which is suppressed by the addition of IgE. Therefore,elevation of tissue IgE during allergic diseases, such as the lung inasthma or the skin in AD, could contribute to increased mast cellnumbers by promoting cytokine release and mast cell survival. Wetherefore tested whether FceRIβ exon skipping would also elim-inate the prosurvival effect of IgE on mast cells. BMMCs weredeprived of the culture growth-promoting cytokine, IL-3, for 24 hafter transfection with AONs. Over the course of 72 h, IgE almostcompletely protected mast cells from cell death and apoptosisafter withdrawal of IL-3 in control AON-treated BMMCs (Fig.S1). However, treatment of BMMCs with FceRIβ AON, whichresulted in loss of surface FceRI expression (Fig. S1), eliminatedthe protective effect of IgE after IL-3 withdrawal (Fig. S1). Theapparent decrease in FceRI expression in the presence of IgE webelieve reflects the decreased affinity of MAR-1 for FceRI once itis occupied by IgE (32). Taken together, FceRIβ exon skippingcould potentially suppress the prosurvival effect of elevated IgEin vivo and thus the increase in mast cell population as well asreduce IgE-dependent degranulation in allergic disease.We next examined the effects of FceRIβ exon skipping (day 0)
on mast cell proliferation in the presence of IL-3, while moni-toring both surface FceRI expression (Fig. S2) and proliferationusing a CellTrace Violet dilution assay (Fig. S2). The majority ofthe proliferation occurred between day 5 and day 7 with apopulation of cells appearing with diluted CellTrace dye at day7 (Fig. S2). There was no difference in proliferation in the ab-sence or presence of FceRIβ exon skipping at either 5 or 7 d.There was a population of BMMCs treated with FceRIβ AONthat began to express FceRI on the surface at day 7, despite all ofthe cells at day 5 being negative for surface FceRI (Fig. S2),suggesting that these cells were regaining FceRI expression.Gating the populations of cells based on surface FceRI expres-sion and plotting CellTrace Violet fluorescence demonstratedthat the cells expressing surface FceRI were the cells that hadproliferated (Fig. S2). These data most likely indicate that, al-though overall proliferation is not affected by FceRIβ AONtreatment, BMMCs that do proliferate dilute the FceRIβ AONbetween daughter cells, thus reducing exon-skipping efficacy.
In Vivo Passive Cutaneous Anaphylaxis. As proof-of-concept oftherapeutic utility, we sought evidence that the FceRIβ exon-skippingapproach is effective in vivo, having in mind the possibility of
Fig. 2. Transfection of FceRIβ AONs eliminates mast cell responsiveness to IgE. (A)Transfection of FceRIβ AON in mouse BMMCs results in a dose-dependent re-duction in degranulation in response to antigen challenge. Data are the mean ±SEM from five experiments. (B) Degranulation of BMMCs in response to eitherantigen (DNP, Left) or thapsigargin (Right). Standard control AON is in blue, andFceRIβ AON is in red. (C) Ratiometric calcium signalingwas performed on the samecells as in B. The arrowhead denotes the time of the stimulant addition. Trans-fection of FceRIβ AON (red lines) eliminates both degranulation (B, Left) and thecalcium response (C, Left) to antigen without affecting the degranulatory or cal-cium responses to thapsigargin (B and C, Right). Data are the mean ± SEM fromfour experiments. Statistical analyses were carried out as ANOVA with Tukey’sposttest, comparing each DNP concentration to the corresponding DNP concen-tration with 0 μMAON (A) or FceRIβ AON to Std Con AON for each concentration(B) or time point (C). *P < 0.05, **P < 0.01, ****P < 0.0001.
Fig. 3. Transfection of FceRIβ AONs eliminates IgE-dependent cell signaling, cytokine production, andmigration. (A) Immunoblots from mouse BMMCstransfected with either standard control AON (leftfour lanes) or FceRIβ AON (right four lanes) andstimulated with the indicated molecules. (B–D)Combined quantification of phosphorylation datafor standard control AON (blue bars)- or the FceRIβAON (red bars)-transfected BMMCs. (E) ELISA datafor GM-CSF release after 6 h. (F) BMMC migrationwith FceRIβ AON treatment. Data are the mean ± SEMfrom three (B–E) or four (F) experiments. *P < 0.05,**P < 0.01, ****P < 0.0001.
