Embed Size (px)
Citation preview

Evaluating the Potential of Scaling due to
Calcium Compounds in Hydrometallurgical Processes
by
Ghazal Azimi
A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy
Graduate Department of Chemical Engineering and Applied Chemistry University of Toronto
© Copyright by Ghazal Azimi 2010

ii
Evaluating the Potential of Scaling due to
Calcium Compounds in Hydrometallurgical Processes
Ghazal Azimi
Degree of Doctor of Philosophy
Graduate Department of Chemical Engineering and Applied Chemistry University of Toronto
2010
ABSTRACT
A fundamental theoretical and experimental study on calcium sulphate scale formation in
hydrometallurgical solutions containing various minerals was conducted. A new database for the
Mixed Solvent Electrolyte (MSE) model of the OLI Systems® software was developed through
fitting of existing literature data such as mean activity, heat capacity and solubility data in
simple binary and ternary systems. Moreover, a number of experiments were conducted to
investigate the chemistry of calcium sulphate hydrates in laterite pressure acid leach (PAL)
solutions, containing Al2(SO4)3, MgSO4, NiSO4, H2SO4, and NaCl at 25–250ºC. The database
developed, utilized by the MSE model, was shown to accurately predict the solubilities of all
calcium sulphate hydrates (and hence, predict scaling potential) in various multicomponent
hydrometallurgical solutions including neutralized zinc sulphate leach solutions, nickel
sulphate–chloride solutions of the Voisey’s Bay plant, and laterite PAL solutions over a wide
temperature range (25–250°C).
The stability regions of CaSO4 hydrates (gypsum, hemihydrate and anhydrite) depend on
solution conditions, i.e., temperature, pH and concentration of ions present. The transformation
between CaSO4 hydrates is one of the common causes of scale formation. A systematic study

iii
was carried out to investigate the effect of various parameters including temperature, acidity,
seeding, and presence of sulphate/chloride salts on the transformation kinetics. Based on the
results obtained, a mechanism for the gypsum–anhydrite transformation below 100°C was
proposed.
A number of solutions for mitigating calcium sulphate scaling problems throughout the
processing circuits were recommended: (1) operating autoclaves under slightly more acidic
conditions (~0.3–0.5 M acid); (2) mixing recycled process solutions with seawater; and (3)
mixing the recycling stream with carbonate compounds to reject calcium as calcium carbonate.
Furthermore, aging process solutions, saturated with gypsum, with anhydrite seeds at moderate
temperatures (~80°C) would decrease the calcium content, provided that the solution is slightly
acidic.

iv
Acknowledgments
I wish to express my sincere gratitude to all those who have helped make this thesis possible.
Foremost, I would like to thank my supervisor, Professor Vladimiros G. Papangelakis for his
continuous support, motivation, enthusiasm, guidance and immense knowledge. His endless
encouragement has been a major contribution in achieving the goals setout for this work.
Many thanks are due to my Supervisory Committee, Professor Donald Kirk, Professor Roger
Newman, and Professor Honghi Tran for their advice, feedback and comments. In addition, I
would like to thank Professor Alison Lewis for acting as the external examiner in my final
defense.
Dr. John Dutrizac is greatly acknowledged and thanked for all the support and constructive
guidance he has provided with my work, experiments and publications.
The contribution of Dr. Andre Anderko and Dr. Peiming Wang of the OLI Systems Inc. to this
work has been extensive. They are greatly acknowledged and thanked for their support,
guidance and help and of course for providing the OLI software.
Many thanks are due to Anglo American Plc., Barrick Gold Corp., Norilsk Nickel, NSERC,
OGS, Sherritt International Corp., and Vale Inco Ltd. for their contribution, and the financial
support provided for this project.
Special and sincere thanks are due to my dear friend, Ilya Perederiy, for his continuous support
and encouragement with all aspects of my work, experiments, publications, and thesis.
Many thanks go to Ramanpal Saini for his help and suggestions in writing this thesis. In
addition, the former and current members of the APEC group, in particular, Haixia Liu,
Matthew Jones, Sam Roshdi, and Sammy Peters are greatly acknowledged for their support over
the past four years.
Dr. John Graydon and Mr. Mark Berkley are also greatly acknowledged for their constructive
feedback.

v
Special thanks go Dr. Mike Gorton and Mr. George Kretschmann at the Department of
Geology for their countless help with the Scanning Electron Microscope (SEM) and Powder
X-Ray Diffraction (XRD) facilities.
I also would like to thank my former supervisor, Professor Cyrus Ghotbi, for his always help
and guidance and for introducing the beauty of thermodynamics to me.
I wish to pay a very special thank to my family and all my friends, in particular, my mom, my
dad and my sister, for their endless inspiration, encouragement and love throughout my life
which was the basis of making me who I am now.
Finally, I would like to thank my very best friend and my husband, Navid, without whom none
of these were possible. His endless love, continuous motivation and always support and
understanding over the past ten years provided me with the strength to move forward and
achieve my goals. I cannot imagine any of these without him. This dissertation is dedicated to
him.
Z{tétÄ Té|Å| ]tÇâtÜç ECDC

vi
Table of Contents ACKNOWLEDGMENTS ...................................................................................................................................... IV
TABLE OF CONTENTS........................................................................................................................................ VI
LIST OF TABLES ....................................................................................................................................................X
LIST OF FIGURES ................................................................................................................................................ XI
CHAPTER 1 INTRODUCTION .........................................................................................................................1
1.1 SCALE FORMATION OF CALCIUM SULPHATE..............................................................................................1 1.2 PREVIOUS STUDIES ....................................................................................................................................4
1.2.1 Experimental Studies of Calcium Sulphate Solubilities........................................................................4 1.2.2 Theoretical Studies of Calcium Sulphate Solubilities...........................................................................5
1.3 OBJECTIVES ...............................................................................................................................................7 1.4 THESIS OVERVIEW.....................................................................................................................................8
CHAPTER 2 MODELLING OF CALCIUM SULPHATE SOLUBILITY IN MULTICOMPONENT
SULPHATE SOLUTIONS......................................................................................................................................10
2.1 INTRODUCTION ........................................................................................................................................10 2.2 MODELLING METHODOLOGY...................................................................................................................11
2.2.1 Chemical Equilibria ...........................................................................................................................11 2.2.2 Equilibrium Constant .........................................................................................................................13 2.2.3 Activity Coefficient Model ..................................................................................................................13 2.2.4 Evaluation of the Model Parameters..................................................................................................17 2.2.5 Standard State Gibbs Free Energy and Entropy of Formation ..........................................................18
2.3 RESULTS AND DISCUSSION ......................................................................................................................19 2.3.1 Binary Systems (Metal Sulphate–H2O)...............................................................................................21
2.3.1.1 CaSO4–H2O System..................................................................................................................................21 2.3.1.2 Calcium Sulphate–Water Solubility Diagram...........................................................................................22 2.3.1.3 MnSO4–H2O System ................................................................................................................................23 2.3.1.4 NiSO4–H2O System..................................................................................................................................24 2.3.1.5 Fe2(SO4)3–H2O System.............................................................................................................................25
2.3.2 Ternary (Metal sulphate–H2SO4–H2O) Systems.................................................................................25 2.3.2.1 CaSO4–H2SO4–H2O System .....................................................................................................................25 2.3.2.2 NiSO4–H2SO4–H2O System .....................................................................................................................27 2.3.2.3 MnSO4–H2SO4–H2O System....................................................................................................................28 2.3.2.4 Al2(SO4)3–H2SO4–H2O System ................................................................................................................29
2.3.3 Ternary (CaSO4–Metal sulphate–H2O) Systems ................................................................................30 2.3.3.1 CaSO4–ZnSO4–H2O System.....................................................................................................................30 2.3.3.2 CaSO4–Na2SO4–H2O System ...................................................................................................................31

vii
2.3.3.3 CaSO4–NiSO4–H2O System .....................................................................................................................32 2.3.3.4 CaSO4–MgSO4–H2O System....................................................................................................................35 2.3.3.5 CaSO4–MnSO4–H2O System....................................................................................................................37
2.3.4 Effect of Divalent Cations on the Solubility of CaSO4........................................................................38 2.3.5 Industrial Implications of the Model in Zinc Producing Industries ...................................................39
2.3.5.1 CaSO4–ZnSO4–H2SO4 (0.1 M)–H2O System ...........................................................................................39 2.3.5.2 CaSO4–H2SO4–ZnSO4 (1.5 M)–H2O System ...........................................................................................40 2.3.5.3 CaSO4–MgSO4–H2SO4 (0.1 M)–ZnSO4 (1.15 M)–H2O System...............................................................41 2.3.5.4 CaSO4–H2SO4–ZnSO4 (2.5 M)–MgSO4 (0.41 M)–MnSO4 (0.18 M)–H2O System..................................41 2.3.5.5 CaSO4–(NH4)2SO4–ZnSO4 (2.5M)–MgSO4(0.41M)–H2SO4(pH=3.8)–MnSO4(0.18M)–H2O System ........42 2.3.5.6 CaSO4–Na2SO4–ZnSO4(2.5M)–MgSO4(0.41M)–MnSO4(0.18M)–H2SO4(pH=3.8)–H2O System...........43 2.3.5.7 CaSO4–Fe2(SO4)3–H2SO4 (0.3 M)–ZnSO4 (1.15M)–H2O System............................................................44 2.3.5.8 CaSO4–ZnSO4–H2SO4–H2O System ........................................................................................................45
2.4 SUMMARY................................................................................................................................................46
CHAPTER 3 MODELLING OF CALCIUM SULPHATE SOLUBILITY IN CHLORIDE/SULPHATE
SOLUTIONS 47
3.1 INTRODUCTION ........................................................................................................................................47 3.2 MODELLING STRATEGY ...........................................................................................................................50 3.3 RESULTS AND DISCUSSION ......................................................................................................................52
3.3.1 Evaluation of the Model Parameters..................................................................................................52 3.3.1.1 CaCl2–H2O System...................................................................................................................................52 3.3.1.2 CaSO4-CaCl2-H2O/CaSO4-HCl-H2O/CaSO4-NaCl-H2O/CaSO4-MgCl2-H2O Systems .............................52 3.3.1.3 CaSO4–AlCl3–H2O System.......................................................................................................................57 3.3.1.4 CaSO4–FeCl3–HCl–H2O System..............................................................................................................58
3.3.2 Industrial Implications of the Model in Nickel Hydrometallurgy.......................................................59 3.3.2.1 CaSO4–H2SO4–Fe2(SO4)3 (0.2 M)–NiSO4 (1.3 M)–LiCl (0.3 M)–H2O System........................................59 3.3.2.2 CaSO4–Fe2(SO4)3–H2SO4 (0.15 M)–NiSO4 (1.3 M)–LiCl (0.3 M)–H2O System .....................................60 3.3.2.3 CaSO4–NiSO4–Fe2(SO4)3 (0.2 M)–H2SO4 (0.15 M)–LiCl (0.3 M)–H2O System......................................61 3.3.2.4 CaSO4–LiCl–H2SO4 (0.15 M)–NiSO4 (1.3 M)–Fe2(SO4)3 (0.2 M)–H2O System .....................................62 3.3.2.5 CaSO4–Na2SO4–H2SO4 (0.15 M)–NiSO4 (1.3 M)–LiCl (0.3 M)–H2O System ........................................63
3.3.3 Predictive Capacity of the Model Parameters in Mixed Chloride Solutions......................................64 3.3.3.1 CaSO4–CaCl2–HCl–H2O System..............................................................................................................64 3.3.3.2 CaSO4–MgCl2–HCl–H2O System ............................................................................................................66 3.3.3.3 CaSO4–CaCl2–MgCl2–HCl–H2O System .................................................................................................67 3.3.3.4 CaSO4–Na2SO4–NaCl–H2O System.........................................................................................................68 3.3.3.5 CaSO4–Na2SO4–MgCl2–H2O System.......................................................................................................69 3.3.3.6 CaSO4–MgSO4–HCl–H2O / CaSO4–NiSO4–H2SO4–H2O Systems ..........................................................70
3.4 SUMMARY................................................................................................................................................72

viii
CHAPTER 4 SOLUBILITY OF GYPSUM AND ANHYDRITE IN LATERITE PRESSURE ACID
LEACH SOLUTIONS .............................................................................................................................................73
4.1 INTRODUCTION ........................................................................................................................................73 4.2 EXPERIMENTAL PROCEDURE....................................................................................................................76 4.3 RESULTS AND DISCUSSION ......................................................................................................................78
4.3.1 Reproducibility Experiments in CaSO4–H2O System .........................................................................78 4.3.2 Experimental Measurements and Model Predictions in Laterite PAL Solutions.......................................79
4.3.2.1 Effect of H2SO4 Concentration .................................................................................................................79 4.3.2.2 Effect of NiSO4 Concentration .................................................................................................................81 4.3.2.3 Effect of MgSO4 Concentration................................................................................................................82 4.3.2.4 Effect of the Chloride Concentration ........................................................................................................84
4.4 PROCESS IMPLICATIONS OF THE RESULTS................................................................................................85 4.5 SUMMARY................................................................................................................................................89
CHAPTER 5 TRANSFORMATION OF GYPSUM INTO ANHYDRITE IN AQUEOUS
ELECTROLYTE SOLUTIONS .............................................................................................................................91
5.1 INTRODUCTION ........................................................................................................................................91 5.2 EXPERIMENTAL SECTION .........................................................................................................................93 5.3 RESULTS AND DISCUSSION ......................................................................................................................96
5.3.1 Gypsum–Anhydrite Transformation in Water ....................................................................................96 5.3.2 Theoretical Determination of the Transformation Temperature ........................................................97 5.3.3 Effect of Sulphuric Acid on the Gypsum Transformation ...................................................................99 5.3.4 Theoretical and Practical Stability Regions of Gypsum in H2SO4 Solutions....................................101 5.3.5 Effect of Temperature on the Transformation Kinetics ....................................................................102 5.3.6 Effect of Seeding on Gypsum–Anhydrite Transformation ................................................................105 5.3.7 Effect of Sulphate and Chloride Salts on the Transformation Process ............................................107 5.3.8 Mechanism of Gypsum–Anhydrite Transformation..........................................................................109
5.3.8.1 In the Presence of H2SO4 ........................................................................................................................109 5.3.8.2 Transformation Mechanism in Pure Water .............................................................................................115
5.3.9 Industrial Implication: Precipitation due to Super-saturation.........................................................115 5.4 SUMMARY..............................................................................................................................................117
CHAPTER 6 CONCLUSIONS........................................................................................................................119
CHAPTER 7 RECOMMENDATIONS FOR FUTURE WORK..................................................................122
REFERENCES.......................................................................................................................................................124

ix
APPENDIX A: LITERATURE REVIEW ON THE SOLUBILITIES OF CALCIUM SULPHATE
HYDRATES IN VARIOUS ELECTROLYTE SYSTEMS ................................................................................133
APPENDIX B: REGRESSED MODEL PARAMETERS...................................................................................138
APPENDIX C: EXPERIMENTAL MEASUREMENTS IN LATERITE PAL SOLUTIONS........................140
APPENDIX D: X-RAY DIFFRACTION PATTERNS.......................................................................................145
APPENDIX E: SCHEMATIC DIAGRAMS OF THE EXPERIMENTAL SET-UP .......................................150
APPENDIX F: EXPERIMENTAL MEASUREMENTS FOR DH-AH TRANSFORMATION.....................151
APPENDIX G: ADDITIONAL SEM IMAGES ..................................................................................................153
APPENDIX H: THE RIETVELD METHOD (FULL-PATTERN ANALYSIS)..............................................155

x
List of Tables Table 2.1–Binary and ternary systems studied for the parameterization purpose ......................................................19 Table 2.2–Multicomponent systems studied for validating the model along with AARD% between experimental
data and predicted results ...........................................................................................................................................20 Table 3.1–Systems studied for the parameterization purpose ....................................................................................51 Table 3.2–Multicomponent systems studied for validating the model along with AARD% between experimental
data and predicted results ...........................................................................................................................................51 Table 5.1–Detailed experimental matrix studied in this chapter ................................................................................94 Table 6.1–Applicability regions of the model..........................................................................................................119 Table B.1–Regressed MSE middle-range interaction parameters (OLI-version 8.1.3)............................................138 Table B.2–Regressed standard state Gibbs free energy and entropy of formation of various solids .......................139 Table C.1 – Solubility of CaSO4 dihydrate (gypsum) in water at various NiSO4 concentrations ............................140 Table C.2 – Solubility of CaSO4 anhydrite in water at various NiSO4 concentrations ............................................140 Table C.3 – Solubility of CaSO4 anhydrite in water at various MgSO4 concentrations...........................................140 Table C.4 – Composition of laterite leach solutions (Huang, 2007) ........................................................................141 Table C.5 – Solubility of CaSO4 dihydrate (gypsum) in 0.23M MgSO4–0.07M NiSO4–0.004M Al2(SO4)3 solutions
at various H2SO4 concentrations ..............................................................................................................................141 Table C.6 – Solubility of CaSO4 anhydrite in 0.22M MgSO4–0.06M NiSO4–0.005M Al2(SO4)3 solutions at various
H2SO4 concentrations...............................................................................................................................................141 Table C.7 – Solubility of CaSO4 dihydrate (gypsum) in 0.2M H2SO4–0.22M MgSO4–0.005M Al2(SO4)3 solutions at
various NiSO4 concentrations ..................................................................................................................................141 Table C.8 – Solubility of CaSO4 anhydrite in 0.3M H2SO4–0.22M MgSO4–0.005M Al2(SO4)3 solutions at various
NiSO4 concentrations ...............................................................................................................................................142 Table C.9 – Solubility of CaSO4 dihydrate (gypsum) in 0.2M H2SO4–0.05M NiSO4–0.005M Al2(SO4)3 solutions at
various MgSO4 concentrations.................................................................................................................................142 Table C.10 – Solubility of CaSO4 anhydrite in 0.3M H2SO4–0.06M NiSO4–0.005M Al2(SO4)3 solutions at various
MgSO4 concentrations..............................................................................................................................................142 Table C.11 – Solubility of CaSO4 anhydrite in 0.25M H2SO4–0.2M MgSO4–0.005M Al2(SO4)3–0.05M NiSO4
solutions at 0.0 and 0.5M NaCl concentrations........................................................................................................143 Table C.12 – Solubility of CaSO4 dihydrate (gypsum) in 0.5M H2SO4 solutions at various NaCl concentrations..143 Table C.13 – Solubility of CaSO4 dihydrate in 0.5M HCl solutions at various MgSO4 concentrations ..................144 Table C.14 – Solubility of CaSO4 dihydrate in 0.5M H2SO4 solutions at various NiSO4 concentrations................144 Table F.1– Gypsum–anhydrite transformation at 90°C in water..............................................................................151 Table F.2– Concentration of CaSO4 and composition of saturating solid phases at various temperatures and
residence times in 0.5 and 1.0 M H2SO4 solutions...................................................................................................151 Table F.3– Concentration of CaSO4 and composition of saturating solid phases at various temperatures and
residence times in 1.5 and 2.0 M H2SO4 solutions...................................................................................................152

xi
List of Figures Figure 1.1 Solubility diagram of CaSO4 in water. Experimental data are from Dutrizac, 2002; Templeton and
Rodgers, 1967; Marshall et al., 1964; Sborgi and Bianchi, 1940; Hill and Wills, 1938; Posnjak, 1938; Partridge and
White, 1929..................................................................................................................................................................2 Figure 1.2 Process flow diagram of pressure acid leaching of ore concentrates. .........................................................3 Figure 2.1 Chemical modelling algorithm applied in this work. ................................................................................18 Figure 2.2 Gypsum solubility in H2O vs. temperature. Experimental data are from Dutrizac, 2002; Power et al.,
1966; Marshall and Slusher, 1966; Marshall et al., 1964; Posnjak, 1938; Hill and Wills, 1938; Hill and Yanick,
1935; Hulett and Allen, 1902. The curve is determined from the OLI default database............................................21 Figure 2.3 Hemihydrate solubility in H2O vs. temperature. Experimental data are from Sborgi and Bianchi, 1940;
Partridge and White, 1929. The curve represents the regressed model results...........................................................22 Figure 2.4 Anhydrite solubility in H2O at various temperatures. Experimental data are from Templeton and
Rodgers, 1967; Marshall et al., 1964; Bock, 1961; Posnjak, 1938; Straub, 1932; Partridge and White, 1929. The
curve is the OLI default database results....................................................................................................................22 Figure 2.5 Solubility diagram of CaSO4 in H2O. The solid and dashed curves show the stable and metastable
phases, respectively, at a given temperature. .............................................................................................................23 Figure 2.6 Solubility of MnSO4 in H2O. Experimental data are from Linke and Seidell (1958); curve shows the
model results. .............................................................................................................................................................24 Figure 2.7 Solubility of NiSO4 in H2O at various temperatures. Experimental data are from Linke and Seidell
(1958) and Bruhn et al. (1965); curve shows the fitted model results........................................................................24 Figure 2.8 CaSO4 solubility in ternary system of CaSO4–H2SO4–H2O. Curves show the regressed model results.
Experimental data are from (Dutrizac, 2002; Zdanovskii et al., 1968; Marshall and Jones, 1966)............................26 Figure 2.9 Solubility diagram of CaSO4 in H2SO4–H2O solutions; the surfaces were obtained from the model.......26 Figure 2.10 Transition diagram of CaSO4 hydrates in CaSO4–H2SO4–H2O system. Region I: gypsum stable, Region
II: anhydrite stable, gypsum metastable, Region III: anhydrite stable, hemihydrate metastable. Experimental data
are from Zdanovskii et al. (1968), Ling and Demopoulos (2004)..............................................................................27 Figure 2.11 NiSO4 solubility in aqueous H2SO4 solutions below 100ºC; experimental data are from Kudryashov
(1989), and Girich (1986). The curves are the regressed model results. ....................................................................28 Figure 2.12 NiSO4 solubility in aqueous H2SO4 solutions above 200ºC; experimental data are from Marshall et al.
(1962). The curves are the regressed model results....................................................................................................28 Figure 2.13 MnSO4 solubility in aqueous H2SO4 solutions; experimental data are from Linke and Seidell (1958),
and the curves are the regressed model results...........................................................................................................29 Figure 2.14 Aluminum sulphate solubility in H2SO4 solutions; experimental data are from Linke and Seidell (1958)
and the curves are the fitted model.............................................................................................................................29 Figure 2.15 CaSO4 solubility in ZnSO4 solutions below 100ºC; experimental data are from Umetsu et al. (1989) and
Zatonskaya et al. (1988), and the curves are fitted model results...............................................................................30

xii
Figure 2.16 CaSO4 solubility in ZnSO4 solutions above 100ºC; experimental data are from Umetsu et al. (1989),
and the curves are fitted model results. ......................................................................................................................31 Figure 2.17 CaSO4 solubility in Na2SO4 solutions below 100ºC; experimental data are from Block and Waters
(1968), Denman (1961), Hill and Wills (1938). The curves are the fitted model. .....................................................32 Figure 2.18 CaSO4 solubility in Na2SO4 solutions above 100ºC; experimental data are from Block and Waters
(1968), Templeton and Rodgers (1967), Hill and Wills (1938), Straub (1932). The curves are the fitted model. .....32 Figure 2.19 Gypsum solubility in NiSO4 solutions below 100ºC. Experimental data are from Azimi and
Papangelakis (2010b), Wollmann and Voigt (2008) and Campbell and Yanick (1932); the curves are the fitted
model..........................................................................................................................................................................33 Figure 2.20 Anhydrite solubility in NiSO4 solutions above 100ºC. Experimental data are from Azimi and
Papangelakis (2010b); the curves are the fitted model...............................................................................................34 Figure 2.21 Total concentration of Ca along with Ca2+ and CaSO4(aq) concentrations in CaSO4–NiSO4–H2O system
at 90ºC. Calculated values of ( 22)( 42
4wCaSOSO
am ⋅⋅ ±− γ ) and ( 2)(4 wCaSO a
aq⋅γ ) are also presented. .....................................35
Figure 2.22 Gypsum solubility in aqueous MgSO4 solutions. Experimental data are from Tanji (1969), Arslan and
Dutt (1993), Umetsu et al. (1989), Linke and Seidell (1958); the curves are the fitted model...................................36 Figure 2.23 Hemihydrate solubility in aqueous MgSO4 solutions. Experimental data are from Umetsu et al. (1989);
the curves are the fitted model. ..................................................................................................................................36 Figure 2.24 Anhydrite solubility in aqueous MgSO4 solutions. Experimental data are from Azimi and Papangelakis
(2010b); the curves are the fitted model.....................................................................................................................37 Figure 2.25 CaSO4 solubility in MnSO4 solutions; experimental data are from Wollmann and Voigt (2008) and
Zhelnin et al. (1973), and the curves are the fitted model. .........................................................................................38 Figure 2.26 CaSO4 solubility in MSO4 (M=Ni, Mg, Mn) solutions. Experimental data are from Azimi, Papangelakis
(2010b); Wollmann, Voigt (2008); Arslan, Dutt (1993); Zhelnin et al. (1973); Tanji (1969); Campbell,Yanick
(1932). Curves represent the model predictions.........................................................................................................39 Figure 2.27 CaSO4 solubility in CaSO4–ZnSO4–H2SO4 (0.1 M)–H2O solutions. Experimental data are from
Dutrizac (2002); the curves are the predicted results. ................................................................................................40 Figure 2.28 CaSO4 solubility in CaSO4–H2SO4–ZnSO4 (1.5M)–H2O solutions. Experimental data are from Dutrizac
(2002); the curves are the predicted results. ...............................................................................................................40 Figure 2.29 CaSO4 solubility in CaSO4–MgSO4–ZnSO4 (1.15 M)–H2SO4 (0.1 M)–H2O solutions; experimental data
are from Dutrizac (2002); curves represent model predictions. .................................................................................41 Figure 2.30 CaSO4 solubility in CaSO4–H2SO4–ZnSO4 (2.5M)–MgSO4 (0.41M)–MnSO4 (0.18M)–H2O solutions
vs. pH. Experimental data are from Dutrizac (2002); curves represent the predicted values.....................................42 Figure 2.31 CaSO4 solubility in CaSO4–(NH4)2SO4–ZnSO4(2.5M)–MgSO4(0.41M)–MnSO4(0.18M)–H2SO4(pH=3.8)–
H2O solutions; experimental data are from Dutrizac (2002); curves are model predictions..........................................43 Figure 2.32 CaSO4 solubility in CaSO4–Na2SO4–ZnSO4(2.5M)–MgSO4(0.41M)–MnSO4(0.18M)–H2SO4 (pH=3.8)–
H2O solutions. Experimental data are from Dutrizac (2002). Curves are the predicted values..................................44 Figure 2.33 CaSO4 solubility in CaSO4–Fe2(SO4)3–H2SO4 (0.3M)–ZnSO4 (1.15M)–H2O solutions. Experimental
data are from Dutrizac (2002); the curves are fitted model results. ...........................................................................45

xiii
Figure 2.34 CaSO4 solubility in CaSO4–ZnSO4–H2SO4–H2O solutions; experimental data are from Mutalala et al.
(1988); surface shows model prediction results. ........................................................................................................45 Figure 3.1 Schematic flowsheet of the Vale Inco developed hydrometallurgical process for the recovery of Ni and
Co values from sulphide concentrates (Kerfoot et al., 2002). ....................................................................................48 Figure 3.2 Gypsum solubility in CaCl2 solutions at different temperatures. Experimental data are from Li and
Demopoulos (2002, 2005) and Cameron and Seidell (1901). The curves represent the fitted model. .......................53 Figure 3.3 Anhydrite solubility in CaCl2 solutions at different temperatures. Experimental data are from Li and
Demopoulos (2005), Templeton and Rogers (1967), Gromova (1960). Curves are the fitted model. .......................54 Figure 3.4 Gypsum solubility as a function of HCl concentration. Experimental data are from Li and Demopoulos
(2002, 2005), Gupta (1968), Linke and Seidell (1958), Silcock (1979). Curves represent the fitted model. .............54 Figure 3.5 Anhydrite solubility in aqueous HCl solutions; experimental data are from Li and Demopoulos (2005),
and curves represent the fitted model.........................................................................................................................55 Figure 3.6 Hemihydrate solubility in aqueous HCl solutions; experimental data are from Li and Demopoulos
(2005), and lines are the fitted model.........................................................................................................................55 Figure 3.7 Gypsum solubility in aqueous NaCl solutions. Experimental data are from Marshall and Slusher (1966),
Ostroff and Metler (1966), Marshall et al. (1964), Linke and Seidell (1958), Silcock (1979); curves represent the
fitted model. ...............................................................................................................................................................56 Figure 3.8 Anhydrite solubility in aqueous NaCl solutions; experimental data are from Templeton and Rogers
(1967), Marshall et al. (1964), Bock (1961) and Silcock (1979); curves represent the fitted model. ........................56 Figure 3.9 Hemihydrate solubility as a function of NaCl concentration in aqueous solutions. Experimental data are
from Marshall et al. (1964), and the curve represents the fitted model. .....................................................................57 Figure 3.10 Gypsum solubility in aqueous AlCl3 solutions. Experimental data are from Li and Demopoulos (2006a)
and curves represent the fitted model.........................................................................................................................58 Figure 3.11 Gypsum solubility vs. FeCl3 concentration in CaSO4–FeCl3–HCl–H2O solutions. Experimental data are
from Li and Demopoulos (2006a); curves represent the fitted model. .......................................................................58 Figure 3.12 Gypsum solubility vs. FeCl3 concentration in CaSO4–FeCl3–HCl–H2O solutions. Experimental data are
from Li and Demopoulos (2006a), and curves represent the predicted values...........................................................59 Figure 3.13 CaSO4 solubility vs. H2SO4 concentration in CaSO4–H2SO4–Fe2(SO4)3(0.2M)–NiSO4(1.3M)–
LiCl(0.3M)–H2O solutions. Experimental data are from Dutrizac and Kuiper (2006). Curves represent the predicted
values. ........................................................................................................................................................................60 Figure 3.14 CaSO4 solubility vs. Fe2(SO4)3 concentration in CaSO4–Fe2(SO4)3–NiSO4(1.3M)–H2SO4 (0.15M)–
LiCl(0.3M)–H2O solutions; experimental data are from Dutrizac and Kuiper (2006), and curves represent the
predicted values..........................................................................................................................................................61 Figure 3.15 CaSO4 solubility vs. NiSO4 concentration in CaSO4–NiSO4–Fe2(SO4)3(0.2M)–H2SO4 (0.15M)–
LiCl(0.3M)–H2O solutions; experimental data are from Dutrizac and Kuiper (2006), and curves represent the model
results. ........................................................................................................................................................................62

xiv
Figure 3.16 CaSO4 solubility vs. LiCl concentration in CaSO4–LiCl–H2SO4(0.15M)–Fe2(SO4)3(0.2M)–
NiSO4(1.3M)–H2O solutions; experimental data are from Dutrizac and Kuiper (2006), and curves represent the
predicted values..........................................................................................................................................................63 Figure 3.17 CaSO4 solubility vs. Na2SO4 concentration in CaSO4–Na2SO4–NiSO4(1.3M)–H2SO4(0.15M)–
LiCl(0.3M)–H2O solutions; experimental data are from Dutrizac and Kuiper (2006), and curves represent the
predicted values..........................................................................................................................................................64 Figure 3.18 Gypsum solubility as a function of CaCl2 concentration in CaSO4–CaCl2–HCl–H2O solutions.
Experimental data are from Li and Demopoulos (2002, 2005) and Silcock (1979); curves represent the predicted
values. ........................................................................................................................................................................65 Figure 3.19 Anhydrite solubility vs. CaCl2 concentration in CaSO4–CaCl2–HCl–H2O solutions; experimental data
are from Li and Demopoulos (2005), and curves represent the predicted values.......................................................65 Figure 3.20 Hemihydrate solubility vs. CaCl2 concentration in CaSO4–CaCl2–HCl–H2O solutions; the experimental
data are from Li and Demopoulos (2005), and curves represent the predicted values. ..............................................66 Figure 3.21 Gypsum solubility vs. MgCl2 concentration in CaSO4–MgCl2–HCl–H2O solutions; the experimental
data are from Li and Demopoulos (2006a), and curves represent the predicted values. ............................................66 Figure 3.22 Gypsum solubility vs. CaCl2 concentration in CaSO4–CaCl2–MgCl2–HCl–H2O solutions; the
experimental data are from Li and Demopoulos (2006a), and the curve shows model predictions. ..........................67 Figure 3.23 Gypsum solubility vs. Na2SO4 concentration in CaSO4–Na2SO4–NaCl–H2O solutions; the experimental
data are from Block and Waters (1968), and the curves represent the predicted values. ...........................................68 Figure 3.24 Anhydrite solubility vs. Na2SO4 concentration in CaSO4–Na2SO4–NaCl–H2O solutions; the
experimental data are from Templeton and Rodgers (1967), and curves represent the predicted values...................69 Figure 3.25 Gypsum solubility vs. Na2SO4 concentration in CaSO4–Na2SO4–MgCl2–H2O solutions; the
experimental data are from Barba et al. (1984), and curves represent the predicted values.......................................69 Figure 3.26 CaSO4 solubility in CaSO4–MgSO4–HCl (0.5M)–H2O solutions; experimental data are from Azimi and
Papangelakis (2010a), and curves are the model predictions. ....................................................................................70 Figure 3.27 CaSO4 solubility in CaSO4–NiSO4–H2SO4 (0.5M)–H2O solutions; experimental data are from Azimi
and Papangelakis (2010a), and curves are the model predictions. .............................................................................71 Figure 4.1 Solubility diagram of CaSO4 in water. Experimental data are from this work and the literature (Dutrizac,
2002; Hill and Wills, 1938; Posnjak, 1938; Marshall et al., 1964; Partridge and White, 1929; Templeton and
Rodgers, 1967). The solid and dashed curves show the stable and metastable phases, respectively. ........................78 Figure 4.2 Gypsum solubility vs. H2SO4 concentration in CaSO4–H2SO4–NiSO4(0.07M)–MgSO4(0.23M)–
Al2(SO4)3(0.004M)–H2O solutions; experimental data are from Azimi and Papangelakis (2010b). The curves are the
predicted values..........................................................................................................................................................80 Figure 4.3 Anhydrite solubility vs. H2SO4 concentration in CaSO4–H2SO4–NiSO4(0.06M)–MgSO4(0.22M)–
Al2(SO4)3(0.005M)–H2O solutions; experimental data are from Azimi and Papangelakis (2010b) and the curves are
the model prediction results. ......................................................................................................................................80

xv
Figure 4.4 Gypsum solubility vs. NiSO4 concentration in CaSO4–NiSO4–H2SO4(0.2M)–MgSO4(0.22M)–
Al2(SO4)3(0.005M)–H2O solutions; experimental data are from Azimi and Papangelakis (2010b). The curves are the
predicted values..........................................................................................................................................................81 Figure 4.5 Anhydrite solubility vs. NiSO4 concentration in CaSO4–NiSO4–H2SO4(0.3M)–MgSO4(0.22M)–
Al2(SO4)3(0.005M)–H2O solutions; experimental data are from Azimi and Papangelakis (2010b). The curves are the
predicted values..........................................................................................................................................................82 Figure 4.6 Gypsum solubility vs. MgSO4 concentration in CaSO4–MgSO4–H2SO4(0.2M)–NiSO4(0.05M)–
Al2(SO4)3(0.005M)–H2O solutions; experimental data are from Azimi and Papangelakis (2010b). The curves are the
predicted values..........................................................................................................................................................83 Figure 4.7 Anhydrite solubility vs. MgSO4 concentration in CaSO4–MgSO4–H2SO4(0.3M)–NiSO4(0.06M)–
Al2(SO4)3(0.005M)–H2O solutions; experimental data are from Azimi and Papangelakis (2010b). The curves are the
predicted values..........................................................................................................................................................83 Figure 4.8 Anhydrite solubility vs. temperature in CaSO4–NaCl–MgSO4(0.2M)–H2SO4(0.25M)–NiSO4(0.05M)–
Al2(SO4)3(0.004M)–H2O solutions; experimental data are from Azimi and Papangelakis (2010b). The curves are the
predicted values..........................................................................................................................................................84 Figure 4.9 Gypsum solubility vs. NaCl concentration in CaSO4–NaCl– H2SO4(0.5M)– H2O solutions; experimental
data are from Azimi and Papangelakis (2010b). The curves are the predicted values. ..............................................85 Figure 4.10 Gypsum solubility as a function of temperature at various NiSO4 concentrations in comparison with
that in pure water; the curves are the model prediction results. .................................................................................87 Figure 4.11 Anhydrite solubility vs. temperature in pure water and in 0.22 M H2SO4 solution in comparison with
that in laterite PAL solutions containing MgSO4(0.2M)–H2SO4(0.22M)–NiSO4(0.05M)–Al2(SO4)3 (0.005M) at
various chloride concentrations. Solid curves are model prediction results for anhydrite; the dashed line shows
gypsum saturation level in pure water at 25°C...........................................................................................................88 Figure 4.12 Anhydrite solubility vs. temperature in pure water and in 0.22 M H2SO4 solutions compared to that in
laterite PAL solutions. Solid curves are model prediction results for anhydrite; the dashed line shows gypsum
saturation level in pure water at 25°C. .......................................................................................................................89 Figure 5.1 Solubility diagram of CaSO4 in water. Experimental data are from Dutrizac, 2002; Templeton and
Rodgers, 1967; Marshall et al., 1964; Sborgi and Bianchi, 1940; Hill and Wills, 1938; Posnjak, 1938; Partridge and
White, 1929. Curves obtained from the OLI/MSE model (Azimi et al., 2007)..........................................................96 Figure 5.2 Percentage of gypsum present in the equilibrating solid phase based on XRD results at various retention
times for gypsum–anhydrite transformation in water at 90°C. ..................................................................................97 Figure 5.3 Theoretical transformation temperature of gypsum into anhydrite as a function of the activity of water.
Solid curve derived from Hardie (1967); dashed curve obtained from the OLI/MSE model. ...................................98 Figure 5.4 Dissolution–precipitation profiles for CaSO4 along with the composition of saturating solids at different
temperatures after various retention times obtained on heating and subsequent cooling in: (a) 0.5 M H2SO4; (b) 1.0
M H2SO4; (c) 1.5 M H2SO4; (d) 2.0 M H2SO4 solutions. .........................................................................................100 Figure 5.5 Concentration of CaSO4 in 1.0 M H2SO4 solutions as a function of temperature: (▲) this work, heating;
(∆) this work, cooling; (■) Dutrizac (2002), heating; (□) Dutrizac (2002), cooling. ...............................................101

xvi
Figure 5.6 Theoretical and practical stability regions of gypsum at various H2SO4 concentrations. Solid curve
represents the theoretical transformation temperature obtained from the MSE thermodynamic model. Regions (I):
theoretical stability region of gypsum; (II): practical stability region of gypsum. ...................................................102 Figure 5.7 Kinetics of gypsum–anhydrite transformation at various temperatures in 1.5 M H2SO4 solutions in the
absence of anhydrite seeds. ......................................................................................................................................103 Figure 5.8 Variation of the ln(tin) vs. 1/T at various temperatures in 1.5 M H2SO4 solutions with no seeds present.
..................................................................................................................................................................................105 Figure 5.9 Kinetics of gypsum–anhydrite transformation at 70°C in 1.5 M H2SO4 solutions in the presence of an
initial 5 g/L of anhydrite seeds compared to no seeding case. .................................................................................106 Figure 5.10 CaSO4 solubility in 1.5 M H2SO4 solutions at 70°C at various residence times in the presence of 5 g/L
anhydrite seeds compared to no seeding case. .........................................................................................................107 Figure 5.11 Kinetics of gypsum–anhydrite transformation at 80°C in: (–■–) acid only (1.5 M H2SO4); (–▲–) 1.0 M
NiSO4–1.5 M H2SO4; (– –) 0.5 M NaCl–1.5 M H2SO4 solutions...........................................................................108 Figure 5.12 Calcium sulphate concentrations vs. retention time at 80°C: (–■–) in acid only (1.5 M H2SO4); (–▲–)
in 1.0 M NiSO4–1.5 M H2SO4; (– –) in 0.5 M NaCl–1.5 M H2SO4 solutions. .......................................................108 Figure 5.13 Kinetics of gypsum dissolution at 80°C: (–■–) in 1.5 M H2SO4; (–▲–) in 1.0 M NiSO4–1.5M H2SO4;
(– –) in 0.5 M NaCl–1.5M H2SO4 solutions; dashed lines represent the gypsum saturation level. ........................109 Figure 5.14 SEM images of a) gypsum feed; b) equilibrating solid phase after 3 h; c) solid phase after 12 days; and
d) transformed anhydrite crystals after 20 days in 1.5 M H2SO4 media at 70°C......................................................110 Figure 5.15 XRD patterns of AII–anhydrite: 072-0916 (orthorhombic) and γ–anhydrite: 037-0184 (tetragonal)
obtained from ICDD database. The characteristic line of AII–AH is marked with an asterisk. ...............................111 Figure 5.16 SEM images of saturating solids in 1.5 M H2SO4 media at 80°C after various retention times. ..........114 Figure 5.17 CaSO4 concentration at various retention times in 1.5 M H2SO4 solutions initially saturated with
gypsum at 80°C after adding 10 g of anhydrite seeds. .............................................................................................116 Figure D.1 X-ray diffraction pattern of the gypsum feed.........................................................................................145 Figure D.2 X-ray diffraction pattern of the anhydrite feed (AII)..............................................................................145 Figure D.3 X-ray diffraction pattern of hemihydrate*. .............................................................................................146 Figure D.4 X-ray diffraction pattern of soluble (AIII or γ) anhydrite ......................................................................146 Figure D.5 X-ray diffraction pattern of the equilibrating solid phase in H2SO4 media at 70°C (retention time=3 h):
(a) 2θ = 10–55° (b) 2θ = 37–44°. SEM image of this solid is presented in Fig. 5.14 (b). ........................................147 Figure D.6 X-ray diffraction pattern of the equilibrating solid phase in H2SO4 media at 70°C (retention time=12
days): (a) 2θ = 10–60° (b) 2θ = 31–45°. SEM image of this solid is presented in Fig. 5.14 (c). .............................148 Figure D.7 XRD patterns of solid samples in H2SO4 media at 25°C after a) 9h; b) 24h; c) 19 days. ......................149 Figure E.1 Schematic diagram of the glass reactors utilized in this work................................................................150 Figure E.2 Schematic diagram of the titanium autoclave utilized in this work........................................................150 Figure G.1 SEM images of solid samples in 1.5 M H2SO4 media at 25°C after a) 9h; b) 24h; c) and d) 19 days. ..153 Figure G.2 SEM images of saturating solid samples in pure water at 90°C after various retention times...............154

xvii
NOMENCLATURE
List of symbols ai Activity of species i aij UNIQUAC interaction parameter between i and j aij
(k) UNIQUAC adjustable parameter between i and j aw Activity of water A Arrhenius (pre-exponential) constant Ax Debye–Hückel parameter Bij Middle-range interaction parameters between i and j bij Middle-range adjustable parameters between i and j cij Middle-range adjustable parameters between i and j (c–cs) Absolute super-saturation Cp Heat capacity ds Solvent density (mol/m3) e Electron charge (1.60218×10-19 C) Ea Activation energy GE Excess Gibbs free energy ΔGf
º Standard state Gibbs free energy of formation of the solid I Ionic strength Ix Mole fraction-based ionic strength Iº
x,i Ionic strength for a pure component i K Equilibrium constant Ksp Solubility product Ka Association constant kB Boltzmann constant (1.38066×10−23 J·K-1) kc Rate constant of anhydrite crystallization m Molality (mol/kg of water) M Molarity (mol/L) ni Number of moles of species i NA Avogadro number (6.022×1023 mol-1) P Pressure q Pure-component area parameter r Pure-component size parameter R Gas constant (8.314 J·mol-1·K-1) Sf
º Standard state entropy of formation of the solid

xviii
T Temperature (K) tind Induction time xi Mole fraction of species i
Greek symbols εo Permittivity of vacuum (8.854×10-12 C2·J-1·m-1) εs Dielectric constant of the solvent
iγ Activity coefficient of species i
±γ Mean activity coefficient of the electrolyte
ϕi Segment fraction
iν Stoichiometric coefficient
θi Area fraction
Subscripts aq Aqueous phase s Solid phase g Gaseous phase
Abbreviations AARD Absolute Average Relative Deviation AII–anhydrite Stable (insoluble) anhydrite AIII– or γ–anhydrite Metastable (soluble) anhydrite AH Anhydrous calcium sulphate (anhydrite) DH Calcium sulphate dihydrate (gypsum) HH Calcium sulphate hemihydrate (hemihydrate) HKF Helgeson–Kirkham–Flowers model ICP–OES Inductively coupled plasma–optical emission spectrometer LR Long-range interactions MR Middle-range interactions MSE Mixed solvent electrolyte PAL Pressure acid leaching SEM Scanning electron microscopy SR Short-range interactions XRD X-ray diffraction

1
CHAPTER 1 INTRODUCTION
caling or precipitation fouling is the formation of a solid layer on equipment surfaces or
piping networks. Scale forms primarily on localized hot surfaces or in poorly agitated
regions. It is a persistent problem encountered in many industrial processes such as oil and gas
production, desalination, steam generation operations and hydrometallurgical processes. The
formation of scale is affected by several parameters including temperature, pressure, flow rate,
solution composition and pH. Scaling causes production losses by reducing the volume of
equipment and heat transfer capacity of heat exchangers. It also leads to emergency shutdowns
due to blocked pipelines, increased corrosion and fatigue in metal parts. Periodic shutdowns of
plants for mechanical removal of scales are necessary. Costs involved in maintenance and
frequent shutdowns of these plants are high; hence, scaling prevention measures and techniques
for evaluating scaling tendencies in these processes are of great interest.
1.1 Scale Formation of Calcium Sulphate
Calcium sulphate, with its high scaling potential, is one of the most common inorganic salts
encountered in many industrial processes including wastewater treatment, oil and gas
production, desalination, sulphur dioxide removal from coal-fired power plant flue gas (Lee et
al., 2006; Dathe et al., 2006) and in hydrometallurgical processes (Azimi and Papangelakis,
2010b; Dutrizac and Kuiper, 2008, 2006; Dutrizac, 2002). Calcium sulphate exists as three
different hydrates: dihydrate or gypsum (DH: CaSO4•2H2O); hemihydrate (HH: CaSO4•0.5H2O)
and anhydrite (AH: CaSO4). The stability regions of the CaSO4 hydrates depend on the solution
conditions. Each crystalline phase can be stable, metastable or unstable at certain temperatures
and compositions. Figure 1.1 presents the solubility diagram of CaSO4 in water. As is clear,
gypsum is the stable phase at temperatures below 45–50°C, and above that it transforms into
anhydrite. Hemihydrate is metastable at all temperatures.
The transformation of gypsum (DH) to anhydrite (AH) results in a significant decrease in the
solubility level and makes the prediction and control of calcium sulphate formation complicated.
Therefore, understanding the chemistry of CaSO4 phase equilibria and being able to estimate its
S

2
scaling potential in industrial processes involving electrolytes is of great theoretical
significance and practical importance.
0 50 100 150 200 250 3000.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
CaSO4(s)
CaSO4.2H2O
CaS
O4 so
lubi
lity,
mol
al
Temperature, oC
Gypsum Exp data Anhydrite Exp data Hemihydrate Exp data
CaSO4.0.5H
2O
Figure 1.1 Solubility diagram of CaSO4 in water. Experimental data are from Dutrizac, 2002; Templeton and Rodgers, 1967; Marshall et al., 1964; Sborgi and Bianchi, 1940; Hill and Wills, 1938; Posnjak, 1938; Partridge and White, 1929.
Hydrometallurgical processes, dealing with complex multicomponent solutions, are most
susceptible to scaling. A typical hydrometallurgical process has an ore leaching stage, followed
by solution neutralization. Sulphuric acid is the most common leachant used. Pressure acid
leaching of the concentrate feed is carried out in autoclaves at high temperatures between 150°C
and 250°C. In order to increase the leach solution pH and precipitate the soluble impurities, the
slurry leaving the autoclave is oxidized and subsequently neutralized by limestone (CaCO3).
After filtration, the solution containing base metals is further treated by a number of methods
including solvent extraction and electrowinning to refine and extract the base metals from the
solution. The raffinate continues to a final neutralization stage to further increase pH and
precipitate the remaining metal ions, providing an environmentally safe solution for disposal.
The upstream process solution from the neutralization stage is recycled to the beginning of the
circuit for further usage. A process flow diagram is shown in Figure 1.2.

