Embed Size (px)
Citation preview

OpenMP
Méthode Fine Grain Versus
Coarse Grain

Architectures parallèles
Définition :
Un ordinateur est dit parallèle quand il est constitué de plusieurs processeurs homogènes ou non. Cet ensemble permet de réaliser en simultanée une opération sur l’ensemble (ou un sous ensemble)de la machine.
Single Instruction Single Data : machine séquentielle (Von Neumann)
Single Instruction Multiple Data : machine vectorielle. Plusieurs données traitées en
même temps par une seule instruction. Exemple machine Cray, ou famille des SX chez Nec
Multiple Instruction Single Data : Machine Architecture pipeline. L’idée est de faire en
sorte que les calculs successifs se recouvrent. Exemple IMB 360/91.
Multiple Instruction Multiple Data : Exécution d’une instruction / processeur et
pour pour différentes données. Souvent on exécute la même application pour tous les processeurs , ce qui amène à un type d’exécution particulière Single Program Multiple Data.
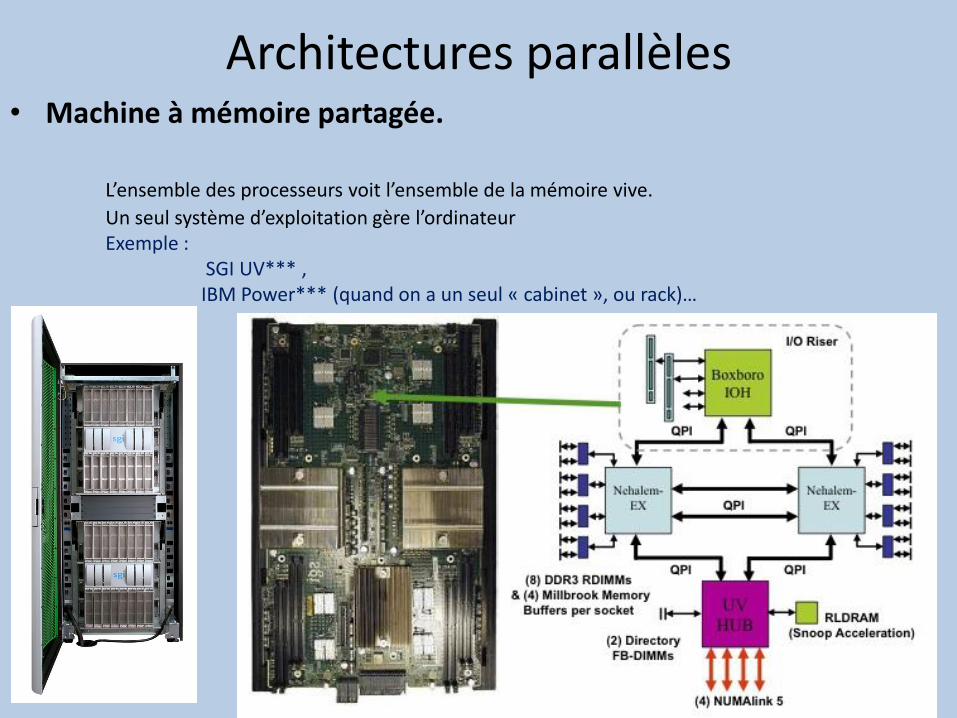
• Machine à mémoire partagée.
L’ensemble des processeurs voit l’ensemble de la mémoire vive.
Un seul système d’exploitation gère l’ordinateur Exemple : SGI UV*** , IBM Power*** (quand on a un seul « cabinet », ou rack)…
Architectures parallèles

• Machine à mémoire distribuée La machine est divisée en (p+q) nœuds de calcul, qui ont un ou plusieurs CPU avec
une certaine quantité de mémoire. Autant de système d’exploitation (identique ou non) que de nœuds.
Exemple : Les gros - cluster X86 (CINES), BGene …
Architectures parallèles

Présentation succincte de OpenMP
• Interface (bibliothèque) de programmation pour le calcul parallèle sur machine à mémoire partagée (ou un sous ensemble d’une machine à mémoire distribuée).
• Basé sur des directives (Pragma) à insérer dans le code source (C, C++, Fortran)
• La norme OpenMP est définie par un ensemble de fonctions et de variables d’environnement qui doit être intégré dans le compilateur. Tous les compilateurs récents l’intégrent plus ou moins intégralement (gcc (4.*), icc, gfortran (4.*), ifort, PGI …