Cruse et al. PNAS Early Edition | 3 of 6
IMMUNOLO
GYAND
INFLAMMATION
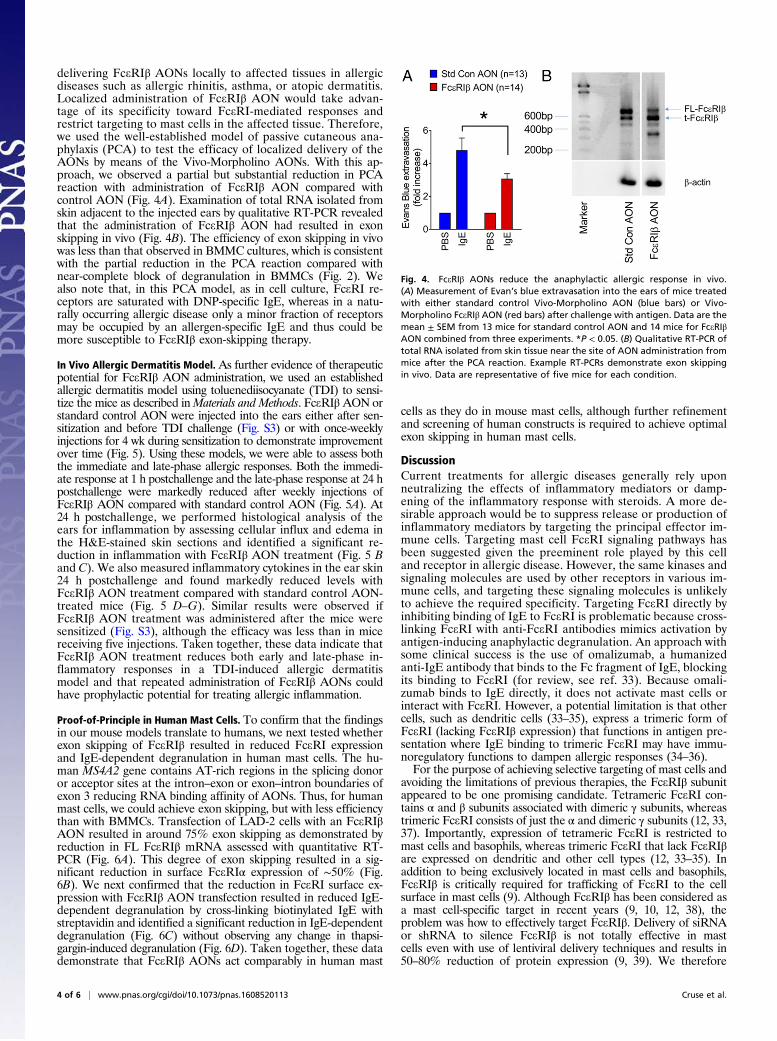
delivering FceRIβ AONs locally to affected tissues in allergicdiseases such as allergic rhinitis, asthma, or atopic dermatitis.Localized administration of FceRIβ AON would take advan-tage of its specificity toward FceRI-mediated responses andrestrict targeting to mast cells in the affected tissue. Therefore,we used the well-established model of passive cutaneous ana-phylaxis (PCA) to test the efficacy of localized delivery of theAONs by means of the Vivo-Morpholino AONs. With this ap-proach, we observed a partial but substantial reduction in PCAreaction with administration of FceRIβ AON compared withcontrol AON (Fig. 4A). Examination of total RNA isolated fromskin adjacent to the injected ears by qualitative RT-PCR revealedthat the administration of FceRIβ AON had resulted in exonskipping in vivo (Fig. 4B). The efficiency of exon skipping in vivowas less than that observed in BMMC cultures, which is consistentwith the partial reduction in the PCA reaction compared withnear-complete block of degranulation in BMMCs (Fig. 2). Wealso note that, in this PCA model, as in cell culture, FceRI re-ceptors are saturated with DNP-specific IgE, whereas in a natu-rally occurring allergic disease only a minor fraction of receptorsmay be occupied by an allergen-specific IgE and thus could bemore susceptible to FceRIβ exon-skipping therapy.