3
Autoclave
T = 150-250 ºC
Feed tankNeutralization
tank
S1S3 S2
Neutralization tank
H2SO4(aq)CaCO3(s)
T = 90 ºC
Extracted Metal +Solvents
Organic SolventsCaO(s)
CaSO4.2H2O(s) +trace metals
Water and reagents
Residue
Wash waterCounter Current DecantationCCD
Solvent Extraction
Oreconcentrate
T=95 oC
T=50 oC
Figure 1.2 Process flow diagram of pressure acid leaching of ore concentrates.
Calcium enters the sulphate refining electrolytes in different ways. In some cases, the ore itself
contains calcium (Whittington and Muir, 2000). Also, the addition of calcium containing bases,
i.e., lime and limestone, in the neutralization stage increases the concentration of calcium in the
process circuit. In addition, in some refineries, process water is a source of calcium ions.
Calcium sulphate hydrates (DH, HH, and AH) are relatively insoluble and are formed wherever
calcium and sulphate occur together in aqueous solutions. Many processes operate with very
low solution bleeds and as a result, calcium sulphate accumulates in the refining electrolyte.
Furthermore, transformation of gypsum into anhydrite is another common cause of CaSO4 scale
formation, particularly in the solvent extraction circuit or in hot zones of plants (i.e., autoclaves
and heat exchangers).
Calcium sulphate scale formation has the potential to create severe operational problems, as was
the case with the Bulong Nickel/Cobalt Plant. The Bulong plant commenced production in
1999. Shortly after start-up, massive precipitation of gypsum occurred in the nickel solvent
extraction circuit. Furthermore, recycling of process solutions saturated with gypsum at ambient
temperature resulted in significant anhydrite scaling of the pre-heaters (Nofal et al., 2001). The
extent of the problem was such that weekly shutdowns were required for effective scale
removal.

4
In hydrometallurgical processing circuits, temperature and solution composition change over
broad ranges. These variations make it hard to predict the formation of CaSO4 scales in these
processes. Moreover, during operation at higher temperatures, transformation between the
calcium sulphate hydrates has a complex effect on solubility, making the behaviour of calcium
sulphate difficult to predict and control. Therefore, having a thorough understanding of the
phase behaviour of calcium sulphate, and being able to accurately estimate the scaling potential
in such processes is of great practical importance.
1.2 Previous Studies
A review of the literature reveals that no previous theoretical or experimental work has been
undertaken to study the simultaneous effects of coexisting metal sulphates and chlorides on the
solubilities of CaSO4 hydrates over broad temperature and concentration ranges in industrial
systems, particularly in laterite pressure acid leach (PAL) solutions.
1.2.1 Experimental Studies of Calcium Sulphate Solubilities
A considerable amount of experimental work has been conducted to study calcium sulphate
solubilities under atmospheric pressure from 25°C to 95°C in water or in H2SO4 and HCl acidic
solutions as well as in multicomponent metal sulphate–chloride systems (Farrah et al., 2007;
Dutrizac and Kuiper, 2006; Li and Demopoulos, 2002, 2005, 2006a; Dutrizac, 2002; Block and
Waters, 1968; Zdanovskii et al., 1968; Bock, 1961; Hill and Wills, 1938; Posnjak, 1938; Hulett
and Allen, 1902).
Several experimental studies have also been undertaken at elevated temperatures, up to 350°C,
on the solubility of calcium sulphate in water or in H2SO4 media as well as in ternary or
quaternary solutions containing NaCl, Na2SO4, and MgCl2 (Blount and Dickson, 1969; Furby et
al., 1968; Marshall and Slusher, 1966, 1968; Templeton and Rodgers, 1967; Marshall and Jones,
1966; Marshall et al., 1964; Partridge and White, 1929). However, no previous work has been
carried out to account for the effect of metal sulphates/chlorides on calcium sulphate hydrates
solubilities in multicomponent solutions over a wide temperature range, particularly at elevated
temperatures, under acidic conditions. A comprehensive list of the related literature on the
solubilities of calcium sulphate hydrates along with the range of conditions investigated is
presented in Appendix A.

5
1.2.2 Theoretical Studies of Calcium Sulphate Solubilities
In terms of theoretical modelling, most of the previous studies focused on CaSO4 solubility in
water or in ternary and quaternary aqueous solutions containing H2SO4, MgSO4, Na2SO4, etc.
Marshall and Slusher (1966) proposed an empirical model based on an extended Debye-Hückel
expression with only one parameter, referred to as “ion size” parameter. In this model, the
variation of the solubility product with ionic strength and temperature was obtained by assuming
complete dissociation of CaSO4 to Ca2+ and SO42- in the solution. This empirical model was
shown to predict the solubility of gypsum at 60°C and 95°C, and of hemihydrate and anhydrite
at 100–200°C in synthetic sea salt solutions containing CaCl2, KC1, MgC12, MgSO4, NaC1, and
Na2SO4 over a concentration range of 0 to 6.0 molal (Marshall and Slusher, 1968).
A computational method based on the Guggenheim–Davies correlation (Guggenheim, 1955) for
activity coefficient model was proposed by Tanji and Doneen (1966) to predict gypsum
solubility in aqueous salt solutions of NaCl, MgCl2, CaCl2, and MgSO4 over a concentration
range of 0–1.5 molal. The main purpose of this study was to evaluate the scaling potential of
gypsum in semi-arid and arid regions, where gypsum occurs in agricultural soils and contributes
to salinity. Moreover, gypsum is sometimes added as a soil amendment for reclamation of sodic
soil and as a water amendment to reduce the sodium content of irrigation waters. This improves
soil permeability by increasing electrolyte concentration.
A thermodynamic model has been developed by Barba et al. (1982) to describe the solubility of
gypsum in saline water. In this model, the excess Gibbs energy consists of three terms of the
Debye–Hückel limiting law, Born model contribution, and the NRTL model. The first two terms
account for the long-range interactions between charged species, whereas, the last term accounts
for short-range interactions between non-charged species in the solution. This model is capable
of predicting gypsum solubility in seawater at 25°C using only binary parameters. However, for
concentrated multicomponent solutions with high sodium chloride contents, a new set of binary
parameters must be regressed to improve the calculation.
Zemaitis et al. (1986) have applied a theoretical thermodynamic method to calculate the
solubility of gypsum in NaCl, CaC12 and HC1 aqueous solutions to evaluate various activity
coefficient models such as Bromley (Bromley, 1972, 1973), Meissner (Kusik and Meissner,

6
1978) and Pitzer (Pitzer, 1972, 1973, 1980). In their calculations, complete dissociation was
assumed for CaSO4 and other electrolytes. It was shown that the prediction results for gypsum
solubility in multicomponent electrolyte solutions based on the interaction parameters obtained
in the gypsum–water binary system were not accurate and additional parameters were required
to improve the predictions in such systems.
Demopoulos et al. (1987) also used the Meissner model (Kusik and Meissner, 1978) to simulate
the gypsum solubility in concentrated aqueous NaCl solutions at 25ºC. In their study, a new
Meissner parameter for CaSO4 was used. The model was shown to successfully predict the
solubility of gypsum in the systems studied.
Arslan and Dutt (1993) developed a computer program to determine the solubility of gypsum in
various salt solutions containing Ca, Mg, Na, Cl, and SO4 at 25ºC. The Guggenheim and Davis
activity coefficient model (Guggenheim, 1955) based on extended Debye–Hückel theory, was
employed in their calculations. In their study, the association between Ca2+ and SO42- and the
formation of calcium sulphate neutral species, CaSO4(aq), was taken into account. By regressing
the available experimental data, a new set of parameters for the activity model used was
calculated. It was shown that the model can acceptably predict gypsum solubility only at low
concentrations (less than 1 molal) of solutes.
Most of the studies indicated above assumed complete electrolyte dissociation. This assumption
is based on the non-speciation approach by utilizing different correlations for the activity
coefficient, such as the extended Debye-Hückel and Guggenheim-Davies expressions as well as
the Bromley, Meissner or Pitzer models. The non-speciation approach usually gives comparable
results to those of the speciation approach for simple electrolyte systems. However, in
multicomponent systems with complex solution chemistries, speciation becomes an important
factor in the prediction of solid solubilities (Anderko et al., 2002). This is attributable to the fact
that the distribution of species in multicomponent systems may be different from that in simple
single-salt systems, and this may in turn affect the solubility along with other properties.
The speciation modelling approach was used by Adams (2004) to predict the solubility of
gypsum and its scaling potential in sulphate systems over the temperature range of 25–90ºC.
The Mixed Solvent Electrolyte (MSE) (Wang et al., 2002, 2004, 2006) activity coefficient
model of the OLI® software was employed. This model was capable of predicting the solubility

7
of gypsum over the indicated temperature range. However, other forms of calcium sulphate
such as anhydrite and hemihydrate were not taken into account in the study.
Li and Demopoulos (2002, 2005, 2006a) measured the solubilities of all the calcium sulphate
hydrates (DH, HH, and AH) in HCl and in HCl–based aqueous solutions containing various
metal chloride salts, such as AlCl3, CaCl2, FeCl3, MgCl2, and NaCl over the temperature range
of 10–100ºC. Subsequently, they used the experimental data to develop a model for the
solubility of calcium sulphate in multicomponent aqueous chloride solutions (Li and
Demopoulos, 2006b, 2007). The Bromley–Zemaitis activity coefficient model (Zemaitis, 1980)
was employed and the regression of the experimental data was carried out with the aid of the
OLI® software package. Their model successfully estimated the solubility of all calcium
sulphate hydrates in mixed chloride HCl-containing solutions up to 100ºC.
The above review indicates that most of the previous studies focused on the modelling of
gypsum solubility under atmospheric pressure below 100°C. Although mixed multicomponent
systems are present in various industrial processes including hydrometallurgical solutions, no
theoretical work had been formally undertaken to model the solubilities of the calcium sulphate
hydrates in such systems.
1.3 Objectives
The overall aim of this work is to investigate the solution chemistry and phase equilibria of
calcium sulphate hydrates (gypsum, hemihydrate and anhydrite), both theoretically and
experimentally, in multicomponent hydrometallurgical solutions containing various minerals
over a wide temperature and composition range. The ultimate goal is to identify systematic
trends in solubility behaviour of calcium sulphate hydrates, with an aim to provide practical
guidelines that might reduce calcium sulphate scaling in such processes.
The specific objectives are: (1) to model the chemistry (solubility) of calcium sulphate hydrates
(DH, HH, and AH) to identify the conditions that might lead to scale formation; (2) to perform
systematic solubility measurements of calcium sulphate in laterite pressure acid leach (PAL)
solutions over the temperature range of 25–250ºC; (3) to identify the mechanism of gypsum–
anhydrite transformation and to investigate the effect of temperature, acidity, and addition of

8
seeds on the transformation kinetics; (4) to propose means of mitigating, or at least controlling,
calcium sulphate scaling in such processes, particularly inside autoclaves.
1.4 Thesis Overview
The present thesis is composed of a number of chapters, which are structured as follows:
• Chapter 2 presents the chemical modelling strategy utilized in this work to develop a
database for the Mixed Solvent Electrolyte (MSE) model of the OLI software, capable of
accurately predicting calcium sulphate solubility and scaling potential during the
neutralization stage of zinc sulphate hydrometallurgical processes.
• Chapter 3 focuses on further extending the database developed in Chapter 2, such that it
is applicable to complex multicomponent chloride–sulphate solutions containing CaSO4,
CaCl2, Fe2(SO4)3, FeCl3, H2SO4, HCl, LiCl, MgSO4, MgCl2, Na2SO4, NaCl, and NiSO4.
The database, utilized by the MSE model, provides a valuable tool for predicting the
solubility of calcium sulphate in the neutralization stage of nickel sulphate-chloride
processing solutions of the Voisey’s Bay plant from 20°C to 95°C.
• Chapter 4 describes the experimental measurements of the solubility of gypsum at 25–
90°C and that of anhydrite at 150–250°C in simulated laterite pressure acid leach (PAL)
solutions. In this chapter, the predictive capacity of the model, utilizing the developed
database, was tested against the measured experimental data for both solids over wide
ranges of composition and temperature.
• Chapter 5 focuses on the transformation of gypsum into anhydrite. The effects of
temperature, addition of acid and sulphate/chloride salts as well as anhydrite seeding on
the transformation kinetics are investigated. Based on the results obtained, a mechanism
for the gypsum–anhydrite transformation is proposed in this chapter.
• Finally, Chapter 6 summarizes the major conclusions drawn from this work; Chapter 7
outlines recommendations for future work.
The thesis was prepared based on the following refereed journal publications, some of which
have already been published in the course of the investigation; others are either in press or

9
submitted. In the beginning of each chapter, there is a statement indicating the paper(s) based
on which the chapter is constructed.
Azimi G., Papangelakis V.G., Dutrizac J.E., 2007. Modelling of calcium sulphate solubility in concentrated multicomponent sulphate solutions. Fluid Phase Equilibria, 260(2), 300–315.
Azimi G., Papangelakis V.G., Dutrizac J.E., 2008. Development of an MSE-based chemical model for the solubility of calcium sulphate in mixed chloride-sulphate solutions. Fluid Phase Equilibria, 266, 172–186.
Azimi G., Papangelakis V.G., 2010a. Thermodynamic modeling and experimental measurement of calcium sulphate solubility in complex aqueous solutions. Fluid Phase Equilibria, 290, 88–94.
Azimi G., Papangelakis V.G., Dutrizac J.E., 2010. Development of a chemical model for the solubility of calcium sulphate in zinc processing solutions. Can. Met. Quarter. 49(1), 1–8.
Azimi G., Papangelakis V.G., 2010b. Gypsum and anhydrite solubility in simulated laterite pressure acid leach solutions up to 250°C. Hydrometallurgy, in press.
Azimi G., Papangelakis V.G., 2010c. Mechanism and kinetics of transformation between calcium sulphate hydrates in aqueous electrolyte solutions. Crystal Growth & Design, submitted.

10
CHAPTER 2 MODELLING OF CALCIUM SULPHATE SOLUBILITY IN MULTICOMPONENT SULPHATE SOLUTIONS
his chapter presents the chemical modelling strategy utilized in this work to develop a
database for the Mixed Solvent Electrolyte (MSE) model of the OLI software, capable of
accurately predicting calcium sulphate solubility and scaling potential during the neutralization
stage of zinc sulphate hydrometallurgical processes. The present chapter is based on the
following publications:
- Azimi G., Papangelakis V.G., Dutrizac J.E., 2007. Fluid Phase Equilibria, 260 (2), 300–315.
- Azimi G., Papangelakis V.G., Dutrizac J.E., 2010. Can. Met. Quarter. 49 (1), 1–8.
2.1 Introduction
Most of the world’s zinc metal is produced by hydrometallurgical processes, in which zinc
concentrates are leached in sulphuric acid media. The resulting zinc sulphate solution is purified
and zinc metal is produced electrolytically. The feed used in the zinc industry usually contains
calcium, particularly when concentrates originate from sedimentary ores containing calcite
(CaCO3) or dolomite (CaMg(CO3)2) (Dutrizac, 2002).
Calcium sulphate scale formation occurs during acid leaching or during the neutralization of
free sulphuric acid where sulphates are removed from the solution by the addition of calcium-
containing bases such as lime or limestone. Depending on the process conditions, such as pH or
temperature, calcium sulphate can form three different hydrates: dihydrate (DH: CaSO4•2H2O),
hemihydrate (HH: CaSO4•0.5H2O), and anhydrite (AH: CaSO4). Although, the formation of
CaSO4 is beneficial in that it restricts the accumulation of calcium and sulphate in the
processing circuit, it is also undesirable because CaSO4 scale formation results in reducing the
production capacity and process efficiency because of decreased volume of the equipment and
reduced heat transfer capacity, blocked pipelines and reduction of material flow. The
precipitation of calcium sulphate in solvent extraction operations could create serious crud
formation problems (Dutrizac, 2002).
T

11
In zinc processing hydrometallurgy, temperature and concentration of sulphuric acid and zinc
sulphate change over broad ranges. These variations make it hard to predict the formation of
CaSO4 in these solutions. As a result, developing a chemical model to describe and predict the
behaviour of CaSO4 in these processes is highly desirable. The purpose of the present chapter is
to develop a chemical model for estimating the solubility of calcium sulphate hydrates (DH,
HH, and AH) in multicomponent sulphate solutions including simulated zinc sulphate
processing solutions for which experimental data are available in the literature (Dutrizac, 2002).
A review of published modelling studies shows that no previous work has been formally
undertaken to study the simultaneous effects of coexisting metal sulphates on the solubility of
the three phases of CaSO4 over broad ranges of temperature and concentration, particularly in
industrial solutions. As indicated in the previous chapter, most of the previous studies attempted
to theoretically model the solubilities of calcium sulphate compounds in water or in ternary and
quaternary aqueous solutions containing H2SO4, MgSO4, Na2SO4, etc. (Marshall and Slusher,
1966; Barba et al., 1982; Arslan and Dutt, 1993).
In this chapter, a new database for the Mixed Solvent Electrolyte (MSE) model of the OLI®
software (Wang et al., 2002, 2004, 2006) was developed by regressing binary activity, heat
capacity, and solubility data, as well as ternary solubility data. The model interaction parameters
for free calcium ions and associated calcium sulphate neutral species with other dominant
species in the solution were determined. The predictive capacity of the model containing new
interaction parameters was tested with reference to the solubility measurements made in
simulated zinc sulphate processing solutions containing ZnSO4, H2SO4, MgSO4, MnSO4,
Fe2(SO4)3, Na2SO4, and (NH4)2SO4 (Dutrizac, 2002). The details of the procedures followed are
described in the next section and are also available in the literature (Azimi et al., 2007, 2010).
2.2 Modelling Methodology
2.2.1 Chemical Equilibria
The solubility of calcium sulphate hydrates is equal to the sum of the molalities of the free
calcium ion (Ca2+) and the associated calcium sulphate neutral species (CaSO4(aq)).
Consequently, the solubility of calcium sulphate hydrates is governed by following equilibria:

12
OnHSOCaOnHCaSO s 224
2)(24 . ++= −+ (2.1)
)(424
2aqCaSOSOCa =+ −+ (2.2)
where n = 0, 0.5 and 2 correspond to anhydrite, hemihydrate and dihydrate, respectively. The
thermodynamic equilibrium constants for reactions (2.1) and (2.2) are:
nwCaSOSOCa
nwSOSOCaCaSP ammammK )())(())()(( 2
)( 424
224
24
22 ±−+−−++ == γγγ (2.3)
))(( 24
24
22
)(4)(4
−−++
=SOSOCaCa
CaSOCaSOa mm
mK aqaq
γγ
γ (2.4)
where SPK is the solubility product, Ka is the association constant of calcium sulphate neutral
species, m is the molality of calcium ions, sulphate ions and calcium sulphate neutral species
(mol·kg-1), )( 4CaSO±γ is the mean activity coefficient of CaSO4, )(4 aqCaSOγ is the activity coefficient
of calcium sulphate neutral species and wa is the activity of water.
After re-arranging equations (2.3) and (2.4), the molalities of the free calcium ion, Ca2+, and the
calcium sulphate neutral species, CaSO4(aq), become:
nwCaSOSO
SPCa am
Km)(2
)( 424
2
±−
+ = γ (2.5)
)(4
24
24
22
)(4
))((
aq
aqCaSO
SOSOCaCaaCaSO
mmKm
γ
γγ −−++
= (2.6)
The solubility of calcium sulphate hydrates is equal to the sum of the molalities of free calcium
ions and associated calcium sulphate neutral species, as follows:
42][ CaSOCatotal mmCa += + (2.7)
After substituting +2Cam and
)(4 aqCaSOm in equation (2.7), the solubility of calcium sulphate
hydrates is:

13
nwCaSO
aSPn
wCaSOSO
SP
CaSO
SOSOCaCaa
nwCaSOSO
SPtotal
aKK
amK
mmK
amKCa
aq
aq
⋅+
⋅⋅=
+=
±
±
−
−−++
−
)(4424
)(4
24
24
22
424
2)(
2)(
))((][
γγ
γ
γγ
γ (2.8)
To calculate the solubility of calcium sulphate hydrates, the solubility product (Ksp) and the
association constant of calcium sulphate neutral species (Ka) as well as the mean activity
coefficient of CaSO4, the activity coefficient of calcium sulphate neutral species and the activity
of water need to be determined.
2.2.2 Equilibrium Constant
To obtain the equilibrium constants in Eqs. (2.3) and (2.4) at temperature T and pressure P, the
standard state chemical potentials of products and reactants must be known. These data are
widely available in standard thermodynamic compilations. The Helgeson–Kirkham–Flowers
(HKF) model, developed by Helgeson et al. (1981) and revised by Tanger and Helgeson (1988),
is embedded in the OLI software to calculate the standard state thermodynamic properties at
high temperatures and pressures, up to 1000°C and 5 kbar. The general equation is as follows:
),,,,,,,,( 214321, ωccaaaaPTXX PT =o (2.9)
where X denotes a thermodynamic property such as chemical potential (μ), partial molal
enthalpy (H), entropy (S), volume (V), or heat capacity (Cp), and ω,,,,,, 214321 ccaaaa are HKF
parameters.
2.2.3 Activity Coefficient Model
The activity coefficient is a parameter which accounts for the non-ideality (excess properties) of
electrolyte solutions, and is defined by the excess Gibbs free energy of the solution, GE:
ijnPTi
E
i nRTG
≠
⎟⎟⎠
⎞⎜⎜⎝
⎛∂
∂=
,,
)/(lnγ (2.10)
where ni is the number of moles of the solution constituents (species i), and j is any other
species. The pursuit of an expression for GE to calculate γ has been ongoing for decades.

14
Numerous models have been proposed and some of them have been incorporated into
commercial software and applied in industry (Liu et al., 2005).
The more recently developed Mixed Solvent Electrolyte (MSE) model (Wang et al., 2002, 2004,
2006) is capable of accurately calculating the thermodynamic properties of electrolyte solutions
in water and/or organic solvent(s) over the entire concentration range from infinite dilution to
pure fused salt electrolytes. The application of the MSE model within the OLI® software
platform for hydrometallurgical processes has already proven its efficiency and accuracy in
predicting the properties of multicomponent solutions (Liu et al., 2005; Liu and Papangelakis,
2006; Azimi et al., 2006, 2007, 2010).
The most important advantage of the MSE model over other speciation-based models, such as
the Bromley–Zemaitis model, is that the MSE model treats the electrolyte systems to a limit of
no water. That is, it is valid from infinite dilution to the pure solute limit which is the situation
for molten salts or pure acids (e.g., HF or H2SO4) or even pure bases (e.g., NaOH). The MSE
model treats non-electrolyte systems of any composition (mole fractions from 0 to 1 for any
component), but the Bromley–Zemaitis model considers only aqueous systems in a limited
concentration range (Wang et al., 2002). Finally, the MSE model requires fewer parameters to
model a system compared to the Bromley–Zemaitis model, because it has a more powerful ion
interaction handling capability.
In this work, the MSE activity coefficient model (Wang et al., 2002, 2004, 2006), embedded in
the OLI® software platform, is employed to establish the desired chemical model for
investigating the chemistry (solubility) of calcium sulphate in multicomponent electrolyte
solutions. The MSE model was established by combining an excess Gibbs energy model for
mixed-solvent electrolyte systems with a comprehensive treatment of chemical equilibria. In this
framework, the excess Gibbs energy is expressed as (Wang et al., 2002, 2004):
RTG
RTG
RTG
RTG E
SREMR
ELR
E
++= (2.11)
where ELRG represents the contribution of long-range electrostatic interactions caused by the
Coulomb electrostatic forces and mainly describes the direct effect of charge interactions, EMRG
accounts for the middle-range ionic interactions resulting from the indirect effect of charge

15
interactions such as charge-dipole interactions and charge-induced dipole interactions not
included in the long-range term, and ESRG is the short-range contribution term resulting from
intermolecular interactions which are identical between non-electrolyte species; the term is
calculated by the UNIQUAC model.
The long-range interaction contribution is obtained from the Pitzer–Debye–Hückel formula
expressed as follows (Pitzer, 1980):
⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛
+
+⎟⎠
⎞⎜⎝
⎛−=∑∑
iixi
xxx
ii
ELR
IxIIAn
RTG
])(1[1
ln42/1
,
2/1
oρρ
ρ (2.12)
where the sum is over all species, Ix is the mole fraction-based ionic strength, Iºx,i is the ionic
strength for a pure component i, i.e., 2x,i 2
1I iZ=o , ρ is an empirical constant related to the hard-
core collision diameter and Ax is the Debye–Hückel constant which is given by:
2/3
0
22/1
4)2(
31
⎟⎟⎠
⎞⎜⎜⎝
⎛=
TkedNA
BsSAx επε
π (2.13)
where ds and εs are the molar density and the dielectric constant of the solvent, respectively. All
calculations related to the long-range contribution are handled by the OLI software, and there is
no adjustable parameter in the long-range contribution.
The middle-range interaction is the contribution of indirect effects of charge interactions on the
excess Gibbs free energy. This term is calculated from an ionic-strength dependent, second
coefficient-type expression:
∑∑∑ ⎟⎠
⎞⎜⎝
⎛−=
i jxijji
ii
EMR IBxxn
RTG )( (2.14)
where x is the mole fraction of the species and ijB is a binary interaction parameter between the
species i and j (ion or molecule) and is similar to the second virial coefficient, which is a
function of ionic strength according to the following equation:

16
)()01.0exp(.)( jiijxijijxij BBIcbIB =+−+= (2.15)
where bij and cij are adjustable parameters. In general, the bij and cij parameters are calculated as
functions of temperature:
TbTbT
bTbbb ijij
ijijijij ln,4
2,3
,2,1,0 ++++= (2.16)
TcTcT
cTccc ijij
ijijijij ln,4
2,3
,2,1,0 ++++= (2.17)
where ijkb , (k=0,…,4) and ijkc , (k=0,…,4) are adjustable parameters between species i and j that
can be obtained by regressing experimental data such as the mean activity coefficient, activity of
water, osmotic coefficient, heat capacity and solubility.
In the MSE model, the UNIQUAC model is used to account for the short-range interactions.
The excess Gibbs energy in the UNIQUAC model is calculated as sum of a combinatorial and a
residual term:
RT
GRT
GRT
G Eresidual
Eialcombinator
EUNIQUAC += (2.18)
The expression for GE (combinatorial) contains two composition variables, i.e., the average area
fraction (θ) and the average segment fraction (ϕ):
⎥⎦
⎤⎢⎣
⎡+⎟
⎠
⎞⎜⎝
⎛= ∑ ∑∑i i i
iii
i
ii
ii
Eialcombinator xqZ
xxn
RTG
φθφ ln
2ln (2.19)
The expression for GE (residual) contains only one composition variable (the average area
fraction θ):
⎥⎦
⎤⎢⎣
⎡⎟⎠
⎞⎜⎝
⎛−= ∑ ∑∑
i jijjii
ii
Eresidual xqnRT
G )ln( τθ (2.20)

17
where
∑=
jjj
iii xq
xqθ (2.21)
∑=
jjj
iii xr
xrφ (2.22)
)exp(RTaij
ij −=τ (2.23)
In the above equations, qi and ri are the surface and size parameters for species i, respectively,
which are equal to 1.0 for ionic species; for inorganic neutral species, they are assigned to be
equal to those of water (q = 1.4; r = 0.92), and for organic species their values can be found
from the literature (Abrams and Prausnitz, 1975). Z is the coordination number with a value of
10, xi is the mole fraction of species i, θi the average area fraction and ϕi the average segment
fraction, which are calculated using Equations (2.21) and (2.22). In the UNIQUAC model, there
are only two binary interaction parameters (aij and aji) between species i and j, which are only
functions of temperature:
2)2()1()0( TaTaaa ijijijij ++= (2.24)
2)2()1()0( TaTaaa jijijiji ++= (2.25)
The regression parameters in the MSE framework are those of the UNIQUAC and middle-range
parameters. The UNIQUAC parameters are primarily for non-electrolyte species and the
middle-range parameters are primarily for ion–ion and ion–molecule species. In this work, ions
are the dominant species in the systems of interest, and as a result, only middle-range
interactions between dominant ions or between ions and molecules have been considered.
2.2.4 Evaluation of the Model Parameters
Evaluation of the model parameters and validation of the regressed parameters requires a large
amount of experimental data of various types, such as the activity coefficients in completely

18
dissociated aqueous systems, the activity of water, the solubility of salts in water or in mixed
solvent solutions and heat capacities. Model parameters are determined by utilizing available
experimental data in binary and ternary systems, and minimizing differences between
experimental and calculated properties. Validation of the regressed parameters was
accomplished by comparing model results with experimental data in multicomponent systems
beyond the range used for parameterization. Figure 2.1 shows the modelling algorithm used in
this work.
Figure 2.1 Chemical modelling algorithm applied in this work.
2.2.5 Standard State Gibbs Free Energy and Entropy of Formation
The adjustment of the standard state Gibbs free energy (ΔGfº) and entropy of formation (Sf
º) is a
practical method to determine the optimum solubility products of various solids, which has
previously been used by Wang et al. (2004, 2006). Since the exponent function of the Gibbs free
energy of reaction is used to calculate the reaction equilibrium constant (Keq), the error in Keq
increases very rapidly with an error in Gibbs free energy of reaction. It has been shown by Rafal
et al. (1994) that the ratio of “error” Keq to “true” Keq is 30 when there is a –2 kcal/mol error in
Gibbs free energy of reaction. Such an error in Keq would propagate into error in the calculated
solubility of the solids. For example, the ratio of “error” solubility to “true” solubility is 5.4 for
–2 kcal/mol error in Gibbs free energy of reaction (Rafal et al., 1994). Therefore, small
adjustments of ΔGfº and Sf
º of solids would result in remarkable improvements. This approach

19
gives better extrapolation behavior for the model with respect to temperature, as compared to
fitting empirical parameters in the solubility product equation, the practice which has been used
in previous studies (Li and Demopoulos, 2007, 2006b; Adams, 2004). In the present work, ΔGfº
and Sfº of several solids were adjusted by fitting the experimental solubility data over the entire
temperature range; the regressed values are presented in Appendix B (Table B.2).
2.3 Results and Discussion
Zinc processing solutions typically contain ZnSO4, H2SO4, Fe2(SO4)3, MgSO4, Na2SO4, and
(NH4)2SO4. In order to model the solubility of calcium sulphate hydrates in such solutions, the
solubility of various metal sulphates in water (binary systems) was first verified to determine
whether the default databank (ver. 8.1.3) of the OLI software is capable of reproducing the
available experimental data, or whether it was necessary to perform an estimation of the
parameters through the OLI built-in regression feature. Then, the solubility of CaSO4 in ternary
systems of CaSO4–MSO4–H2O, where M is Zn, Fe(III), Mg, Na, and Mn, and in the system of
CaSO4–H2SO4–H2O was investigated. In most cases, interactions between the various dominant
species are significant and need to be taken into account. Therefore, the MSE interaction
parameters were regressed for better performance of the model. A list of the various systems
studied in this work along with the typical range of conditions investigated is given in Table 2.1.
The obtained model parameters as well as regressed values for the standard state Gibbs free
energy and entropy of the different solids studied are presented in Appendix B.
Table 2.1–Binary and ternary systems studied for the parameterization purpose
System Data type Temperature range, ºC Solid phase
MnSO4-H2O γ± - aw - Cp - solubility 0-180 MnSO4•7H2O, MnSO4•5H2O, MnSO4•1H2O
MgSO4-H2O γ± - aw - solubility 0-250 MgSO4•7H2O, MgSO4•6H2O, MgSO4•1H2O
Na2SO4-H2O γ± - aw - solubility 0-240 Na2SO4•10H2O, Na2SO4
ZnSO4-H2O γ± - aw - solubility 0-300 ZnSO4•7H2O, ZnSO4•6H2O, ZnSO4•1H2O
NiSO4-H2O γ± - solubility 0-300 NiSO4•7H2O, NiSO4•6H2O, NiSO4•1H2O
CaSO4-H2O solubility 0-300 CaSO4•2H2O, CaSO4•0.5H2O, CaSO4 Bin
ary
syst
ems
Fe2(SO4)3-H2O γ± - aw 25 –

20
System Data type Temperature range, ºC Solid phase
CaSO4-(NH4)2SO4-H2O solubility 25-100 CaSO4•2H2O, CaSO4
Al2(SO4)3-H2SO4-H2O solubility 25-60 Al2(SO4)3•16H2O
Fe2(SO4)3-H2SO4-H2O solubility 25-140 Fe2(SO4)3•9H2O, Fe2(SO4)3•6H2O, Fe2(SO4)3
CaSO4-MnSO4-H2O solubility 25-100 CaSO4•2H2O
CaSO4-MgSO4-H2O solubility 25-175 CaSO4•2H2O, CaSO4•0.5H2O, CaSO4
CaSO4-Na2SO4-H2O solubility 25-300 CaSO4•2H2O, CaSO4
NiSO4-H2SO4-H2O solubility 20-300 NiSO4•6H2O, NiSO4•1H2O
MnSO4-H2SO4-H2O solubility 25-65 MnSO4•1H2O
CaSO4-H2SO4-H2O solubility 25-300 CaSO4•2H2O, CaSO4•0.5H2O, CaSO4
CaSO4-ZnSO4-H2O solubility 25-200 CaSO4•2H2O, CaSO4•0.5H2O, CaSO4
CaSO4-NiSO4-H2O solubility 25-175 CaSO4•2H2O, CaSO4
Ter
nary
syst
ems
ZnSO4-H2SO4-H2O solubility 15-70 ZnSO4•7H2O, ZnSO4•6H2O, ZnSO4•1H2O
A list of multicomponent zinc processing systems used for validation purpose is summarized in
Table 2.2. Predicted model results, utilizing the newly regressed parameters obtained in the
binary and ternary systems, are in good agreement with these data, without additional fitting
over the temperature range studied, i.e., 25–90ºC. The Absolute Average Relative Deviations
(AARD%1) between the experimental data and predicted results obtained from the model are
also presented Table 2.2.
Table 2.2–Multicomponent systems studied for validating the model along with AARD% between experimental data and predicted results
System Temperature Range, ºC Solid Phase AARD%
CaSO4-ZnSO4-H2SO4(0.1M)-H2O 25-90 CaSO4•2H2O 5.8 CaSO4-H2SO4-ZnSO4(1.5M)-H2O 25-90 CaSO4•2H2O 5.6 CaSO4-MgSO4-ZnSO4(1.15M)-H2SO4(0.1M)-H2O 25-90 CaSO4•2H2O 5.2 CaSO4-MgSO4-ZnSO4(1.15M)-H2SO4(0.3M)-H2O 25-90 CaSO4•2H2O 8.6 CaSO4-Fe2(SO4)3-ZnSO4(1.15M)-H2SO4(0.3M)-H2O 25-90 CaSO4•2H2O 6.4 CaSO4-Na2SO4-ZnSO4(2.5M)-MgSO4(0.41M)-MnSO4(0.18M)-H2SO4(pH=3.8)-H2O 25-90 CaSO4•2H2O 5.9
CaSO4-H2SO4-ZnSO4(2.5M)-MgSO4(0.41M)-MnSO4(0.18M)-H2O 25-90 CaSO4•2H2O 6.5 CaSO4-(NH4)2SO4-ZnSO4(2.5M)-MgSO4(0.41M)-MnSO4(0.18M)-H2SO4(pH=3.8)-H2O 25-90 CaSO4•2H2O 7.0
1
∑−
=NP
i valueExp
valueCalculatedvalueExp
NPAARD
.
.100(%) , NP: total number of experimental points