Présentation succincte de OpenMP
Ce qu’il permet de gérer :
Création de processus légers (thread)
Partage du travail entre ces processus
Synchronisation entre ces processus
Gestion des variables (public ou privé)

Présentation succincte de OpenMP
Quelques limites :
Extensibilité limitée à la taille de la machine SMP.
Limitation de la bande passante mémoire cumulée / nbre de cœurs. En gros, on peut pouvoir utiliser p+q cœurs sur un nœud, mais l’efficacité du code n’est optimale que pour un nombre plus petit de cœurs.
Surcoût implicite d’OpenMP. Lors de la création de région parallèle, ou à la synchronisation des données.
On ne peut pas localiser les données avec OpenMP
L’accélération finale est limitée par le coût de la partie séquentielle (Loi de d’Amdahl). Vrai pour l’ensemble des paradigmes …

Loi d’Amdalh
Elle « prédit » l’accélération théorique maximale obtenue en parallélisant une application donnée pour un problème de taille fixe :
Acc(Proc) = Temps_seq/Temps_para(Proc)
= 1/(partie_seq+(1-partie_seq)/Proc)
< 1/partie_seq (si Proc -> infini)

Problème
Soit un domaine borné Ω, on cherche la solution u(x,y) de l’équation de Poisson :
Avec
La solution exacte de ce problème est :

Résolution
DF d’ordre 2 centré :
Méthodes utilisées :
Jacobi
G.S.
S.S.O.R
G.C
Condition initiale : U(x,y) est initialisé via bruit blanc (Random)

Séquentiel, résidu :f(itérations)

Séquentiel : erreur relative
Jacobi G.S
S.S.O.R G.C

Profiling du code sequentiel : l’outil Gprof
Makefile type en Fortran (sinon voir doc Intel): Ajouter : - pg dans la ligne de compilation. Exemple Méthodologie pour utiliser gprof :
1. Compiler le code avec l’option –pg (pour intel)
2. Exécuter une première fois l’exécutable comme d’habitude
3. Gprof mon_exe > fichier_profil.txt

Profiling du code séquentiel : l’outil Gprof
Flat profile: % cumulative self self total time seconds seconds calls s/call s/call name 45.54 5.87 5.87 50 0.12 0.12 calcul_gc_ 16.99 8.06 2.19 cvtas_t_to_a 5.43 8.76 0.70 cvt_ieee_t_to_text_ex 5.12 9.42 0.66 for_write_seq_fmt_xmit 5.04 10.07 0.65 __intel_ssse3_rep_memcpy 4.19 10.61 0.54 __intel_memset 3.65 11.08 0.47 for_write_seq_fmt 3.18 11.49 0.41 for__format_value 1.94 11.74 0.25 for__desc_ret_item 1.47 11.93 0.19 for__put_sf 1.16 12.08 0.15 for__acquire_lun 1.09 12.22 0.14 for__interp_fmt 0.93 12.34 0.12 1 0.12 6.02 MAIN__

Profiling du code séquentiel : Partie du code à paralléliser
do j= 1,ny
do i= 1,nx u_new(i,j)= c0 * ( c1*(u(i+1,j)+u(i-1,j)) & + c2*(u(i,j+1)+u(i,j-1)) - f(i,j)) enddo enddo
Jacobi
res=0.D0
do j=1,ny do i=1,nx r = (u(i+1,j)-2.*u(i,j)+u(i-1,j))/hx**2& + (u(i,j+1)-2.*u(i,j)+u(i,j-1))/hy**2& - f(i,j) res=r*r+res enddo enddo

OpenMP approche classique : Fine grain (Grain fin)
Définition : Méthode la plus simple & commune pour utiliser openMP, en utilisant la directive Do pour partager le travail entre les threads (processus légers). Exemple :
!$OMP PARALLEL DO PRIVATE(i,j) & !$OMP SHARED (residu) do j= 1,ny do i= 1,nx residu(i,j)=0.d0 enddo enddo !$OMP END PARALLEL DO

OpenMP approche classique : Fine grain (Grain fin)
Jacobi Facile à implémenter, il n’y a aucune difficulté, le code devient :
do j= 1,ny
do i= 1,nx u_new(i,j)= c0 * ( c1*(u(i+1,j)+u(i-1,j)) & + c2*(u(i,j+1)+u(i,j-1)) - f(i,j)) enddo enddo
!$OMP PARALLEL DO PRIVATE(i,j) & !$OMP SHARED (ny,nx,u,u_new,f,c0,c1,c2) do j= 1,ny do i= 1,nx u_new(i,j)= c0 * ( c1*(u(i+1,j)+u(i-1,j)) & + c2*(u(i,j+1)+u(i,j-1)) - f(i,j)) enddo enddo !$OMP END PARALLEL DO
Jacobi version sequentiel Jacobi version OpenMP FG