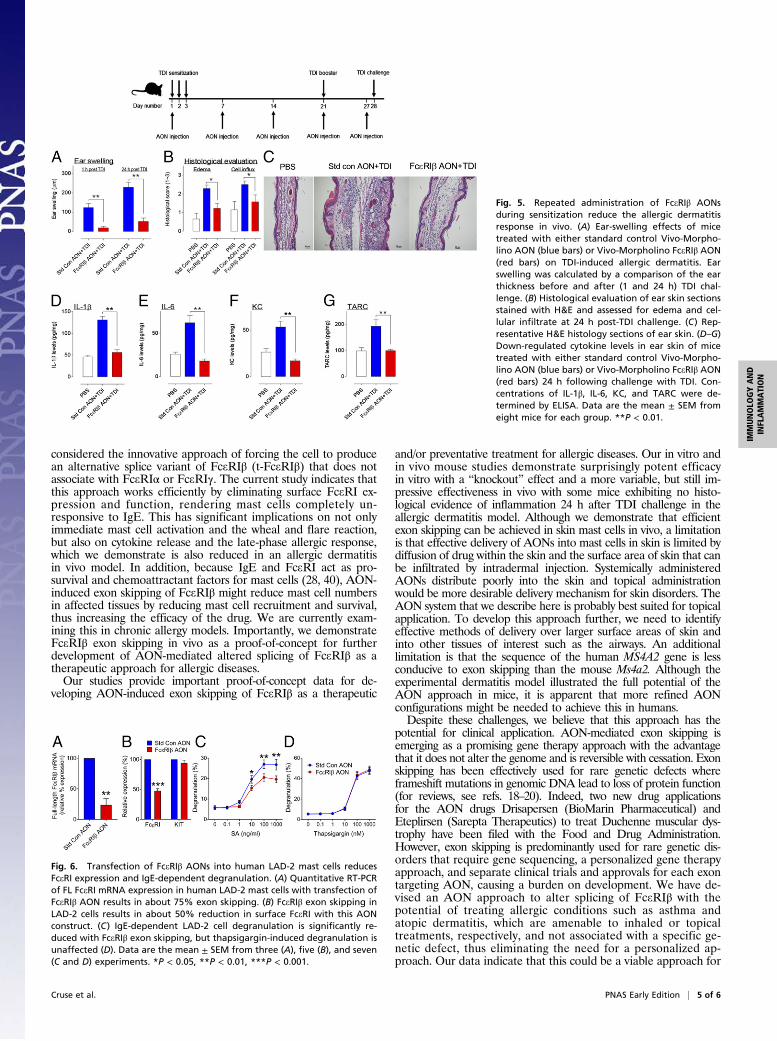
In Vivo Allergic Dermatitis Model. As further evidence of therapeuticpotential for FceRIβ AON administration, we used an establishedallergic dermatitis model using toluenediisocyanate (TDI) to sensi-tize the mice as described inMaterials and Methods. FceRIβ AON orstandard control AON were injected into the ears either after sen-sitization and before TDI challenge (Fig. S3) or with once-weeklyinjections for 4 wk during sensitization to demonstrate improvementover time (Fig. 5). Using these models, we were able to assess boththe immediate and late-phase allergic responses. Both the immedi-ate response at 1 h postchallenge and the late-phase response at 24 hpostchallenge were markedly reduced after weekly injections ofFceRIβ AON compared with standard control AON (Fig. 5A). At24 h postchallenge, we performed histological analysis of theears for inflammation by assessing cellular influx and edema inthe H&E-stained skin sections and identified a significant re-duction in inflammation with FceRIβ AON treatment (Fig. 5 Band C). We also measured inflammatory cytokines in the ear skin24 h postchallenge and found markedly reduced levels withFceRIβ AON treatment compared with standard control AON-treated mice (Fig. 5 D–G). Similar results were observed ifFceRIβ AON treatment was administered after the mice weresensitized (Fig. S3), although the efficacy was less than in micereceiving five injections. Taken together, these data indicate thatFceRIβ AON treatment reduces both early and late-phase in-flammatory responses in a TDI-induced allergic dermatitismodel and that repeated administration of FceRIβ AONs couldhave prophylactic potential for treating allergic inflammation.