21
2.3.1 Binary Systems (Metal Sulphate–H2O)
2.3.1.1 CaSO4–H2O System
The solubility of CaSO4 solid phases (dihydrate, hemihydrate, and anhydrite) has been
extensively measured (dihydrate: (Dutrizac, 2002; Power et al., 1966; Marshall and Slusher,
1966; Marshall et al., 1964; Posnjak, 1938; Hill and Wills, 1938; Hill and Yanick, 1935; Hulett
and Allen, 1902); hemihydrate: (Sborgi and Bianchi, 1940; Partridge and White, 1929);
anhydrite: (Templeton and Rodgers, 1967; Marshall et al., 1964; Bock, 1961; Posnjak, 1938;
Straub, 1932; Partridge and White, 1929)). Most of the measurements are in fairly good
agreement with each other. These experimental solubility data were used to verify the OLI®
default databank.
Although the solubility of CaSO4 dihydrate (gypsum) in H2O at 0–110ºC (Figure 2.2) can be
calculated accurately with the OLI default database (version 8.1.3) using the MSE model, there
is no data for hemihydrate (CaSO4.0.5H2O) in the OLI default database. Therefore, literature
solubility data (Sborgi and Bianchi, 1940; Partridge and White, 1929) were used to adjust the
standard state Gibbs free energy, and entropy of formation of the solid as a function of
temperature, up to 200ºC. The regressed solubility curve is presented in Figure 2.3.
0 20 40 60 80 100 1200.010
0.011
0.012
0.013
0.014
0.015
0.016
0.017
Gyp
sum
Sol
ubili
ty, m
olal
Temperature, oC
OLI default database Exp. data
Figure 2.2 Gypsum solubility in H2O vs. temperature. Experimental data are from Dutrizac, 2002; Power et al., 1966; Marshall and Slusher, 1966; Marshall et al., 1964; Posnjak, 1938; Hill and Wills, 1938; Hill and Yanick, 1935; Hulett and Allen, 1902. The curve is determined from the OLI default database.
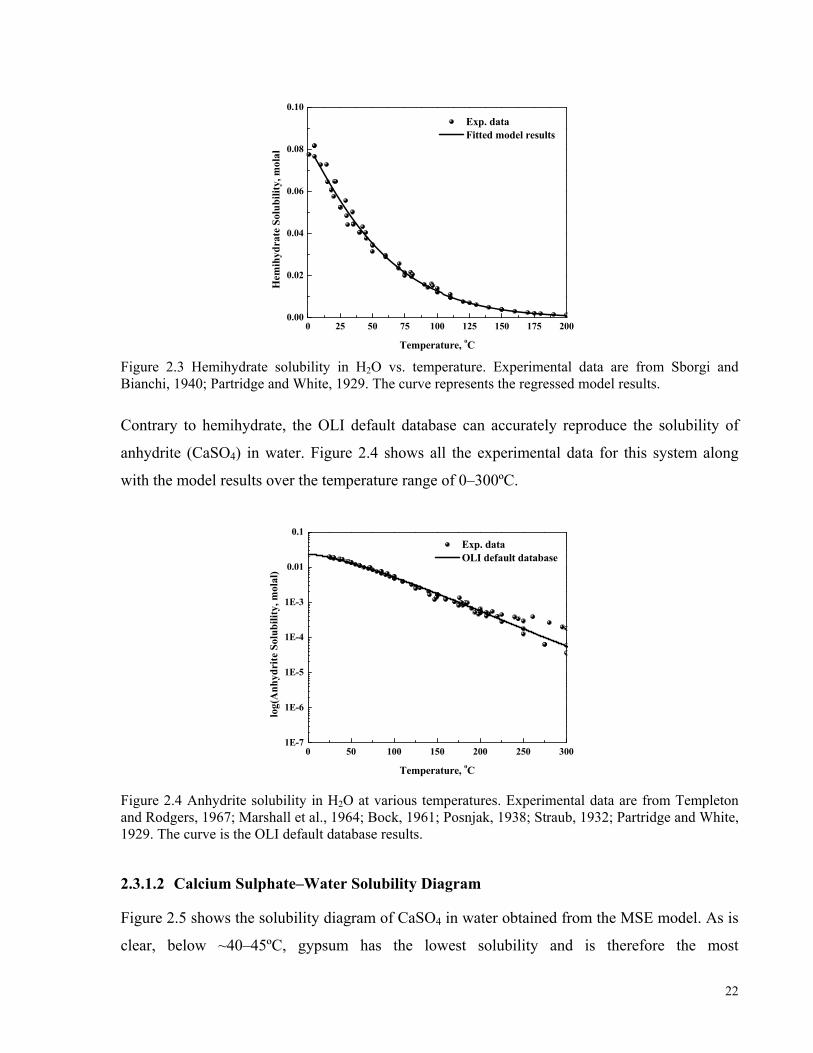
22
0 25 50 75 100 125 150 175 2000.00
0.02
0.04
0.06
0.08
0.10
Hem
ihyd
rate
Sol
ubili
ty, m
olal
Temperature, oC
Exp. data Fitted model results
Figure 2.3 Hemihydrate solubility in H2O vs. temperature. Experimental data are from Sborgi and Bianchi, 1940; Partridge and White, 1929. The curve represents the regressed model results.
Contrary to hemihydrate, the OLI default database can accurately reproduce the solubility of
anhydrite (CaSO4) in water. Figure 2.4 shows all the experimental data for this system along
with the model results over the temperature range of 0–300ºC.
0 50 100 150 200 250 3001E-7
1E-6
1E-5
1E-4
1E-3
0.01
0.1
log(
Anh
ydri
te S
olub
ility
, mol
al)
Temperature, oC
Exp. data OLI default database
Figure 2.4 Anhydrite solubility in H2O at various temperatures. Experimental data are from Templeton and Rodgers, 1967; Marshall et al., 1964; Bock, 1961; Posnjak, 1938; Straub, 1932; Partridge and White, 1929. The curve is the OLI default database results.
2.3.1.2 Calcium Sulphate–Water Solubility Diagram
Figure 2.5 shows the solubility diagram of CaSO4 in water obtained from the MSE model. As is
clear, below ~40–45ºC, gypsum has the lowest solubility and is therefore the most

23
thermodynamically stable phase. The transition point of gypsum to anhydrite lies at 40±5ºC,
and that of gypsum to hemihydrate lies at 99±5ºC.
0 50 100 150 200 250 3000.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
gypsum
anhydrite
Cal
cium
Sul
phat
e So
lubi
lity,
mol
al
Temperature, oC
hemihydrate
Figure 2.5 Solubility diagram of CaSO4 in H2O. The solid and dashed curves show the stable and metastable phases, respectively, at a given temperature.
In the region between these two temperatures, gypsum is metastable, although the degree of
metastability in dilute aqueous solutions is significant. Thus, gypsum–water slurries can be
heated to 100ºC without transforming gypsum to anhydrite or hemihydrate. In contrast, gypsum
transforms rapidly to anhydrite in concentrated acid–salt solutions at temperatures above 70–
80ºC (Dutrizac, 2002). More details regarding the gypsum transformation in such systems are
available in Chapter 5.
2.3.1.3 MnSO4–H2O System
There are no relevant data for MnSO4 in the OLI default database. Therefore, experimental data
on the solubility of MnSO4 in H2O (Linke and Seidell, 1958), the mean activity coefficient (γ±)
(Guendouzi et al., 2003), the activity of water (awater) (Guendouzi et al., 2003) and heat capacity
(Cp) (Aseyev, 1996) were used to regress model parameters including the standard state Gibbs
free energy and entropy of formation of the solids; i.e., MnSO4•7H2O, MnSO4•5H2O, and
MnSO4•1H2O as a function of temperature. In addition, MSE middle-range interaction
parameters between Mn2+ and SO42- ions were regressed. The solubility curve of this system is
shown in Figure 2.6.

24
0 30 60 90 120 150 1800.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
MnSO4.7H
2O
MnSO4.5H
2O
MnS
O4 so
lubi
lity,
mol
al
Temperature, oC
MnSO4.7H
2O Exp. Data
MnSO4.5H2O Exp. Data MnSO
4.1H
2O Exp. Data
MnSO4.1H2O
Figure 2.6 Solubility of MnSO4 in H2O. Experimental data are from Linke and Seidell (1958); curve shows the model results.
2.3.1.4 NiSO4–H2O System
Experimental data on the mean activity coefficient, the activity of water, and the solubility of
NiSO4 (Linke and Seidell, 1958; Robinson and Stokes, 2002; Bruhn et al., 1965) were used to
fit the MSE middle-range interaction parameters between Ni2+ and SO42- ions, as well as the
standard state Gibbs free energy and entropy of the solids, NiSO4•7H2O, NiSO4•6H2O, and
NiSO4•1H2O, as a function of temperature. Figure 2.7 shows the solubility of NiSO4 in H2O up
to 300°C.
0 50 100 150 200 250 3000.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
NiSO4.1H2O
NiSO4.6H2O
NiS
O4 so
lubi
lity,
mol
al
Temperature, oC
NiSO4.7H2O Exp data NiSO4.6H2O Exp data NiSO4.1H2O Exp data
NiSO4.7H2O
Figure 2.7 Solubility of NiSO4 in H2O at various temperatures. Experimental data are from Linke and Seidell (1958) and Bruhn et al. (1965); curve shows the fitted model results.

25
No additional fitting was carried out on the MgSO4, ZnSO4 and Na2SO4 aqueous metal
sulphate systems, because the OLI default database (ver. 8.1.3) was found to predict these
systems accurately.
2.3.1.5 Fe2(SO4)3–H2O System
No reliable data are available on the solubility of Fe2(SO4)3 in water, the mean activity
coefficient of Fe2(SO4)3 solutions and the activity of water. The solubility in this system is
difficult to study mainly because of the pronounced tendency of Fe2(SO4)3 to hydrolyse in
aqueous solutions and form a variety of precipitates. The solutions formed are a yellow-brown
colour, as a result of the presence of Fe(III)–hydroxyl ions (hydrated Fe3+ ions are nearly
colourless) (Kononova and Redzhepov, 1996).
2.3.2 Ternary (Metal sulphate–H2SO4–H2O) Systems
2.3.2.1 CaSO4–H2SO4–H2O System
The solubility of CaSO4 hydrates in H2SO4 solutions has been measured by Ling and
Demopoulos (2004), Dutrizac (2002), Zdanovskii and Vlasov (1968), Zdanovskii et al. (1968)
and Marshall and Jones (1966). The OLI default database does not predict the solubility
behaviour in this system accurately. Consequently, experimental data for gypsum, hemihydrate
and anhydrite were used to regress the MSE middle range interaction parameters between the
Ca2+–HSO4¯ and CaSO4(aq)–HSO4
¯ species over the temperature of 25–300ºC. Figure 2.8 shows
the experimental data along with the fitted model results for gypsum at 25–95ºC and that of
anhydrite at 150–250ºC. As can be seen, the model fits the experimental data accurately for both
solids over the whole temperature range (AARD%=6.0). Figure 2.9 shows the three–
dimensional solubility diagram of this system.
At low temperatures (25–60°C), the addition of H2SO4 increases the solubility of gypsum
moderately, whereas at higher temperatures, the solubility increases strongly with increasing
acid concentration. This behaviour is due to the decrease of the second dissociation constant of
H2SO4 with increasing temperature. Thus, the addition of H2SO4 to saturated CaSO4–H2O
solutions reduces the SO42- concentration and allows an increase in the solubility of CaSO4 to
satisfy the solubility product (Marshall and Jones, 1966).

26
0.0 0.5 1.0 1.5 2.0 2.5 3.00.00
0.02
0.04
0.06
0.08
0.10
0.12Solid phase: CaSO4
Exp data, 150 oC Exp data, 200 oC Exp data, 250 oC
Solid phase: CaSO4.2H2O
Exp data, 25 oC Exp data, 50 oC Exp data, 75 oC Exp data, 90 oC
CaS
O4 so
lubi
lity,
mol
al
H2SO4, molal
Figure 2.8 CaSO4 solubility in ternary system of CaSO4–H2SO4–H2O. Curves show the regressed model results. Experimental data are from (Dutrizac, 2002; Zdanovskii et al., 1968; Marshall and Jones, 1966).
01
23
4 3040
5060
7080
900.02
0.04
0.06
0.08
0.10
0.12
gypsum
anhydrite
CaS
O4 so
lubi
lity,
mol
al
Tempera
ture, o CH
2 SO4 , molal
hemihydrate
Figure 2.9 Solubility diagram of CaSO4 in H2SO4–H2O solutions; the surfaces were obtained from the model.
Moreover, as the H2SO4 concentration increases from pure water, the solubility of gypsum
increases gradually. After passing a maximum, the solubility decreases smoothly with further
increasing acid concentration. The initial increase is due to the formation of bisulphate ions. The
solubility decrease in concentrated H2SO4 solutions can be explained by the concept of
solvation: as the concentration of electrolyte increases, fewer water molecules can participate in
the dissolution process because they are tightly held (solvated) by cations and anions in the
solution, this is known as the salting-out effect (Görgényi et al., 2006; Kessler et al., 1963).

27
The phase transition between gypsum–anhydrite and gypsum–hemihydrate was determined on
the basis of phase solubilities. At the transition point, where there is equilibrium between two
phases, the solubilities of both phases are equal. Figure 2.10 shows the phase transition
diagrams of gypsum–anhydrite and gypsum–hemihydrate in H2SO4–H2O solutions obtained on
the basis of solubility curves calculated from the new model.
0 1 2 3 4 5 6 70
102030405060708090
100110120130140
Anhydrite stable
IIIHemihydrate metastable
IIGypsum metastable
Tem
pera
ture
, o C
H2SO4, molal
Zdanovskii et al., 1968 Zdanovskii et al., 1968, Ling and Demopoulos, 2004
IGypsum stable
Figure 2.10 Transition diagram of CaSO4 hydrates in CaSO4–H2SO4–H2O system. Region I: gypsum stable, Region II: anhydrite stable, gypsum metastable, Region III: anhydrite stable, hemihydrate metastable. Experimental data are from Zdanovskii et al. (1968), Ling and Demopoulos (2004).
To validate the diagram, available experimental data, measured by Zdanovskii et al. (1968) up
to 95°C and by Ling and Demopoulos (2004) at 100°C, are also presented on the graph. The
phase diagram was constructed using a procedure similar to the one suggested by Li and
Demopoulos (2006c). Based on the diagram, the gypsum–anhydrite and gypsum–hemihydrate
transformation in water takes place at around 40ºC and 100ºC, respectively. However, the
kinetics of these transformations is slow, resulting in gypsum retention as a metastable phase for
longer periods of time. Transformation kinetics was investigated more thoroughly and more
details are available in Chapter 5.
2.3.2.2 NiSO4–H2SO4–H2O System
The solubility of NiSO4 in aqueous sulphuric acid solutions was studied by Kudryashov and
Lebedev (1989), Girich and Buchinskii (1986) and Marshall et al. (1962). Additional fittings
were performed for the Ni2+–HSO4¯ MSE middle range parameters to improve the prediction of
the chemistry for the system. The results obtained from the fitting compared with the

28
experimental data over a temperature range of 20–300ºC are shown in Figures 2.11 and 2.12.
As is clear, the model results are in good agreement with the experimental data (AARD%=9 for
80 points).
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.50
1
2
3
4
5
6
7
8
60 oC
40 oC
Solid phase: NiSO4.6H2O
Exp data, 20 oC Exp data, 40 oC Exp data, 60 oC
NiS
O4 so
lubi
lity,
mol
al
H2SO
4, molal
20 oC
Figure 2.11 NiSO4 solubility in aqueous H2SO4 solutions below 100ºC; experimental data are from Kudryashov (1989), and Girich (1986). The curves are the regressed model results.
0.0 0.2 0.4 0.6 0.8 1.0 1.2 1.4 1.60.0
0.5
1.0
1.5
2.0
2.5
3.0
250 oC
300 oC275 oC
200 oC
Solid phase: NiSO4.1H2O
Exp data, 200 oC Exp data, 250 oC Exp data, 275 oC Exp data, 300 oC
NiS
O4 so
lubi
lity,
mol
al
H2SO
4, molal
Figure 2.12 NiSO4 solubility in aqueous H2SO4 solutions above 200ºC; experimental data are from Marshall et al. (1962). The curves are the regressed model results.
2.3.2.3 MnSO4–H2SO4–H2O System
The experimental data for this system were selected from the Linke and Seidell (1958) solubility
data collection. Regression was performed on the model parameters between the Mn2+–HSO4¯
and MnSO4(aq)–HSO4¯ species to obtain an acceptable model prediction. The fitted results

29
corresponding to the experimental data are shown in Figure 2.13, which are in good agreement
(AARD%=5.8).
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.50.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
65 oC
45 oC
MnS
O4 so
lubi
lity,
mol
al
H2SO4, molal
Solid phase: MnSO4.1H2O
Exp data, 25 oC Exp data, 45 oC Exp data, 65 oC
25 oC
Figure 2.13 MnSO4 solubility in aqueous H2SO4 solutions; experimental data are from Linke and Seidell (1958), and the curves are the regressed model results.
2.3.2.4 Al2(SO4)3–H2SO4–H2O System
The experimental data for this system were reported by Linke and Seidell (1958). The default
MSE database (ver. 8.1.3) of the OLI does not predict the solubility behaviour of this system
accurately. Consequently, regression was performed on the MSE middle-range interaction
parameters between the Al(SO4)2¯–H3O+ and AlSO4
+–HSO4¯ species to allow an acceptable
model prediction (presented in Appendix B). The fitted results along with the experimental data
are shown in Figure 2.14; the results accurately reflect the experimental data (AARD%=2.4).
0.0 0.5 1.0 1.5 2.00.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
2.0
60 oC
50 oC
Al 2(S
O4) 3 so
lubi
lity,
mol
al
H2SO
4, molal
Solid phase: Al2(SO
4)
3.16H
2O
Exp data, 25 oC Exp data, 50 oC Exp data, 60 oC
25 oC
Figure 2.14 Aluminum sulphate solubility in H2SO4 solutions; experimental data are from Linke and Seidell (1958) and the curves are the fitted model.

30
No additional fitting was carried out on the MgSO4, ZnSO4, Na2SO4, and Fe2(SO4)3 in H2SO4
aqueous systems, because the OLI default database (ver.8.1.3) was found to predict these
systems accurately.
2.3.3 Ternary (CaSO4–Metal sulphate–H2O) Systems
2.3.3.1 CaSO4–ZnSO4–H2O System
The solubility of calcium sulphate in the CaSO4–ZnSO4–H2O system was studied by Umetsu et
al. (1989) and Zatonskaya et al. (1988) over the temperature range of 25–150ºC. Their
experimental solubility data were used to regress the MSE middle range interaction parameters
between Zn2+ and Ca2+ as well as Zn2+ and CaSO4(aq). In this system, Ca2+ is the dominant
calcium species at lower concentrations of ZnSO4, whereas at higher concentrations, the neutral
calcium sulphate ion pair (CaSO4(aq)) becomes dominant. Figures 2.15 and 2.16 show the model
results compared with the experimental data.
As is clear, the model accurately reflects the experimental data (AARD%=5.5). Also, it is clear
that calcium sulphate dihydrate is the stable solid phase up to 90ºC and calcium sulphate
hemihydrate is the stable phase from 100°C to 150ºC. These results are consistent with the XRD
data reported by Umetsu et al. (1989).
0.0 0.4 0.8 1.2 1.6 2.0 2.40.00
0.01
0.02
0.03
0.04
0.05
0.06
90 oC70 oC
40 oC
CaS
O4 so
lubi
lity,
mol
al
ZnSO4, molal
Solid phase: CaSO4.2H2O
Exp data, 25 oC Exp data, 40 oC Exp data, 70 oC Exp data, 90 oC
25 oC
Figure 2.15 CaSO4 solubility in ZnSO4 solutions below 100ºC; experimental data are from Umetsu et al. (1989) and Zatonskaya et al. (1988), and the curves are fitted model results.
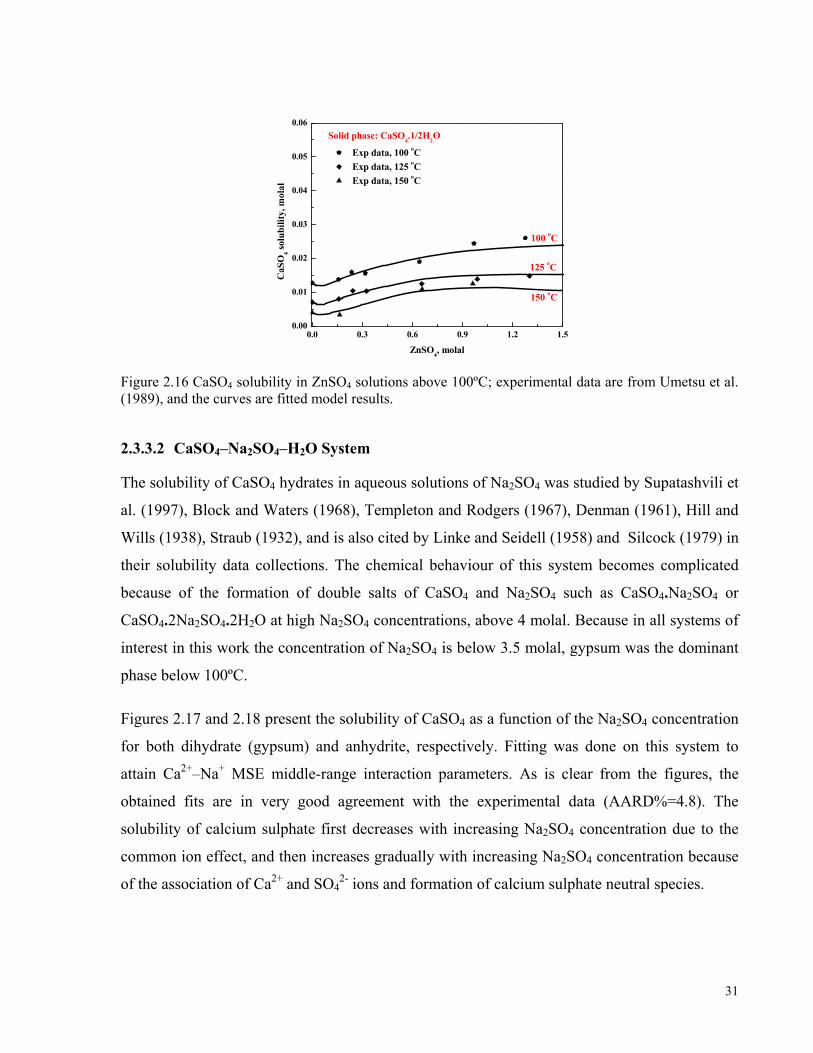
31
0.0 0.3 0.6 0.9 1.2 1.50.00
0.01
0.02
0.03
0.04
0.05
0.06
150 oC
125 oC
Solid phase: CaSO4.1/2H
2O
Exp data, 100 oC Exp data, 125 oC Exp data, 150 oC
CaS
O4 so
lubi
lity,
mol
al
ZnSO4, molal
100 oC
Figure 2.16 CaSO4 solubility in ZnSO4 solutions above 100ºC; experimental data are from Umetsu et al. (1989), and the curves are fitted model results.
2.3.3.2 CaSO4–Na2SO4–H2O System
The solubility of CaSO4 hydrates in aqueous solutions of Na2SO4 was studied by Supatashvili et
al. (1997), Block and Waters (1968), Templeton and Rodgers (1967), Denman (1961), Hill and
Wills (1938), Straub (1932), and is also cited by Linke and Seidell (1958) and Silcock (1979) in
their solubility data collections. The chemical behaviour of this system becomes complicated
because of the formation of double salts of CaSO4 and Na2SO4 such as CaSO4.Na2SO4 or
CaSO4.2Na2SO4.2H2O at high Na2SO4 concentrations, above 4 molal. Because in all systems of
interest in this work the concentration of Na2SO4 is below 3.5 molal, gypsum was the dominant
phase below 100ºC.
Figures 2.17 and 2.18 present the solubility of CaSO4 as a function of the Na2SO4 concentration
for both dihydrate (gypsum) and anhydrite, respectively. Fitting was done on this system to
attain Ca2+–Na+ MSE middle-range interaction parameters. As is clear from the figures, the
obtained fits are in very good agreement with the experimental data (AARD%=4.8). The
solubility of calcium sulphate first decreases with increasing Na2SO4 concentration due to the
common ion effect, and then increases gradually with increasing Na2SO4 concentration because
of the association of Ca2+ and SO42- ions and formation of calcium sulphate neutral species.

32
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.50.00
0.01
0.02
0.03
0.04
0.05
75 oC
50 oC
CaS
O4 so
lubi
lity,
mol
al
Na2SO4, m
Solid phase: CaSO4.2H2O
Exp data, 25 oC Exp. data, 50 oC Exp. data, 75 oC
25 oC
Figure 2.17 CaSO4 solubility in Na2SO4 solutions below 100ºC; experimental data are from Block and Waters (1968), Denman (1961), Hill and Wills (1938). The curves are the fitted model.
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.00.000
0.005
0.010
0.015
0.020
0.025
0.030
0.035
0.040
100 oC
75 oC
CaS
O4 so
lubi
lity,
mol
al
Na2SO
4, m
Solid phase: CaSO4
Exp. data, 50 oC Exp. data, 75 oC Exp. data, 100 oC Exp. data, 207 oC Exp. data, 300 oC
50 oC
Figure 2.18 CaSO4 solubility in Na2SO4 solutions above 100ºC; experimental data are from Block and Waters (1968), Templeton and Rodgers (1967), Hill and Wills (1938), Straub (1932). The curves are the fitted model.
2.3.3.3 CaSO4–NiSO4–H2O System
The solubility of CaSO4 dihydrate in aqueous solutions of NiSO4 has been measured by
Campbell and Yanick (1932) from 45°C to 95ºC and also by Wollmann and Voigt (2008) at
25ºC and 45ºC. The lowest NiSO4 concentration examined by Campbell and Yanick (1932) was
0.4 molal, and therefore, they missed the minimum solubility of gypsum due to the common ion
effect. The solubility data measured by Wollmann and Voigt (2008) at 45ºC agreed with
0.00 0.05 0.10 0.15 0.20 0.25 0.30 0.3500
05
0
5
20
25
30
35
40
45
50
300 oC
207 oC

33
Campbell’s results up to 1.5 molal NiSO4, however, at higher concentrations, the solubility
data measured by Wollmann and Voigt (2008) were lower than those measured by Campbell
and Yanick (1932). This is due to the method of calcium analysis used by Campbell and Yanick
(1932). In their work, calcium was analyzed gravimetrically as calcium oxalate monohydrate.
Because nickel oxalate dihydrate, Ni(C2O4)•2H2O(s), has a similar solubility as calcium oxalate,
it creates a bias towards higher apparent calcium sulfate concentrations.
To obtain reliable data, the solubility of gypsum in aqueous NiSO4 systems was measured at 25–
95ºC in this work. Also, anhydrite solubility was measured inside an autoclave at elevated
temperatures of 150°C and 175ºC. The experimental procedure is discussed in detail in Chapter
4. Tables C.1 and C.2 in Appendix C summarize the newly measured data for gypsum and
anhydrite in this system. These data were combined with those measured by Campbell and
Yanick (1932) (below 1.5 molal NiSO4) and those of Wollmann and Voigt (2008) and were
used to regress the MSE middle range interaction parameters between calcium species, Ca2+ and
CaSO4(aq), and Ni2+ ions. The regressed model results are shown in Figures 2.19 and 2.20. As
can be seen, the model fits the experimental data accurately with an absolute average relative
deviation (AARD%) of 5.6 for 116 points.
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.50.000
0.005
0.010
0.015
0.020
0.025
0.030
0.035
0.040
0.045
90 oC
45 oC
Solid: CaSO4.2H2O
This work+Wollmann (2008) Exp data, 25oC This work+Campbell (1932) Exp data, 45oC This work+Campbell (1932) Exp data, 90oC
CaS
O4 so
lubi
lity,
mol
al
NiSO4, molal
25 oC
Figure 2.19 Gypsum solubility in NiSO4 solutions below 100ºC. Experimental data are from Azimi and Papangelakis (2010b), Wollmann and Voigt (2008) and Campbell and Yanick (1932); the curves are the fitted model.

34
0.0 0.2 0.4 0.6 0.80.000
0.002
0.004
0.006
0.008
150 oC
175 oC
Solid phase: CaSO4
This work, 150 oC This work, 175 oC
CaS
O4 so
lubi
lity,
mol
al
NiSO4, molal
Figure 2.20 Anhydrite solubility in NiSO4 solutions above 100ºC. Experimental data are from Azimi and Papangelakis (2010b); the curves are the fitted model.
As the NiSO4 concentration increases from pure water, the solubility of gypsum initially drops
and then increases gradually with increasing NiSO4 concentration. After passing a maximum,
the solubility decreases smoothly with further increasing NiSO4 concentration. The initial drop
is due to the common ion effect, shifting the dissolution reaction (equation 2.1) to the left; the
subsequent increase is attributable to the association of Ca2+ and SO42- ions and formation of
calcium sulphate neutral species (equation 2.2). The solubility decrease in concentrated NiSO4
solutions can be explained by the salting-out effect, which is a result of reduced number of free
water molecules in the solution to dissolve calcium sulphate.
According to equations (2.7) and (2.8), the solubility of calcium sulphate is equal to the sum of
+2Cam and
)(4 aqCaSOm which are inversely proportional to )( 2)( 4
24
nwCaSOSO
am ⋅⋅ ±− γ and )()(4
nwCaSO a
aq⋅γ ,
respectively. Figure 2.21 presents the solubility of gypsum (dihydrate) along with +2Cam and
)(4 aqCaSOm as well as )( 22)( 4
24
wCaSOSO am ⋅⋅ ±− γ and )( 2)(4 wCaSO a
aq⋅γ terms calculated by the aid of the MSE
model containing regressed interaction parameters as a function of the NiSO4 concentration at
90ºC. As is clear, the value of )( 2)(4 wCaSO a
aq⋅γ term decreases with an increase in NiSO4
concentration, reaching a minimum at ~2 molal which corresponds to the maximum of the
solubility curve. Above this point, the value of )( 2)(4 wCaSO a
aq⋅γ term increases gradually as a result

35
of the increase in )(4 aqCaSOγ with concentration due to the salting-out effect, resulting in a decrease
in the concentration of CaSO4(aq) and consequently, the decrease in gypsum solubility.
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.50.00
0.01
0.02
0.03
0.04
0.05
0.06
mSO4
2- * aw2 * γ±
2
Solid phase: CaSO4.2H2O
Con
cent
ratio
n, m
olal
NiSO4, molal
γCaSO4(a)
* aw2 * 10-1
T= 90 oC
solubility curve
[Ca2+]
[CaSO4(aq)
]
[Catotal
]
Figure 2.21 Total concentration of Ca along with Ca2+ and CaSO4(aq) concentrations in CaSO4–NiSO4–H2O system at 90ºC. Calculated values of ( 22
)( 424
wCaSOSOam ⋅⋅ ±− γ ) and ( 2
)(4 wCaSO aaq⋅γ ) are also presented.
2.3.3.4 CaSO4–MgSO4–H2O System
The solubility of gypsum in aqueous MgSO4 solutions at different temperatures was measured
by different researchers (Wollmann and Voigt, 2008; Arslan and Dutt, 1993; Umetsu et al.,
1989; Tanji, 1969; Novikova, 1957) and was also cited by Linke and Seidell (1958) in their
solubility data compilation. Umetsu et al. (1989) also measured the solubility of calcium
sulphate at elevated temperatures at 100–175ºC. They observed that the solid phases
corresponding to their solubility data were hemihydrate between 100°C and 150ºC and
anhydrite above 150ºC. These data were used to regress the MSE middle range interaction
parameters between Ca2+–Mg2+ as well as CaSO4(aq)–Mg2+ species. The modelling results
showed that the experimental data measured by Umetsu et al. (1989) above 150ºC, closely
match the solubility of hemihydrate instead of that of anhydrite. Therefore, in this work, the
solubility of anhydrite was measured in an autoclave at 150°C and 175ºC in aqueous MgSO4
solutions which are summarized in Appendix C (Table C.3). Details regarding the experimental
procedure are available in Chapter 4. The newly measured data were compiled along with other
experimental data in a data regression file to regress the interaction parameters between Mg2+
and the calcium species. Figures 2.22 to 2.24 show the model results in comparison with the
experimental data for gypsum, hemihydrate and anhydride, respectively.

36
0.0 0.5 1.0 1.5 2.0 2.5 3.00.00
0.01
0.02
0.03
0.04
0.05
0.06
75 oC
45 oC
CaS
O4 so
lubi
lity,
mol
al
MgSO4, molal
Solid phase: CaSO4.2H2O
Exp data, 25 oC Exp data, 45 oC Exp data, 75 oC
25 oC
Figure 2.22 Gypsum solubility in aqueous MgSO4 solutions. Experimental data are from Tanji (1969), Arslan and Dutt (1993), Umetsu et al. (1989), Linke and Seidell (1958); the curves are the fitted model.
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.90.000
0.005
0.010
0.015
0.020
0.025
0.030
0.035
0.040
100 oC
125 oC
CaS
O4 so
lubi
lity,
mol
al
MgSO4, molal
Solid phase: CaSO4. 1/2 H2O
Exp data, 100 oC Exp data, 125 oC Exp data, 175 oC
175 oC
Figure 2.23 Hemihydrate solubility in aqueous MgSO4 solutions. Experimental data are from Umetsu et al. (1989); the curves are the fitted model.

37
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.70.000
0.001
0.002
0.003
0.004
0.005
0.006
0.007
175 oC
Solid phase: CaSO4
This work, 150 oC This work, 175 oC
CaS
O4 so
lubi
lity,
mol
al
MgSO4, molal
150 oC
Figure 2.24 Anhydrite solubility in aqueous MgSO4 solutions. Experimental data are from Azimi and Papangelakis (2010b); the curves are the fitted model.
As in the case with NiSO4, the solubility of gypsum first decreases with increasing MgSO4
concentration due to the common ion effect of added SO42- ions and then increases gradually
with increasing MgSO4 concentration because of the association of Ca2+ and SO42- ions and
formation of calcium sulphate neutral species. For concentrated solutions containing more than
1.0–1.5 molal MgSO4, the salting-out effect becomes dominant, and as a result, the solubility
drops accordingly.
2.3.3.5 CaSO4–MnSO4–H2O System
Zhelnin et al. (1973) measured the solubility of CaSO4 in the CaSO4–MnSO4–H2O system from
25°C to 100ºC, and their observations show that in this temperature range, even up to 3.5 molal
MnSO4, gypsum is the only solid in equilibrium with the solution. Wollmann and Voigt (2008)
also measured CaSO4 solubility in this system at 25ºC and 40ºC. All the experimental data were
used to fit Ca2+–Mn2+ and also CaSO4(aq)–Mn2+ MSE interaction parameters, because at higher
concentrations of MnSO4, CaSO4(aq) is more abundant than the Ca2+ species. The fitted results
for this system are shown in Figure 2.25, and the calculated solubilities are consistent with the
experimental data (AARD%=5.9).

38
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.50.00
0.01
0.02
0.03
0.04
0.05
0.06
100 oC
50 oC
75 oC
CaS
O4 so
lubi
lity,
mol
al
MnSO4, molal
Solid phase: CaSO4.2H
2O
Exp data, 25 oC Exp data, 50 oC Exp data, 75 oC Exp data, 100 oC
25 oC
Figure 2.25 CaSO4 solubility in MnSO4 solutions; experimental data are from Wollmann and Voigt (2008) and Zhelnin et al. (1973), and the curves are the fitted model.
2.3.4 Effect of Divalent Cations on the Solubility of CaSO4
Figure 2.26 presents a comparison between the solubility of CaSO4 dihydrate (gypsum) in
ternary solutions of CaSO4–NiSO4–H2O, CaSO4–MgSO4–H2O and CaSO4–MnSO4–H2O along
with the model predictions at two different temperatures of 25ºC and 75°C. It is clear that the
difference between CaSO4 solubilities in these systems at both temperatures is always less than
15% indicating that the cation type does not have a dramatic effect on the CaSO4 solubility. For
all three cations, as the metal sulphate concentration increases from pure water, the solubility
initially drops and then increases gradually. After passing a maximum, the solubility decreases
smoothly with further increasing the MSO4 (M=Mg, Mn, Ni) concentration. The initial drop is
due to the common ion effect which shifts the dissolution reaction (Eq. 2.1) to the left; the
subsequent increase is attributable to the association of Ca2+ and SO42- ions and formation of
calcium sulfate neutral species (Eq. 2.2). The solubility decrease in concentrated solutions is due
to the salting-out effect. Moreover, for all three cations, gypsum solubility increases with
increasing temperature due to the fact that the association constant (Ka) of CaSO4(aq) increases
with temperature, which would shift Eq. 2.2 to the right.
In industrial applications, particularly in hydrometallurgy, solutions usually contain several
cations for which there are no experimental data available. By knowing that cation type does not
have a significant effect on the solubility behaviour, all divalent cations can be substituted with
a certain cation for which experimental data are available during the simulation of the process.

39
0.00 0.25 0.50 0.75 1.00 1.25 1.50 1.75 2.000.00
0.01
0.02
0.03
0.04
0.05 MgSO4 solubility MnSO
4 solubility
NiSO4 solubility
T=25 oC
CaS
O4 so
lubi
lity,
mol
al
MSO4 (M=Ni, Mg, Mn), molal
Solid phase: CaSO4.2H2OExp. data:
MgSO4, 25oC
MnSO4, 25oC
NiSO4, 25oC
MgSO4, 75oC
MnSO4, 75oC
NiSO4, 75oC
T=75 oC
Figure 2.26 CaSO4 solubility in MSO4 (M=Ni, Mg, Mn) solutions. Experimental data are from Azimi, Papangelakis (2010b); Wollmann, Voigt (2008); Arslan, Dutt (1993); Zhelnin et al. (1973); Tanji (1969); Campbell,Yanick (1932). Curves represent the model predictions.
2.3.5 Industrial Implications of the Model in Zinc Producing Industries
So far, it has been shown that the MSE model is effective for fitting the solubility of CaSO4
hydrates in binary or ternary electrolyte solutions. In order to validate the model parameters, the
solubility of CaSO4 hydrates was calculated in multicomponent zinc sulphate processing
solutions containing ZnSO4, H2SO4, Fe2(SO4)3, MgSO4, and (NH4)2SO4, for which no fitting
was carried out. As will be seen later in this section, the model is capable of predicting the
chemistry of all the multicomponent systems studied. This fact illustrates the usefulness of the
model, utilizing the developed database, in assessing the scaling potential of calcium sulphate in
a variety of complex aqueous processing streams where no experimental data are available.
2.3.5.1 CaSO4–ZnSO4–H2SO4 (0.1 M)–H2O System
Dutrizac (2002) has studied the effect of ZnSO4 concentration on the solubility of calcium
sulphate in solutions containing 0.1 M H2SO4 as a function of temperature. The solubility of
calcium sulphate decreases steadily as the ZnSO4 concentration increases from 0.0 to 0.5 M
ZnSO4 because of the common ion effect. The experimental data were obtained on heating to
95ºC and on subsequent cooling. In this system, because the acid concentration used is relatively
low, the dehydration of gypsum to anhydrite does not practically occur at temperatures below
95ºC. Figure 2.27 shows the experimental solubility data of CaSO4 vs. ZnSO4 concentration in

40
0.1 M H2SO4 media at various temperatures along with the model predictions. The predictions
are in good agreement with the experimental data (AARD%=5.8).
0.0 0.5 1.0 1.5 2.00.00
0.01
0.02
0.03
0.04
0.05
0.06
90 oC
70 oC
40 oC
CaS
O4 so
lubi
lity,
mol
al
ZnSO4, molal
Solid phase: CaSO4.2H2O
Exp data, 25 oC Exp data, 40 oC Exp data, 70 oC Exp data, 90 oC
25 oC
H2SO4=0.1 M
Figure 2.27 CaSO4 solubility in CaSO4–ZnSO4–H2SO4 (0.1 M)–H2O solutions. Experimental data are from Dutrizac (2002); the curves are the predicted results.
2.3.5.2 CaSO4–H2SO4–ZnSO4 (1.5 M)–H2O System
The solubility of CaSO4 as a function of acid concentration in solutions containing 1.5 M ZnSO4
was also measured by Dutrizac (2002). Figure 2.28 shows the experimental data and the
predicted results from the model. The model predictions are in close agreement with the
experimental data (AARD%=5.6). It is also clear that acid concentration has a relatively minor
effect on the solubility of CaSO4 when the solution contains 1.5 M of ZnSO4. This effect is due
to the free sulphate ions released from the dissociation of ZnSO4.
0.0 0.5 1.0 1.5 2.0 2.50.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
90 oC
70 oC
40 oC
CaS
O4 so
lubi
lity,
mol
al
H2SO4, molal
Solid phase: CaSO4.2H2O
Exp data, 25 oC Exp data, 40 oC Exp data, 70 oC Exp data, 90 oC
25 oC
ZnSO4=1.5 M
Figure 2.28 CaSO4 solubility in CaSO4–H2SO4–ZnSO4 (1.5M)–H2O solutions. Experimental data are from Dutrizac (2002); the curves are the predicted results.

41
2.3.5.3 CaSO4–MgSO4–H2SO4 (0.1 M)–ZnSO4 (1.15 M)–H2O System
Zinc processing solutions typically contain modest concentrations of MgSO4 and MnSO4
(Dutrizac, 2002). The effect of MgSO4 on the solubility of CaSO4 was studied because its
impact on CaSO4 chemistry was unknown. For this purpose, the solubility of CaSO4 in a system
containing MgSO4, ZnSO4 and H2SO4 was modelled using the new database. As shown in
Figure 2.29, the model is capable of accurately predicting the experimental data in this system
(AARD%=5.2).
0.0 0.2 0.4 0.6 0.8 1.0 1.20.00
0.01
0.02
0.03
0.04
0.05
0.06
ZnSO4=1.5MH2SO4=0.1M
25 oC
40 oC70 oC
CaS
O4 so
lubi
lity,
mol
al
MgSO4, molal
Solid phase: CaSO4.2H
2O
Exp data, 25 oC Exp data, 40 oC Exp data, 70 oC Exp data, 90 oC
90 oC
Figure 2.29 CaSO4 solubility in CaSO4–MgSO4–ZnSO4 (1.15 M)–H2SO4 (0.1 M)–H2O solutions; experimental data are from Dutrizac (2002); curves represent model predictions.
2.3.5.4 CaSO4–H2SO4–ZnSO4 (2.5 M)–MgSO4 (0.41 M)–MnSO4 (0.18 M)–H2O System
To ascertain the effect of pH on the solubility of calcium sulphate under weakly acidic
conditions, a series of solubility measurements was carried out by Dutrizac (2002) at various
temperatures in solutions containing 2.5 mol/L ZnSO4, 0.41 mol/L MgSO4 and 0.18 mol/L
MnSO4. The pH was varied from 3.6 to 4.6. The experimental solubility data for this system are
shown in Figure 2.30 along with the model predictions, which are in good agreement
(AARD%=6.5). Also, it is clear that CaSO4 solubility does not change significantly with
changing pH in a weakly acidic solution.