OpenMP approche classique : Fine grain (Grain fin)
G.S and S.S.O.R Problème : Il y a un nid de boucle avec plusieurs dépendances. Dans ce cas la méthode Fine Grain n’est pas aisée et l’algorithme perd de sa lisibilité entre sa version scalaire et parallèle. Méthode hyperplan, pipeline … Soit utiliser la méthode Coarse Grid; Cependant elle dénature aussi le code. Quoiqu’il en soit la méthode FG n’est pas utilisable simplement avec ce genre d’algorithme
do j= 1,ny do i= 1,nx u(i,j)= c0 * ( c1*(u(i+1,j)+u(i-1,j)) & + c2*(u(i,j+1)+u(i,j-1)) - f(i,j)) enddo enddo
do j= 1,ny do i= 1,nx u(i,j)= omega*(c0 * ( c1*(u(i+1,j)+u(i-1,j)) & + c2*(u(i,j+1)+u(i,j-1)) - f(i,j)))+ (1.-omega)*u(i,j) enddo enddo do j= ny,1,-1 do i= nx,1,-1 u(i,j)= omega*(c0 * ( c1*(u(i+1,j)+u(i-1,j)) & + c2*(u(i,j+1)+u(i,j-1)) - f(i,j)))+ (1.-omega)*u(i,j) enddo enddo

OpenMP approche classique : Fine grain (Grain fin)
G.C Dans sa formulation la plus simple, sans pré-conditionnement, le G.C est très simple à paralléliser en OpenMP … A noter les opérations de « réduction » sont l’aspect le plus « difficile » du passage sequentiel -> openmp
!$OMP PARALLEL DO PRIVATE(i,j,somme,sommeold) &
!$OMP SHARED (ny,nx,residu,residu_old) & !$OMP REDUCTION(+:sommer) & !$OMP REDUCTION(+:sommeoldr) do j=1,ny do i=1,nx somme= residu(i,j)*residu(i,j) sommeold= residu_old(i,j)*residu_old(i,j) sommer=sommer+somme sommeoldr=sommeoldr+sommeold enddo enddo !$OMP END PARALLEL DO

FG, scalabilité : Jacobi & GC

OpenMP approche coarse grain (gros grain)
Définition : Cette méthode englobe du code dans une seule région parallèle et distribue manuellement le travail aux processus légers… Elle s’appuie sur une décomposition de domaine, cependant toute l’univers de MPI lié à la topologie n’existe pas dans ce cas. L’utilisateur doit donc tout gérer par lui-même! Exemple simple :
!$OMP PARALLEL & !$OMP PRIVATE (rang,jdeb,jfin) rang=0 nbproc=1 rang=OMP_GET_THREAD_NUM() nbproc=OMP_GET_NUM_THREADS()
nty=ny*nbproc !ny : nbre d’élement par sous domaine jdeb=1+(rang*ny) jfin=(ny+rang*ny) !$OMP PARALLEL

OpenMP approche coarse grain (gros grain)
Une fois la décomposition de domaine réalisée, il ne reste plus qu’à modifier les boucles ? Non! Il faut ensuite gérer la synchronisation des threads, donc la mise à jour de la mémoire! Exemple sur Jacobi:
do j= jdeb,jfin do i= 1,nx u_new(i,j) = c0 * ( c1*(u(i+1,j)+u(i-1,j)) & + c2*(u(i,j+1)+u(i,j-1)) & - f(i,j)) enddo enddo do j= jdeb,jfin do i= 1,nx u(i,j)=u_new(i,j) enddo enddo !$omp barrier !$omp flush

CG versus FG versus MPI scalabilité : Jacobi

Conclusion
OpenMP (FG) est une excellente approche pour aller vite sur des cas simples! En quelques heures! On a des résultats… OpenMP (CG) est une excellent approche sur les cas complexes ou quand on veut passer à des problèmes de taille importante ou programmation hybride. Utiliser des outils de profiling comme gprof, Scalasca permet de mieux comprendre le comportement de son code MAIS ne donne pas nécessairement de réponse à la question cruciale : Pourquoi mon code passe mal à l’échelle ?!





