Proof-of-Principle in Human Mast Cells. To confirm that the findingsin our mouse models translate to humans, we next tested whetherexon skipping of FceRIβ resulted in reduced FceRI expressionand IgE-dependent degranulation in human mast cells. The hu-man MS4A2 gene contains AT-rich regions in the splicing donoror acceptor sites at the intron–exon or exon–intron boundaries ofexon 3 reducing RNA binding affinity of AONs. Thus, for humanmast cells, we could achieve exon skipping, but with less efficiencythan with BMMCs. Transfection of LAD-2 cells with an FceRIβAON resulted in around 75% exon skipping as demonstrated byreduction in FL FceRIβ mRNA assessed with quantitative RT-PCR (Fig. 6A). This degree of exon skipping resulted in a sig-nificant reduction in surface FceRIα expression of ∼50% (Fig.6B). We next confirmed that the reduction in FceRI surface ex-pression with FceRIβ AON transfection resulted in reduced IgE-dependent degranulation by cross-linking biotinylated IgE withstreptavidin and identified a significant reduction in IgE-dependentdegranulation (Fig. 6C) without observing any change in thapsi-gargin-induced degranulation (Fig. 6D). Taken together, these datademonstrate that FceRIβ AONs act comparably in human mast
cells as they do in mouse mast cells, although further refinementand screening of human constructs is required to achieve optimalexon skipping in human mast cells.
DiscussionCurrent treatments for allergic diseases generally rely uponneutralizing the effects of inflammatory mediators or damp-ening of the inflammatory response with steroids. A more de-sirable approach would be to suppress release or production ofinflammatory mediators by targeting the principal effector im-mune cells. Targeting mast cell FceRI signaling pathways hasbeen suggested given the preeminent role played by this celland receptor in allergic disease. However, the same kinases andsignaling molecules are used by other receptors in various im-mune cells, and targeting these signaling molecules is unlikelyto achieve the required specificity. Targeting FceRI directly byinhibiting binding of IgE to FceRI is problematic because cross-linking FceRI with anti-FceRI antibodies mimics activation byantigen-inducing anaphylactic degranulation. An approach withsome clinical success is the use of omalizumab, a humanizedanti-IgE antibody that binds to the Fc fragment of IgE, blockingits binding to FceRI (for review, see ref. 33). Because omali-zumab binds to IgE directly, it does not activate mast cells orinteract with FceRI. However, a potential limitation is that othercells, such as dendritic cells (33–35), express a trimeric form ofFceRI (lacking FceRIβ expression) that functions in antigen pre-sentation where IgE binding to trimeric FceRI may have immu-noregulatory functions to dampen allergic responses (34–36).For the purpose of achieving selective targeting of mast cells and
avoiding the limitations of previous therapies, the FceRIβ subunitappeared to be one promising candidate. Tetrameric FceRI con-tains α and β subunits associated with dimeric γ subunits, whereastrimeric FceRI consists of just the α and dimeric γ subunits (12, 33,37). Importantly, expression of tetrameric FceRI is restricted tomast cells and basophils, whereas trimeric FceRI that lack FceRIβare expressed on dendritic and other cell types (12, 33–35). Inaddition to being exclusively located in mast cells and basophils,FceRIβ is critically required for trafficking of FceRI to the cellsurface in mast cells (9). Although FceRIβ has been considered asa mast cell-specific target in recent years (9, 10, 12, 38), theproblem was how to effectively target FceRIβ. Delivery of siRNAor shRNA to silence FceRIβ is not totally effective in mastcells even with use of lentiviral delivery techniques and results in50–80% reduction of protein expression (9, 39). We therefore
Fig. 4. FceRIβ AONs reduce the anaphylactic allergic response in vivo.(A) Measurement of Evan’s blue extravasation into the ears of mice treatedwith either standard control Vivo-Morpholino AON (blue bars) or Vivo-Morpholino FceRIβ AON (red bars) after challenge with antigen. Data are themean ± SEM from 13 mice for standard control AON and 14 mice for FceRIβAON combined from three experiments. *P < 0.05. (B) Qualitative RT-PCR oftotal RNA isolated from skin tissue near the site of AON administration frommice after the PCA reaction. Example RT-PCRs demonstrate exon skippingin vivo. Data are representative of five mice for each condition.