42
3.8 3.9 4.0 4.1 4.2 4.3 4.4 4.5 4.60.00
0.01
0.02
0.03
0.04
0.05
0.06
90 oC
75 oC
45 oC
CaS
O4 so
lubi
lity,
mol
al
pH
Solid phase: CaSO4.2H2O
Exp data, 25 oC Exp data, 45 oC Exp data, 75 oC Exp data, 90 oC
ZnSO4= 2.5 MMgSO4= 0.41 MMnSO
4= 0.18 M
25 oC
Figure 2.30 CaSO4 solubility in CaSO4–H2SO4–ZnSO4 (2.5M)–MgSO4 (0.41M)–MnSO4 (0.18M)–H2O solutions vs. pH. Experimental data are from Dutrizac (2002); curves represent the predicted values.
2.3.5.5 CaSO4–(NH4)2SO4–ZnSO4 (2.5M)–MgSO4(0.41M)–H2SO4(pH=3.8)–MnSO4(0.18M)–H2O System
High concentrations of Fe2(SO4)3 are commonly generated in the hot acid leaching circuits of
hydrometallurgical zinc operations (Dutrizac, 2002). At some point in the process, the dissolved
iron must be eliminated, and the removal is most commonly carried out by the precipitation of
jarosite-type compounds (MFe3(SO4)2(OH)6, where M represents K, Na, NH4, etc.). To form the
jarosite precipitate, Na+ or NH4+ ions are added to the solution, and inevitably, a circulating load
of Na2SO4 or (NH4)2SO4 results. The experimental measurements to investigate the effect of the
concentration of (NH4)2SO4 on the solubility of CaSO4 were performed by Dutrizac (2002). The
experimental data and model predictions for this system are shown in Figure 2.31, where the
prediction results accurately reflect the solubility of CaSO4 in this multicomponent acid-
containing system (AARD%=7.0).
The presence of low concentrations of NH4+ ions, as (NH4)2SO4, has a minimal effect on the
solubility of calcium sulphate; consequently, the ammonium additions required for the
precipitation of jarosite-type compounds do not have a significant effect on the solubility of
calcium sulphate.

43
0.00 0.05 0.10 0.15 0.200.00
0.01
0.02
0.03
0.04
0.05
0.06
40 oC
70 oC
90 oC
MnSO4= 0.18 M
pH=3.8
ZnSO4= 2.5 M
MgSO4= 0.41 M
CaS
O4 so
lubi
lity,
mol
al
(NH4)
2SO
4, molal
Solid phase: CaSO4.2H
2O
Exp data, 40 oC Exp data, 70 oC Exp data, 90 oC
Figure 2.31 CaSO4 solubility in CaSO4–(NH4)2SO4–ZnSO4(2.5M)–MgSO4(0.41M)–MnSO4(0.18M)–H2SO4(pH=3.8)–H2O solutions; experimental data are from Dutrizac (2002); curves are model predictions.
2.3.5.6 CaSO4–Na2SO4–ZnSO4(2.5M)–MgSO4(0.41M)–MnSO4(0.18M)–H2SO4(pH=3.8)–H2O System
As mentioned above, Na+ ions (as Na2SO4) are sometimes added to hydrometallurgical zinc
solutions to precipitate Fe2(SO4)3 as a jarosite-type compound. Therefore, the influence of
sodium sulphate on the solubility of CaSO4 is of some practical relevance.
Dutrizac (2002) measured the solubility of CaSO4 as a function of Na2SO4 in multicomponent
solutions containing ZnSO4, MgSO4, MnSO4, and H2SO4. It was shown that increasing Na
concentration from 0 to 12 g/L has only a small effect on the solubility of CaSO4, which
decreases slightly with increasing sodium sulphate concentration. Generally, hydrometallurgical
Zn processing solutions contain 1 to 5 g/L Na, and such concentrations will have a negligible
effect on the solubility of CaSO4 (Dutrizac, 2002). The experimental data and the model
predictions are shown in Figure 2.32, in which the model shows a near-perfect prediction of
solution chemistry.

44
0.00 0.05 0.10 0.15 0.20 0.25 0.30 0.35 0.400.00
0.01
0.02
0.03
0.04
0.05
0.06
90 oC
75 oC
50 oC
25 oC
ZnSO4= 2.5 MMgSO4= 0.41 MMnSO4= 0.18 MpH=3.8
CaS
O4 so
lubi
lity,
mol
al
Na2SO
4, molal
Solid phase: CaSO4.2H2O
Exp data, 25 oC Exp data, 50 oC Exp data, 75 oC Exp data, 90 oC
Figure 2.32 CaSO4 solubility in CaSO4–Na2SO4–ZnSO4(2.5M)–MgSO4(0.41M)–MnSO4(0.18M)–H2SO4 (pH=3.8)–H2O solutions. Experimental data are from Dutrizac (2002). Curves are the predicted values.
2.3.5.7 CaSO4–Fe2(SO4)3–H2SO4 (0.3 M)–ZnSO4 (1.15M)–H2O System
In the dominant roast-leach-electrolysis zinc process, iron present in the concentrate feed is
oxidized to the ferric state in the roaster and is subsequently solubilized as ferric sulphate in the
hot acid leaching areas of the process. Accordingly, knowledge of the influence of dissolved
ferric sulphate on the solubility of CaSO4 is of some commercial importance. Generally, the
presence of ferric sulphate in the solution has only a modest effect on the solubility of CaSO4.
At higher temperatures, increasing Fe2(SO4)3 concentrations cause a slight increase in the
solubility of calcium sulphate (Dutrizac, 2002).
There were no experimental data available in the literature for the CaSO4–Fe2(SO4)3–H2O
system. Consequently, the experimental solubility data of CaSO4 in aqueous solutions of
Fe2(SO4)3–H2SO4–ZnSO4 measured by Dutrizac (2002) were used to regress the MSE middle-
range interaction parameters of Fe3+–Ca2+ and Fe3+–CaSO4(aq) to predict the chemistry of this
system. The regressed MSE model parameters are presented in Appendix B. The fitted results
obtained for the solubility of CaSO4 vs. Fe2(SO4)3 concentration are shown in Figure 2.33 along
with the experimental data. The model results accurately reflect the experimental data
(AARD%=6.4).

45
0.0 0.2 0.4 0.6 0.8 1.0 1.2 1.40.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
0.09
25 oC
45 oC
70 oC
ZnSO4= 1.15 M
H2SO4= 0.3 M
Solid: CaSO4.2H2O
Exp data, 25 oC Exp data, 40 oC Exp data, 70 oC Exp data, 90 oC
CaS
O4 so
lubi
lity,
mol
al
Fe2(SO4)3, molal
90 oC
Figure 2.33 CaSO4 solubility in CaSO4–Fe2(SO4)3–H2SO4 (0.3M)–ZnSO4 (1.15M)–H2O solutions. Experimental data are from Dutrizac (2002); the curves are fitted model results.
2.3.5.8 CaSO4–ZnSO4–H2SO4–H2O System
The solubility of CaSO4 in aqueous solutions of ZnSO4 (0.0–1.4 M) and H2SO4 (0.0–2.2 M) was
measured by Mutalala et al. (1988) over the temperature range of 25–60°C. Their results show
that CaSO4 solubility in ZnSO4 media increases with increasing H2SO4 concentrations to ~1.0 M
H2SO4 because of the formation of bisulphate ions, but decreases at higher acid concentrations
due to the salting-out effect. Increasing ZnSO4 depresses the CaSO4 solubility because of the
common ion effect. In their study, the temperature was limited to 60°C to ensure that gypsum
was always the saturating solid phase. Figure 2.34 shows the experimental data and the model
prediction results for this system at 60°C as a function of H2SO4 and ZnSO4 concentrations. The
model reflects the experimental data closely without any further fitting (AARD%=8.0).
0.0 0.20.4
0.60.8
1.01.2
1.4
0.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.00.5
1.01.5
2.02.5
CaS
O4 so
lubi
lity,
mol
al
H 2SO 4
, molalZnSO
4 , molal
Solid phase: CaSO4.2H2O
T=60 oC
Figure 2.34 CaSO4 solubility in CaSO4–ZnSO4–H2SO4–H2O solutions; experimental data are from Mutalala et al. (1988); surface shows model prediction results.

46
2.4 Summary
The solubility of all common phases of calcium sulphate was successfully modelled by
developing a new database for the Mixed Solvent Electrolyte model of the OLI® software.
Modelling involved fitting of binary activity, heat capacity, and solubility data as well as ternary
solubility data. New interaction parameters for calcium ions and associated calcium sulphate
neutral species with other dominant species in the solution were determined. The model
containing new interaction parameters was shown to accurately predict the solubility of calcium
sulphate in zinc sulphate hydrometallurgical solutions, containing ZnSO4, H2SO4, MgSO4,
MnSO4, Fe2(SO4)3, Na2SO4 and (NH4)2SO4 from 25°C to 95ºC. The solubility of calcium
sulphate in water reaches a maximum at ~40ºC, followed by a slight decrease in the solubility at
higher temperatures. The addition of H2SO4 up to ~1.5 mol/L increases the calcium sulphate
solubility. Furthermore, it was found that in ZnSO4–H2SO4 media, increasing MgSO4,
Fe2(SO4)3, and (NH4)2SO4 concentrations does not have a significant effect on the solubility of
calcium sulphate.
As a result of the above, process solutions operating under atmospheric pressure, and are
saturated with calcium sulphate during an upstream neutralization step, have the potential for
gypsum scale formation when cooled down to lower temperatures. Moreover, gypsum solubility
decreases at temperatures above 100°C, where gypsum transforms into anhydrite, which has
lower solubility levels.
As it is not practical to measure solubility data under all possible conditions, because of the
large number of components involved, the database developed, utilized by the OLI/MSE model
is a valuable tool to assess calcium sulphate solubility and predict the scaling potential in a wide
variety of complex aqueous processing streams.

47
CHAPTER 3 MODELLING OF CALCIUM SULPHATE SOLUBILITY IN CHLORIDE/SULPHATE SOLUTIONS
he focus of the current chapter is on extending the database developed in Chapter 2, such
that it is applicable to complex multicomponent chloride–sulphate solutions containing
CaSO4, CaCl2, Fe2(SO4)3, FeCl3, H2SO4, HCl, LiCl, MgSO4, MgCl2, Na2SO4, NaCl, and NiSO4.
The database, utilized by the MSE model, provides a valuable tool for predicting the solubility
of calcium sulphate in the neutralization stage of nickel sulphate-chloride processing solutions
of the Voisey’s Bay plant. This chapter is prepared based on the following publications:
- Azimi G., Papangelakis V.G., Dutrizac J.E., 2008. Fluid Phase Equilibria, 266, 172–186.
- Azimi G., Papangelakis V.G., 2010a. Fluid Phase Equilibria, 290, 88–94.
3.1 Introduction
The nickel industry worldwide has traditionally smelted concentrates produced from nickel,
copper and cobalt sulphide ores to make an intermediate sulphide product, matte. Produced
matte has been further refined utilizing hydrometallurgical processes to produce high purity
nickel, copper and cobalt for marketing. Thus, traditionally production of these metals has
occurred in two steps: smelting and refining.
Recently, Vale Inco has developed a new hydrometallurgical process to treat the nickel sulphide
concentrates produced from the Voisey's Bay Ni–Cu–Co deposits directly to metal products
without first having to smelt the concentrate. It is more economical and environmentally
friendly since the sulphur dioxide and dust emissions associated with a smelter are eliminated.
The process consists of an O2–Cl2 gas preleach at ambient pressure, followed by an oxidative
pressure leach at 150ºC in a sulphate–chloride leaching medium. It was found that the presence
of 5–10 g/L chloride ions in the autoclave accelerates the rate of base metal dissolution and
leaching kinetics. Furthermore, addition of chloride ions results in a major reduction in the
amount of sulphide oxidized to sulphates, and therefore, recovery of almost all the iron content
of the concentrate as hematite in the solid residue. This would result in a significant decrease in
the amount of oxygen consumed during the oxidative pressure leaching. With the chloride
addition, the leach solution typically has a pH value of 2.5 to 3.0, and the concentration of iron
T

48
is less than 1 g/L. Therefore, the costs of subsequently treating the oxidative pressure leach
solution, i.e., neutralization and iron removal processes, are reduced (Kerfoot et al., 2002).
The leach slurry is flashed to atmospheric pressure, and liquid–solids separation is effected by
countercurrent decantation (CCD). Most of the iron initially present in the concentrate feed
precipitates as hematite in the autoclave, but some iron remains in the discharged leach solution.
Accordingly, the solution is oxidized and neutralized with lime to precipitate the soluble iron.
After filtration, the solution fraction is treated in a series of three solvent extraction operations to
remove copper, various solution impurities, and cobalt, respectively. The purified solution is
then electrolyzed to produce nickel metal and to evolve an O2–Cl2 gas at the anode. The O2–Cl2
gas is recycled to the preleaching operation to consume all the chlorine generated during
electrolysis (Dutrizac and Kuiper, 2006). Figure 3.1 presents a schematic flowsheet of the
process.
Figure 3.1 Schematic flowsheet of the Vale Inco developed hydrometallurgical process for the recovery of Ni and Co values from sulphide concentrates (Kerfoot et al., 2002).
Because lime is employed for the neutralization of free sulphuric acid and iron removal, calcium
sulphate is generated. As mentioned in the previous chapters, calcium sulphate has a limited
solubility in aqueous media; therefore, it would precipitate throughout the circuit as the
temperature and solution composition changes, result in several operational problems including
reduced heat transfer capacity and process efficiency. Therefore, periodic shut-downs of the

49
plant for mechanical removal of precipitated calcium sulphate hydrates from the processing
circuit are necessary.
In Chapter 2, a new database for the mixed solvent electrolyte (MSE) model of the OLI®
software was developed, and shown to accurately predict the solubility of calcium sulphate in
various multicomponent sulphate solutions, including neutralized zinc sulphate hydromet
solutions containing ZnSO4, MgSO4, MnSO4, Fe2(SO4)3, Na2SO4, (NH4)2SO4 and H2SO4 at 25–
95ºC (Azimi et al., 2007, 2010).
The purpose of the current chapter is to further extend the developed database such that it can be
successfully applied to complex multicomponent chloride–sulphate solutions containing Ca,
Fe(III), Li, Mg, Na, Ni, H2SO4, and HCl. This database, utilized by the MSE model, is a
valuable tool for assessing the calcium sulphate scaling potential in various industrial processes
in which unwanted calcium sulphate precipitation occurs.
A review of published modelling studies shows that no previous work has been undertaken to
study the simultaneous effect of various metal sulphate salts, as well as metal chloride salts, on
the solubility of the three phases of calcium sulphate in H2SO4 or HCl media, over a significant
temperature and concentration range. The detailed literature review was presented in Chapter 1.
To extend the applicability of the developed database from multicomponent sulphate solutions
to mixed sulphate–chloride systems, interaction parameters between Cl¯–SO42−, Cl¯–HSO4
¯, and
Cl¯–CaSO4(aq) were obtained by fitting the experimental data for ternary systems. The interaction
parameters were subsequently validated by predicting the solubility of calcium sulphate in the
neutralization stage of nickel sulphate–chloride processing solutions of the Voisey’s Bay
hydrometallurgical plant, containing NiSO4, Fe2(SO4)3, Na2SO4, H2SO4, and LiCl from 20°C to
95°C for which experimental data are available in the literature (Dutrizac and Kuiper, 2006).
The model was also used to predict the solubility of calcium sulphate in multicomponent
chloride solutions containing Ca, Fe(III), HCl, Mg, Na, Cl¯, and SO42− over wide ranges of
temperature and composition.
The approach utilized in this work is superior to that used by Li and Demopoulos (2006b, 2007).
They measured the solubility of calcium sulphate hydrates (DH, HH, and AH) in HCl and in
HCl-based aqueous solutions containing various metal chloride salts, such as CaCl2, FeCl3,

50
MgCl2, and NaCl under atmospheric pressure at 10–100ºC (Li and Demopoulos, 2002, 2005,
2006a). Subsequently, they used these experimental data to develop a model for the solubility of
calcium sulphate in multicomponent aqueous chloride solutions. In their approach, the
interaction parameters between Ca2+–SO42− or Ca2+–HSO4
¯ were obtained utilizing the
experimental data of a quaternary system (CaSO4–CaCl2–HCl–H2O). In contrast, in the present
work, interaction parameters for free calcium ions and associated calcium sulphate neutral
species with other dominant species were determined in binary and ternary solutions and then
validated in multicomponent solutions. That is, instead of regressing Ca2+–SO42− or Ca2+–HSO4
¯
interaction parameters using a quaternary system (e.g., CaSO4–CaCl2–HCl–H2O), binary
CaSO4–H2O and ternary CaSO4–H2SO4–H2O systems were used, respectively. Those interaction
parameters, along with other regressed parameters generated during the parameterization step,
were subsequently used to predict the solubility of gypsum, hemihydrate, and anhydrite in
multicomponent solutions similar to the above-mentioned quaternary system. In fact, the
interaction parameters between two species determined in a multicomponent system, such as
those determined by Li and Demopoulos (2006b, 2007), do not guarantee accurate results in
simple binary or ternary solutions that consist of the same species. One of the most important
features of a chemical model is its adherence to the additivity principle by being able to predict
the properties of complex multicomponent systems utilizing parameters derived from
experimental data generated for simpler systems (usually binary and sometimes ternary systems)
(Wang et al., 2004).
In this chapter, all available experimental data for the entire temperature and concentration
range were used during the parameterization step and the predictive capacity of the obtained
parameters was examined over the temperature range of 25–250°C, or even higher, from dilute
to concentrated solutions in mixed chloride–sulphate media for which experimental data were
available. The procedures followed were similar to those described in Chapter 2 and more
details are available in the literature (Azimi et al., 2007, 2008).
3.2 Modelling Strategy
Because the database developed in this chapter is an extension of the database previously
mentioned in Chapter 2, only a few model parameters needed to be regressed utilizing the
experimental data available in mixed chloride–sulphate systems. A list of the various systems

51
studied in this work along with the typical range of conditions investigated is given in Table
3.1. The obtained model parameters are presented in Appendix B.
Table 3.1–Systems studied for the parameterization purpose
System Data Type Temperature Range, ºC Solid Phases
CaSO4-CaCl2-H2O solubility 22-300 DH, HH, AH
CaSO4-HCl-H2O solubility 10-80 DH, HH, AH
CaSO4-NaCl-H2O solubility 10-300 DH, HH, AH
CaSO4-MgCl2-H2O solubility 25-250 DH, AH
CaSO4-AlCl3-H2O solubility 25-250 DH
CaSO4-FeCl3-HCl (0.5M)-H2O solubility 20-80 DH
Note: DH: CaSO4•2H2O, HH: CaSO4•0.5H2O, AH: CaSO4
Validation of the parameters was performed by predicting the chemistry of quaternary or
multicomponent systems that were not used in the regression stage. A list of systems used for
validation purpose is summarized in Table 3.2. The predicted model results, utilizing the newly
regressed parameters, are in good agreement with these data, without additional fitting. The
Absolute Average Relative Deviations (AARD%2) between the experimental data and predicted
results obtained from the model are also presented Table 3.2.
Table 3.2–Multicomponent systems studied for validating the model along with AARD% between experimental data and predicted results
System Temperature Range, ºC Solid Phases AARD
%
CaSO4-CaCl2-HCl-H2O 22-80 DH, HH, AH 7.7
CaSO4-MgCl2-HCl-H2O 25-80 DH, AH 5.4
CaSO4-CaCl2-MgCl2-HCl-H2O 50-60 DH, HH 3.3
CaSO4-FeCl3-HCl (3.0M)-H2O 20-50 DH 8.5
CaSO4-CaCl2-NaCl-H2O 25-300 DH, AH 8.4
CaSO4-MgCl2-NaCl-H2O 28-250 DH, AH 4.6
CaSO4-Na2SO4-NaCl-H2O 25-300 DH, AH 6.5
CaSO4-Na2SO4-MgCl2-H2O 40 DH 8.0
CaSO4-NaCl-MgSO4-MgCl2-H2O 25-100 DH 8.5
2
∑−
=NP
i valueExp
valueCalculatedvalueExp
NPAARD
.
.100(%) , NP: total number of experimental points

52
System Temperature Range, ºC Solid Phases AARD
%
CaSO4-NiSO4-H2SO4 (0.15M)-Fe2(SO4)3 (0.2M)-LiCl (0.3M)-H2O 30-90 DH 6.1
CaSO4-H2SO4-NiSO4 (1.3M)-Fe2(SO4)3 (0.2M)-LiCl (0.3M)-H2O 30-90 DH 8.0
CaSO4- Fe2(SO4)3-H2SO4 (0.15M)-NiSO4 (1.3M)-LiCl (0.3M)-H2O 30-90 DH 4.3
CaSO4-LiCl-H2SO4 (0.15M)-NiSO4 (1.3M)-Fe2(SO4)3 (0.2M)-H2O 30-90 DH 5.6
CaSO4-Na2SO4-NiSO4 (1.3M)-H2SO4 (0.15M)-LiCl (0.3M)-H2O 30-90 DH 4.9
Note: DH: CaSO4•2H2O, HH: CaSO4•0.5H2O, AH: CaSO4
3.3 Results and Discussion
3.3.1 Evaluation of the Model Parameters
The solubilities of calcium chloride in water and of calcium sulphate hydrates in mixed
chloride–sulphate systems (shown in Table 3.1) were verified to determine whether the default
databank of the OLI® software (ver. 8.1.3) is capable of reproducing the available experimental
data, or whether it would be necessary to regress the model parameters through the OLI built-in
regression feature. The obtained model parameters are presented in Appendix B.
3.3.1.1 CaCl2–H2O System
The OLI® default database (ver. 8.1.3) was tested using the mean activity coefficient (γ±) (Rard
and Clegg, 1997; Robinson and Stokes, 2002), the activity of water (awater) (Rard and Clegg,
1997; Robinson and Stokes, 2002), and the solubility of CaCl2 in water (Garvin et al., 1987;
Clynne and Potter, 1979; Linke and Seidell, 1958). The database was shown to describe this
system accurately, in accordance with the experimental data, at 0–260°C. This confirmed that
the existing interaction parameters between calcium species and chloride ion in the OLI default
database can predict the system accurately.
3.3.1.2 CaSO4-CaCl2-H2O/CaSO4-HCl-H2O/CaSO4-NaCl-H2O/CaSO4-MgCl2-H2O Systems
The ability of the OLI default database to predict the solubility of CaSO4 hydrates in different
ternary aqueous chloride solutions, containing CaCl2, HCl, NaCl and MgCl2, was evaluated
using available experimental data. Some deviations were found to exist in the case of gypsum
(CaSO4.2H2O) and anhydrite (CaSO4), particularly at temperatures above 60ºC and for solutions
having HCl concentrations greater than 3 molal. Also, as was mentioned in Chapter 2, there are
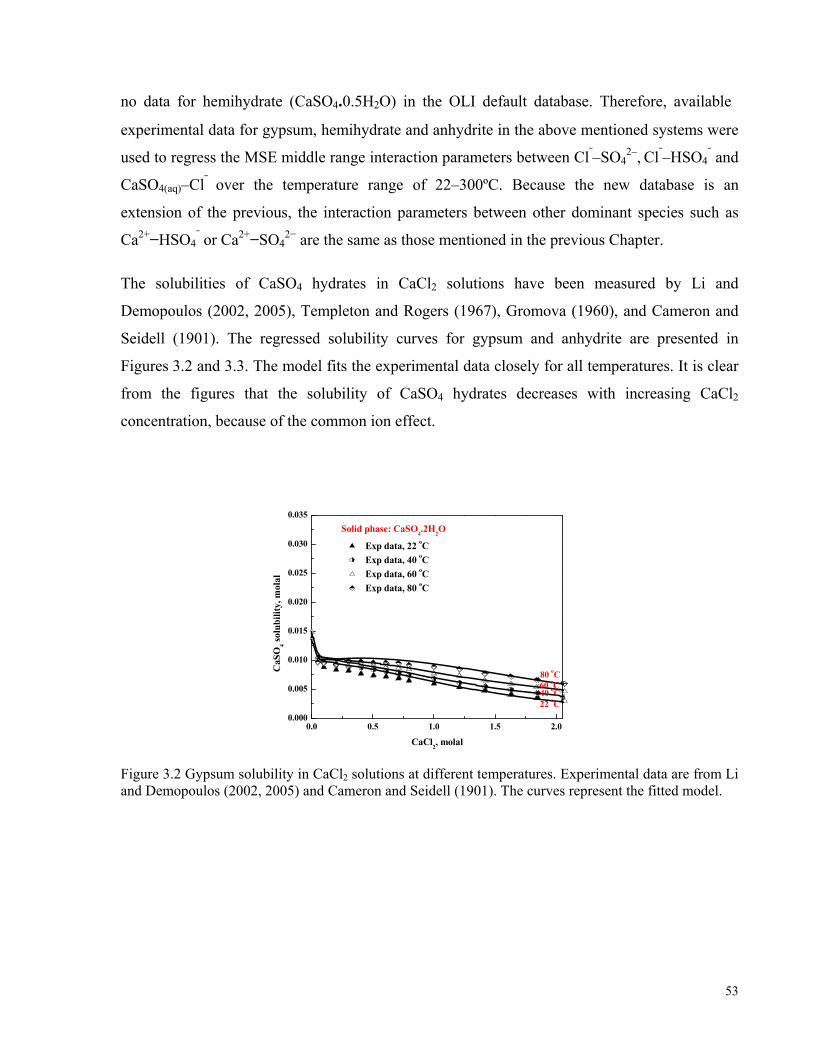
53
no data for hemihydrate (CaSO4.0.5H2O) in the OLI default database. Therefore, available
experimental data for gypsum, hemihydrate and anhydrite in the above mentioned systems were
used to regress the MSE middle range interaction parameters between Cl¯–SO42–, Cl¯–HSO4
¯ and
CaSO4(aq)–Cl¯ over the temperature range of 22–300ºC. Because the new database is an
extension of the previous, the interaction parameters between other dominant species such as
Ca2+−HSO4¯ or Ca2+−SO4
2− are the same as those mentioned in the previous Chapter.
The solubilities of CaSO4 hydrates in CaCl2 solutions have been measured by Li and
Demopoulos (2002, 2005), Templeton and Rogers (1967), Gromova (1960), and Cameron and
Seidell (1901). The regressed solubility curves for gypsum and anhydrite are presented in
Figures 3.2 and 3.3. The model fits the experimental data closely for all temperatures. It is clear
from the figures that the solubility of CaSO4 hydrates decreases with increasing CaCl2
concentration, because of the common ion effect.
0.0 0.5 1.0 1.5 2.00.000
0.005
0.010
0.015
0.020
0.025
0.030
0.035
80 oC60 oC40 oC
CaS
O4 so
lubi
lity,
mol
al
CaCl2, molal
Solid phase: CaSO4.2H2O
Exp data, 22 oC Exp data, 40 oC Exp data, 60 oC Exp data, 80 oC
22 oC
Figure 3.2 Gypsum solubility in CaCl2 solutions at different temperatures. Experimental data are from Li and Demopoulos (2002, 2005) and Cameron and Seidell (1901). The curves represent the fitted model.

54
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.00.000
0.005
0.010
0.015
0.020
0.025
0.030
0.035
0.040
0.045
0.050
50 oC80 oC110 oC
CaS
O4 so
lubi
lity,
mol
al
CaCl2, molal
Solid phase: CaSO4 (s)
Exp data, 25 oC Exp data, 50 oC Exp data, 80 oC Exp data, 110 oC Exp data, 250 oC Exp data, 300 oC
25 oC
Figure 3.3 Anhydrite solubility in CaCl2 solutions at different temperatures. Experimental data are from Li and Demopoulos (2005), Templeton and Rogers (1967), Gromova (1960). Curves are the fitted model.
The CaSO4–HCl–H2O system was studied by Li and Demopoulos (2002, 2005), Gupta (1968)
and is also cited by Linke and Seidell (1958) and Silcock (1979) in their solubility data
compilations over the temperature range of 10–80ºC. As shown in Figures 3.4 to 3.6, the model
fits the experimental data closely at all temperatures and for all three CaSO4 hydrates.
0 1 2 3 4 50.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
80 oC
60 oC
40 oC
22 oC
CaS
O4 so
lubi
lity,
mol
al
HCl, molal
Solid phase: CaSO4.2H
2O
Exp data, 22 oC Exp data, 40 oC Exp data, 60 oC Exp data, 80 oC
Figure 3.4 Gypsum solubility as a function of HCl concentration. Experimental data are from Li and Demopoulos (2002, 2005), Gupta (1968), Linke and Seidell (1958), Silcock (1979). Curves represent the fitted model.
0.00 0.02 0.04 0.06 0.08 0.10 0.12 0.14 0.16 0.180.0000
0.0002
0.0004
0.0006
0.0008
0.0010
300 oC
250 oC

55
The solubility of gypsum and anhydrite increases with increasing HCl concentration in the
range of 0.0–3.0 molal due to the formation of bisulphate ions which reduces the SO42-
concentration and allows an increase in the solubility of CaSO4 to satisfy the solubility product.
This effect is nullified at higher HCl concentrations due to the salting-out effect.
0 1 2 3 4 5 60.0
0.1
0.2
0.3
0.4
0.5
0.6
60 oC
80 oC
40 oC
CaS
O4 so
lubi
lity,
mol
al
HCl, molal
Solid phase: CaSO4
Exp data, 25 oC Exp data, 40 oC Exp data, 60 oC Exp data, 80 oC
25 oC
Figure 3.5 Anhydrite solubility in aqueous HCl solutions; experimental data are from Li and Demopoulos (2005), and curves represent the fitted model.
8.5 9.0 9.5 10.0 10.5 11.00.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
CaS
O4 so
lubi
lity,
mol
al
HCl, molal
Solid phase: CaSO4.1/2H
2O
Exp data, 25 oC Exp data, 50 oC
Figure 3.6 Hemihydrate solubility in aqueous HCl solutions; experimental data are from Li and Demopoulos (2005), and lines are the fitted model.
Similarly, the solubility of CaSO4 hydrates in aqueous solutions of NaCl was studied by
Templeton and Rogers (1967), Marshall and Slusher (1966), Ostroff and Metler (1966),
Marshall et al. (1964), Bock (1961) and also cited by Linke and Seidell (1958) and Silcock

56
(1979) in their solubility data compilations over the temperature range of 10–300ºC. The fitted
model results for the solubility of gypsum, anhydrite and hemihydrate in this ternary solution
are shown in Figures 3.7 to 3.9.
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.00.01
0.02
0.03
0.04
0.05
0.06
CaS
O4 so
lubi
lity,
mol
al
NaCl, molal
Solid phase: CaSO4.2H
2O
Exp data, 25 oC Exp data, 50 oC Exp data, 70 oC Exp data, 95 oC
Figure 3.7 Gypsum solubility in aqueous NaCl solutions. Experimental data are from Marshall and Slusher (1966), Ostroff and Metler (1966), Marshall et al. (1964), Linke and Seidell (1958), Silcock (1979); curves represent the fitted model.
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.50.00
0.02
0.04
0.06
0.08
0.10
0.12
0.14
0.16
250 oC175 oC125 oC
50 oC
CaS
O4 so
lubi
lity,
mol
al
NaCl, molal
Solid phase: CaSO4
Exp data, 25 oC Exp data, 50 oC Exp data, 125 oC Exp data, 175 oC Exp data, 250 oC
25 oC
Figure 3.8 Anhydrite solubility in aqueous NaCl solutions; experimental data are from Templeton and Rogers (1967), Marshall et al. (1964), Bock (1961) and Silcock (1979); curves represent the fitted model.
The solubility of all three solid phases increases slightly with increasing NaCl at low
concentrations due to the complexation effect of chloride ions and formation of calcium chloride
complexes (Williams-Jones and Seward, 1989).

57
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.00.00
0.02
0.04
0.06
0.08
0.10
CaS
O4 so
lubi
lity,
mol
al
NaCl, molal
Solid phase: CaSO4.1/2H2O
Exp data, 125 oC
Figure 3.9 Hemihydrate solubility as a function of NaCl concentration in aqueous solutions. Experimental data are from Marshall et al. (1964), and the curve represents the fitted model.
Cameron and Seidell (1901), Templeton and Rogers (1967), Ostroff and Metler (1966), and
Zdanovskii and Chernova (1976) measured the solubility of CaSO4 in the CaSO4–MgCl2–H2O
system from 25°C to 250ºC. Linke and Seidell (1958) also presented some data on this system
in their solubility data compilation. These data were also used, in addition to the experimental
data mentioned above (i.e., the solubility of all three CaSO4 hydrates in aqueous solutions of
CaCl2 and HCl, as well as in solutions of NaCl) to obtain interaction parameters between
Cl¯−SO42–, Cl¯−HSO4
¯ and CaSO4(aq)− Cl¯.
3.3.1.3 CaSO4–AlCl3–H2O System
The solubility of gypsum in aqueous AlCl3 solutions was measured by Li and Demopoulos
(2006a) from 25°C to 80°C. Additional fitting was performed on this system to attain the
interaction parameters between the AlSO4+−Ca2+ and Al3+−Ca2+ species (Appendix B). As is
clear from Figure 3.10, the obtained fits are in good agreement (AARD%=4.6) with the
experimental data. The solubility of gypsum first increases with increasing AlCl3 concentration
due to the complexation effect of chloride ions, however, at higher concentrations of AlCl3
(above 1 molal), this effect in nullified because of the decreased number of free water molecules
to participate in the dissolution process (the salting-out effect).

58
0.0 0.2 0.4 0.6 0.8 1.0 1.20.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
80 oC
50 oC
CaS
O4 so
lubi
lity,
mol
al
AlCl3, molal
Solid phase: CaSO4.2H
2O
Exp data, 25 oC Exp data, 50 oC Exp data, 80 oC
25 oC
Figure 3.10 Gypsum solubility in aqueous AlCl3 solutions. Experimental data are from Li and Demopoulos (2006a) and curves represent the fitted model.
3.3.1.4 CaSO4–FeCl3–HCl–H2O System
The solubility of gypsum in aqueous solutions containing FeCl3 and HCl (0.5 and 3.0 mol/L)
was measured by Li and Demopoulos (2006a). For this system, the MSE middle-range
interaction parameters between Ca2+ and FeCl2+ were regressed using the experimental data on
the solubility of gypsum in FeCl3 and 0.5 M of HCl system. Then, the obtained parameters were
employed to predict (without further regression) gypsum solubility in aqueous solutions
containing FeCl3 and 3 M of HCl. Figures 3.11 and 3.12 present regressed and predicted results
for these systems along with the experimental data, which are in good agreement.
0.0 0.5 1.0 1.5 2.0 2.50.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
80 oC
CaS
O4 so
lubi
lity,
mol
al
FeCl3, molal
Solid phase: CaSO4.2H2O
Exp data, 50 oC Exp data, 80 oC
[HCl]=0.5 M
50 oC
Figure 3.11 Gypsum solubility vs. FeCl3 concentration in CaSO4–FeCl3–HCl–H2O solutions. Experimental data are from Li and Demopoulos (2006a); curves represent the fitted model.

59
0.0 0.5 1.0 1.5 2.0 2.5 3.00.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
0.55
20 oC
50 oC
CaS
O4 so
lubi
lity,
mol
al
FeCl3, molal
Solid phase: CaSO4.2H
2O
Exp data, 20 oC Exp data, 50 oC
[HCl]=3 M
Figure 3.12 Gypsum solubility vs. FeCl3 concentration in CaSO4–FeCl3–HCl–H2O solutions. Experimental data are from Li and Demopoulos (2006a), and curves represent the predicted values.
3.3.2 Industrial Implications of the Model in Nickel Hydrometallurgy
As indicated in the previous section, the MSE activity coefficient model is effective for fitting
the solubility behaviour of the three solid phases of CaSO4 (gypsum, hemihydrate and
anhydrite) in mixed chloride-sulphate media. In order to validate the model parameters, the
solubilities of CaSO4 hydrates were calculated in multicomponent mixed chloride-sulphate
nickel hydromet processing solutions, containing NiSO4, Fe2(SO4)3, H2SO4, Na2SO4, and LiCl.
As will be seen later in this section, the model containing newly regressed parameters accurately
predicts (without additional fitting) the solubility behaviour of calcium sulphate in the systems
studied.
3.3.2.1 CaSO4–H2SO4–Fe2(SO4)3 (0.2 M)–NiSO4 (1.3 M)–LiCl (0.3 M)–H2O System
The effect of H2SO4 concentration from 0.0 to 0.7 mol/L, the maximum range of acid
concentration anticipated in the Voisey’s Bay Hydrometallurgical process, on the solubility of
calcium sulphate in simulated nickel sulphate-chloride processing solutions containing NiSO4,
H2SO4, Fe2(SO4)3 and LiCl at 25–95°C was studied by Dutrizac and Kuiper (2006). Figure 3.13
presents the experimental data and the solubilities predicted by the model. As is clear, the model
predictions are in good agreement with the experimental data (AARD%=8.0). Acid
concentration has a relatively minor effect on the solubility of CaSO4 in solutions containing 1.3
M NiSO4, 0.2 M Fe2(SO4)3 and 0.3 M LiCl. This dependence is due to the fact that the calcium

60
sulphate solubility in this system is mostly affected by the concentration of total free sulphate
ions released from dissociation of NiSO4 and Fe2(SO4)3. As a result, the presence of modest
concentrations of H2SO4 does not change the solubility of CaSO4 significantly. In this system,
gypsum was consistently the saturating solid phase in the experiments carried out using acid
concentrations less than 0.2 M H2SO4. But at higher acid concentrations, gypsum transformed
into anhydrite at temperatures above 90°C, with a consequential abrupt decrease in the solubility
of calcium sulphate.
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.90.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
90 oC
60 oC
[Fe2(SO4)3]=0.2M[NiSO
4]=1.3 M
[LiCl]=0.3 M
CaS
O4 so
lubi
lity,
mol
al
H2SO
4, molal
Solid Phase: CaSO4.2H
2O
Exp data, 30 oC Exp data, 60 oC Exp data, 90 oC
30 oC
Figure 3.13 CaSO4 solubility vs. H2SO4 concentration in CaSO4–H2SO4–Fe2(SO4)3(0.2M)–NiSO4(1.3M)–LiCl(0.3M)–H2O solutions. Experimental data are from Dutrizac and Kuiper (2006). Curves represent the predicted values.
3.3.2.2 CaSO4–Fe2(SO4)3–H2SO4 (0.15 M)–NiSO4 (1.3 M)–LiCl (0.3 M)–H2O System
In nickel pressure leaching processes, iron is dissolved in the autoclave along with nickel.
Although most of the dissolved iron re-precipitates in the autoclave as ferric oxide, ferric
oxyhydroxide or jarosite, some remains in the discharged solution. The dissolved iron is
subsequently oxidized and precipitated when the solution is neutralized with lime. Thus, various
iron concentrations are encountered in different parts of the process, for this reason, the effect of
the concentration of ferric sulphate on the solubility of calcium sulphate is important.
The solubility of CaSO4 as a function of ferric sulphate concentration in solutions containing 1.3
M NiSO4, 0.15 M H2SO4 and 0.3 M LiCl was also measured by Dutrizac and Kuiper (2006).
The predicted results obtained for the solubility of CaSO4 vs. Fe2(SO4)3 concentration are shown

61
in Figure 3.14 along with the experimental data. The model predictions accurately reflect the
experimental data (AARD%=4.3). Generally, the presence of ferric sulphate in the solution has
only a modest effect on the solubility of CaSO4. At 90°C, increasing Fe2(SO4)3 concentrations
causes a slight increase in the solubility of calcium sulphate, whereas at 30 and 60°C, solubility
is nearly independent of the concentration of Fe2(SO4)3.
0.0 0.2 0.4 0.6 0.8 1.00.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
30 oC
60 oC
[H2SO
4]=0.15 M
[NiSO4]=1.3 M
[LiCl]=0.3 M
Solid phase: CaSO4.2H
2O
Exp data, 30 oC Exp data, 60 oC Exp data, 90 oC
CaS
O4 so
lubi
lity,
mol
al
Fe2(SO
4)
3, molal
90 oC
Figure 3.14 CaSO4 solubility vs. Fe2(SO4)3 concentration in CaSO4–Fe2(SO4)3–NiSO4(1.3M)–H2SO4 (0.15M)–LiCl(0.3M)–H2O solutions; experimental data are from Dutrizac and Kuiper (2006), and curves represent the predicted values.
3.3.2.3 CaSO4–NiSO4–Fe2(SO4)3 (0.2 M)–H2SO4 (0.15 M)–LiCl (0.3 M)–H2O System
In the Voisey’s Bay Hydrometallurgical process, the maximum concentration of NiSO4 is
anticipated to be around 1.4 mol/L. Dutrizac and Kuiper (2006) studied the effect of NiSO4
concentration from 0.0 to 1.4 mol/L on the solubility of calcium sulphate in nickel sulphate-
chloride processing solutions containing 0.15 M H2SO4, 0.2 M Fe2(SO4)3, and 0.3 M LiCl at 25–
95°C. Figure 3.15 presents the experimental data along with the model predictions. The
predicted results are in good agreement with the experimental data (AARD%=6.1). The
solubility of calcium sulphate initially decreases with increasing NiSO4 concentration up to
0.2 M because of the common ion effect. This effect is diminished at higher NiSO4
concentrations (above 0.2 M) because of the association of Ca2+ and SO42- and formation of
calcium sulphate neutral species.