4 of 6 | www.pnas.org/cgi/doi/10.1073/pnas.1608520113 Cruse et al.
considered the innovative approach of forcing the cell to producean alternative splice variant of FceRIβ (t-FceRIβ) that does notassociate with FceRIα or FceRIγ. The current study indicates thatthis approach works efficiently by eliminating surface FceRI ex-pression and function, rendering mast cells completely un-responsive to IgE. This has significant implications on not onlyimmediate mast cell activation and the wheal and flare reaction,but also on cytokine release and the late-phase allergic response,which we demonstrate is also reduced in an allergic dermatitisin vivo model. In addition, because IgE and FceRI act as pro-survival and chemoattractant factors for mast cells (28, 40), AON-induced exon skipping of FceRIβ might reduce mast cell numbersin affected tissues by reducing mast cell recruitment and survival,thus increasing the efficacy of the drug. We are currently exam-ining this in chronic allergy models. Importantly, we demonstrateFceRIβ exon skipping in vivo as a proof-of-concept for furtherdevelopment of AON-mediated altered splicing of FceRIβ as atherapeutic approach for allergic diseases.Our studies provide important proof-of-concept data for de-
veloping AON-induced exon skipping of FceRIβ as a therapeutic
and/or preventative treatment for allergic diseases. Our in vitro andin vivo mouse studies demonstrate surprisingly potent efficacyin vitro with a “knockout” effect and a more variable, but still im-pressive effectiveness in vivo with some mice exhibiting no histo-logical evidence of inflammation 24 h after TDI challenge in theallergic dermatitis model. Although we demonstrate that efficientexon skipping can be achieved in skin mast cells in vivo, a limitationis that effective delivery of AONs into mast cells in skin is limited bydiffusion of drug within the skin and the surface area of skin that canbe infiltrated by intradermal injection. Systemically administeredAONs distribute poorly into the skin and topical administrationwould be more desirable delivery mechanism for skin disorders. TheAON system that we describe here is probably best suited for topicalapplication. To develop this approach further, we need to identifyeffective methods of delivery over larger surface areas of skin andinto other tissues of interest such as the airways. An additionallimitation is that the sequence of the human MS4A2 gene is lessconducive to exon skipping than the mouse Ms4a2. Although theexperimental dermatitis model illustrated the full potential of theAON approach in mice, it is apparent that more refined AONconfigurations might be needed to achieve this in humans.Despite these challenges, we believe that this approach has the
potential for clinical application. AON-mediated exon skipping isemerging as a promising gene therapy approach with the advantagethat it does not alter the genome and is reversible with cessation. Exonskipping has been effectively used for rare genetic defects whereframeshift mutations in genomic DNA lead to loss of protein function(for reviews, see refs. 18–20). Indeed, two new drug applicationsfor the AON drugs Drisapersen (BioMarin Pharmaceutical) andEteplirsen (Sarepta Therapeutics) to treat Duchenne muscular dys-trophy have been filed with the Food and Drug Administration.However, exon skipping is predominantly used for rare genetic dis-orders that require gene sequencing, a personalized gene therapyapproach, and separate clinical trials and approvals for each exontargeting AON, causing a burden on development. We have de-vised an AON approach to alter splicing of FceRIβ with thepotential of treating allergic conditions such as asthma andatopic dermatitis, which are amenable to inhaled or topicaltreatments, respectively, and not associated with a specific ge-netic defect, thus eliminating the need for a personalized ap-proach. Our data indicate that this could be a viable approach for
Fig. 5. Repeated administration of FceRIβ AONsduring sensitization reduce the allergic dermatitisresponse in vivo. (A) Ear-swelling effects of micetreated with either standard control Vivo-Morpho-lino AON (blue bars) or Vivo-Morpholino FceRIβ AON(red bars) on TDI-induced allergic dermatitis. Earswelling was calculated by a comparison of the earthickness before and after (1 and 24 h) TDI chal-lenge. (B) Histological evaluation of ear skin sectionsstained with H&E and assessed for edema and cel-lular infiltrate at 24 h post-TDI challenge. (C) Rep-resentative H&E histology sections of ear skin. (D–G)Down-regulated cytokine levels in ear skin of micetreated with either standard control Vivo-Morpho-lino AON (blue bars) or Vivo-Morpholino FceRIβ AON(red bars) 24 h following challenge with TDI. Con-centrations of IL-1β, IL-6, KC, and TARC were de-termined by ELISA. Data are the mean ± SEM fromeight mice for each group. **P < 0.01.
Fig. 6. Transfection of FceRIβ AONs into human LAD-2 mast cells reducesFceRI expression and IgE-dependent degranulation. (A) Quantitative RT-PCRof FL FceRI mRNA expression in human LAD-2 mast cells with transfection ofFceRIβ AON results in about 75% exon skipping. (B) FceRIβ exon skipping inLAD-2 cells results in about 50% reduction in surface FceRI with this AONconstruct. (C) IgE-dependent LAD-2 cell degranulation is significantly re-duced with FceRIβ exon skipping, but thapsigargin-induced degranulation isunaffected (D). Data are the mean ± SEM from three (A), five (B), and seven(C and D) experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
Cruse et al. PNAS Early Edition | 5 of 6
IMMUNOLO
GYAND
INFLAMMATION

treatment of allergic diseases and a mast cell-specific therapeuticstrategy that, we believe, has the potential to translate into the clinic.
Materials and MethodsFor additional information on methods, see SI Materials and Methods.
Experiments on mice carried out at NIH were conducted under a protocol ap-proved by the Animal Care and Use Committee at National Institute of Allergy andInfectious Diseases (NIAID), NIH. BMMCs were developed from bone marrowobtained from femurs of C57BL/6J mice (The Jackson Laboratory), as previouslydescribed (41).
Transfection of Mast Cells with AONs. Transfection was achieved using theNucleofector II and Cell Line Kit V (Lonza) as described (9), and program X-001was used. AONs were purchased from Gene-Tools LLC.
Allergic Dermatitis TDI Model. Female 6-wk-old BALB/cAnN were purchasedfrom Charles River Laboratories. All aspects of the current study were con-ducted in accordance with the Animal Care and Use Program of the NorthCarolina State University (Institutional Animal Care and Use Committee Pro-tocol No. 13-111-B).
ACKNOWLEDGMENTS.We thank Dr. Ana Olivera (NIAID, NIH) for discussionsand Sarah Ehling (Department of Molecular Biomedical Sciences, NorthCarolina State University) for technical assistance. Financial support wasprovided by the Division of Intramural Research of National Institute ofAllergy and Infectious Diseases and National Heart, Lung, and BloodInstitute within the NIH, and from departmental start-up funds (to G.C.)(Department of Molecular Biomedical Sciences, College of Veterinary Med-icine, North Carolina State University).
1. Cruse G, et al. (2010) Counterregulation of β2-adrenoceptor function in human mastcells by stem cell factor. J Allergy Clin Immunol 125(1):257–263.e1–5.
2. Akdis CA (2012) Therapies for allergic inflammation: Refining strategies to inducetolerance. Nat Med 18(5):736–749.
3. Schneider L, et al. (2013) Atopic dermatitis: A practice parameter update 2012.J Allergy Clin Immunol 131(2):295–299.e1–27.
4. Sandford AJ, et al. (1993) Localisation of atopy and β subunit of high-affinity IgEreceptor (FceRI) on chromosome 11q. Lancet 341(8841):332–334.
5. Stafford AN, Rider SH, Hopkin JM, Cookson WO, Monaco AP (1994) A 2.8 Mb YACcontig in 11q12-q13 localizes candidate genes for atopy: FceRIβ and CD20. Hum MolGenet 3(5):779–785.
6. Liang Y, Buckley TR, Tu L, Langdon SD, Tedder TF (2001) Structural organization of thehuman MS4A gene cluster on chromosome 11q12. Immunogenetics 53(5):357–368.
7. Liang Y, Tedder TF (2001) Identification of a CD20-, FceRIbeta-, and HTm4-related genefamily: Sixteen new MS4A family members expressed in human and mouse. Genomics72(2):119–127.