62
0.0 0.2 0.4 0.6 0.8 1.0 1.2 1.40.00
0.02
0.04
0.06
0.08
0.10
0.12
30 oC60 oC
[Fe2(SO4)3]=0.2M[H2SO4]=0.15 M[LiCl]=0.3 M
CaS
O4 so
lubi
lity,
mol
al
NiSO4, molal
Solid Phase: CaSO4.2H2O
Exp data, 30 oC Exp data, 60 oC Exp data, 90 oC
90 oC
Figure 3.15 CaSO4 solubility vs. NiSO4 concentration in CaSO4–NiSO4–Fe2(SO4)3(0.2M)–H2SO4 (0.15M)–LiCl(0.3M)–H2O solutions; experimental data are from Dutrizac and Kuiper (2006), and curves represent the model results.
3.3.2.4 CaSO4–LiCl–H2SO4 (0.15 M)–NiSO4 (1.3 M)–Fe2(SO4)3 (0.2 M)–H2O System
The addition of chloride ions to a sulphate processing solution could affect the solubility of
calcium sulphate. To ascertain the influence of chloride concentration on the solubility of
calcium sulphate, a systematic series of solubility measurements was carried out by Dutrizac
and Kuiper (2006) at various temperatures from 30°C to 90°C in solutions containing 1.3 M
NiSO4, 0.15 M H2SO4 and 0.2 M Fe2(SO4)3. LiCl was used as the chloride source, rather than
NaCl, to avoid the precipitation of sodium jarosite at temperatures above 80°C.
The experimental solubility data for this system are shown in Figure 3.16 along with the model
predictions for the system. As can be seen, the agreement is good at all temperatures studied
(AARD%=5.6). Also, it is clear that the solubility of CaSO4 decreases slightly as the
concentration of chloride ions in the solution increases because of the salting-out effect. At zero
concentration of LiCl, the solution already contains 1.3 M NiSO4, 0.2 M Fe2(SO4)3 and 0.15 M
H2SO4. Addition of other electrolytes (such as LiCl) results in a reduction in the number of free
water molecules participating in the dissolution process because they are tightly held by cations
and anions in the solution.

63
0.0 0.2 0.4 0.6 0.8 1.0 1.20.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
90 oC
60 oC
[Fe2(SO
4)
3]=0.2 M
[H2SO
4]=0.15 M
[NiSO4]=1.3 M
CaS
O4 so
lubi
lity,
mol
al
LiCl, molal
Solid Phase: CaSO4.2H
2O
Exp data, 30 oC Exp data, 60 oC Exp data, 90 oC
30 oC
Figure 3.16 CaSO4 solubility vs. LiCl concentration in CaSO4–LiCl–H2SO4(0.15M)–Fe2(SO4)3(0.2M)–NiSO4(1.3M)–H2O solutions; experimental data are from Dutrizac and Kuiper (2006), and curves represent the predicted values.
3.3.2.5 CaSO4–Na2SO4–H2SO4 (0.15 M)–NiSO4 (1.3 M)–LiCl (0.3 M)–H2O System
In nickel hydrometallurgy, sodium carbonate is sometimes used, directly or indirectly, for pH
control. This practice results in the presence of sodium ions in the circulating sulphate–chloride
processing solutions. The sodium ions can be considered to be present, formally, as either
Na2SO4 or NaCl. To investigate the effect of sodium ions on the solubility of calcium sulphate
in nickel sulphate–chloride processing solutions, at a constant chloride concentration, various
experiments were carried out by Dutrizac and Kuiper (2006) wherein Na2SO4 was added to the
simulated nickel processing solutions containing NiSO4, H2SO4 and LiCl.
The experimental data and the model predictions are shown in Figure 3.17, it is clear the model
closely predicts the solution chemistry (AARD%=4.9), without need for additional fitting. The
presence of modest concentrations of Na2SO4 in the 1.3 M NiSO4–0.15 M H2SO4–0.3 M LiCl
base solution has a minimal effect on the solubility of calcium sulphate at all temperatures
studied. This reflects the fact that calcium sulphate solubility is predominantly affected by the
total sulphate concentration. Accordingly, for concentrated NiSO4 solutions, the presence of the
modest concentrations (up to 0.5 M) of Na2SO4 does not affect the solubility of CaSO4
significantly.

64
0.0 0.1 0.2 0.3 0.4 0.5 0.60.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
90 oC
60 oC
[H2SO
4]=0.15 M
[NiSO4]=1.3 M
[LiCl]=0.3 M
CaS
O4 so
lubi
lity,
mol
al
Na2SO
4, molal
Solid Phase: CaSO4.2H2O
Exp data, 30 oC Exp data, 60 oC Exp data, 90 oC
30 oC
Figure 3.17 CaSO4 solubility vs. Na2SO4 concentration in CaSO4–Na2SO4–NiSO4(1.3M)–H2SO4(0.15M)–LiCl(0.3M)–H2O solutions; experimental data are from Dutrizac and Kuiper (2006), and curves represent the predicted values.
3.3.3 Predictive Capacity of the Model Parameters in Mixed Chloride Solutions
To further validate the predictive capacity of the model containing new interaction parameters,
the solubilities of calcium sulphate hydrates were calculated in multicomponent solutions
containing various chloride–sulphate electrolytes such as CaCl2, HCl, NaCl, MgCl2, Na2SO4,
and MgSO4 for which experimental data are available in the literature. As will be seen in the
following section, the model predictions are in good agreement with the experimental data
without performing additional fittings. This shows the usefulness of the model in predicting the
chemistry of complex systems for which no experimental data are available.
3.3.3.1 CaSO4–CaCl2–HCl–H2O System
The solubility of CaSO4 in mixed CaCl2 and HCl (1, 3, 5 M) aqueous solutions was measured
by Li and Demopoulos (2002, 2005) over the temperature range of 22–80°C. Silcock (1979)
also reported some data on this system in his solubility data compilation. Figures 3.18 to 3.20
show the experimental solubility data for all three solid phases of CaSO4 as a function of CaCl2
concentration at fixed HCl concentrations.

65
0.0 0.5 1.0 1.5 2.0 2.50.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
60 oC
40 oC
22 oC
[HCl]=6 M
CaS
O4 so
lubi
lity,
mol
al
CaCl2, molal
Solid phase: CaSO4.2H
2O
Exp data 22 oC Exp data 40 oC Exp data 60 oC
Figure 3.18 Gypsum solubility as a function of CaCl2 concentration in CaSO4–CaCl2–HCl–H2O solutions. Experimental data are from Li and Demopoulos (2002, 2005) and Silcock (1979); curves represent the predicted values.
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.50.00
0.02
0.04
0.06
0.08
0.10
0.12
0.14
0.16
50 oC80 oC
25 oC
[HCl]=6 M
CaS
O4 so
lubi
lity,
mol
al
CaCl2, molal
Solid phase: CaSO4
Exp data, 25 oC Exp data, 50 oC Exp data, 80 oC
Figure 3.19 Anhydrite solubility vs. CaCl2 concentration in CaSO4–CaCl2–HCl–H2O solutions; experimental data are from Li and Demopoulos (2005), and curves represent the predicted values.
Even at high concentrations of CaCl2 and HCl, the model closely predicts the solubility of all
three CaSO4 hydrates (AARD%=7.7 for 137 points). The trend shows that the addition of
calcium chloride to concentrated chloride solutions causes the solubility to decrease sharply
because of the common ion effect. Also, increasing HCl concentrations from 3 M to 6 M
reduces the number of free water molecules participating in the dissolution process, which
results in decreasing CaSO4 solubilities due to the salting-out effect.

66
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.50.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
[HCl]=3 M
[HCl]=6 M
CaS
O4 so
lubi
lity,
mol
al
CaCl2, molal
Solid phase: CaSO4.1/2H2O
Exp data, 60 oC Exp data, 80 oC
Figure 3.20 Hemihydrate solubility vs. CaCl2 concentration in CaSO4–CaCl2–HCl–H2O solutions; the experimental data are from Li and Demopoulos (2005), and curves represent the predicted values.
3.3.3.2 CaSO4–MgCl2–HCl–H2O System
Several experiments were carried out by Li and Demopoulos (2002, 2006a) to investigate the
effect of magnesium chloride concentration on the solubility of gypsum and anhydrite at
constant concentrations of hydrochloric acid, over the temperature range of 25–80°C. The
experimental solubility data for gypsum in this system at 3 M HCl are shown in Figure 3.21
along with the model predictions.
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.50.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
25 oC
[HCl]=3 M
CaS
O4 so
lubi
lity,
mol
al
MgCl2, molal
Solid Phase: CaSO4.2H
2O
Exp data, 25 oC Exp data, 50 oC
50 oC
Figure 3.21 Gypsum solubility vs. MgCl2 concentration in CaSO4–MgCl2–HCl–H2O solutions; the experimental data are from Li and Demopoulos (2006a), and curves represent the predicted values.
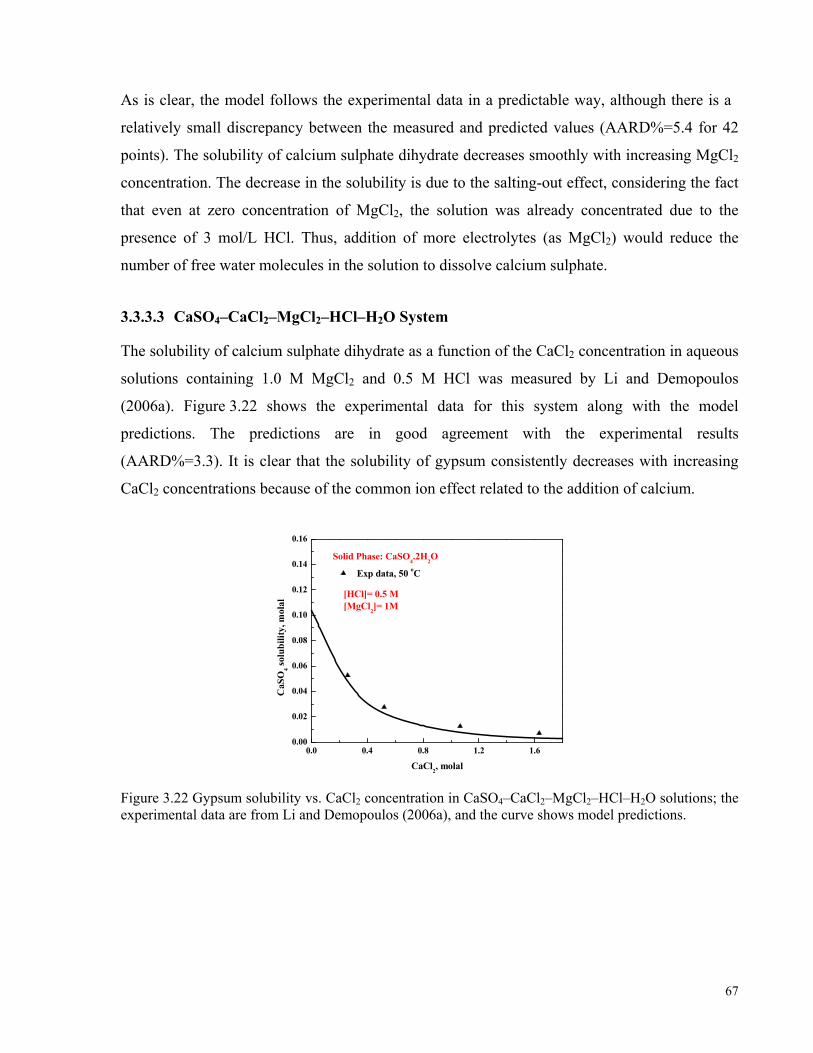
67
As is clear, the model follows the experimental data in a predictable way, although there is a
relatively small discrepancy between the measured and predicted values (AARD%=5.4 for 42
points). The solubility of calcium sulphate dihydrate decreases smoothly with increasing MgCl2
concentration. The decrease in the solubility is due to the salting-out effect, considering the fact
that even at zero concentration of MgCl2, the solution was already concentrated due to the
presence of 3 mol/L HCl. Thus, addition of more electrolytes (as MgCl2) would reduce the
number of free water molecules in the solution to dissolve calcium sulphate.
3.3.3.3 CaSO4–CaCl2–MgCl2–HCl–H2O System
The solubility of calcium sulphate dihydrate as a function of the CaCl2 concentration in aqueous
solutions containing 1.0 M MgCl2 and 0.5 M HCl was measured by Li and Demopoulos
(2006a). Figure 3.22 shows the experimental data for this system along with the model
predictions. The predictions are in good agreement with the experimental results
(AARD%=3.3). It is clear that the solubility of gypsum consistently decreases with increasing
CaCl2 concentrations because of the common ion effect related to the addition of calcium.
0.0 0.4 0.8 1.2 1.60.00
0.02
0.04
0.06
0.08
0.10
0.12
0.14
0.16
[HCl]= 0.5 M[MgCl
2]= 1M
CaS
O4 so
lubi
lity,
mol
al
CaCl2, molal
Solid Phase: CaSO4.2H
2O
Exp data, 50 oC
Figure 3.22 Gypsum solubility vs. CaCl2 concentration in CaSO4–CaCl2–MgCl2–HCl–H2O solutions; the experimental data are from Li and Demopoulos (2006a), and the curve shows model predictions.

68
3.3.3.4 CaSO4–Na2SO4–NaCl–H2O System
The solubility of gypsum and anhydrite in quaternary aqueous solutions of CaSO4, Na2SO4 and
NaCl, over the temperature range of 25–300°C, was determined by various researchers (Yeatts
and Marshall, 1972; Block and Waters, 1968; Furby et al., 1968; Templeton and Rodgers, 1967;
Cameron et al., 1907). It was shown that the solubility of gypsum first decreases sharply and
then increases gradually with increasing Na2SO4 concentration in the presence of up to 1 molal
NaCl. The initial decrease is due to the common ion effect of the added sulphate; the subsequent
increase is attributable to the association of Ca2+ and SO42- ions and formation of calcium
sulphate neutral species. For solutions containing 2.0 molal NaCl and above, the salting-out
effect becomes dominant, as a result, the increase in the solubility of gypsum is not observed
(Block and Waters, 1968; Templeton and Rodgers, 1967).
Similar trends are observed for the solubility of anhydrite, although solubilities are significantly
lower than those of gypsum at a given concentration of NaCl and Na2SO4. The new database,
utilized by the OLI/MSE model, is capable of making accurate predictions in these complex
systems (AARD%=6.5) as illustrated in Figures 3.23 and 3.24.
0.0 0.2 0.4 0.6 0.8 1.0 1.2 1.4 1.6 1.8 2.0 2.20.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
NaCl=4.0 mNaCl=2.0 m
NaCl=1.0 m
CaS
O4 so
lubi
lity,
mol
al
Na2SO
4, molal
Solid Phase: CaSO4.2H2O Exp data, [NaCl]=0.0 m Exp data, [NaCl]=1.0 m Exp data, [NaCl]=2.0 m Exp data, [NaCl]=4.0 m
NaCl=0.0 m
T= 40 oC
Figure 3.23 Gypsum solubility vs. Na2SO4 concentration in CaSO4–Na2SO4–NaCl–H2O solutions; the experimental data are from Block and Waters (1968), and the curves represent the predicted values.

69
0.00 0.05 0.10 0.15 0.20 0.25 0.300.0000
0.0005
0.0010
0.0015
0.0020
0.0025
0.0030
0.0035
0.0040
300 oC
250 oC
300 oC
I=0.5 m
CaS
O4 so
lubi
lity,
mol
al
Na2SO
4, molal
Solid phase: CaSO4
Exp data, 300 oC Exp data, 250 oC Exp data, 300 oC
I=0.9 m
Figure 3.24 Anhydrite solubility vs. Na2SO4 concentration in CaSO4–Na2SO4–NaCl–H2O solutions; the experimental data are from Templeton and Rodgers (1967), and curves represent the predicted values.
3.3.3.5 CaSO4–Na2SO4–MgCl2–H2O System
The solubility of gypsum in aqueous solutions of CaSO4, Na2SO4 and MgCl2 was measured by
Barba et al. (1984). Figure 3.25 shows the predicted results obtained from the model utilizing
the newly developed database compared with the experimental data. As can be seen, the model
predicts the experimental data closely (AARD%=8.0).
0.0 0.2 0.4 0.6 0.8 1.0 1.2 1.4 1.6 1.8 2.00.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
[MgCl2]=0.0 M
[MgCl2]=0.2 M
[MgCl2]=0.4 M
CaS
O4 so
lubi
lity,
mol
al
Na2SO
4, molal
Solid phase: CaSO4.2H
2O
Exp data, [MgCl2]=0.0 M Exp data, [MgCl2]=0.2 M Exp data, [MgCl2]=0.4 M Exp data, [MgCl2]=0.6 M
T=40 oC
[MgCl2]=0.6 M
Figure 3.25 Gypsum solubility vs. Na2SO4 concentration in CaSO4–Na2SO4–MgCl2–H2O solutions; the experimental data are from Barba et al. (1984), and curves represent the predicted values.

70
In this system, the solubility of calcium sulphate initially decreases with increasing Na2SO4
concentration at a fixed MgCl2 molality because of the common ion effect (SO42- is added and
the addition shifts the dissolution reaction to the left). However, for Na2SO4 concentrations
above ~0.4 molal, this effect is nullified by the association between Ca2+ and SO42− and the
consequential formation of calcium sulphate neutral species. Also, at a relatively low fixed
Na2SO4 concentration (below 0.4 molal) the solubility of gypsum increases with increasing
MgCl2 molality due to the complexation effect of chloride ions. However, the reverse trend is
observed for Na2SO4 concentrations above 0.4 molal where gypsum solubility decreases slightly
with increasing MgCl2 concentration due to the salting-out effect.
3.3.3.6 CaSO4–MgSO4–HCl–H2O / CaSO4–NiSO4–H2SO4–H2O Systems
To compare the effect of H2SO4 and HCl on the solubility of calcium sulfate, gypsum solubility
was measured in MgSO4 solutions containing 0.5 M HCl, as well as in NiSO4 solutions
containing 0.5 M H2SO4 in this work. The measured solubility data along with the model
prediction results from 25°C to 90°C are shown in Figures 3.26 and 3.27. Details regarding the
experimental procedure are discussed in Chapter 4.
0.0 0.2 0.4 0.6 0.8 1.0 1.2 1.4 1.60.00
0.05
0.10
0.15
0.20
0.25
0.30
Solid phase: CaSO4.2H
2O
Exp data, 25 oC Exp data, 45 oC Exp data, 70 oC Exp data, 90 oC
CaS
O4 so
lubi
lity,
mol
al
MgSO4, molal
[HCl]=0.5 M
Figure 3.26 CaSO4 solubility in CaSO4–MgSO4–HCl (0.5M)–H2O solutions; experimental data are from Azimi and Papangelakis (2010a), and curves are the model predictions.
In both systems, the solubility first decreases with increasing MSO4 (M=Ni, Mg) concentration
due to the common ion effect of the added SO42- ions. This effect is nullified by further
increasing MSO4 concentration because of the association of Ca2+ and SO42- ions and formation
of calcium sulfate neutral species. The solubility of gypsum is significantly higher in the
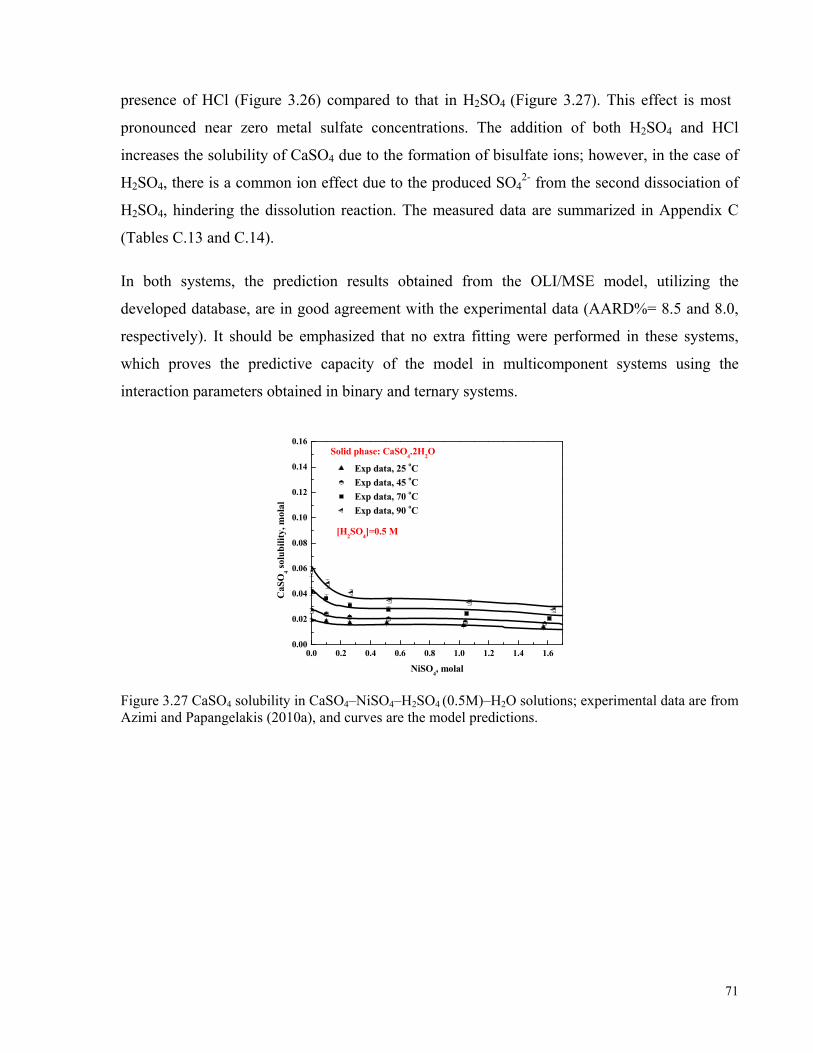
71
presence of HCl (Figure 3.26) compared to that in H2SO4 (Figure 3.27). This effect is most
pronounced near zero metal sulfate concentrations. The addition of both H2SO4 and HCl
increases the solubility of CaSO4 due to the formation of bisulfate ions; however, in the case of
H2SO4, there is a common ion effect due to the produced SO42- from the second dissociation of
H2SO4, hindering the dissolution reaction. The measured data are summarized in Appendix C
(Tables C.13 and C.14).
In both systems, the prediction results obtained from the OLI/MSE model, utilizing the
developed database, are in good agreement with the experimental data (AARD%= 8.5 and 8.0,
respectively). It should be emphasized that no extra fitting were performed in these systems,
which proves the predictive capacity of the model in multicomponent systems using the
interaction parameters obtained in binary and ternary systems.
0.0 0.2 0.4 0.6 0.8 1.0 1.2 1.4 1.60.00
0.02
0.04
0.06
0.08
0.10
0.12
0.14
0.16
[H2SO4]=0.5 M
CaS
O4 so
lubi
lity,
mol
al
NiSO4, molal
Solid phase: CaSO4.2H2O
Exp data, 25 oC Exp data, 45 oC Exp data, 70 oC Exp data, 90 oC
Figure 3.27 CaSO4 solubility in CaSO4–NiSO4–H2SO4 (0.5M)–H2O solutions; experimental data are from Azimi and Papangelakis (2010a), and curves are the model predictions.

72
3.4 Summary
The recently developed database for the mixed-solvent electrolyte (MSE) model on calcium
sulphate chemistry in sulphate electrolyte solutions was extended to be applicable in complex
multicomponent chloride-sulphate solutions. To improve the predictive capacity and self-
consistency of the new model, interaction parameters were obtained by fitting the experimental
data for ternary systems, and the interaction parameters were subsequently validated in
multicomponent systems. This approach is superior to that taken in other studies, wherein
interaction parameters were adjusted using multicomponent solutions, because the latter
approach does not guarantee consistent and accurate results in binary, ternary and other
multicomponent systems. The fact that modelling in binary and ternary systems is sufficient to
predict the behaviour of more complex multicomponent systems is a substantive contribution to
the existing field.
The new database, utilized by the OLI/MSE model, accurately predicts the solubility of calcium
sulphate, and therefore the scaling potential, in the neutralization stage of nickel sulphate-
chloride processing solutions of the Voisey’s Bay hydrometallurgical plant, containing NiSO4,
Fe2(SO4)3, Na2SO4, H2SO4, and LiCl at 20–95°C. The model was also shown to successfully
predict the solubilities of all three phases of calcium sulphate (DH, HH and AH) in mixed
chloride-sulphate solutions containing HCl, Ca, Fe(III), Mg, Na, and SO42- and over wide ranges
of temperature and composition. These facts illustrate the usefulness of the model in gaining
comprehensive insight for process improvements and optimization of conditions. The developed
database is utilized by mining companies such as Vale Inco to assess calcium sulphate scaling
potential in processing streams over wide ranges of temperature and concentration.

73
CHAPTER 4 SOLUBILITY OF GYPSUM AND ANHYDRITE IN LATERITE PRESSURE ACID LEACH SOLUTIONS
he focus of this chapter is on the experimental measurements of the solubility of gypsum
at 25–90°C and that of anhydrite at 150–250°C in simulated laterite pressure acid leach
(PAL) solutions. In this chapter, the predictive capacity of the model, utilizing the developed
database, was tested against the measured experimental data for both solids over wide ranges of
composition and temperature. This chapter is based on the following publication:
- Azimi, G., Papangelakis, V.G., 2010b. Hydrometallurgy, in press.
4.1 Introduction
In recent years, industrial processes at elevated temperatures have become of great interest. This
is particularly true in the hydrometallurgical nickel industry where sulphuric acid pressure
leaching of laterite ores has become the process of choice because of its fast kinetics, selective
extraction of valuable metals (Ni and Co) and simultaneous rejection of impurities (Al and Fe)
(Whittington and Muir, 2000).
Pressure acid leaching of the concentrate feed is carried out in autoclaves at high temperatures
between 150°C and 250°C. After digestion, the slurry is flashed to approximately 100°C and the
solids are separated from the liquid phase in a counter-current decantation (CCD) thickener
circuit. Most of impurities in the form of iron and aluminum initially present in the concentrate
feed precipitate in the autoclave, but a small amount remains in the discharged leach solution. In
order to increase the leach solution pH and precipitate the soluble impurities, the solution is
oxidized and subsequently neutralized by limestone (CaCO3). After filtration, the solution
containing the base metals is further treated by a number of methods including solvent
extraction and electrowinning to refine and extract the base metals from the solution. The
extracted metals are sent for further processing to recover pure metal or metal compound
products. The raffinate continues to a final neutralization stage to further increase pH and
precipitate the remaining metal ions, providing an environmentally safe solution for disposal.
The upstream process solution from the neutralization stage is saturated with gypsum at ambient
T

74
temperature and recycled to the beginning of the circuit for further usage. A process flow
diagram is presented in Chapter 1 (Figure 1.2).
The formation of undesirable calcium sulphate byproducts, mostly as scale, during pressure acid
leaching of laterite ores is one of the significant problems encountered in these processes. As
the scale layer becomes increasingly thicker, it reduces the production capacity and process
efficiency because of decreased volume of the equipment and reduced heat transfer capacity,
blocked pipelines and reduction of material flow, corrosion and wearing out of construction
materials. Also, calcium sulphate precipitation in the purification stage of base metals,
particularly in the solvent extraction stage, could create severe operational problems because of
the formation of a third solid phase (crud) (Nofal et al., 2001). Therefore, periodic shut-downs
of the plant for mechanical removal of precipitated calcium sulphate hydrates from the
processing circuit are necessary.
Calcium enters the sulphate refining electrolytes in different ways. Sometimes, the ore itself
contains calcium (Whittington and Muir, 2000). Also, the addition of calcium-containing bases
in the neutralization stage increases the concentration of calcium in the process circuit.
Moreover, in some refineries, the process water is a source of calcium ions. Calcium sulphate
hydrates (gypsum (CaSO4•2H2O), hemihydrate (CaSO4•0.5H2O) and anhydrite (CaSO4)) are
relatively insoluble and they are formed wherever calcium and sulphate occur together in
aqueous solutions. Many processes operate with very low solution bleeds and as a result,
calcium sulphate accumulates in the refining electrolyte. Furthermore, the transformation of
gypsum, the stable phase below 100°C, to anhydrite, the stable one at higher temperatures,
decreases the solubility significantly and makes the prediction and control of calcium sulphate
formation in these processes complicated. Therefore, having a thorough knowledge of the phase
behaviour of calcium sulphate and being able to accurately estimate the scaling potential in
these systems at various temperatures is of great practical importance.
A review of the literature reveals that no previous theoretical or experimental work has been
undertaken to study the simultaneous effects of coexisting metal sulphates and chlorides on the
solubility of CaSO4 hydrates over a broad temperature and concentration range in industrial
systems, particularly in laterite pressure acid leach (PAL) solutions. In terms of theoretical
modelling, most of the previous studies focused on CaSO4 solubility in water or in ternary and

75
quaternary aqueous solutions containing H2SO4, MgSO4, Na2SO4, etc. (Li and Demopoulos,
2006a; Arslan and Dutt, 1993; Barba et al., 1982).
A considerable amount of experimental work has been conducted to study calcium sulphate
solubilities under atmospheric pressure from 25°C to 95°C in water or in H2SO4 and HCl acidic
solutions as well as in multicomponent metal sulphate-chloride solutions (Farrah et al., 2007;
Dutrizac and Kuiper, 2006; Li and Demopoulos, 2005, 2006a; Dutrizac, 2002; Block and
Waters, 1968; Zdanovskii et al., 1968; Bock, 1961; Hill and Wills, 1938; Posnjak, 1938; Hulett
and Allen, 1902). At elevated temperatures, several experimental studies have also been
performed on solubility of calcium sulphate in water or in H2SO4 media as well as in ternary or
quaternary solutions containing NaCl, Na2SO4, MgCl2 up to 350°C (Blount and Dickson, 1969;
Furby et al., 1968; Marshall and Slusher, 1966, 1968; Templeton and Rodgers, 1967; Marshall
and Jones, 1966; Marshall et al., 1964; Partridge and White, 1929). However, no previous work
has been carried out to take into account the effect of metal sulphates on calcium sulphate
hydrates solubilities in multicomponent solutions over wide temperature ranges, particularly at
elevated temperatures, under acidic conditions.
In Chapters 2 and 3, a new database for the Mixed Solvent Electrolyte (MSE) model of the OLI
software was developed to predict calcium sulphate solubilities in multicomponent sulphate-
chloride electrolyte solutions (Azimi et al., 2007, 2008). The model was shown to accurately
predict gypsum and anhydrite solubility data in various industrial solutions including the nickel
sulphate-chloride processing solutions of the Voisey’s Bay plant (Azimi et al., 2008) and the
neutralized zinc sulphate leach solutions (Azimi et al., 2010) for which the experimental data
were measured by Dutrizac and Kuiper (2006) and Dutrizac (2002), respectively.
In this chapter, a number of experiments were conducted to measure the solubility of gypsum
and that of anhydrite in synthetic laterite pressure acid leach solutions containing NiSO4, H2SO4,
MgSO4, Al2(SO4)3 and NaCl in the respective temperature stability ranges, i.e., 25–90°C for
gypsum and 150–250°C for anhydrite. Then, the developed database, utilized by the OLI/MSE
model, was used to predict gypsum and anhydrite solubilities in the systems studied.

76
4.2 Experimental Procedure
All solutions used in this study were prepared by dissolving reagent grade chemicals directly
without further purification. Calcium sulphate dihydrate (gypsum) reagent was from J.T. Baker
with 99.4% purity and was used as one of the saturating solid phases. Calcium sulphate
anhydrite was also from J.T. Baker with 100% purity and was used as the other saturating solid
phase in this work. X-ray diffraction analysis was carried out on both solids using a Philips
PW3719 diffractometer. The diffractograms showed 100% gypsum and anhydrite, respectively
(Appendix D, Figures D.1 and D.2). No traces of hemihydrate or anhydrite were found in the
gypsum solid powder. For comparison purpose, the diffractogram of hemihydrate is also
presented in Appendix D.
Low temperature experiments under atmospheric pressure were performed inside 1 L double
layer reactors where heating was provided through a circulating oil jacket. Temperature was
controlled within ±1°C of the set-point. The reactor slurry was kept suspended by a shaft stirrer.
Samples were withdrawn through a dip tube using preheated syringes and filtrations were
performed using 0.22 μm PTFE syringe filters from Fisher Scientific. To avoid solution
evaporation during the runs, the stirrer bushings were fully sealed using Dow Corning® high
vacuum grease. Moreover, the concentration of elements other than Ca was monitored
throughout the experiments to confirm that the solution composition remained unchanged.
High temperature experiments were carried out in a 600 mL titanium autoclave, manufactured
by the Parr Instrument Company. Agitation was provided by a motor-driven titanium shaft
impeller. Temperature was controlled by manipulating an electrical heating mantle and a
cooling water stream, maintaining the autoclave temperature within ±1°C of the set-point. The
solution was placed inside a glass liner to protect the interior wall of the bomb from corrosion
and metal deposition. The autoclave was equipped with a dip tube for sample withdrawal
through an in situ 2 μm porous titanium filter. Schematic diagrams of the glass reactor and
autoclave are presented in Appendix E.
Solutions of known composition were placed in the reactors with an excess of saturating solid
phase. Experiments were started by heating the charged reactors to temperature and allowing
sufficient time to reach equilibrium while the reactor contents were agitated thoroughly.

77
Equilibration time in solubility measurements can vary over a wide range, from several hours
to several days, depending on the dissolution rate of the solid phase under the applied
conditions. In the present study, several kinetic tests were conducted at various temperatures and
the results showed that ~24 h was necessary to achieve complete saturation for low temperature
experiments, whereas 4–6 h was sufficient to reach equilibrium in high temperature experiments
inside the autoclave. In each test, two to three samples were taken towards the end of the run at
time intervals of 0.5–1 h to ensure that the calcium concentration reached a plateau as an
indication of equilibration.
Along with time-stable solubilities, the solubility data were also determined on heating and
cooling methods, based on dissolution and precipitation, as confirmatory indications of
equilibration. As long as the saturating solid phase remained unchanged, the various data
measured on heating and cooling were consistent. Reproducibility tests showed that the
experimentally measured data are accurate to within ±5%.
Withdrawn solution samples were diluted with 5% HNO3 and stored in sealed plastic test tubes
at room temperature. The Ca concentration was determined by ICP–OES analysis using the
317.933 nm emission line. The densities of the corresponding solutions were determined using a
portable density meter (DMA 35N) from Anton Paar. For the tests performed at temperatures
below 100°C, densities were measured “at temperature”, whereas for high temperature
experiments, they were measured at room temperature.
Samples of the equilibrating solid phase were also withdrawn, filtered, and washed with a small
amount of denatured alcohol, containing 85% ethanol and 15% methanol, to replace the solution
and dried below 40°C in an oven under vacuum. Powder X-ray diffraction patterns of the solid
samples were collected on a Philips PW3719 diffractometer utilizing Cu Ka radiation in the
range 10–60° 2θ with a step size of 0.02° and a collection time of 1.25 s/step. The generated
patterns were matched against the International Centre for Diffraction Data® files (JCPDF-
ICDD file numbers 070–0982 for gypsum and 072–0916 for anhydrite).
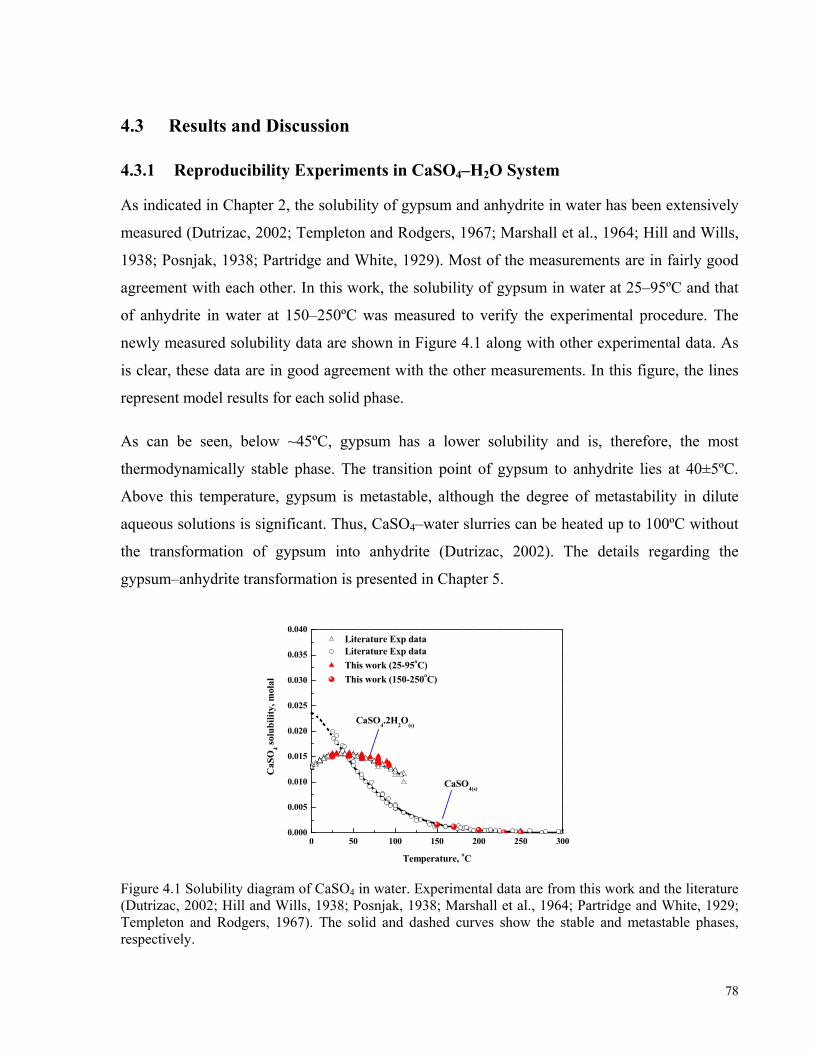
78
4.3 Results and Discussion
4.3.1 Reproducibility Experiments in CaSO4–H2O System
As indicated in Chapter 2, the solubility of gypsum and anhydrite in water has been extensively
measured (Dutrizac, 2002; Templeton and Rodgers, 1967; Marshall et al., 1964; Hill and Wills,
1938; Posnjak, 1938; Partridge and White, 1929). Most of the measurements are in fairly good
agreement with each other. In this work, the solubility of gypsum in water at 25–95ºC and that
of anhydrite in water at 150–250ºC was measured to verify the experimental procedure. The
newly measured solubility data are shown in Figure 4.1 along with other experimental data. As
is clear, these data are in good agreement with the other measurements. In this figure, the lines
represent model results for each solid phase.
As can be seen, below ~45ºC, gypsum has a lower solubility and is, therefore, the most
thermodynamically stable phase. The transition point of gypsum to anhydrite lies at 40±5ºC.
Above this temperature, gypsum is metastable, although the degree of metastability in dilute
aqueous solutions is significant. Thus, CaSO4–water slurries can be heated up to 100ºC without
the transformation of gypsum into anhydrite (Dutrizac, 2002). The details regarding the
gypsum–anhydrite transformation is presented in Chapter 5.
0 50 100 150 200 250 3000.000
0.005
0.010
0.015
0.020
0.025
0.030
0.035
0.040
CaSO4(s)
CaSO4.2H2O(s)
CaS
O4 so
lubi
lity,
mol
al
Temperature, oC
Literature Exp data Literature Exp data This work (25-95oC) This work (150-250oC)
Figure 4.1 Solubility diagram of CaSO4 in water. Experimental data are from this work and the literature (Dutrizac, 2002; Hill and Wills, 1938; Posnjak, 1938; Marshall et al., 1964; Partridge and White, 1929; Templeton and Rodgers, 1967). The solid and dashed curves show the stable and metastable phases, respectively.

79
4.3.2 Experimental Measurements and Model Predictions in Laterite PAL Solutions
Table 4.C in Appendix C shows the composition of a typical laterite PAL solution that would be
produced during the processing of a tropical limonite ore at 33% solids, containing about 0.7%
nickel, 1% magnesium and less than 3% aluminum, which was selected as a baseline in this
work. The effect of NiSO4, MgSO4 and H2SO4 concentrations on the solubility of gypsum and
anhydrite was investigated over their stability temperature ranges, which is 25–95°C for gypsum
and 150–250°C for anhydrite. Because seawater with various salinities has been used in the
Australian plants (Murrin Murrin, Ravensthorpe, Cawse and Bulong), the effect of NaCl
concentration on the solubility of calcium sulphate was also investigated in this work. As will be
seen later in this chapter, the model predicts the chemistry of CaSO4 hydrates in the systems
studied without performing additional fitting.
4.3.2.1 Effect of H2SO4 Concentration
The free acid concentration of leach solutions in the operating pressure acid leach processing
plants, which are the Cuban Moa Bay and the Australian Murrin Murrin processing plants,
ranges between 20–40 g/L (Whittington and Muir, 2000). Therefore, the effect of acid
concentration on the solubility of calcium sulphate is an important process parameter. Figures
4.2 and 4.3 present the measured solubility data for gypsum and anhydrite as a function of
H2SO4 concentration at different temperatures along with the model predictions. For both solids,
model predictions reflect the experimental data accurately. Tables C.5 and C.6 in Appendix C
summarize the measured data for gypsum and anhydrite, respectively. X-ray diffraction analysis
of the equilibrating solid phase showed gypsum at temperatures below 100°C and anhydrite at
150–250°C inside the autoclave.
As can be seen in Figure 4.2, at low temperatures (25–45°C), the addition of H2SO4 increases
the solubility of gypsum moderately, whereas at higher temperatures, the solubility increases
strongly with increasing acid concentration. The behaviour in dilute to moderately concentrated
acid is due to the decrease of the second dissociation constant of H2SO4 with increasing
temperature. Therefore, the addition of H2SO4 results in a reduction of the SO42- concentration
and allows an increase in the solubility of CaSO4 to satisfy the solubility product. Regardless of
the acid concentration, the solubility of gypsum increases monotonically with increasing
temperature. Figure 4.3 shows the solubility of anhydrite in similar solutions at elevated

80
temperatures. Under these conditions, anhydrite solubility increases significantly with
increasing acid concentration from 0.2 to 0.4 M. However, calcium sulphate solubility decreases
with increasing temperature. This is due to the fact that the dielectric constant of water decreases
with increasing temperature above 100°C, rendering water to be a deficient solvent for
dissolving polar compounds (Helgeson, 1967).
0.00 0.05 0.10 0.15 0.20 0.25 0.30 0.350.00
0.01
0.02
0.03
0.04
0.05
0.06
AARD%=7.5
Solid phase: CaSO4.2H2O
Exp data, 25 oC Exp data, 45 oC Exp data, 70 oC Exp data, 90 oC
CaS
O4 so
lubi
lity,
mol
al
H2SO
4, molal
[Al2(SO
4)
3]=0.004 M
[NiSO4]=0.07 M
[MgSO4]=0.23 M
Figure 4.2 Gypsum solubility vs. H2SO4 concentration in CaSO4–H2SO4–NiSO4(0.07M)–MgSO4(0.23M)–Al2(SO4)3(0.004M)–H2O solutions; experimental data are from Azimi and Papangelakis (2010b). The curves are the predicted values.
0.20 0.25 0.30 0.35 0.40 0.450.000
0.005
0.010
0.015
0.020
0.025
0.030
[Al2(SO4)3]=0.005 M[NiSO
4]=0.06 M
[MgSO4]=0.22 M
Solid phase: CaSO4 (s)
Exp data, 150 oC Exp data, 175 oC Exp data, 200 oC Exp data, 250 oC
CaS
O4 so
lubi
lity,
mol
al
H2SO
4, molal
AARD%=7.0
Figure 4.3 Anhydrite solubility vs. H2SO4 concentration in CaSO4–H2SO4–NiSO4(0.06M)–MgSO4(0.22M)–Al2(SO4)3(0.005M)–H2O solutions; experimental data are from Azimi and Papangelakis (2010b) and the curves are the model prediction results.