8. Tedder TF, Engel P (1994) CD20: A regulator of cell-cycle progression of B lympho-cytes. Immunol Today 15(9):450–454.
9. Cruse G, et al. (2013) A truncated splice-variant of the FceRIβ receptor subunit iscritical for microtubule formation and degranulation in mast cells. Immunity 38(5):906–917.
10. Cruse G, Kaur D, Leyland M, Bradding P (2010) A novel FceRIβ-chain truncation reg-ulates human mast cell proliferation and survival. FASEB J 24(10):4047–4057.
11. Gilfillan AM, Tkaczyk C (2006) Integrated signalling pathways for mast-cell activation.Nat Rev Immunol 6(3):218–230.
12. Kraft S, Rana S, Jouvin MH, Kinet JP (2004) The role of the FcepsilonRI β-chain in al-lergic diseases. Int Arch Allergy Immunol 135(1):62–72.
13. Donnadieu E, Jouvin MH, Kinet JP (2000) A second amplifier function for the allergy-associated FceRI-β subunit. Immunity 12(5):515–523.
14. Singleton TE, Platzer B, Dehlink E, Fiebiger E (2009) The first transmembrane regionof the beta-chain stabilizes the tetrameric FceRI complex. Mol Immunol 46(11-12):2333–2339.
15. On M, Billingsley JM, Jouvin MH, Kinet JP (2004) Molecular dissection of the FcRβsignaling amplifier. J Biol Chem 279(44):45782–45790.
16. Laprise C, Boulet LP, Morissette J, Winstall E, Raymond V (2000) Evidence for associ-ation and linkage between atopy, airway hyper-responsiveness, and the beta subunitGlu237Gly variant of the high-affinity receptor for immunoglobulin E in the French-Canadian population. Immunogenetics 51(8-9):695–702.
17. Donnadieu E, Cookson WO, Jouvin MH, Kinet JP (2000) Allergy-associated polymor-phisms of the FceRI β subunit do not impact its two amplification functions. J Immunol165(7):3917–3922.
18. Siva K, Covello G, Denti MA (2014) Exon-skipping antisense oligonucleotides to cor-rect missplicing in neurogenetic diseases. Nucleic Acid Ther 24(1):69–86.
19. Touznik A, Lee JJ, Yokota T (2014) New developments in exon skipping and splicemodulation therapies for neuromuscular diseases. Expert Opin Biol Ther 14(6):809–819.
20. Veltrop M, Aartsma-Rus A (2014) Antisense-mediated exon skipping: Taking advan-tage of a trick from Mother Nature to treat rare genetic diseases. Exp Cell Res 325(1):50–55.
21. Mendell JR, et al.; Eteplirsen Study Group (2013) Eteplirsen for the treatment ofDuchenne muscular dystrophy. Ann Neurol 74(5):637–647.
22. Kole R, Krieg AM (2015) Exon skipping therapy for Duchenne muscular dystrophy.Adv Drug Deliv Rev 87:104–107.
23. Maeyama K, Hohman RJ, Metzger H, Beaven MA (1986) Quantitative relationshipsbetween aggregation of IgE receptors, generation of intracellular signals, and his-tamine secretion in rat basophilic leukemia (2H3) cells. Enhanced responses withheavy water. J Biol Chem 261(6):2583–2592.
24. Isersky C, Metzger H, Buell DN (1975) Cell cycle-associated changes in receptors for IgEduring growth and differentiation of a rat basophilic leukemia cell line. J Exp Med141(5):1147–1162.
25. Coleman JW, Godfrey RC (1981) The number and affinity of IgE receptors on dis-persed human lung mast cells. Immunology 44(4):859–863.
26. Ogawa M, et al. (1983) Suspension culture of human mast cells/basophils from um-bilical cord blood mononuclear cells. Proc Natl Acad Sci USA 80(14):4494–4498.
27. Ma HT, Beaven MA (2011) Regulators of Ca2+ signaling in mast cells: Potential targetsfor treatment of mast cell-related diseases? Adv Exp Med Biol 716:62–90.
28. Kitaura J, et al. (2005) IgE− and IgE+Ag-mediated mast cell migration in an autocrine/paracrine fashion. Blood 105(8):3222–3229.