81
4.3.2.2 Effect of NiSO4 Concentration
In laterite pressure acid leach processes, the nickel concentration varies between 5 and 20 g/L.
The effect of this variation on the solubility of calcium sulphate in solutions containing
0.2–0.3 M H2SO4, 0.22 M MgSO4 and 0.005 M Al2(SO4)3 was investigated both at atmospheric
pressure at 25–90°C and at elevated pressures inside an autoclave at 150–250°C. X-ray
diffraction analysis showed gypsum as the saturating solid phase below 100°C and anhydrite
above 150°C. Figures 4.4 and 4.5 present the measured solubility data for gypsum and
anhydrite. It is clear from Figure 4.4 that the solubility of gypsum increases steadily with
increasing temperature due to the increase of the association constant (Ka) of CaSO4(aq) with
temperature. In contrast, as can be seen in Figure 4.5, the solubility of anhydrite decreases with
an increase in temperature above 150°C due to the reduction of the dielectric constant of water.
The measured data are presented in Appendix C (Tables C.7, C.8).
0.00 0.05 0.10 0.15 0.20 0.25 0.30 0.350.00
0.01
0.02
0.03
0.04
0.05
Solid phase: CaSO4.2H2O
Exp data, 25 oC Exp data, 45 oC Exp data, 70 oC Exp data, 90 oC
CaS
O4 so
lubi
lity,
mol
al
NiSO4, molal
[Al2(SO
4)
3]=0.005 M
[H2SO
4]=0.2 M
[MgSO4]=0.22 M
AARD%=5.4
Figure 4.4 Gypsum solubility vs. NiSO4 concentration in CaSO4–NiSO4–H2SO4(0.2M)–MgSO4(0.22M)–Al2(SO4)3(0.005M)–H2O solutions; experimental data are from Azimi and Papangelakis (2010b). The curves are the predicted values.
Both the solubility of gypsum and anhydrite first decreases moderately with increasing NiSO4
concentration due to the common ion effect. This effect is diminished at higher NiSO4
concentrations (above 0.2 M) due to the formation of calcium sulphate neutral species. The
MSE model containing the interaction parameters obtained in this work was used to predict the
solubility of both gypsum and anhydrite in these solutions over the temperature range studied.
The model predictions along with the experimental data are shown in Figures 4.4 and 4.5, which
are in good agreement (AARD%= 5.4 and 7.3).

82
0.05 0.10 0.15 0.20 0.25 0.30 0.350.000
0.004
0.008
0.012
0.016
[Al2(SO
4)
3]=0.005 M
[H2SO
4]=0.3 M
[MgSO4]=0.22 M
Solid phase: CaSO4
Exp data, 150 oC Exp data, 175 oC Exp data, 200 oC
CaS
O4 so
lubi
lity,
mol
al
NiSO4, molal
AARD%=7.3
Figure 4.5 Anhydrite solubility vs. NiSO4 concentration in CaSO4–NiSO4–H2SO4(0.3M)–MgSO4(0.22M)–Al2(SO4)3(0.005M)–H2O solutions; experimental data are from Azimi and Papangelakis (2010b). The curves are the predicted values.
4.3.2.3 Effect of MgSO4 Concentration
The concentration of MgSO4 in the laterite PAL solutions varies within the range of 0.05 to
0.5 M depending on the ore type (Baghalha, 1999). Limonitic ores contain low-Mg fractions
(about 1 wt%) compared to saprolite ores which contain high-Mg fractions between 10–20 wt%.
The variation in Mg content of laterite leach solutions has a significant impact on the solution
chemistry, particularly on the sulphuric acid consumption during the leaching process. This, in
turn, affects the Ni–Co leaching kinetics and the solubility of metal sulphates and impurities.
The impact of these variations on the solubility of calcium sulphate has not been studied before.
To provide some information on this aspect, the effect of MgSO4 on the solubilities of gypsum
and anhydrite in solutions containing 0.2–0.3 M H2SO4, 0.06 M NiSO4 and 0.005 M Al2(SO4)3
over the temperature range of 25–90°C and 150–250°C was investigated. The measured data are
summarized in Appendix C (Tables C.9, C.10). Saturating solid phases at all temperatures were
analyzed by X-ray diffraction. The results showed gypsum at temperatures below 100°C and
anhydrite above 150°C. Figure 4.6 shows the gypsum solubility data obtained at 25–90°C. In
this system, gypsum solubility first decreases as the MgSO4 concentration increases due to the
common ion effect; this effect is more pronounced at temperatures above 70°C. However, in the
presence of modest MgSO4 concentrations (above 0.25 M), the common ion effect is less
significant because of the association of Ca2+ and SO42- ions and formation of CaSO4(aq).

83
0.10 0.15 0.20 0.25 0.30 0.35 0.400.000
0.005
0.010
0.015
0.020
0.025
0.030
0.035
0.040
0.045[Al
2(SO
4)
3]=0.005 M
[H2SO
4]=0.2 M
[NiSO4]=0.05 M
Solid phase: CaSO4.2H
2O
Exp data, 25 oC Exp data, 45 oC Exp data, 70 oC Exp data, 90 oC
CaS
O4 so
lubi
lity,
mol
al
MgSO4, molal
AARD%=6.0
Figure 4.6 Gypsum solubility vs. MgSO4 concentration in CaSO4–MgSO4–H2SO4(0.2M)–NiSO4(0.05M)–Al2(SO4)3(0.005M)–H2O solutions; experimental data are from Azimi and Papangelakis (2010b). The curves are the predicted values.
The solubility of anhydrite over the temperature range of 150–250°C is presented in Figure 4.7.
Increasing MgSO4 concentration from 0.05 to 0.35 M in the solution containing 0.3 M H2SO4,
0.06 M NiSO4 and 0.005 Al2(SO4)3 results in a systematic decrease in the solubility due to the
common ion effect of SO42-. The model was used to predict the solubilities of both gypsum and
anhydrite in these systems over the temperature and concentration ranges studied. The curves
presented in Figures 4.6 and 4.7 are model prediction results which are in good agreement with
the measured data (AARD%=6.0 and 8.8).
0.10 0.15 0.20 0.25 0.30 0.350.000
0.005
0.010
0.015
0.020
0.025
0.030
0.035
[Al2(SO
4)
3]=0.005 M
[H2SO4]=0.3 M[NiSO
4]=0.06 M
Solid phase: CaSO4
Exp data, 150 oC Exp data, 175 oC Exp data, 200 oC Exp data, 250 oC
CaS
O4 so
lubi
lity,
mol
al
MgSO4, molal
AARD%=8.8
Figure 4.7 Anhydrite solubility vs. MgSO4 concentration in CaSO4–MgSO4–H2SO4(0.3M)–NiSO4(0.06M)–Al2(SO4)3(0.005M)–H2O solutions; experimental data are from Azimi and Papangelakis (2010b). The curves are the predicted values.
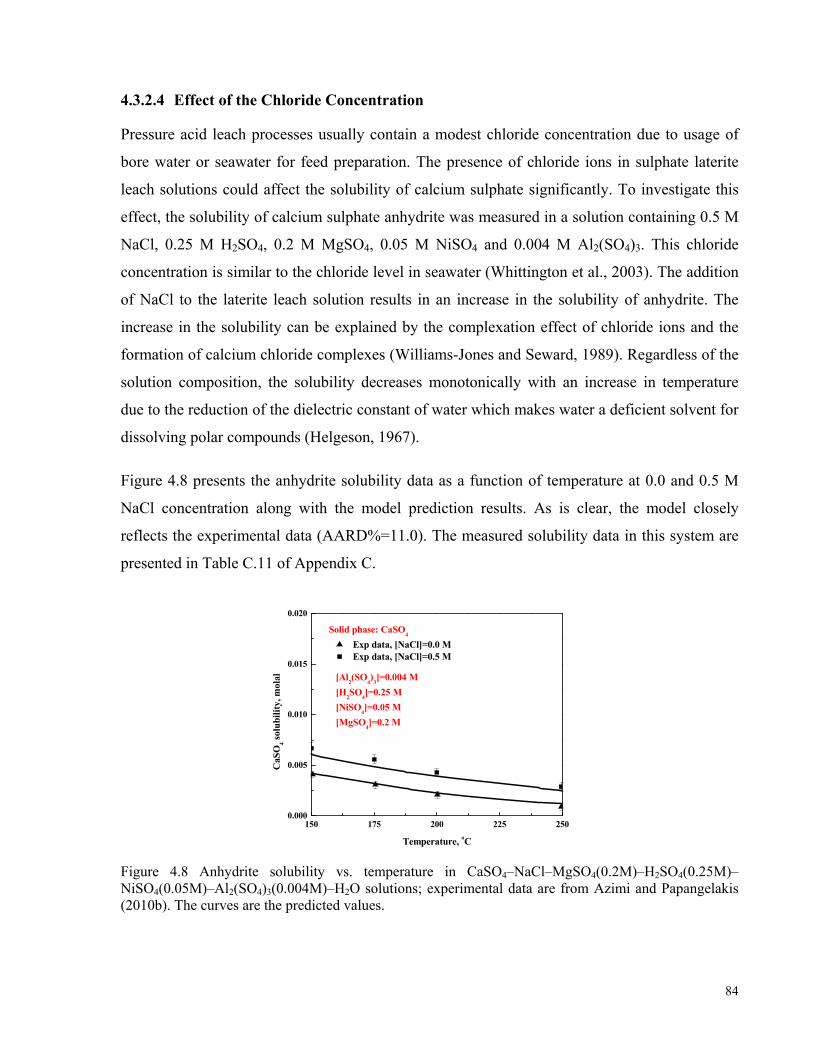
84
4.3.2.4 Effect of the Chloride Concentration
Pressure acid leach processes usually contain a modest chloride concentration due to usage of
bore water or seawater for feed preparation. The presence of chloride ions in sulphate laterite
leach solutions could affect the solubility of calcium sulphate significantly. To investigate this
effect, the solubility of calcium sulphate anhydrite was measured in a solution containing 0.5 M
NaCl, 0.25 M H2SO4, 0.2 M MgSO4, 0.05 M NiSO4 and 0.004 M Al2(SO4)3. This chloride
concentration is similar to the chloride level in seawater (Whittington et al., 2003). The addition
of NaCl to the laterite leach solution results in an increase in the solubility of anhydrite. The
increase in the solubility can be explained by the complexation effect of chloride ions and the
formation of calcium chloride complexes (Williams-Jones and Seward, 1989). Regardless of the
solution composition, the solubility decreases monotonically with an increase in temperature
due to the reduction of the dielectric constant of water which makes water a deficient solvent for
dissolving polar compounds (Helgeson, 1967).
Figure 4.8 presents the anhydrite solubility data as a function of temperature at 0.0 and 0.5 M
NaCl concentration along with the model prediction results. As is clear, the model closely
reflects the experimental data (AARD%=11.0). The measured solubility data in this system are
presented in Table C.11 of Appendix C.
150 175 200 225 2500.000
0.005
0.010
0.015
0.020
[Al2(SO
4)
3]=0.004 M
[H2SO4]=0.25 M[NiSO
4]=0.05 M
[MgSO4]=0.2 M
Solid phase: CaSO4
Exp data, [NaCl]=0.0 M Exp data, [NaCl]=0.5 M
CaS
O4 so
lubi
lity,
mol
al
Temperature, oC
Figure 4.8 Anhydrite solubility vs. temperature in CaSO4–NaCl–MgSO4(0.2M)–H2SO4(0.25M)–NiSO4(0.05M)–Al2(SO4)3(0.004M)–H2O solutions; experimental data are from Azimi and Papangelakis (2010b). The curves are the predicted values.

85
The solubility of gypsum as a function of NaCl concentration was also measured in solutions
containing 0.5 M H2SO4 at temperatures below 100°C. Table C.12 in Appendix C summarizes
these data. Similar to the high temperature results, gypsum solubility increases with increasing
the NaCl concentration due to the complexation effect of chloride ions. Figure 4.9 shows the
gypsum solubility as a function of NaCl concentration in comparison with the predicted results
obtained from the model. As can be seen, the model results are in good agreement with the
experimental data (AARD%=4.1).
0.0 0.2 0.4 0.6 0.8 1.0 1.2 1.4 1.60.00
0.03
0.06
0.09
0.12
0.15
0.18
CaS
O4 so
lubi
lity,
mol
al
NaCl, molal
Solid phase: CaSO4.2H
2O
Exp data, 25 oC Exp data, 45 oC Exp data, 70 oC Exp data, 90 oC
[H2SO
4]=0.5M
AARD%=4.1
Figure 4.9 Gypsum solubility vs. NaCl concentration in CaSO4–NaCl– H2SO4(0.5M)– H2O solutions; experimental data are from Azimi and Papangelakis (2010b). The curves are the predicted values.
4.4 Process Implications of the Results
The database developed in this work, utilized by the OLI/MSE model, was shown to accurately
predict the solubility behaviour of calcium sulphate (gypsum and anhydrite) in multicomponent
solutions containing Al2(SO4)3, H2SO4, MgSO4, NiSO4, and NaCl from ambient temperature to
250ºC. The model can be used as a valuable tool to map the behaviour of calcium sulphate in
aqueous industrial solutions and to assess the potential for scaling in various aqueous streams.
This, in turn, will provide different industries with the opportunity to investigate the effect of
different variables such as temperature and composition and aid them in finding solutions to
minimize scale formation in the processing circuits. The following implications can be drawn
from this study:

86
1. Calcium sulphate dihydrate (gypsum) is the practically stable solid phase below 100ºC,
due to slow kinetics of phase transformation, although the true upper temperature is
about 40ºC. Above 100ºC anhydrous calcium sulphate (anhydrite) becomes the stable
phase, and its solubility decreases with temperature because the dielectric constant of
water decreases with increasing temperature above 100°C (Helgeson, 1967).
2. The solubility of anhydrite is much lower than that of gypsum (approximately one order
of magnitude lower). As a result, process solutions which are saturated with gypsum at
ambient temperature and recycled to the autoclave need to be processed to decrease their
calcium content and make it less than the saturation level of anhydrite inside the
autoclave. This can be done by mixing the recycling stream with carbonate compounds
to reject calcium as calcium carbonate, provided that the solution is not acidic.
Otherwise, scale formation in the hot zones of the plants will be unavoidable. In fact,
such scaling issues have been reported in pre-heaters during the Bulong plant operation
(Nofal et al., 2001).
3. The addition of H2SO4 has a strong positive effect on the solubility of calcium sulphate
in water (up to 10 times increase) over the temperature range of 25–250ºC. The addition
of acid up to around 1.0–2.0 M increases the solubility of calcium sulphate hydrates due
to the formation of HSO4- ions. Above this concentration, calcium sulphate solubility
reaches a plateau, and upon further acid addition, the solubility decreases due to the
salting-out effect.
4. The addition of sulphate electrolytes such as NiSO4 and MgSO4 first has a negative
effect on the solubility of calcium sulphate due to the common ion effect. However, in
the presence of modest sulphate concentration (above 0.2–0.3 molal), this effect is
nullified due to the association of Ca2+ and SO42- ions and formation of calcium sulphate
neutral species which results in an increase in the solubility. After passing a maximum,
the solubility decreases at still higher sulphate concentrations (above ~1.5–2.0 molal) as
a result of the salting-out effect.
5. The solubility of gypsum in water reaches a maximum at ~45–50ºC, followed by a slight
decrease in the solubility at higher temperatures. However, in multicomponent process
solutions, gypsum is increasingly soluble with temperature. This effect is shown in

87
Figure 4.10 at different NiSO4 concentrations from 0.1 to 1.0 molal in comparison with
that in pure water. As can be seen, the solubility, which decreases with increasing
temperatures above ~40ºC in water, becomes positively related to temperature at high
sulphate concentrations. As a result, process solutions saturated with gypsum during a
hot upstream neutralization step have the potential for scale formation when cooled to
lower temperatures.
30 40 50 60 70 80 900.000
0.005
0.010
0.015
0.020
0.025
pure water
[NiSO4]=0.2 m
[NiSO4]=0.1 m
[NiSO4]=0.5 m
C
aSO
4 Sol
ubili
ty, m
olal
Temperature, oC
[NiSO4]=1 mSolid phase: CaSO4.2H2O
Figure 4.10 Gypsum solubility as a function of temperature at various NiSO4 concentrations in comparison with that in pure water; the curves are the model prediction results.
6. The addition of chloride ions (up to ~1.5–2.0 molal) increases the solubility of calcium
sulphate in the systems studied. Above this concentration, the solubility reaches a
plateau. The increase in the solubility is due to the complexation effect of the chloride
ions. However, this effect is less significant at higher salt concentrations due to the
salting-out effect. Figure 4.11 depicts the predicted anhydrite solubility in pure water as
well as in an aqueous solution of 0.22 M H2SO4 in comparison with that in laterite PAL
solutions containing 0.22 M H2SO4, 0.2 M MgSO4, 0.05 M NiSO4 and 0.005 M
Al2(SO4)3 at various chloride concentrations equivalent to those found in tap water,
seawater, saline and hyper–saline water (Whittington et al., 2003). For comparison, the
saturation level of gypsum in pure water at 25ºC is also presented. As is clear,
transformation between gypsum, the “practically” stable solid phase below 100ºC, and
anhydrite, the stable phase above 150ºC, results in a significant decrease (~90%) in the
solubility of calcium sulphate in pure water. However, the addition of H2SO4 has a
positive effect on the solubility of anhydrite in the solution. In this figure, anhydrite

88
solubility in a solution containing 0.22 M H2SO4 was increased by almost 70%
compared to that in pure water. In contrast, the addition of metal sulphates, i.e., MgSO4,
NiSO4 and Al2(SO4)3 results in a decrease in anhydrite solubility in 0.22 M H2SO4
solutions. The addition of chloride ions has a positive effect on the solubility of
anhydrite in the laterite PAL solutions.
150 175 200 225 2500.000
0.002
0.004
0.006
0.008
0.010
0.012
0.014
0.016
0.018
PALhypersaline, Cl-=74550 ppm
saline, Cl-=35500 ppmseawater, Cl-=18000 ppm
tapwater, Cl-=140 ppm
CaS
O4 so
lubi
lity,
mol
al
Temperature, oC
gypsum saturated water at 25 oC
anhydrite in pure water
in pure H2SO4=0.22 M
Figure 4.11 Anhydrite solubility vs. temperature in pure water and in 0.22 M H2SO4 solution in comparison with that in laterite PAL solutions containing MgSO4(0.2M)–H2SO4(0.22M)–NiSO4(0.05M)–Al2(SO4)3 (0.005M) at various chloride concentrations. Solid curves are model prediction results for anhydrite; the dashed line shows gypsum saturation level in pure water at 25°C.
7. To indicate the effect of H2SO4 concentration, the predicted anhydrite solubility in pure
water as well as in 0.22 M H2SO4 solutions along with that in laterite PAL solutions
containing 0.2 M MgSO4, 0.05 M NiSO4 and 0.005 M Al2(SO4)3 at three different H2SO4
concentrations, i.e., 0.22, 0.33, and 0.44 M are presented in Figure 4.12. The dashed line
represents the saturation level of gypsum in pure water at 25ºC. As discussed above, the
transformation of gypsum to anhydrite results in a ~90% decrease in the solubility of
CaSO4 in pure water. The addition of 0.22 M H2SO4 increases anhydrite solubility in
water by ~70%. However, in PAL solutions, the solubility decreases due to the common
ion effect. Increasing acid concentration from 0.22 M to 0.33 M results in ~60% increase
in the anhydrite solubility in PAL solutions at 250ºC. Therefore, higher acidities, higher
water salinity and lower sulphate concentrations are favorable conditions for minimizing
anhydrite scaling inside an autoclave. These parameters must be optimized to target
capital and operating costs, material consumption, and environmental regulations.

89
150 175 200 225 2500.000
0.002
0.004
0.006
0.008
0.010
0.012
0.014
0.016
0.018
PAL
PAL
[H2SO4]=0.44 M
[H2SO4]=0.33 M
PAL
CaS
O4 so
lubi
lity,
mol
al
Temperature, oC
gypsum saturated water at 25 oC
anhydrite in pure water
in pure H2SO4=0.22 M
[H2SO4]=0.22 M
Figure 4.12 Anhydrite solubility vs. temperature in pure water and in 0.22 M H2SO4 solutions compared to that in laterite PAL solutions containing MgSO4(0.2M)–NiSO4(0.05M)–Al2(SO4)3 (0.005M) at various H2SO4 concentrations. Solid curves are model prediction results for anhydrite; the dashed line shows gypsum saturation level in pure water at 25°C.
4.5 Summary
In hydrometallurgical processing of metals such as nickel, cobalt and copper, the limited
solubility of calcium sulphate results in very low concentrations of calcium in the circuit.
However, as the temperature and solution composition vary, calcium sulphate scaling may occur
in the process. During processing at higher temperatures, transformation between the calcium
sulphate hydrates has a complex effect on the solubility, making the behaviour of calcium
sulphate difficult to predict and control. Most of the industries dealing with calcium sulphate
scale formation employ regular removal of precipitated calcium sulphate because the build-up of
calcium concentration in these process streams is unavoidable.
The solubilities of gypsum and anhydrite in synthetic pressure acid leach solutions containing
NiSO4, H2SO4, Al2(SO4)3, and MgSO4 were measured from 25°C to 250ºC. Gypsum is the
stable solid phase at low temperatures; however, at high temperatures, above 100ºC, anhydrite
becomes the stable phase and its solubility decreases with temperature. Furthermore, the
solubility of anhydrite in pure water above 100ºC is roughly one tenth of that of dihydrate below
100ºC. Therefore, process solutions which are saturated with gypsum at room temperature and
recycled to the autoclave have the potential to form anhydrite scales. The addition of H2SO4 up

90
to ~2.0 M results in a significant increase in the solubility of both gypsum and anhydrite.
Above this concentration, calcium sulphate solubility decreases with increasing acid
concentration.
The addition of metal sulphates such as NiSO4, and MgSO4 has a negative effect on the
solubility of calcium sulphate hydrates in laterite PAL solutions over the temperature range
studied due to the common ion effect. It was also found that the quality of the process water
affects the solubility of CaSO4 in these solutions. The addition of chlorides from tap water
(~140 ppm) to hyper-saline water (~75000 ppm) increases the solubility of anhydrite by almost
20%. Therefore, mixing recycled process solutions with seawater is favorable for decreasing
anhydrite scale formation in these systems provided that chloride-corrosion issues can be
controlled by utilizing chloride-resistant materials and metal alloys.
The experimentally measured solubility data were successfully modelled using the newly
developed database utilized by the OLI/MSE model. The model was shown to accurately predict
the solubility behaviour of calcium sulphate in solutions under the conditions studied. The
model can be used to map calcium sulphate chemistry in the sulphuric acid pressure leaching of
laterite ores over wide ranges of temperature and concentration. This, in turn, results in a
comprehensive insight for process improvements and optimization.

91
CHAPTER 5 TRANSFORMATION OF GYPSUM INTO ANHYDRITE IN AQUEOUS ELECTROLYTE SOLUTIONS
his chapter focuses on the transformation of gypsum into anhydrite. The effect of
temperature, sulphuric acid concentration, anhydrite seeding as well as addition of
sulphate and chloride salts on the transformation kinetics is discussed. Based on the results
obtained, a mechanism for the gypsum–anhydrite transformation is proposed. The contents of
this chapter have been submitted to the Crystal Growth & Design for publication (Submission #:
cg-2010-00172b).
5.1 Introduction
Calcium sulphate dihydrate (CaSO4•2H2O) occurs naturally as the mineral gypsum and is the
most common sulphate mineral in the environment that has been traditionally extracted from the
ground (Hand, 1997). In the modern world, gypsum is widely used as a constituent in
construction materials such as cement or gypsum wallboards (Christensen et al., 2008; Solberg
et al., 2002) as well as in the production of biocompatible materials such as bone void fillers
(Doadrio et al., 2004). However, calcium sulphate appears as an undesirable byproduct, mostly
as discard solids and/or as scale, in many industrial processes including wastewater treatment,
oil and gas production, desalination, sulphur dioxide removal from coal-fired power plants flue
gas (Lee et al., 2006; Dathe et al., 2006) and during neutralization of free sulphuric acid in
hydrometallurgical processes (Azimi and Papangelakis, 2010b; Dutrizac and Kuiper, 2008,
2006; Adams and Papangelakis, 2007; Dutrizac, 2002). Calcium sulphate scale formation in
industrial plants results in reduced production capacity and process efficiency due to decreased
equipment volume, heat transfer capacity, and material flow, blocked pipelines and corrosion.
Therefore, periodic shut-downs of the plant for maintenance and mechanical removal of
precipitated calcium sulphate hydrates are necessary.
Five crystalline phases in the form of hydrates or polymorphs exist in the CaSO4–H2O system:
dihydrate or gypsum (CaSO4•2H2O, DH), hemihydrate in the two polymorphs of
α–CaSO4•0.5H2O and β–CaSO4•0.5H2O, and anhydrite in the two polymorphs of soluble and
T

92
insoluble, denoted as AIII or γ–CaSO4 and AII–CaSO4, respectively (Christensen et al., 2008).
The difference between polymorphs and hydrates is that polymorphs are different crystal
structures of the same stoichiometry, whereas, hydrates are crystals of a compound incorporated
with a different number of water molecules (Zupancic et al., 2005).
One of the most common causes of CaSO4 scale formation in various industries is the
transformation of gypsum into insoluble anhydrite (AII–CaSO4), as was the case with the
Bulong Nickel/Cobalt Plant (Nofal et al., 2001). Theoretically, gypsum is the stable solid phase
in water up to ~45–50°C, and above that it transforms into anhydrite (Farrah et al., 2004; Freyer
and Voigt, 2003; Dutrizac, 2002; Nývlt, 1997). However, the transformation does not practically
occur up to ~80–90°C due to the slow kinetics in the absence of anhydrite seeding. Because the
solubility of anhydrite is lower than that of gypsum, the transformation of gypsum into
anhydrite generates a supersaturated solution, which results in anhydrite scale formation. In
industrial practice, this means that during sulphuric acid neutralization with calcium-containing
bases, first gypsum forms as a metastable phase because of its higher solubility, according to the
Ostwald step rule (Santen, 1984), then gradually transforms into anhydrite. Thus, a thorough
understanding of the mechanism and kinetics of the solid phase transformation in addition to the
solution chemistry of CaSO4 is of great practical importance to accurately evaluate the scaling
potential in various electrolyte solutions.
The thermodynamics of transformation between gypsum and anhydrite in water at one
atmosphere has been studied by several researchers (Knacke and Gans, 1977; Hardie, 1967;
Power at al., 1964; Posnjak, 1938; Hill, 1937; Partridge and White, 1929; Van’t Hoff et al.,
1903). The effect of salt solutions on the gypsum–anhydrite transformation has also been
investigated (Ostroff, 1964; Bock, 1961; Posnjak, 1940; Hill and Wills, 1938; Van’t Hoff et al.,
1903). More recently, Li and Demopoulos (2006c) constructed thermodynamic phase diagrams
of calcium sulphate hydrates in HCl–CaCl2–H2O solutions up to 100°C and derived theoretical
stability regions of various hydrates as a function of temperature and composition. Most of these
studies have focused on the thermodynamics of transformation and determination of the
theoretical transition temperature between gypsum and anhydrite.
The kinetics of transformation between various calcium sulphate hydrates, particularly under the
conditions resembling those in industrial processes, has also been studied over the present

93
decade. Transformation of gypsum into anhydrite in hot acidic manganese sulphate solutions
has been studied by Farrah et al. (2004), where an autocatalytic process was suggested to fit the
transformation kinetics. The gypsum–anhydrite transformation in simulated nickel sulphate–
chloride and copper sulphate electrorefining solutions has also been studied by Dutrizac and
Kuiper (2006, 2008). In a recent study (Dutrizac, 2002) on the solubility of gypsum in sulphuric
acid solutions, it has been realized that the equilibration kinetic curves in the case of heating and
cooling were not coincident for H2SO4 concentrations above 0.6 M, i.e., the solubility increased
up to ~80–90°C and then dropped abruptly as a result of the transformation of gypsum into
anhydrite. In all previous studies, the concentration of sulphuric acid has been found to play an
important role in the kinetics of the transformation. That is, a higher acid concentration lowers
the time required to complete the transformation.
In the previous chapters, the solubilities of gypsum and anhydrite in various multicomponent
industrial systems was studied and a new database for the Mixed Solvent Electrolyte (MSE)
model of the OLI® software was developed, which is capable of predicting the solubility
behaviour (and hence the scaling potential) of CaSO4 in such processes up to 250°C (Azimi and
Papangelakis, 2010a, 2010b; Azimi et al., 2010, 2008, 2007). In this chapter, a systematic study
of the kinetics of gypsum–anhydrite transformation was carried out over the temperature range
of 25–90°C, which is the temperature range where neutralization processes occur in industry. A
number of experiments were conducted to study the effects of temperature, acid concentration
from 0.5 to 2.0 M, anhydrite seeding and addition of sulphate/chloride salts (NiSO4 and NaCl)
on the transformation kinetics. The transformation of gypsum into anhydrite was monitored
closely both in liquid and the solid phases.
5.2 Experimental Section
All solutions were prepared by dissolving reagent grade chemicals directly without further
purification. Gypsum reagent with 99.4% purity and anhydrite with 100% purity from J.T.
Baker were used as saturating solid phases. X-ray diffraction (XRD) analysis was carried out on
both solids. The diffractograms showed 100% gypsum and anhydrite, respectively (Appendix
D). No traces of hemihydrate or anhydrite were found in the gypsum solid powder.
Experiments were performed inside 1 L double layer glass reactors with tight-fitting lids where
heating was provided through a circulating oil jacket. Temperature was controlled within ±1°C
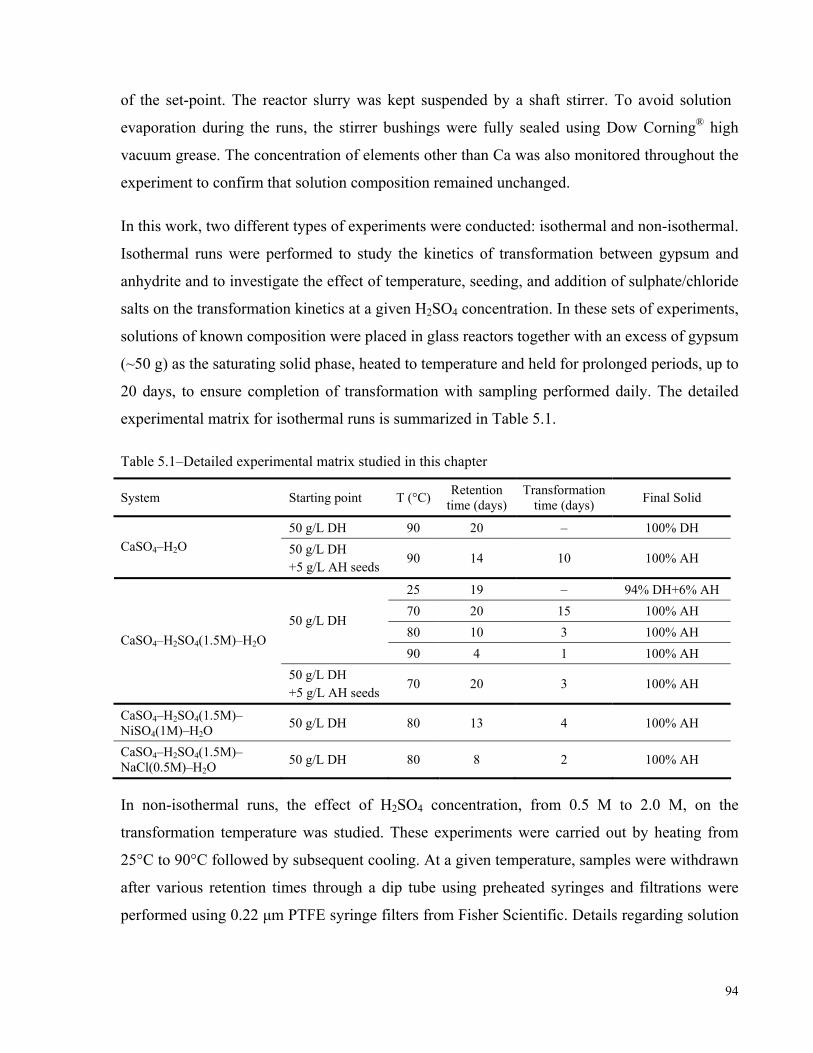
94
of the set-point. The reactor slurry was kept suspended by a shaft stirrer. To avoid solution
evaporation during the runs, the stirrer bushings were fully sealed using Dow Corning® high
vacuum grease. The concentration of elements other than Ca was also monitored throughout the
experiment to confirm that solution composition remained unchanged.
In this work, two different types of experiments were conducted: isothermal and non-isothermal.
Isothermal runs were performed to study the kinetics of transformation between gypsum and
anhydrite and to investigate the effect of temperature, seeding, and addition of sulphate/chloride
salts on the transformation kinetics at a given H2SO4 concentration. In these sets of experiments,
solutions of known composition were placed in glass reactors together with an excess of gypsum
(~50 g) as the saturating solid phase, heated to temperature and held for prolonged periods, up to
20 days, to ensure completion of transformation with sampling performed daily. The detailed
experimental matrix for isothermal runs is summarized in Table 5.1.
Table 5.1–Detailed experimental matrix studied in this chapter
System Starting point T (°C) Retention time (days)
Transformation time (days) Final Solid
50 g/L DH 90 20 – 100% DH CaSO4–H2O 50 g/L DH
+5 g/L AH seeds 90 14 10 100% AH
25 19 – 94% DH+6% AH 70 20 15 100% AH 80 10 3 100% AH
50 g/L DH
90 4 1 100% AH CaSO4–H2SO4(1.5M)–H2O
50 g/L DH +5 g/L AH seeds
70 20 3 100% AH
CaSO4–H2SO4(1.5M)– NiSO4(1M)–H2O 50 g/L DH 80 13 4 100% AH
CaSO4–H2SO4(1.5M)– NaCl(0.5M)–H2O 50 g/L DH 80 8 2 100% AH
In non-isothermal runs, the effect of H2SO4 concentration, from 0.5 M to 2.0 M, on the
transformation temperature was studied. These experiments were carried out by heating from
25°C to 90°C followed by subsequent cooling. At a given temperature, samples were withdrawn
after various retention times through a dip tube using preheated syringes and filtrations were
performed using 0.22 μm PTFE syringe filters from Fisher Scientific. Details regarding solution

95
and solid phase compositions at a given temperature along with the retention times for these
sets of experiments are presented in Appendix F (Tables F.2 and F.3).
Withdrawn solution samples were diluted with 5% HNO3 and stored in sealed plastic test tubes
at room temperature. The Ca concentration was determined by ICP–OES analysis using the
317.933 nm emission line. Reproducibility tests showed that the experimentally measured data
are accurate to within ±5%.
Samples of the equilibrating solid phase were also withdrawn, filtered, and washed with a small
amount of denatured alcohol, containing 85% ethanol and 15% methanol, to replace the solution
and dried below 40°C in an oven under vacuum. Powder X-ray diffraction patterns of the solid
samples were collected on a Philips PW3719 diffractometer utilizing Cu Ka radiation in the
range 10–60° 2θ with a step size of 0.02° and a collection time of 1.25 s/step. The generated
patterns were matched against the International Centre for Diffraction Data® files (JCPDF-
ICDD file numbers 070–0982 for gypsum and 072–0916 for anhydrite). The relative amounts of
gypsum and anhydrite present in the solid samples were estimated by the Rietveld refinement
(details are available in Appendix H). In all experiments carried out in this work, gypsum and
anhydrite were the only crystalline species identified. Hemihydrate was not detected in any of
the samples over the temperature range studied (i.e., 25–90°C). The solid samples were also
analyzed by scanning electronic microscopy (SEM) to study the morphology and structural
changes in CaSO4 crystals during the transformation process. SEM images were obtained on a
JEOL JSM-840 scanning electron microscope. Solid sample powders were mounted rigidly on a
specimen holder, called stub, and coated with gold to facilitate charge removal during
microscope operation. To obtain particles cross-section images, solid powders were dropped
into a resin, and then polished. They were also coated with gold to become electrically
conductive. The time between sample withdrawal and XRD/SEM analysis varied from 1 to 10
days. Meanwhile, samples were preserved in a desiccator, protected against moisture and
humidity.

96
5.3 Results and Discussion
5.3.1 Gypsum–Anhydrite Transformation in Water
The stability regions and temperature of transformation between CaSO4 solid phases, i.e.,
gypsum, hemihydrate and anhydrite, have been studied by various researchers (Freyer and
Voigt, 2003; Dutrizac, 2002; Knacke and Gans, 1977; Hardie, 1967; Bock, 1961; Sborgi and
Bianchi, 1940; Posnjak, 1938; Hill, 1937; Van’t Hoff et al., 1903). Figure 5.1 presents the
solubility diagram of CaSO4 in water.
In solubility diagrams, the solid phase with the lowest solubility is the stable phase at a given
temperature. In Figure 5.1, gypsum is the stable phase at low temperatures, whereas, anhydrite
is stable one above 45–50ºC; hemihydrate is metastable at all temperatures. However, it is
experimentally observed that anhydrite does not crystallize from supersaturated solutions in
water with measurable rates at temperatures below ~80–90ºC, even in the presence of anhydrite
seed crystals (Freyer and Voigt, 2003; Dutrizac, 2002).
0 50 100 150 200 250 3000.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
CaSO4(s)
CaSO4.2H2O
CaS
O4 so
lubi
lity,
mol
al
Temperature, oC
Gypsum Exp data Anhydrite Exp data Hemihydrate Exp data
CaSO4.0.5H
2O
Figure 5.1 Solubility diagram of CaSO4 in water. Experimental data are from Dutrizac, 2002; Templeton and Rodgers, 1967; Marshall et al., 1964; Sborgi and Bianchi, 1940; Hill and Wills, 1938; Posnjak, 1938; Partridge and White, 1929. Curves obtained from the OLI/MSE model (Azimi et al., 2007).
In the present work, the transformation of gypsum into anhydrite in pure water was studied at
90ºC. In the absence of added anhydrite seeds, gypsum remained stable for periods up to 20
days. In contrast, the addition of 5 g/L (10 wt%) of anhydrite seeds resulted in nearly complete
transformation after 10 days. In these sets of experiments, gypsum and anhydrite were the only

97
crystalline species identified, and no intermediate hemihydrate was detected. Figure 5.2 shows
the percentage of the remaining gypsum in the equilibrating solid phase at various retention
times in water at 90°C obtained from the X-ray diffraction patterns, using Rietveld analysis.
The composition of the equilibrating solid phase is summarized in Appendix F (Table F.1).
0 2 4 6 8 10 12 14 16
0
20
40
60
80
100
Gyp
sum
(%)
Time, days
Starting with 50 g/L gypsum +5 g/L anhydrite seeds
T=90 oC
With no seeding
Figure 5.2 Percentage of gypsum present in the equilibrating solid phase based on XRD results at various retention times for gypsum–anhydrite transformation in water at 90°C.
5.3.2 Theoretical Determination of the Transformation Temperature
Based on the solubility diagram of calcium sulphate in water (Figure 5.1), the gypsum–
anhydrite transformation temperature lies at 45–50°C. However, as presented in the previous
section, the kinetics of the transformation in water is slow such that, in the absence of anhydrite
seeds, gypsum remains practically stable up to ~90°C. It has been found that the addition of
electrolytes, particularly acids, has an accelerating effect on the transformation process (Farrah
et al., 2004; Freyer and Voigt, 2003; Dutrizac, 2002; Nývlt, 1997).
The direct dehydration reaction of gypsum to anhydrite and the appropriate expression of the
equilibrium constant are as follows:
CaSO4•2H2O(s) = CaSO4(s) + 2 H2O(l) (5.1)
2)exp( waRTGK =
Δ−=
o
(5.2)

98
The temperature functionality of ΔG˚ for the gypsum dehydration reaction has been proposed
by Hardie (1967) as follows:
TTTTG log44.710262.057.1792890 2 −++−=Δ o (5.3)
where, ΔG˚ is in cal.mol-1 and T is in K. Substituting ΔG˚ from Eq. (5.3) in Eq. (5.2) provides
the relation between the activity of water (aw) and the transformation temperature, which is
shown in Figure 5.3. Using this figure, the gypsum–anhydrite transformation temperature in
water (aw=1.0) lies at ~45°C, in good agreement with the results obtained from the solubility
diagram (Figure 5.1). For comparison, the equilibrium constant of the gypsum dehydration
reaction (Eq. 5.1) at various temperatures was calculated utilizing the OLI/MSE model. Solving
Eq. (5.2), the activity of water was calculated at various temperatures (also presented in Figure
5.3). Based on the curve obtained from the OLI/MSE model, the transformation temperature in
water is ~40°C, close to the accepted values (Figure 5.1).
Figure 5.3 may be used to obtain the “theoretical” temperature of the gypsum–anhydrite
transformation if the activity of water in the solution is known. However, it should be noted that,
in most cases, the kinetics of the transformation near theoretical temperatures are slow, resulting
in gypsum retention as a metastable phase for greater periods of time.
0.4 0.5 0.6 0.7 0.8 0.9 1.00
10
20
30
40
50
60
70
80
MSE model
T/ o C
awater
CaSO4.2H
2O
(s)=CaSO
4(s)+2H
2O
(l)
Hardie (1967)
Figure 5.3 Theoretical transformation temperature of gypsum into anhydrite as a function of the activity of water. Solid curve derived from Hardie (1967); dashed curve obtained from the OLI/MSE model.

99
5.3.3 Effect of Sulphuric Acid on the Gypsum Transformation
A few theoretical studies have been undertaken to determine the effect of H2SO4 addition on the
gypsum transformation (Ling and Demopoulos, 2004; Freyer and Voigt, 2003; Raju and
Atkinson, 1990). The theoretical transformation temperature can be determined using phase
transformation diagrams, Figure 5.3, if the activity of water in the solution is known.
In the present work, the effect of acidity on the kinetics of transformation was studied. Slurries
containing ~50 g/L gypsum in 0.5, 1.0, 1.5 and 2.0 M H2SO4–H2O solutions were heated from
25°C to 90°C, and subsequently cooled to 25°C. Slurries were held for various retention times
between 48 h and 72 h at a given temperature, and samples were taken on a daily basis. No
anhydrite seed was added in these runs.
Figures 5.4 (a) to (d) show the concentrations of CaSO4 at different temperatures for various
retention times. In all cases, the solubility of CaSO4 first increases with increasing temperature
up to 70–90°C, depending on the acid concentration, then drops. X-ray diffraction analysis of
the saturating solid phases shows that, in all cases, gypsum began to transform slowly to
anhydrite. Nevertheless, all solids contained less than 20% anhydrite below ~70°C. At
temperatures above 70°C, however, the transformation of gypsum into anhydrite progressed
significantly. In 0.5 M H2SO4 solutions, complete transformation was obtained at 90°C after 4
days (96 h), whereas in 1.0 M acid, 3 days (72 h) was sufficient at the same temperature (90°C).
In solutions containing 1.5 M and 2.0 M H2SO4, complete transformation was achieved at 80°C
after 3 days and 1 day, respectively. The corresponding solubility data and the composition of
the saturating solids at a given temperature at various retention times are summarized in
Appendix F (Tables F.2, F.3).
Results indicate that gypsum is kinetically stable to at least 80°C for acid concentrations as high
as 2.0 M, confirming the results of other studies (Dutrizac, 2002). During cooling, anhydrite was
detected as the only solid phase above 45°C, but further cooling resulted in the conversion of
anhydrite to gypsum. Higher acid concentrations retain more anhydrite after slurries were
cooled to room temperature.
The implication here is that the conversion of gypsum to less soluble anhydrite would occur
rapidly in many industrial processes containing modest concentrations of sulphuric acid (above
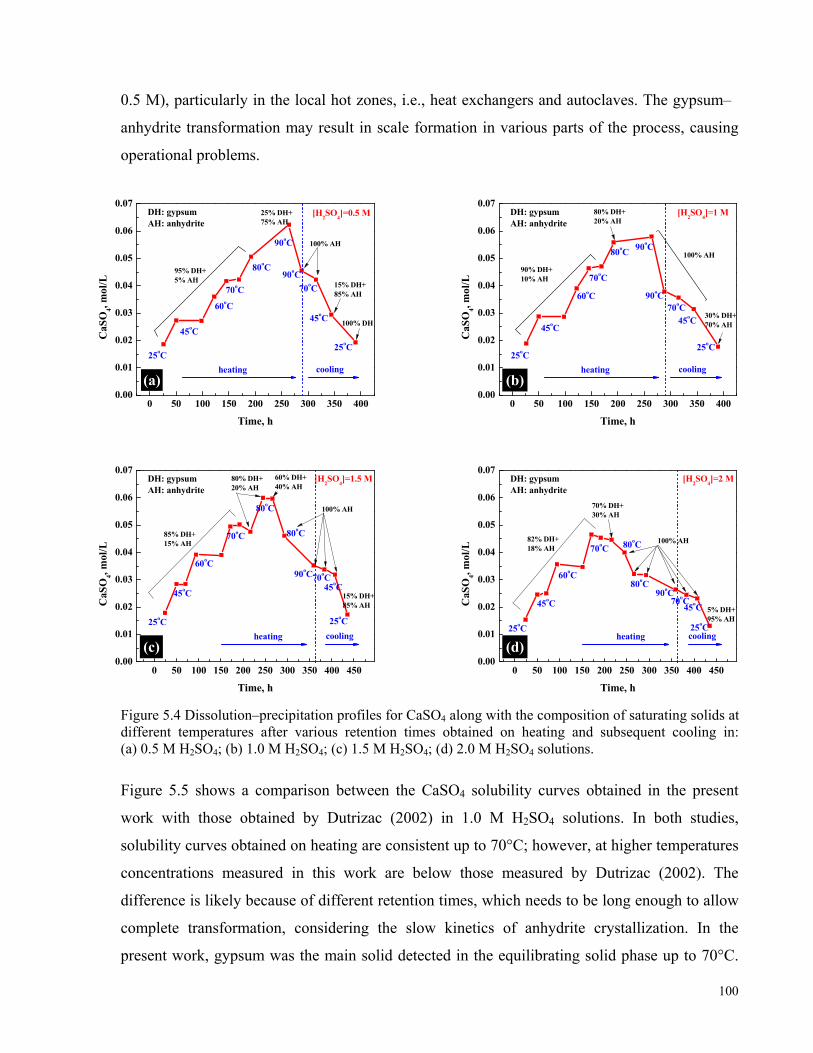
100
0.5 M), particularly in the local hot zones, i.e., heat exchangers and autoclaves. The gypsum–
anhydrite transformation may result in scale formation in various parts of the process, causing
operational problems.
0 50 100 150 200 250 300 350 4000.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
100% DH
15% DH+85% AH
25% DH+75% AH
100% AH
95% DH+5% AH
cooling
25oC
45oC
70oC90oC
90oC
80oC
70oC
60oC
45oC
25oC
CaS
O4, m
ol/L
Time, h
heating
DH: gypsumAH: anhydrite
[H2SO4]=0.5 M
(a)0 50 100 150 200 250 300 350 400
0.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
(b)
[H2SO4]=1 M
30% DH+70% AH
80% DH+20% AH
100% AH
90% DH+10% AH
cooling
25oC
45oC70oC
90oC
90oC80oC
70oC
60oC
45oC
25oC
CaS
O4, m
ol/L
Time, h
heating
DH: gypsumAH: anhydrite
0 50 100 150 200 250 300 350 400 4500.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
(c)
15% DH+85% AH
60% DH+40% AH
80oC85% DH+15% AH
[H2SO4]=1.5 M80% DH+20% AH
100% AH
cooling25oC
45oC70oC90oC
80oC
70oC
60oC
45oC
25oC
CaS
O4, m
ol/L
Time, h
heating
DH: gypsumAH: anhydrite
0 50 100 150 200 250 300 350 400 4500.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
(d)
5% DH+95% AH
80oC
82% DH+18% AH
[H2SO4]=2 M
70% DH+30% AH
100% AH
cooling25oC
45oC70oC
90oC
80oC70oC
60oC
45oC
25oC
C
aSO
4, mol
/L
Time, h
heating
DH: gypsumAH: anhydrite
Figure 5.4 Dissolution–precipitation profiles for CaSO4 along with the composition of saturating solids at different temperatures after various retention times obtained on heating and subsequent cooling in: (a) 0.5 M H2SO4; (b) 1.0 M H2SO4; (c) 1.5 M H2SO4; (d) 2.0 M H2SO4 solutions.
Figure 5.5 shows a comparison between the CaSO4 solubility curves obtained in the present
work with those obtained by Dutrizac (2002) in 1.0 M H2SO4 solutions. In both studies,
solubility curves obtained on heating are consistent up to 70°C; however, at higher temperatures
concentrations measured in this work are below those measured by Dutrizac (2002). The
difference is likely because of different retention times, which needs to be long enough to allow
complete transformation, considering the slow kinetics of anhydrite crystallization. In the
present work, gypsum was the main solid detected in the equilibrating solid phase up to 70°C.

101
At 80°C, the transformation reaction moved forward and complete transformation was
obtained at 90°C after 3 days (72 h). In the experiment performed by Dutrizac (2002), gypsum
was detected as the main solid phase up to 90°C with ~24 h retention time and transformation
completed at 95°C.
20 30 40 50 60 70 80 90 1000.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
30% DH+70% AH
100% AH
100% AH
100% AH80% DH+20% AH
CaS
O4, m
ol/L
Temperature, oC
This work, heating This work, cooling Dutrizac (2002), heating Dutrizac (2002), cooling
[H2SO4]=1 M
Figure 5.5 Concentration of CaSO4 in 1.0 M H2SO4 solutions as a function of temperature: (▲) this work, heating; (∆) this work, cooling; (■) Dutrizac (2002), heating; (□) Dutrizac (2002), cooling.
5.3.4 Theoretical and Practical Stability Regions of Gypsum in H2SO4 Solutions
The kinetics of gypsum–anhydrite transformation is an important factor to be considered in the
solubility determination of systems containing calcium sulphate hydrates. To demonstrate the
significance of such considerations, the “theoretical” and “practical” stability regions of gypsum
and anhydrite are marked in Figure 5.6. The solid curve represents the theoretical transition of
gypsum into anhydrite in H2SO4–H2O solutions obtained from the OLI/MSE model utilizing the
recently developed database (Azimi et al., 2007, 2008). The phase diagram in Figure 5.6 was
constructed using a procedure similar to that suggested by Li and Demopoulos (2006c).
Based on the results obtained in the present work, gypsum was kinetically stable up to 80°C
within the indicated acid range. The exact transformation temperature at a given acid
concentration along with the observed retention times are also presented in Figure 5.6. Region
(II), the area between the equilibrium transition curve and an upper line, is the practical stability
region of gypsum (based on retention time of 1–3 days). That is, in this region, the degree of
metastability of gypsum is significant and an induction period is required to complete the
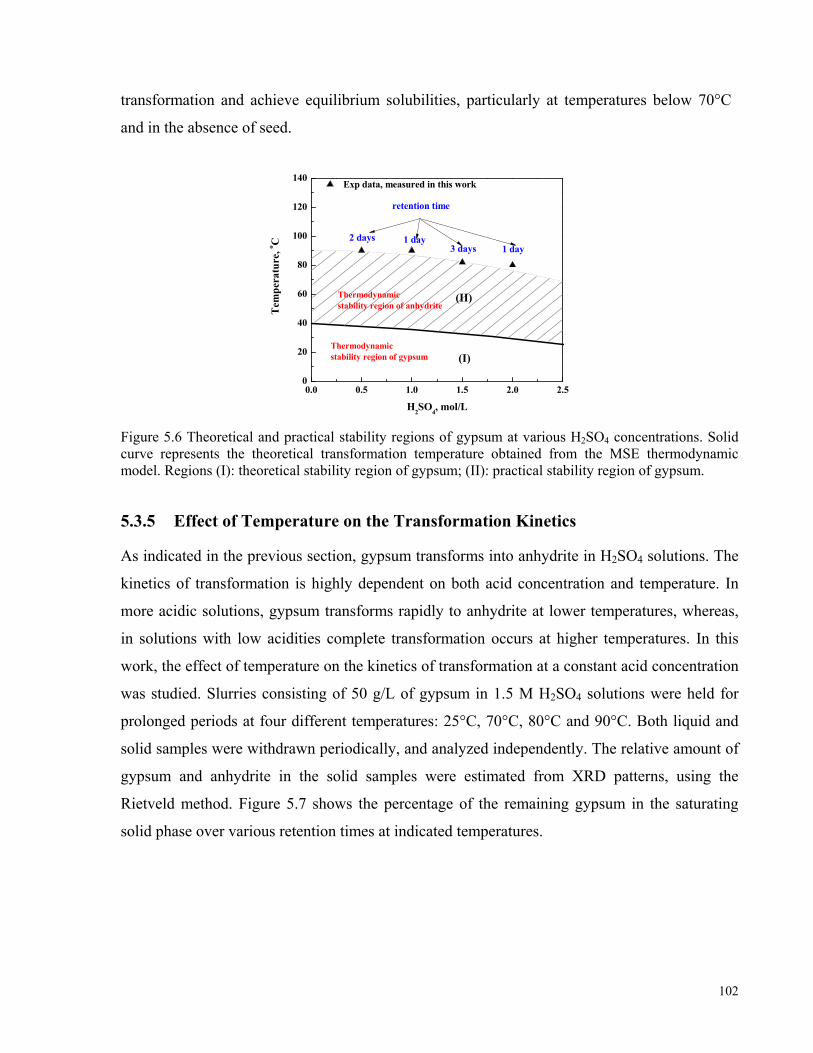
102
transformation and achieve equilibrium solubilities, particularly at temperatures below 70°C
and in the absence of seed.
0.0 0.5 1.0 1.5 2.0 2.50
20
40
60
80
100
120
140
(II)
Exp data, measured in this work
Thermodynamicstability region of anhydrite
1 day3 days1 day2 days
Tem
pera
ture
, o C
H2SO4, mol/L
Thermodynamicstability region of gypsum
retention time
(I)
Figure 5.6 Theoretical and practical stability regions of gypsum at various H2SO4 concentrations. Solid curve represents the theoretical transformation temperature obtained from the MSE thermodynamic model. Regions (I): theoretical stability region of gypsum; (II): practical stability region of gypsum.
5.3.5 Effect of Temperature on the Transformation Kinetics
As indicated in the previous section, gypsum transforms into anhydrite in H2SO4 solutions. The
kinetics of transformation is highly dependent on both acid concentration and temperature. In
more acidic solutions, gypsum transforms rapidly to anhydrite at lower temperatures, whereas,
in solutions with low acidities complete transformation occurs at higher temperatures. In this
work, the effect of temperature on the kinetics of transformation at a constant acid concentration
was studied. Slurries consisting of 50 g/L of gypsum in 1.5 M H2SO4 solutions were held for
prolonged periods at four different temperatures: 25°C, 70°C, 80°C and 90°C. Both liquid and
solid samples were withdrawn periodically, and analyzed independently. The relative amount of
gypsum and anhydrite in the solid samples were estimated from XRD patterns, using the
Rietveld method. Figure 5.7 shows the percentage of the remaining gypsum in the saturating
solid phase over various retention times at indicated temperatures.

103
0 2 4 6 8 10 12 14 16 18 20
0
20
40
60
80
100
Gyp
sum
(%)
Time, days
25 oC 70 oC 80 oC 90 oC
1.5 M H2SO4, 50 g/L gypsum, no anhydrite seeding
Figure 5.7 Kinetics of gypsum–anhydrite transformation at various temperatures in 1.5 M H2SO4 solutions in the absence of anhydrite seeds.
At 25°C, gypsum remained nearly unchanged, with about 5–10% transformation into anhydrite,
for up to 19 days. The XRD patterns of the solid samples withdrawn after 9 h, 24 h and 19 days
are presented in Appendix D. In addition, SEM images of these solid samples are presented in
Appendix G (Figure G.1). At 70°C, gypsum remained the dominant solid phase, with about 10%
anhydrite present, for 8 days, then the transformation accelerated and completed after 15 days of
contact. In runs at 80°C and 90°C, complete transformation occurred after 3 days and 1 day,
respectively. In these experiments, the induction period strongly depended on temperature.
In these sets of experiments, X-ray diffraction analysis of the solid samples indicated an initial
transformation of gypsum into anhydrite in the first few hours. This observation is in contrast
with the results reported by Dutrizac and Kuiper (2008), where gypsum remained unchanged
over a period of time before transforming to stable anhydrite. However, initial transformation
was observed by other researchers. Hardie (1967) has mentioned that in a few of the runs in
which anhydrite was produced from gypsum, a rind, presumed from X-ray diffraction to be
anhydrite, was observed on the surface and along cleavage cracks of gypsum crystals. In his
experiments, about 10–15% of the gypsum grains showed such alterations. Based on these
observations, Hardie (1967) suggested that gypsum directly dehydrates to anhydrite, beginning
at the crystal surfaces where H2O may be transferred to the solution phase.
The discrepancy with Dutrizac and Kuiper’s data is likely due to the solid samples handling. In
all experiments carried out in this work, solids were washed by a small amount of denatured

104
alcohol to replaces the solution, and dried inside an oven at 35–40°C. In the experiments
performed by Dutrizac and Kuiper (2008), solid samples were first washed with a small amount
of water and then with alcohol, and they were air-dried at room temperature. The addition of
water to the solid samples and drying at room temperature, where gypsum is the stable phase,
may have resulted in rehydration of anhydrite formed on the surface of gypsum crystals. The
phenomenon of initial transformation is explained in detail in Section 5.3.8.
The crystallization of stable anhydrite after initial transformation has been found to be surface
controlled, following a rate equation second-order in supersaturation (Nancollas, 1979):
2)( sc cckdtdc
−= (5.4)
where dc/dt is the linear rate of the reaction, kc is the rate constant of anhydrite crystallization,
and (c–cs) is the absolute super-saturation. By considering the fact that the rate constant of
anhydrite crystallization (kc) is inversely proportional to the induction time, i.e., kc=τ/tind, where
τ is a constant in mol-1, and substituting kc in the Arrhenius equation, the following empirical
relation is obtained (Turenne et al., 1999; Liu and Nancollas, 1975):
)exp(1RTEA
ta
ind
−=τ
(5.5)
where tind is the induction time, A is the Arrhenius (pre-exponential) constant, Ea is the
activation energy, R is the gas constant and T is absolute temperature. Equation (5.5) can be
used to calculate the activation energy associated with the induction time for the crystallization
of stable anhydrite. The ln(tin) vs. 1/T is plotted in Figure 5.8 which shows a linear relationship
(R2=0.99). The corresponding apparent activation energy, calculated from the slope of the
straight line, is 35 kcal•mol-1. As expected, this value is higher than that calculated by Liu and
Nancollas (1970, 1975) for gypsum (15 kcal•mol-1).

105
2.75 2.80 2.85 2.90 2.950.00.51.01.52.02.53.03.54.04.55.05.56.06.57.0
T=70 oC
T=80 oC
ln (t
ind)
1000/T (K-1)
Y=17.61 X - 46.29T=90 oC
[H2SO
4]=1.5 M
R2=0.99
Figure 5.8 Variation of the ln(tin) vs. 1/T at various temperatures in 1.5 M H2SO4 solutions with no seeds present.
5.3.6 Effect of Seeding on Gypsum–Anhydrite Transformation
Effect of seeding on the kinetics of the transformation has been studied previously (Dutrizac and
Kuiper, 2008, 2006; Farrah et al., 2004) which indicated the significant role of seeding in the
transformation process. In the present work, the effect of seeding on the gypsum–anhydrite
transformation was also studied in 1.5 M H2SO4 solutions at 70°C. Slurries were prepared by
adding 50 g/L of gypsum, together with 5 g/L of anhydrite seeds into the solutions. Slurries
were periodically sampled, and both the liquid and solid samples were analyzed independently.
Figure 5.9 shows the percentage of the remaining gypsum in the solid samples at various
retention times in the presence of anhydrite seeds compared with that where no seed was added.
As is shown, the addition of 5 g anhydrite seeds to the system containing 50 g of gypsum in
1.5M H2SO4 results in a significantly more rapid transformation of gypsum into anhydrite. In
the absence of anhydrite seeds, complete transformation occurred after 15 days, whereas with
seeding, 2 days was sufficient for near complete transformation.

106
0 2 4 6 8 10 12 14 16 18 20
0
20
40
60
80
100
Gyp
sum
(%)
Time, days
50 g/L gypsum in1.5 M H
2SO
4 solutions
T=70 oC
No seeding
With 5 g/Lanhydrite seeds
Figure 5.9 Kinetics of gypsum–anhydrite transformation at 70°C in 1.5 M H2SO4 solutions in the presence of an initial 5 g/L of anhydrite seeds compared to no seeding case.
Figure 5.10 presents a comparison between the CaSO4 concentration at different retention times
in 1.5 M H2SO4 slurries in the presence of 5 g/L anhydrite seeds compared to no seeding. The
dashed lines represent the saturation levels of gypsum and anhydrite in the system. The
difference between the time for transforming gypsum into anhydrite and for discharging the
supersaturation in the presence of seed can be explained by the slow crystallization kinetics of
anhydrite (Freyer and Voigt, 2003; Nancollas et al., 1973). Similar discrepancy between solid
and solution phase data has been observed by other researchers (Farrah et al., 2004). This
observation indicates that the transformation involves a dissolution–precipitation mechanism.
The end result here is that seeding accelerates the kinetics of the transformation. Therefore,
process solutions saturated with gypsum at temperatures between 60–70°C would reject their
calcium content faster in the presence of anhydrite seeds, which would result in lowering the
risk of anhydrite scale formation inside autoclaves or other hot zones throughout the plant. This
is in contrast to the current industrial practice where gypsum seed is recycled instead.

107
0 2 4 6 8 10 12 14 16 18 200.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
Anhydrite saturation line
CaS
O4 c
once
ntra
tion,
mol
/L
Time, days
No seeding 5 g/L anhydrite seed
50 g/L gypsum in 1.5M H2SO4 solutions
T=70 oC
Gypsum saturation line
Figure 5.10 CaSO4 solubility in 1.5 M H2SO4 solutions at 70°C at various residence times in the presence of 5 g/L anhydrite seeds compared to no seeding case.
5.3.7 Effect of Sulphate and Chloride Salts on the Transformation Process
As indicated in the previous sections, H2SO4 increases the rate of gypsum–anhydrite
transformation. Since many industrial processes contain other electrolytes than acid, the effect
of adding NiSO4 and NaCl on the transformation process was also studied in this work. To
assess the transformation process, slurries consisting of 50 g/L gypsum in 1.0 M NiSO4–1.5 M
H2SO4 and 0.5 M NaCl–1.5 M H2SO4 solutions were held for prolonged periods (up to 14 days).
Results obtained show that the addition of 1.0 M NiSO4 into 1.5 M H2SO4 solution slows the
kinetics of the transformation. In the absence of NiSO4, complete transformation was achieved
within 4 days, whereas, in the presence of 1.0 M NiSO4, one additional day was required. In
contrast, the addition of 0.5 M NaCl results in faster transformation kinetics (completed within 2
days). Figure 5.11 presents the percentage of the remaining gypsum in the saturating solid phase
at various retention times in the systems studied, compared to those in H2SO4 only solutions.

108
0 2 4 6 8 10 12 14
0
20
40
60
80
100
Time, days
T=80 oC
Gyp
sum
(%)
1.5 M H2SO4-1 M NiSO4
1.5 M H2SO4
1.5 M H2SO4-0.5 M NaCl
Figure 5.11 Kinetics of gypsum–anhydrite transformation at 80°C in: (–■–) acid only (1.5 M H2SO4);(–▲–) 1.0 M NiSO4–1.5 M H2SO4; (– –) 0.5 M NaCl–1.5 M H2SO4 solutions.
Figure 5.12 shows the respective concentrations of calcium sulphate. Addition of 1.0 M NiSO4
to a 1.5 M H2SO4 solution decreases the solubility of calcium sulphate by about 60% due to the
common ion effect of added SO42- ions, whereas, the addition of 0.5 M NaCl slightly increases
the solubility (by ~7%). In this figure, the dashed lines represent the saturation levels of gypsum
and anhydrite in the solutions.
0 2 4 6 8 10 12 140.00
0.02
0.04
0.06
0.08
0.10T=80 oC
Anhydrite saturation
Gypsum saturation
Anhydrite saturation
CaS
O4 c
once
ntra
tion,
mol
/L
Time, days
1.5 M H2SO4
1 M NiSO4-1.5 M H2SO4
0.5 M NaCl-1.5 M H2SO
4
Gypsum saturation
Figure 5.12 Calcium sulphate concentrations vs. retention time at 80°C: (–■–) in acid only (1.5 M H2SO4); (–▲–) in 1.0 M NiSO4–1.5 M H2SO4; (– –) in 0.5 M NaCl–1.5 M H2SO4 solutions.
The transformation kinetics depends on both dissolution kinetics of gypsum as well as
nucleation and growth of anhydrite (Freyer and Voigt, 2003; Kontrec et al.; 2002). Figure 5.13
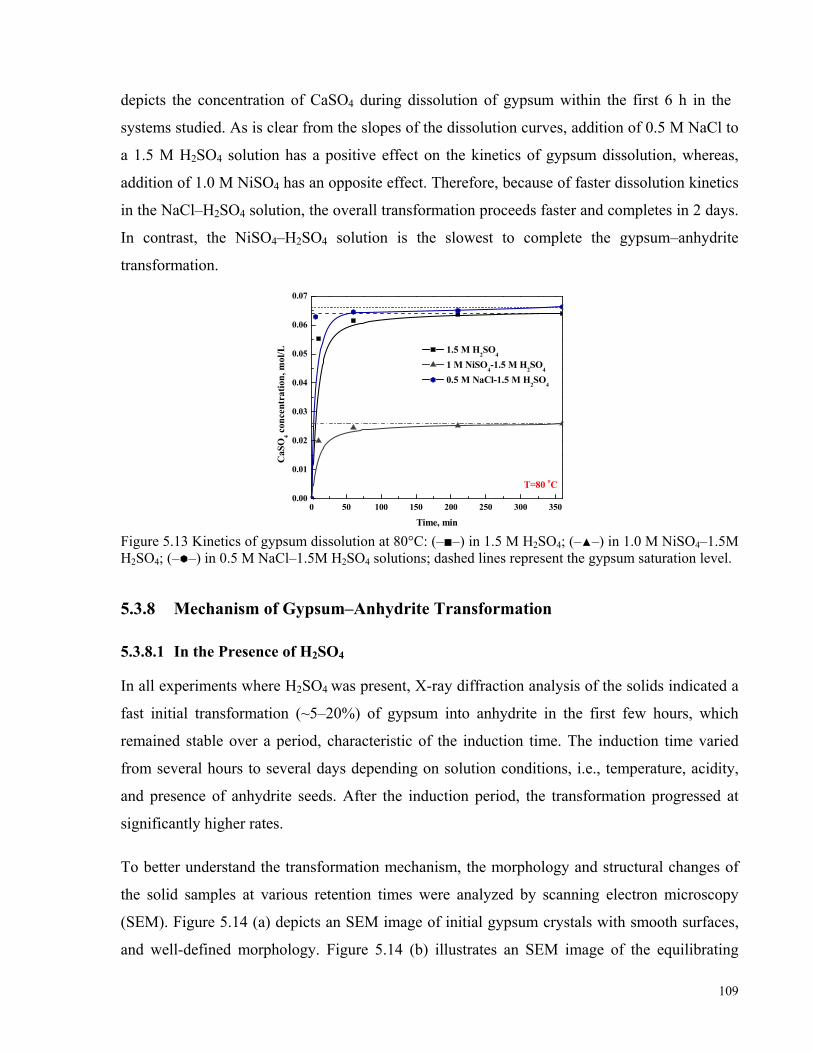
109
depicts the concentration of CaSO4 during dissolution of gypsum within the first 6 h in the
systems studied. As is clear from the slopes of the dissolution curves, addition of 0.5 M NaCl to
a 1.5 M H2SO4 solution has a positive effect on the kinetics of gypsum dissolution, whereas,
addition of 1.0 M NiSO4 has an opposite effect. Therefore, because of faster dissolution kinetics
in the NaCl–H2SO4 solution, the overall transformation proceeds faster and completes in 2 days.
In contrast, the NiSO4–H2SO4 solution is the slowest to complete the gypsum–anhydrite
transformation.
0 50 100 150 200 250 300 3500.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
CaS
O4 c
once
ntra
tion,
mol
/L
Time, min
1.5 M H2SO
4
1 M NiSO4-1.5 M H2SO4
0.5 M NaCl-1.5 M H2SO4
T=80 oC
Figure 5.13 Kinetics of gypsum dissolution at 80°C: (–■–) in 1.5 M H2SO4; (–▲–) in 1.0 M NiSO4–1.5M H2SO4; (– –) in 0.5 M NaCl–1.5M H2SO4 solutions; dashed lines represent the gypsum saturation level.
5.3.8 Mechanism of Gypsum–Anhydrite Transformation
5.3.8.1 In the Presence of H2SO4
In all experiments where H2SO4 was present, X-ray diffraction analysis of the solids indicated a
fast initial transformation (~5–20%) of gypsum into anhydrite in the first few hours, which
remained stable over a period, characteristic of the induction time. The induction time varied
from several hours to several days depending on solution conditions, i.e., temperature, acidity,
and presence of anhydrite seeds. After the induction period, the transformation progressed at
significantly higher rates.
To better understand the transformation mechanism, the morphology and structural changes of
the solid samples at various retention times were analyzed by scanning electron microscopy
(SEM). Figure 5.14 (a) depicts an SEM image of initial gypsum crystals with smooth surfaces,
and well-defined morphology. Figure 5.14 (b) illustrates an SEM image of the equilibrating

110
solid in 1.5 M H2SO4 media at 70°C after 3 h of retention time. X-ray diffraction analysis of
this solid sample revealed ~10% anhydrite formation, seen by SEM as crusts formed on the
surfaces of gypsum crystals and along cleavage cracks. Figure 5.14 (c) shows an SEM image of
the solid sample after 12 days. In this image, the surfaces of gypsum crystals are covered by a
layer of crust, and several needle-shaped crystals are flaked out from the crust layer. X-ray
diffraction analysis confirmed the presence of 45% anhydrite crystals. The last SEM image
(Figure 5.14 (d)) demonstrates the final solid phase (100% anhydrite) obtained after complete
transformation in 20 days.
Figure 5.14 SEM images of a) gypsum feed; b) equilibrating solid phase after 3 h; c) solid phase after 12 days; and d) transformed anhydrite crystals after 20 days in 1.5 M H2SO4 media at 70°C.
As is clear from Figures 5.14 (b) and (c), anhydrite appears in two different crystal forms:
surface crust and detached or semi-detached needles. It has been shown by several researchers
that anhydrite crystals exist as two different polymorphs, soluble anhydrite (known as γ–CaSO4
or AIII phase) and insoluble anhydrite (known as AII–anhydrite) (Ballirano and Melis, 2009;
Christensen et al., 2008; Freyer and Voigt, 2003; Bezou et al., 1995, Bushuev et al., 1983;
Hardie, 1967). Soluble γ–AH is the metastable polymorph of anhydrite, whereas insoluble AII–
AH is the stable one. Metastable γ–AH is the dehydration product of gypsum below 100°C
(a) (b)
(c) (d)

111
(where hemihydrate formation does not occur), or that of hemihydrate at temperatures above
100°C. It has been reported that the crystallization of the stable AII–AH is the slowest compared
to other calcium sulphate phases, and requires a long induction period (Christensen et al., 2008;
Farrah et al., 2004; Freyer and Voigt, 2003; Nancollas et al., 1973). Freyer and Voigt (2003)
concluded that in aqueous solutions, the transformation of gypsum into stable AII–AH is not a
direct reaction.
There are very few published proven structural differences between γ–AH and AII–AH, which
often lead to terminological confusion (Bushuev et al., 1983). In addition, the hydration rate of
γ–AH has been reported to be high (Christensen et al., 2008; Bushuev et al., 1983); therefore,
fast re-hydration of γ–AH may prevent this polymorph from being indentified if samples have
been in contact with water at room temperature prior to XRD/SEM analysis. The situation is
more complicated by the fact that the X-ray diffraction patterns of γ–AH and AII–AH are very
similar (Bushuev et al., 1983) and they can be distinguished only by using high-resolution X-ray
diffractometers. Figure 5.15 shows the XRD patterns for the two anhydrite polymorphs obtained
from International Centre for Diffraction Data® files (JCPDF-ICDD file number 072–0916 for
orthorhombic AII–anhydrite and 037–0184 for tetragonal γ–anhydrite).
Figure 5.15 XRD patterns of AII–anhydrite: 072-0916 (orthorhombic) and γ–anhydrite: 037-0184 (tetragonal) obtained from ICDD database. The characteristic line of AII–AH is marked with an asterisk.
As is clear, the patterns of the two anhydrite polymorphs (soluble and insoluble) are very
similar, except for a characteristic peak distinguishing these two patterns, located at 2θ=41.3°,
* AII–AH (stable)
γ–AH (metastable)

112
which is marked by an asterisk in Figure 5.15. Therefore, below 45°, the XRD patterns for both
anhydrites overlap.
The X-ray diffraction patterns of the intermediate solid samples presented in Figures 5.14 (b)
and (c), collected in the range of 10–55° 2θ, are presented in Appendix D (Figures D.5 and D.6).
The characteristic peak of the stable AII–AH was not detected in the solid sample withdrawn
after 3 h of retention, whereas, it was detected in the solid sample taken after 12 days, where
needle-shaped crystals were also present. Based on these observations, it is likely that the
product of initial dehydration of gypsum is the soluble γ–AH polymorph. However, further
investigations are required to claim this phenomenon without any doubt.
As presented in the previous sections, addition of anhydrite (AII–AH) seeds, both in pure water
and in H2SO4 solutions, shortens the induction time and accelerates the transformation process,
which is evident that the transformation involves a dissolution–precipitation step.
Moreover, no hemihydrate formation was detected in the solid samples analyzed after various
retention times, over the entire temperature and acid concentration ranges studied (i.e., 25–90°C,
and 0–2.0 M H2SO4). Similar behaviour has been observed by others in H2SO4 media at
temperatures below 100°C (Dutrizac and Kuiper, 2008; Farrah et al., 2004; Hardie, 1967).
Based on the above observations, the following mechanism is postulated for the gypsum–
anhydrite transformation in acidic solutions (0.5–2 M H2SO4), over the temperature range of
25–90°C:
(i) Gypsum quickly dehydrates to soluble γ–AH at the crystal–solution interface, along
cleavage cracks, where H2O can be transferred to the aqueous phase (a similar
phenomenon was also assumed to take place by Hardie (1967)). Since the initial
surface dehydration was only observed in the experiments carried out in the presence
of H2SO4 (not in pure water), the reason for that can be attributed to the presence of
H+ ions (from H2SO4), which facilitate stripping out water molecules from the
surface of gypsum crystals, by surface complexation and external H3O+ formation.
(ii) Then, nuclei of stable AII–AH form, likely as a result of polymorphic transition
between γ–AH and AII–AH. If the nucleation of AII–AH occurs on the γ–AH
surface, two possible mechanisms can be envisaged for the formation of AII–AH

113
nuclei, as proposed by Petit and Conquerel (1996). The first mechanism involves
cooperative molecular movements which result in a local deformation of the
structure from the parent phase to the daughter phase. The second possible
mechanism consists of breaking the intermolecular links, followed by a repositioning
of each molecule. Nevertheless, the exact nucleation mechanism of these
polymorphic transitions at molecular level cannot be fully elucidated from the
available experimental data.
(iii) After nucleation of stable AII–AH, the growth of AII–AH nuclei occurs. At first, the
bulk of the solution rejects its super-saturation on the newly-formed stable AII–AH
nuclei. Since the stable AII–AH forms needle-like crystals, resulting in a highly
porous coating, it allows further dissolution of gypsum particles sustaining a super-
saturated solution with respect to AII–AH, and therefore precipitating on and
growing the AII–AH nuclei present on their exterior surface. As the dissolution
proceeds, the interface between the undissolved gypsum (the core) and the outer
layer consisting of AII–AH crystals recedes towards the centre of the gypsum particle
in a fashion similar to the shrinking core model (Hsu et al., 2009; Wen, 1968).
A series of SEM images obtained at various retention times are presented in Figure 5.16 (a)–(e)
to depict the transformation mechanism in 1.5 M H2SO4 solutions at 80°C. In these images, both
particles and cross-sections are presented. Figure 5.16 (a) presents an SEM image of gypsum
feed crystals. Figures 5.16 (b) shows solid samples after 24 h retention, where an exterior layer
of anhydrite appears around the gypsum core on the cross-section image. The SEM images of
solid samples after 34 h and 56 h, are presented in Figures 5.16 (c) and (d), respectively. A
comparison between Figures (b), (c) and (d) reveals that the interface between the undissolved
gypsum and the exterior anhydrite layer moves inward (i.e., gypsum core shrinks) with time. It
also appears that the AII–AH coating starts to disintegrate partially after ~56 h. Figure 5.16 (e)
depicts the final needle-shaped anhydrite particles produced after 80 h.

114
(a) t=0 h 100% DH
(b) t=24 h 85% DH+15% AH
(c) t=34 h 58% DH+42% AH
(d) t=56 h 35% DH+65% AH
(e) t=80 h 100% AH
Figure 5.16 SEM images of saturating solids in 1.5 M H2SO4 media at 80°C after various retention times.

115
5.3.8.2 Transformation Mechanism in Pure Water
Contrary to gypsum behaviour in H2SO4 solutions, gypsum in pure water remained unchanged
(tested for 20 days) at temperatures as high as 90°C in the absence of anhydrite seeds, whereas
addition of 10wt% anhydrite seeds (AII–AH) accelerated the transformation. In these
experiments, the initial dehydration of gypsum to γ–AH was not detected by XRD; therefore,
the transformation is likely to proceed directly through gypsum dissolution followed by AII–AH
precipitation. Several SEM images related to the experiments performed in water are presented
in Appendix G, providing further support to the above conclusions.
More powerful and precise techniques with high resolution detectability such as in situ
synchrotron radiation powder X-ray diffraction (SR-PXD) are required to study the
transformation mechanism rigorously at the atomic level. Utilizing such techniques allows
following all changes in the morphologies and characteristics of the solids in situ over very short
exposure times. In the present work, however, the transformation between gypsum and
anhydrite was investigated at the macroscopic level, with a focus on the effect of various
parameters on the transformation kinetics, to understand the effect of gypsum–anhydrite
transformation on the calcium sulphate scaling problem in the industry. Therefore, the
mechanism proposed is based only on XRD patterns and SEM images of the equilibrating solids
at various retention times, and cannot account for all the changes taking place at the atomic
scale.
5.3.9 Industrial Implication: Precipitation due to Super-saturation
It was shown in the previous sections that in the absence of seeds, gypsum transforms into
insoluble anhydrite in 1.5 M H2SO4 solutions at 80°C after 3 days through gypsum dissolution
followed by nucleation and growth of anhydrite. It is of great practical importance to further
investigate the extent of this transformation in the presence of seed crystals, because it may offer
an opportunity to reduce calcium super-saturation to anhydrite saturation level, which is lower
than that of gypsum, enabling more efficient process water utilization. To this end, a solution of
1.5 M H2SO4 saturated with gypsum at 80°C was prepared. The solution was kept at
temperature for 24 h before 10 g of anhydrite seeds were added. Both liquid and solid samples
were withdrawn periodically and analyzed independently. Figure 5.17 shows the concentration
of CaSO4 as a function of residence time. The first sample was withdrawn before adding

116
anhydrite seeds and represents the saturation level of gypsum. After adding 10 g/L of anhydrite
seeds to the system, the CaSO4 concentration dropped sharply within the first few hours and
after 1 day it almost reached the saturation level of anhydrite in the solution. X-ray diffraction
analysis of the solid samples withdrawn at various retention times detected only anhydrite.
0 20 40 60 80 100 120 140 1600.02
0.03
0.04
0.05
0.06
0.07
0.08
Anhydrite saturation level
CaS
O4, m
ol/L
time, h
Gypsum saturation level
[H2SO
4]=1.5 M
T=80 oC
10 g of anhydrite seed
Figure 5.17 CaSO4 concentration at various retention times in 1.5 M H2SO4 solutions initially saturated with gypsum at 80°C after adding 10 g of anhydrite seeds.
The implication of this study is that process solutions saturated with gypsum at moderate
temperatures (above ~80°C) can be seeded with anhydrite to decrease their calcium content and
lower it to the saturation level of anhydrite, provided that the solution is slightly acidic (~0.5 M
H2SO4). This would result in decreasing the risk of scale formation downstream.

117
5.4 Summary
The kinetics of the gypsum–anhydrite transformation was investigated by monitoring changes in
both liquid and solid phases. The results showed that in pure water, gypsum remained stable up
to 90°C in the absence of anhydrite seeds for a prolonged time (tested for up to 20 days). The
addition of 10 wt% anhydrite seeds accelerated the transformation process in water at similar
temperature (90ºC), resulting in complete transformation after 10 days.
The addition of 0.5 M to 2.0 M H2SO4 was found to promote the transformation process. In
0.5 M H2SO4 solutions, complete transformation was achieved at 90°C after 4 days (96 h),
whereas in 1.0 M acid, 3 days (72 h) was sufficient at the same temperature (90°C). In solutions
containing 1.5 M and 2.0 M H2SO4, complete transformation occurred at 80°C after 3 days and
1 day, respectively. Therefore, gypsum is kinetically stable up to at least 80°C for acid
concentrations as high as 2.0 M. In experiments performed in H2SO4 solutions, anhydrite was
detected as the equilibrating solid phase during cooling process above 45°C, but further cooling
resulted in the conversion of anhydrite to gypsum. Higher acid concentrations made solutions
retain more anhydrite after slurry was cooled to room temperature.
Temperature was shown to have a significant effect on the transformation kinetics. In 1.5 M
H2SO4 solutions, gypsum remained almost unchanged at 25°C over the period tested (i.e., 18
days); at 70°C, gypsum remained the dominant solid phase for 7 or 8 days with less than 10% γ–
anhydrite present; after 8 days, the transformation progressed more rapidly and completed after
15 days of contact. In the runs at 80°C and 90°C, complete transformation occurred after 3 days
and 1 day, respectively. By using induction times at these temperatures and fitting of the
Arrhenius expression, the apparent activation energy for the crystallization of stable AII–
anhydrite was calculated to be 35 kcal•mol-1.
The addition 1.0 M NiSO4 to a 1.5 M H2SO4 solution at 80°C had an impending effect on the
transformation process by causing a 1 day delay. In contrast, the addition of 0.5 M NaCl as a
representative chloride salt to a 1.5 M H2SO4 solution at the same temperature had a positive
effect and accelerated the transformation process by 1 day. This is due to the fact that the
addition of NiSO4 to 1.5 M H2SO4 solutions has a negative effect on the kinetics of gypsum

118
dissolution due to the common ion effect, whereas the addition of NaCl to 1.5 M H2SO4
solutions has an opposite effect.
In all experiments conducted in this work, an initial transformation (5−20%) of gypsum into
anhydrite (likely to be metastable γ–anhydrite polymorph) was observed in the first few hours
which stayed unchanged over an induction time. The duration of induction periods varied from
several hours to several days depending on temperature, acidity and the presence of seeds. After
the induction period, the transformation progressed at higher rates. Hemihydrate was not
detected in any experiment at 25–90°C. Based on these observations, a mechanism was
proposed for the gypsum–anhydrite transformation in H2SO4 solutions consisting of direct
dehydration of gypsum to metastable γ–anhydrite, followed by the nucleation of stable AII–
anhydrite as a result of polymorphic transitions between γ– and AII–anhydrites. The last stage is
the growth of AII–anhydrite nuclei through a dissolution–precipitation process. As the
dissolution proceeds, the interface between the undissolved gypsum (the core) and the layer
consisting of AII–AH crystals moves inward, following the assumptions of a shrinking core
model.
The implication of the results obtained is that the transformation of gypsum into anhydrite,
which results in a significant drop in the solubility (up to one order of magnitude) would occur
rapidly in many industrial processes with modest concentrations of sulphuric acid (above 0.5 M)
at temperatures above 80°C, particularly in the local hot zones of plants, e.g., autoclaves in
hydrometallurgical processes or heat exchangers in other industries. However, the gypsum–
anhydrite transformation can be utilized for mitigating CaSO4 scaling in various industries. That
is, process solutions saturated with gypsum can be aged in the presence of anhydrite seeds at
moderate temperatures (~80°C) to decrease their calcium content, provided that the solution is
slightly acidic (~0.5 M).