29. Asai K, et al. (2001) Regulation of mast cell survival by IgE. Immunity 14(6):791–800.30. Cruse G, Cockerill S, Bradding P (2008) IgE alone promotes human lung mast cell
survival through the autocrine production of IL-6. BMC Immunol 9:2.31. Kalesnikoff J, et al. (2001) Monomeric IgE stimulates signaling pathways in mast cells
that lead to cytokine production and cell survival. Immunity 14(6):801–811.32. Ota T, Aoki-Ota M, Duong BH, Nemazee D (2009) Suppression of IgE B cells and IgE
binding to FceRI by gene therapywith single-chain anti-IgE. J Immunol 182(12):8110–8117.33. Logsdon SL, Oettgen HC (2015) Anti-IgE therapy: Clinical utility and mechanistic in-
sights. Curr Top Microbiol Immunol 388:39–61.34. Platzer B, Stout M, Fiebiger E (2015) Functions of dendritic-cell-bound IgE in allergy.
Mol Immunol 68(2 Pt A):116–119.35. Shin JS, Greer AM (2015) The role of FceRI expressed in dendritic cells and monocytes.
Cell Mol Life Sci 72(12):2349–2360.36. Bieber T (2007) The pro- and anti-inflammatory properties of human antigen-pre-
senting cells expressing the high affinity receptor for IgE (FceRI). Immunobiology212(6):499–503.
37. Kinet JP (1999) The high-affinity IgE receptor (FceRI): From physiology to pathology.Annu Rev Immunol 17:931–972.
38. Cruse G, Bradding P (2016) Mast cells in airway diseases and interstitial lung disease.Eur J Pharmacol 778:125–138.
39. Cruse G, et al. (2015) The CD20 homologue MS4A4 directs trafficking of KIT towardclathrin-independent endocytosis pathways and thus regulates receptor signalingand recycling. Mol Biol Cell 26(9):1711–1727.
40. Kitaura J, et al. (2003) Evidence that IgE molecules mediate a spectrum of effects onmast cell survival and activation via aggregation of the FcepsilonRI. Proc Natl Acad SciUSA 100(22):12911–12916.
41. Jensen BM, Swindle EJ, Iwaki S, Gilfillan AM (2006) Generation, isolation, and main-tenance of rodent mast cells and mast cell lines. Curr Protoc Immunol Unit 3:23.
42. Kirshenbaum AS, et al. (2003) Characterization of novel stem cell factor responsivehuman mast cell lines LAD 1 and 2 established from a patient with mast cell sarcoma/leukemia; activation following aggregation of FceRI or FcγRI. Leuk Res 27(8):677–682.
43. Kuehn HS, Radinger M, Gilfillan AM (2010) Measuring mast cell mediator release. CurrProtoc Immunol Chap 7:38.
44. Tkaczyk C, Beaven MA, Brachman SM, Metcalfe DD, Gilfillan AM (2003) The phos-pholipase Cγ1-dependent pathway of FceRI-mediated mast cell activation is regulatedindependently of phosphatidylinositol 3-kinase. J Biol Chem 278(48):48474–48484.
45. Cruse G, Duffy SM, Brightling CE, Bradding P (2006) Functional KCa3.1 K+ channels arerequired for human lung mast cell migration. Thorax 61(10):880–885.
46. Cruse G, et al. (2011) Functional KCa3.1 K+ channels are required for human fibrocytemigration. J Allergy Clin Immunol 128(6):1303–1309.e2.
47. Smrz D, Iwaki S, McVicar DW, Metcalfe DD, Gilfillan AM (2010) TLR-mediated sig-naling pathways circumvent the requirement for DAP12 in mast cells for the in-duction of inflammatory mediator release. Eur J Immunol 40(12):3557–3569.
48. Fukuyama T, Ehling S, Cook E, Bäumer W (2015) Topically administered Janus-kinase in-hibitors tofacitinib and oclacitinib display impressive antipruritic and anti-inflammatoryresponses in a model of allergic dermatitis. J Pharmacol Exp Ther 354(3):394–405.
6 of 6 | www.pnas.org/cgi/doi/10.1073/pnas.1608520113 Cruse et al.





