119
CHAPTER 6 CONCLUSIONS
he solution chemistry and phase equilibria of calcium sulphate hydrates (gypsum,
hemihydrate and anhydrite) in multicomponent hydrometallurgical solutions containing
various minerals was investigated over wide ranges of temperature and composition. A new
database for the Mixed Solvent Electrolyte (MSE) model of the OLI software was developed
through fitting of existing literature data such as mean activity, heat capacity and solubility in
simple binary and ternary systems, as well as additional solubility data measured in the present
work. Furthermore, a number of experiments were carried out to investigate the effect of various
parameters including temperature, acidity, as well as metal sulphate and chloride concentrations
on the solubilities of calcium sulphate hydrates in laterite pressure acid leach (PAL) processes,
containing Al2(SO4)3, MgSO4, NiSO4, H2SO4, and NaCl, over the temperature range of
25–250ºC. Prior to this work, no previous study had been conducted for such systems and no
reliable (i.e., experimentally verifiable) chemical models existed.
The database developed, utilized by the MSE model, was shown to accurately predict the
solubilities of all calcium sulphate hydrates (and hence, predict the scaling potential) in various
multicomponent hydrometallurgical process solutions including neutralized zinc sulphate leach
solutions in the Zinc Pressure Oxidation process (Zn-POX), nickel sulphate–chloride solutions
from the Vale Inco Ni-POX process currently under implementation to recover Ni from the
Voisey’s Bay deposit in Newfoundland and Labrador, as well as solutions in the High Pressure
Acid Leach process (HPAL) for Ni and Co recovery from laterite ores over a wide temperature
range of 25–250°C. The fact that modelling in binary and ternary systems is sufficient to predict
the behaviour of more complex multicomponent systems is a substantive contribution to the
existing field. The applicability regions of the model with respect to temperature and
composition are summarized in the following table.
Table 6.1–Applicability regions of the model
Compound Range Compound Range HCl 0–6.0 M H2SO4 0–3.0 M NaCl 0–6.0 M Na2SO4 0–3.6 M LiCl 0–1.0 M MgSO4 0–4.0 M CaCl2 0–5.6 M MnSO4 0–3.7 M MgCl2 0–6.0 M NiSO4 0–3.5 M AlCl3 0–1.5 M ZnSO4 0–2.5 M FeCl3 0–2.2 M Fe2(SO4)3 0–1.0 M Temperature range: 25–250˚C
T

120
The results obtained from the model showed that theoretically gypsum transforms into
anhydrite at around 45–50°C. However, the transformation does not practically occur until
about 80–90°C due to slow transformation kinetics. To understand the mechanism of the
gypsum–anhydrite transformation, particularly in industrial solutions, a systematic study was
undertaken to investigate the effects of various parameters including temperature, acidity,
seeding, and the presence of sulphate or chloride salts on the transformation kinetics.
The results obtained from this study led to the following conclusions:
1. The solubility of gypsum in water was confirmed to reach a maximum at ~45–50ºC,
followed by a slight decrease at higher temperatures. However, in all process solutions,
gypsum is increasingly soluble with temperature even beyond 50ºC. As a result, process
solutions saturated with gypsum during a neutralization step at elevated temperatures
have the potential for scale formation when cooled to lower temperatures.
2. Gypsum is the practically stable solid phase up to ~90ºC due to the slow kinetics of
phase transformation, although the transformation temperature at thermodynamic
equilibrium is around 45ºC. At temperatures above 100ºC, anhydrous calcium sulphate
(anhydrite) becomes the stable phase practically and thermodynamically. Anhydrite
solubility decreases with temperature thereafter.
3. The average solubility of anhydrite above 100ºC is lower than that of gypsum below
100ºC (by approximately one order of magnitude). Therefore, process solutions saturated
with gypsum at ambient temperature and recycled to a high temperature reactor (i.e., an
autoclave) have the potential to form anhydrite scales. Such solutions require pre-
processing to decrease their calcium content below the saturation level of anhydrite at
temperature. This can be accomplished, for example, by mixing the recycling stream
with carbonate compounds to reject calcium as calcium carbonate, provided that the
solution is not acidic. Calcium carbonate solubility at room temperature in a neutral
solution is about 80% lower than the anhydrite solubility at 250ºC. Alternatively, the
seeded gypsum–anhydrite transformation could be utilized as another practical method
for reducing the calcium content in processing circuits. Process solutions saturated with
gypsum at moderate temperatures (~80°C) can be aged in the presence of anhydrite
seeds, provided that the solution is slightly acidic (~0.5 mol/L H2SO4).

121
4. The addition of H2SO4, up to around 1.5–2 M, has a strong positive effect on the
solubility of calcium sulphate in water (up to 10 times increase) over the temperature
range of 25–250ºC. Above this concentration, calcium sulphate solubility reaches a
plateau, and upon further acid addition the solubility decreases due to the salting-out
effect. Similar effect can be observed in solutions containing high metal sulphate
concentrations.
5. The chloride content of the process water was also found to affect the solubility of
CaSO4. In the experiments carried out on laterite PAL solutions, the increase of chloride
levels from tap water (140 ppm) to hyper-saline water (75,000 ppm) increased the
solubility of anhydrite by almost 20%. Therefore, in regions where seawater (or saline
bore water) is available, mixing recycled process solutions with chloride-containing
waters is favorable for decreasing anhydrite scale formation in autoclaves, provided that
chloride-corrosion issues can be controlled or tolerated by using chloride-resistant
materials and metal alloys.
Overall, higher acidities, higher water salinity, lower sulphate concentrations and anhydrite
seeding provide favorable conditions for minimizing anhydrite scaling inside high temperature
reactors (autoclaves). Of course, the above provides just general guidelines and must first be
optimized to target capital and operating costs, material consumption, and environmental
regulations.
The results obtained from this study can be utilized to map the behaviour of calcium sulphate in
aqueous industrial solutions and to assess the potential for scaling in various hydrometallurgical
process streams. This, in turn, will provide these industries with the opportunity to investigate
the effect of different variables such as temperature and composition and aid them in finding
solutions for mitigating, or at least, controlling calcium sulphate scale formation in the
processing circuits.

122
CHAPTER 7 RECOMMENDATIONS FOR FUTURE WORK
pon completion of this work, a number of prospective extensions to the project have been
determined:
1. In this work, the kinetics and mechanism of gypsum–anhydrite transformation in water
and in H2SO4-based solutions over the temperature range of 25–90ºC under atmospheric
pressure was studied. It is recommended to further extend the temperature range and to
study the transformation mechanism at higher temperatures and pressures inside an
autoclave. In particular, the temperature range of 100–150ºC, where the formation of
hemihydrate occurs, is of interest. The effect of seeding, acid concentration, addition of
sulphate or chloride salts on the transformation mechanism and kinetics under the
conditions indicated above will provide complementary knowledge on the calcium
sulphate hydrates transformation processes.
2. It was shown in section (5.3.9) that at moderate temperatures (~80°C), solutions
saturated with gypsum (as a metastable phase) would reject their calcium content in the
presence of anhydrite seeds. In the present study, analytical grade anhydrite seeds were
added directly from J.T. Baker bottles. It is recommended to run some additional tests to
confirm that similar phenomenon would occur in the case of recycling the product of
initial seeding into another reactor containing solutions saturated with metastable
gypsum.
3. In this work, a fundamental study on calcium sulphate scale formation during pressure
acid leaching and upstream neutralization in hydrometallurgical processes was
conducted. However, the solvent extraction circuits of the refinery are the other
susceptible areas of the plants for calcium sulphate scale formation, as was the case in
the Bulong plant in Australia, where gypsum scaling in the solvent extraction circuit
created serious operational problems. Since the OLI/MSE model can handle mixed
solvent electrolytes, including organics, it is recommended to further extend the database
developed in this work such that it is also capable of predicting calcium sulphate
solubility (hence the scaling potential) in solvent extraction circuits.
U

123
4. In the present work, transformation between gypsum and anhydrite was studied in the
dissolution direction. It is recommended to further extend this work and study the
transformation between calcium sulphate hydrates and their practical stability regions in
the precipitation direction because in industrial practice, transformation takes place
during precipitation rather than dissolution, i.e., during sulphuric acid neutralization with
calcium-containing bases, first the most metastable phase, with the lowest free energy
barrier of formation, precipitates (Oswald step rule), and then gradually transforms into
the most stable phase over a longer period of time.

124
REFERENCES
Abrams D.S., Prausnitz J.M., 1975. Statistical thermodynamics of liquid mixtures: a new expression for the excess Gibbs energy of partly or completely miscible systems. AIChE Journal, 21, 116–128.
Adams J.F., 2004. Gypsum scale formation and control during continuous sulphuric acid neutralization. Ph.D. thesis, University of Toronto, Toronto.
Adams J.F., Papangelakis, V.G., 2007. Optimum reactor configuration for prevention of gypsum scaling during continuous acid neutralization. Hydrometallurgy, 89, 269–278.
Anderko A., Wang P., Rafal M., 2002. Electrolyte solutions: from thermodynamic and transport property model to the simulation of industrial processes. Fluid Phase Equilib. 123–142.
Arslan A., Dutt G.R., 1993. Solubility of gypsum and its prediction in aqueous solutions of mixed electrolytes. Soil Science, 155 (1), 37–47.
Aseyev G.G., 1996. Thermal properties of electrolyte solutions, methods for calculation of multi-component systems and experimental data. Begell House inc. Publishers, New York, Wallingford (UK).
Azimi G., Adams J.F., Jones M., Liu H., Papangelakis V.G., 2006. Chemical modeling of calcium sulphate solubility in hydrometallurgical process solutions. In: Advanced Processing of Metals and Materials, Sohn International Symposium, Kongoli F., Reddy R.G., Eds., The Minerals, Metals & Materials Society, PA, USA, Vol. 3, 419–426.
Azimi G., Papangelakis V.G., Dutrizac J.E., 2007. Modelling of calcium sulphate solubility in concentrated multi-component sulphate solutions. Fluid Phase Equilib. 260(2), 300–315.
Azimi G., Papangelakis V.G., Dutrizac J.E., 2008. Development of an MSE-based chemical model for the solubility of calcium sulphate in mixed chloride-sulphate solutions. Fluid Phase Equilib. 266 (1-2), 172–186.
Azimi G., Papangelakis V.G., 2010a. Thermodynamic modeling and experimental measurements of calcium sulphate in complex aqueous solutions. Fluid Phase Equilibria, 290, 88–94.
Azimi G., Papangelakis V.G., Dutrizac J.E., 2010. Development of a chemical model for the solubility of calcium sulphate in zinc processing solutions. Canadian Metallurgical Quarterly, 49(1), 1–8.
Azimi G., Papangelakis V.G., 2010b. Gypsum and anhydrite solubility in simulated laterite pressure acid leach solutions up to 250°C. Hydrometallurgy, in press.
Baghalha M., 1999. Aqueous H2SO4–Al2(SO4)3–MgSO4 solutions at 250°C: Identification of chemistry and thermodynamics, application to pressure acid leaching of laterites. Ph.D. thesis, University of Toronto, Toronto.
Ballirano P., Melis E., 2009. The thermal behaviour of γ-CaSO4. Phys. Chem. Minerals. 36, 319–327.
Barba D., Brandani V., Giacomo G., 1982. A thermodynamic model of CaSO4 solubility in multi-component aqueous solutions. Chemical Engineering Journal, 24, 191–200.

125
Barba D., Brandani V., Giacomo G., 1984, Solubility of calcium sulfate dihydrate in the system of Na2SO4–MgCl2–H2O. Journal of Chemical and Engineering Data, 29, 42–45.
Bezou C., Nonat A., Mutin J.C., Lehmann M.S., 1995. Investigation of the crystal structure of γ–CaSO4, CaSO4•0.5H2O and CaSO4•0.6H2O by powder diffraction methods. Journal of Solid Sate Chemistry. 117, 165–176.
Block J., Waters Jr. O.B., 1968. The CaSO4–Na2SO4–NaCl–H2O system at 25 to 100°C. Journal of Chemical and Engineering Data, 13(3), 336–344.
Blount C.W., Dickson F.W., 1969. The solubility of anhydrite (CaSO4) in NaCl–H2O from 100 to 450°C and 1 to 1000 bars. Geochimica et Cosmochimica Acta 33 (2), 227–245.
Bock E., 1961. On the solubility of anhydrous calcium sulphate and of gypsum in concentrated solutions of sodium chloride at 25, 30, 40, and 50ºC. Canadian Journal of Chemistry, 39, 1746–1751.
Bromley L.A., 1972. Approximate individual ion values of β (or B) in extended Debye-Hückel theory for univalent aqueous solutions at 298.15 K. Journal of Chem. Thermodynamics, 4, 669–673.
Bromley L.A., 1973. Thermodynamic properties of strong electrolytes in aqueous solutions. AIChE Journal, 19(2), 313–320.
Bruhn G., Gerlach J., Pawlek F., 1965. Untersuchungen Über dic Löslichkeiten von Salzen und Gasen in Wasser und wässerigen Lösungen bei Temperaturen oberhalb 100ºC. Z. anorg. Allg. Chemie, 337, 68–79.
Bushuev N.N., Maslennikov B.M., Borisov V.M., 1983. Phase transformations in the dehydration of CaSO4•2H2O. Russian Journal of Inorganic Chemistry, 28, 1404–1407.
Cameron F.K., Seidell A., 1901. Solubility of gypsum in aqueous solutions of certain electrolytes. Journal of Physical Chemistry, 5(9), 643–655.
Cameron F.K., Bell J.M., Robinson W.O., 1907. The solubility of certain salts present in alkali soils. Journal of Physical Chemistry, 11(5), 396–420.
Campbell A.N., Yanick N.S., 1932. The system NiSO4–CaSO4–H2O. Transactions of the Faraday Society, 28, 657–61.
Christensen A.N., Olesen M., Cerenius Y., Jensen T.R., 2008. Formation and transformation of five different phases in the CaSO4–H2O system: crystal structure of the subhydrate β-CaSO4•0.5H2O and soluble anhydrite CaSO4. Chemistry of Materials, 20, 2124–2132.
Clynne M.A., Potter II R.W., 1979. Solubility of some alkali and alkaline earth chlorides in water at moderate temperatures. J. Chem. Eng. Data, 24(4), 338–340.
Dathe H., Jentys A., Haider P., Schreier E., Fricke R., Lercher J.A., 2006. On the trapping of SOx on CaO–Al2O3–based novel high capacity sorbents. Physical Chemistry Chemical Physics, 8(13), 1601–1613.
Dean J.A.,1999. Lange's Handbook of Chemistry (15th Edition), McGraw-Hill, New York, NY.
Demopoulos G.P., Kondos P., Papangelakis V.G., 1987. Prediction of solubility in chemical compound precipitation system. In: Crystallization and Precipitation Symposium Proceeding, G.L., Strathdee, M.O., Klein and Melis L.a., Eds., Pergamon Press: Oxford, UK, 231–246.

126
Denman W.L., 1961. Maximum re-use of cooling water based on gypsum content and solubility. Industrial and Engineering Chemistry, 53(10), 817–822.
Doadrio J.C., Arcos D., Cabañas M.V., Vallet-Regí M., 2004. Calcium sulphate-based cement containing cephalexin. Biomaterials, 25 (13), 2629–2635.
Dutrizac J.E., 2002. Calcium sulphate solubilities in simulated zinc processing solutions. Hydrometallurgy, 65 (2-3), 109–135.
Dutrizac J.E., Kuiper A., 2006. The solubility of calcium sulphate in simulated nickel sulphate-chloride processing solutions. Hydrometallurgy, 82, 13–31.
Dutrizac J.E., Kuiper A., 2008. The solubility of calcium sulphate in simulated copper sulphate electro-refining solutions. Hydrometallurgy, 92, 54–68.
Farrah H.E., Lawrance G.A., Wanless E.J., 2004. Gypsum-anhydrite transformation in hot acidic manganese sulphate solution. A comparative kinetic study employing several analytical methods. Hydrometallurgy, 75, 91–98.
Farrah H.E., Lawrance G.A., Wanless E.J., 2007. Solubility of calcium sulphate salts in acidic manganese sulphate solutions from 30 to 105°C. Hydrometallurgy, 86, 13–21.
Freyer D., Voigt W., 2003. Invited review, crystallization and phase stability of CaSO4 and CaSO4-based salts. Monatshefte für Chemie, 134, 693–719.
Furby E., Glueckauf E., McDonald L.A., 1968. The solubility of calcium sulphate in sodium chloride and sea salt solutions. Desalination, 4, 264–276.
Garvin D., Parker V.B., White H.J., 1987. CODATA thermodynamic tables: selections for some compounds of calcium and related mixtures: a prototype set of tables, Hemisphere Pub. Corp. Washington, USA.
Girich T.E., Buchinskii A.K., 1986. Solubility in the system NiSO4–H2SO4–H2O at 333, 343, 353 K. Russian Journal of Applied Chemistry, 59 (4), 814–816.
Golam Mostafa A.T.M., Eakman J.M., 1995. Prediction of standard heats and Gibbs free energies of formation of solid inorganic salts from group contributions. Ind. Eng. Chem. Res., 34, 4577–4582.
Görgényi M., Dewulf J., Langenhove H.V., Héberger K., 2006. Aqueous salting-out effect of inorganic cations and anions on non-electrolytes. Chemosphere 65, 802–810.
Gromova E.T., 1960. The solubility isotherm of the Na, Ca || Cl, SO4–H2O system at 110ºC. Russian Journal of Inorganic Chemistry, 5 (11), 1244–1247.
Guendouzi M.EL, Mounir A., Dinane A., 2003. Water activity, osmotic and activity coefficients of aqueous solutions of Li2SO4, Na2SO4, K2SO4, (NH4)2SO4, MgSO4, MnSO4, NiSO4, CuSO4 and ZnSO4 at T=298.15 K. J. Chem. Thermodynamics, 35, 209–220.
Guggenheim E.A., Turgeon J.C., 1955. Specific interaction of ions. Trans. Faraday Soc. 51, 747–761.
Gupta R.K., 1968. Solubility of calcium sulphate dihydrate in hydrochloric acid solutions. Journal of Applied Chemistry, 18, 49–51.
Hand R.J., 1997. Calcium sulphate hydrates: a review. British Ceramic Transactions, 96(3), 116–120.

127
Hardie L.A., 1967. The gypsum–anhydrite equilibrium at one atmosphere. The American Mineralogist, 52, 171–200.
Helgeson H.C., 1967. Thermodynamics of complex dissociation in aqueous solution at elevated temperatures. Journal of Physical Chemistry, 71 (10), 3121–3136.
Helgeson H.C., Kirkham D.H., Flowers G.C., 1981. Theoretical prediction of the thermodynamic behavior of aqueous electrolytes. American Journal of Science, 281, 1249–1516.
Hill A.E., Yanick N.S., 1935. Ternary systems. XX. Calcium sulfate, ammonium sulfate and water. Journal of the American Chemical Society, 57, 645–651.
Hill A.E., 1937. The transition temperature of gypsum to anhydrite. Journal of the American Chemical Society, 59, 2242–2244.
Hill A.E., Wills J.H., 1938. Ternary systems. XXIV. calcium sulfate, sodium sulfate and water. Journal of the American Chemical Society, 60, 1647–1655.
Hsu W.L., Lin M.J., Hsu J.P., 2009. Dissolution of solid particles in liquids: a shrinking core model. World Academy f Science, Engineering and Technology, 53, 913–918.
Huang M., 2007. Free acidity estimation using conductivity in process solutions up to 250ºC. Ph.D. Thesis, University of Toronto, Toronto.
Hulett G.A., Allen L.E., 1902. The Solubility of gypsum. Journal of the American Chemical Society, 24, 667–679.
Kerfoot D.G.E., Krause E., Love B.J., Singhal A., 2002. Hydrometallurgical process for the recovery of nickel and cobalt values from a sulphidic flotation concentrate. U.S. Patent 6,428,604 B1, August 6, 2002.
Kessler Y.M., Povarov Y.M., Gorbanev A.I., 1963. The salting-out effect. Zhurnal Struktumoi Khimii, 4 (1), 100–102.
Knacke O., Gans W., 1977. The thermodynamics of the system CaSO4–H2O. Z. Phys. Chem. 104, 41–48.
Kononova G.N., Redzhepov B.A., 1996. Solubility in FeSO4–MgSO4–H2O and Fe2(SO4)3–MgSO4–H2O systems at 25 and 90ºC. Russ. J. Inorg. Chem. 41(7), 1173–1177.
Kontrec J., Kralj D., Brecevic L., 2002. Transformation of anhydrous calcium sulphate into calcium sulphate dihydrate in aqueous solutions. J. Crys. Growth, 240, 203–211.
Kudryashov Y.E., Lebedev A.E., 1989. Solubility in the NiSO4–H2SO4–H2O system at 283, 293, and 313 K. Russian Journal of Applied Chemistry, 62 (3), 649–651.
Kusik C.L., Meissner H.P., 1978. Electrolyte activity coefficients in inorganic processing. AIChE Symposium Series, 74 (173), 14–20.
Lee K., Teong B., Subhash M.J., Abdul R., 2006. Preparation and characterization of CaO /CaSO4/coal fly ash sorbent for sulfur dioxide (SO2) removal: Part I. Energy Sources, 28, 1241–1249.
Li Z., Demopoulos G.P., 2002. Calcium sulphate solubilities in concentrated aqueous chloride solutions. In: Peek E., Van Weert G. (Eds.), Chloride Metallurgy 2002, vol. II, CIM, Montreal, Quebec, Canada, 561–574.

128
Li Z., Demopoulos G.P., 2005. Solubility of CaSO4 phases in aqueous HCl+CaCl2 solutions from 283 K to 353 K. J. Chem. Eng. Data, 50(6), 1971–1982.
Li Z., Demopoulos G.P., 2006a. Effect of NaCl, MgCl2, FeCl2, FeCl3, and AlCl3 on solubility of CaSO4 phases in aqueous HCl or HCl+CaCl2 solutions at 298 to 353 K. J. Chem. Eng. Data, 51(2), 569–576.
Li Z., Demopoulos G.P., 2006b. Development of an improved chemical model for the estimation of CaSO4 solubilities in the HCl–CaCl2–H2O system up to 100ºC. Ind. Eng. Chem. Res. 45(9), 2914–2922.
Li Z., Demopoulos G.P., 2006c. Model-based construction of calcium sulphate phase-transition diagrams in the HCl–CaCl2–H2O system between 0 and 100°C. Ind. Eng. Chem. Res. 45(13), 4517–4524.
Li Z., Demopoulos G.P., 2007. Speciation-based chemical equilibrium model of CaSO4 solubility in the H+Na+Ca+Mg+Al+Fe(II)+Cl+H2O system. Ind. Eng. Chem. Res. 46(20), 6385–6392.
Ling Y., Demopoulos G.P., 2004. Solubility of calcium sulphate hydrates in (0 to 3.5) mol.kg-1 sulphuric acid solutions at 100ºC. J. Chem. Eng. Data, 49(5), 1263–1268.
Linke W.F., Seidell A., 1958. Solubilities, inorganic and metal-organic compounds. McGreger & Warner, American Chemical Society, Washington DC, USA, Vol. I: 1958, Vol. II: 1965.
Liu S.T., Nancollas G.H. 1970. The kinetics of crystal growth of calcium sulphate dihydrate. Journal of Crystal Growth, 6, 281–289.
Liu S.T., Nancollas G.H. 1975. A kinetic and morphological study of the seeded growth of calcium sulphate dihydrate in the presence of additives. Journal of Colloid and Interface Science, 52 (3), 593–601.
Liu H., Papangelakis V.G., Adams J.F., 2005. Chemical modeling in hydrometallurgy using OLI. In: Proceeding of the International Symposium on Computational Analysis in Hydrometallurgy, Dixon D.G., Dry M.J. (Eds.) CIM, COM 2005, Calgary, Alberta, Canada, pp. 275–293.
Liu H., Papangelakis V.G., 2006. Solubility of Pb(II) and Ni(II) in mixed sulphate and chloride solutions with the mixed solvent electrolyte model. Ind. Eng. Chem. Res. 45, 39–47.
Marshall W.L., Gill J.S., Slusher R., 1962. Aqueous systems at high temperature, VI. investigation on the system NiO–SO3–H2O and its D2O analogue from 10–4 to 3 m SO3, 150–450ºC. J. Inorg. Nucl. Chem. 24, 889–897.
Marshall W.L., Slusher R., Jones E.V., 1964. Aqueous systems at high temperature, XIV. solubility and thermodynamic relationships for CaSO4 in NaCl–H2SO4 solutions from 40 to 200ºC, 0 to 4 molal NaCl. Journal of Chemical and Engineering Data, 9 (2), 187–191.
Marshall W.L., Jones E.V., 1966. Second dissociation of sulphuric acid from 25 to 350ºC evaluated from solubilities of calcium sulphate in sulphuric acid solutions. Journal of Physical Chemistry, 70, 4028–4040.
Marshall W.L., Slusher R., 1966. Thermodynamics of calcium sulfate dihydrate in aqueous sodium chloride solutions, 0–110ºC. Journal of Physical Chemistry, 70 (12), 4015–27.

129
Marshall W.L., Slusher R., 1968. Aqueous systems at high temperature, solubility to 200ºC of calcium sulphate and its hydrates in seawater and saline-water concentrates, and temperature-concentration limits. J. Chem. Eng. Data, 13(1), 83–93.
Mnyukh Y.V., 1979. Molecular mechanism of polymorphic transitions. Molecular Crystals and Liquid Crystals, 52, 163–200.
Mutalala B.K., Umetsu Y., Tozawa K., 1988. Solubility of calcium sulphate in acidic zinc sulphate solutions over a temperature range of 25 to 60°C. Tohoku Daigaku Senko Seiren Kenkyusho Iho, 44(1), 57–68.
Nancollas G.H., Reddy M.M., Tsai F., 1973. Calcium sulphate dihydrate crystal growth in aqueous solution at elevated temperatures. Journal of Crystal Growth, 20, 125–134.
Nancollas G.H., 1979. Formation and dissolution of high-temperature forms of calcium sulphate scales: the influence of inhibitors. Society of Petroleum Engineers Journal, 19(6), 423–429.
Nofal P., Allen S., Hosking P., Showell T., 2001. Gypsum control at Bulong: the final hurdle? In: ALTA 2001: Nickel/Cobalt-7 Technical Proceedings, ALTA Metallurgical Services, Melbourne, Australia, pp. 1–17.
Novikova L.V., 1957. A study of solubility for the system CaSO4–MgSO4–H2O at 35ºC by the method of tracer atoms. Russian Journal of Inorganic Chemistry, 2 (3), 300–312.
Nývlt J., 1997. On the kinetics of solid–liquid–solid phase transformation. Crystal Research and Technology, 32 (5), 695–699.
Ostroff A.G., 1964. Conversion of gypsum to anhydrite in aqueous salt solutions. Geochimica et Cosmochimica acta, 28, 1363–1372.
Ostroff A.G., Metler A.V., 1966. Solubility of calcium sulphate dihydrate in the system NaCl–MgCl2–H2O from 28 to 70°C. J. Chem. Eng. Data, 11 (3), 346–350.
Partridge E.P., White A.H., 1929. The solubility of calcium sulfate from 0 to 200ºC. Journal of the American Chemical Society, 51 (2), 360–370.
Petit S., Coquerel G., 1996. Mechanism of several solid–solid transformations between dehydrated and anhydrous copper(II) 8-hydroxyquinolinates. Proposition for a Unified Model for the Dehydration of Molecular Crystals. Chem. Mater. 8, 2247–2258.
Pitzer K.S., 1972. Thermodynamic properties of aqueous solutions of bivalent sulphates. Journal of the Chemical Society, Faraday Transactions 2: Molecular and Chemical Physics, 68, 101–113
Pitzer K.S., 1973. Thermodynamics of electrolytes. I. Theoretical basis and general equations. Journal of Physical chemistry, 77(2), 268–277.
Pitzer K.S., 1980. Electrolytes. From dilute solutions to fused salts. Journal of American Chemical Society, 102 (9), 2902–2906.
Posnjak E., 1938. The system, CaSO4–H2O. American Journal of Science, 35A, 247–272.
Posnjak E., 1940. Deposition of calcium sulphate from seawater. American Journal of Science, 238, 559–568.

130
Power W.H., Fabuss B.M., Satterfield C.N., 1964. Transient solubilities in the calcium sulfate–water system. J. Chem. Eng. Data, 9, 437–442.
Power W.H., Fabuss B.M., Satterfield C.N., 1966. Transient solute concentrations and phase changes of calcium sulphate in aqueous sodium chloride. J. Chem. Eng. Data 1966, 11, 149–154.
Rafal M., Berthold J.W., Scrivner N.C., Grise S.L., 1994. Models for electrolyte solutions, in: Sandler S.I., Ed. Models for thermodynamic and phase equilibria calculations. Marcel Dekker, New York, NY, USA, p. 601–670.
Raju K.U.G., Atkinson G., 1990. The thermodynamics of “scale” mineral solubilities. 3. Calcium sulfate in aqueous NaCl. J. Chem. Eng. Data, 35, 361–367.
Rard J.A., Clegg S.L., 1997. Critical evaluation of the thermodynamic properties of aqueous calcium chloride. 1. Osmotic and activity coefficients of 0–10.77 mol/kg aqueous calcium chloride solutions at 298.15 K and correlation with extended Pitzer ion-interaction models. J. Chem. Eng. Data, 42(5), 819–849.
Rietveld H.M., 1969. A profile refinement method for nuclear and magnetic structures. Journal of Applied Crystallography, 2, 65–71.
Robinson R.A., Stokes R.H., 2002. Electrolyte solutions. 2nd revised edition, Dover Publications Inc., New York, NY.
Santen R.A., 1984. The Ostwald step rule. J. Phys. Chem. 88, 5768–5769.
Sborgi U., Bianchi C., 1940. The solubilities, conductivities and X-ray analyses of anhydrous and semihydrated calcium sulphate. Gazzetta Chimica Italiana, 70, 823–835.
Silcock H.L., 1979. Solubilities of inorganic and organic compounds, ternary and multi-component systems of inorganic substances. Pergamon Press, New York, NY, USA.
Solberg C., Evju C., Emanuelson A., Hansen S., 2002. Crystal structures of cementitious compounds part 3: calcium sulphates. ZKG International, 55 (4), 94–97.
Straub F.G., 1932. Solubility of calcium sulfate and calcium carbonate at temperatures between 182 and 316°C. Industrial and Engineering Chemistry, 24 (8), 914–917.
Supatashvili G., Takaishvili N., Macharadze G., 1997. Solubility of gypsum and anhydrite in water and water solutions of electrolytes. Bulletin of the Georgian Academy of Sciences, 155(1), 68–71.
Tanger IV J.C., Helgeson H.C. 1988. Calculation of the thermodynamic and transport properties of aqueous species at high pressures and temperatures: revised equations of state for the “HKF” standard partial molal properties of ions and electrolytes. The American of Journal of Science, 288, 19–98.
Tanji K.K., Doneen L.D., 1966. Predictions on the solubility of gypsum in aqueous salt solutions. Water Resources Research, 2, 543–548.
Tanji K.K., 1969. Solubility of gypsum in aqueous electrolytes as affected by ion association and ionic strengths up to 0.15M and at 25ºC. Environmental Science and Technology, 3 (7), 656–661.
Templeton C.C., Rodgers J.C., 1967. Solubility of anhydrite in several aqueous salt solutions between 250 and 325°C. Journal of Chemical and Engineering Data, 12 (4), 536–547.

131
Turenne C.V., Mahfouz M., Allaf K., 1999. Three models for determining the induction time in the browning kinetics of the Granny Smith apple under static conditions. Journal of Food Engineering, 41, 133–139.
Umetsu Y., Mutalala B.K., Tozawa K., 1989. Solubility of CaSO4 in solutions of zinc, magnesium, copper, and cobalt sulphates over a temperature range of 25 to 200ºC. Journal of Mining and Metallurgy of Japan, 6, 13–22.
Van’t Hoff J.H., Armstrong E.F., Hinrichsen W., Weigert F., Just G., 1903. Gips und Anhydrit. Z. Phys. Chem. 45, 257–306.
Wang P., Anderko A., Young R.D., 2002. A speciation-based model for mixed-solvent electrolyte systems. Fluid Phase Equilibria, 203, 141–176.
Wang P., Springer R.D. Anderko A., Young R.D., 2004. Modeling phase equilibria and speciation in mixed-solvent electrolyte systems. Fluid Phase Equilibria, 222–223, 11–17.
Wang P., Anderko A., Springer R.D., Young R.D., 2006. Modeling phase equilibria and speciation in mixed-solvent electrolyte systems: II. Liquid–liquid equilibria and properties of associating electrolyte solutions. Journal of Molecular Liquids, 125, 37–44.
Wen C.Y., 1968. Non-catalytic heterogeneous solid fluid reaction models. Ind. Eng. Chem. 60, 34–54.
Whittington B.I., Muir D., 2000. Pressure acid leaching of nickel laterites: a review. Mineral Processing and Extractive Metallurgy Review, 21, 527–600.
Whittington B.I., McDonald R.G., Johnson J.A., Muir D., 2003. Pressure acid leaching of acid-region nickel laterite ore. Part I: effect of water quality. Hydrometallurgy, 70, 31–76.
Williams-Jones A.E., Seward T.M., 1989. The stability of calcium chloride ion pairs in aqueous solutions at temperatures between 100 and 360°C. Geochimica et Cosmochimica Acta, 53, 313–318.
Wollmann G., Voigt W., 2008. Solubility of gypsum in MSO4 solutions (M=Mg, Mn, Co, Ni, Cu, Zn) at 298.15K and 313.15K. J. Chem. Eng. Data, 53 (6), 1375–1380.
Yeatts L.B., Marshall W.L., 1972. Solubility of calcium sulfate dihydrate and association equilibria in several aqueous mixed electrolyte salt systems at 25ºC. J. Chem. Eng. Data, 17 (2), 163–168.
Zatonskaya V.M., Volkova N.A., Krashenina S.V., Samoilenko A.I., Burtseva N.F., 1988. Solubility of gypsum in zinc sulfate solutions. Tsvetnye Metally/Non-Ferrous Metals, 29 (2), 25–26.
Zdanovskii A.B., Vlasov G.A., 1968. Determination of the boundaries of the reciprocal transformation of CaSO4.2H2O and γ-CaSO4 in H2SO4 solutions. Russian journal of Inorganic Chemistry, 13 (9), 1318–1319.
Zdanovskii A.B., Vlasov G.A., Sotnikova L.I., 1968. Dehydration of gypsum in sulphuric acid solutions. Russian journal of Inorganic Chemistry, 13, 1418–1420.
Zdanovskii A.B., Chernova Z.S., 1976. Calculation of solubility of calcium sulphate in the CaCl2–CaSO4–MgCl2–H2O System at 25ºC. Russian journal of Inorganic Chemistry, 21(9), 1419–1420.

132
Zemaitis J.F., 1980. Predicting vapour-liquid equilibria in multicomponent aqueous solutions of electrolytes. In: Thermodynamics of Aqueous Systems with Industrial Applications; Newman S.A., Barner H.E., Klein M., Sandler S.I. (Eds.), ACS Symposium Series 133; American Chemical Society: Washington, DC, USA, pp: 227–246.
Zemaitis J.F., Clark D.M., Rafal M., Scrivner N.C., 1986. Handbook of aqueous electrolyte thermodynamics theory & application. AICHE: New York.
Zhelnin B.I., Gorshtein G.I., Bezprozvannaya L.Kh., 1973. Solubility of calcium sulphate dihydrate in manganese sulphate solutions. Journal of Applied Chemistry of the USSR, 46 (3), 534–537.
Zupzncic V., Ograjsek N., Kotar-Jordan B., Vrecer F., 2005. Physical characterization of pantoprazole sodium hydrates. International Journal of Pharmaceutics, 291, 59–68.
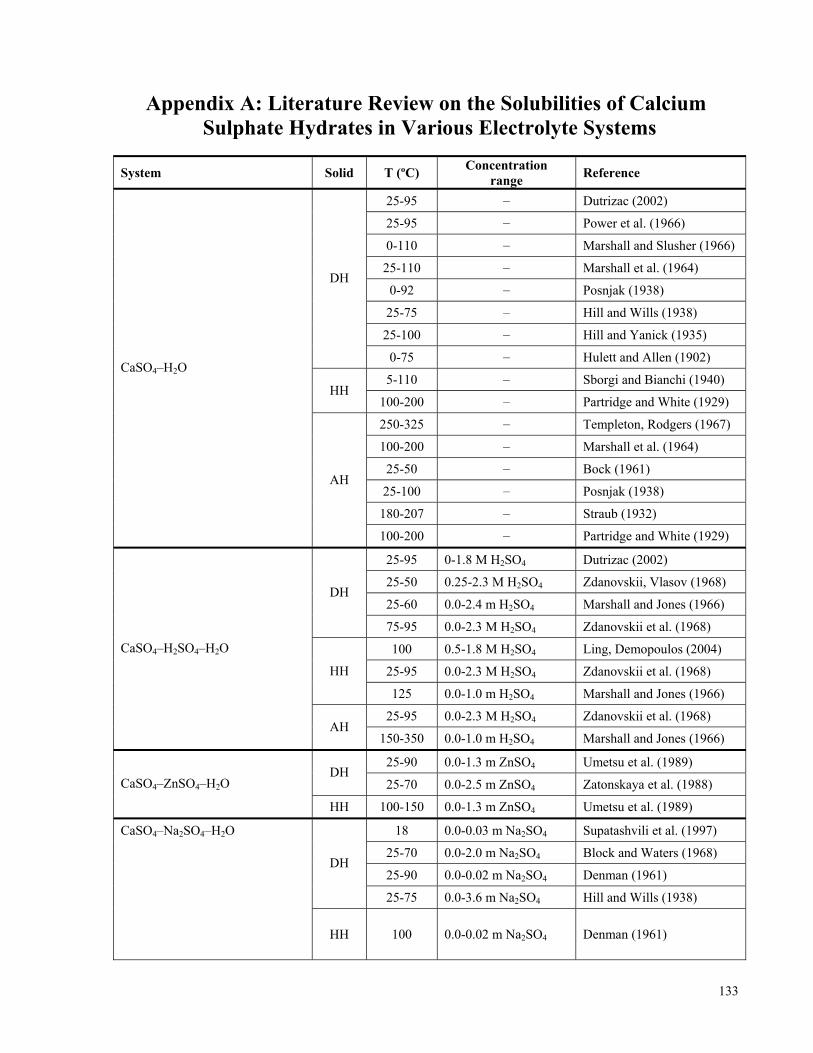
133
Appendix A: Literature Review on the Solubilities of Calcium Sulphate Hydrates in Various Electrolyte Systems
System Solid T (ºC) Concentration range Reference
25-95 – Dutrizac (2002)
25-95 – Power et al. (1966)
0-110 – Marshall and Slusher (1966)
25-110 – Marshall et al. (1964)
0-92 – Posnjak (1938)
25-75 – Hill and Wills (1938)
25-100 – Hill and Yanick (1935)
DH
0-75 – Hulett and Allen (1902)
5-110 – Sborgi and Bianchi (1940) HH
100-200 – Partridge and White (1929)
250-325 – Templeton, Rodgers (1967)
100-200 – Marshall et al. (1964)
25-50 – Bock (1961)
25-100 – Posnjak (1938)
180-207 – Straub (1932)
CaSO4–H2O
AH
100-200 – Partridge and White (1929)
25-95 0-1.8 M H2SO4 Dutrizac (2002)
25-50 0.25-2.3 M H2SO4 Zdanovskii, Vlasov (1968)
25-60 0.0-2.4 m H2SO4 Marshall and Jones (1966) DH
75-95 0.0-2.3 M H2SO4 Zdanovskii et al. (1968)
100 0.5-1.8 M H2SO4 Ling, Demopoulos (2004)
25-95 0.0-2.3 M H2SO4 Zdanovskii et al. (1968) HH
125 0.0-1.0 m H2SO4 Marshall and Jones (1966)
25-95 0.0-2.3 M H2SO4 Zdanovskii et al. (1968)
CaSO4–H2SO4–H2O
AH 150-350 0.0-1.0 m H2SO4 Marshall and Jones (1966)
25-90 0.0-1.3 m ZnSO4 Umetsu et al. (1989) DH
25-70 0.0-2.5 m ZnSO4 Zatonskaya et al. (1988) CaSO4–ZnSO4–H2O
HH 100-150 0.0-1.3 m ZnSO4 Umetsu et al. (1989)
18 0.0-0.03 m Na2SO4 Supatashvili et al. (1997)
25-70 0.0-2.0 m Na2SO4 Block and Waters (1968)
25-90 0.0-0.02 m Na2SO4 Denman (1961) DH
25-75 0.0-3.6 m Na2SO4 Hill and Wills (1938)
CaSO4–Na2SO4–H2O
HH 100 0.0-0.02 m Na2SO4 Denman (1961)
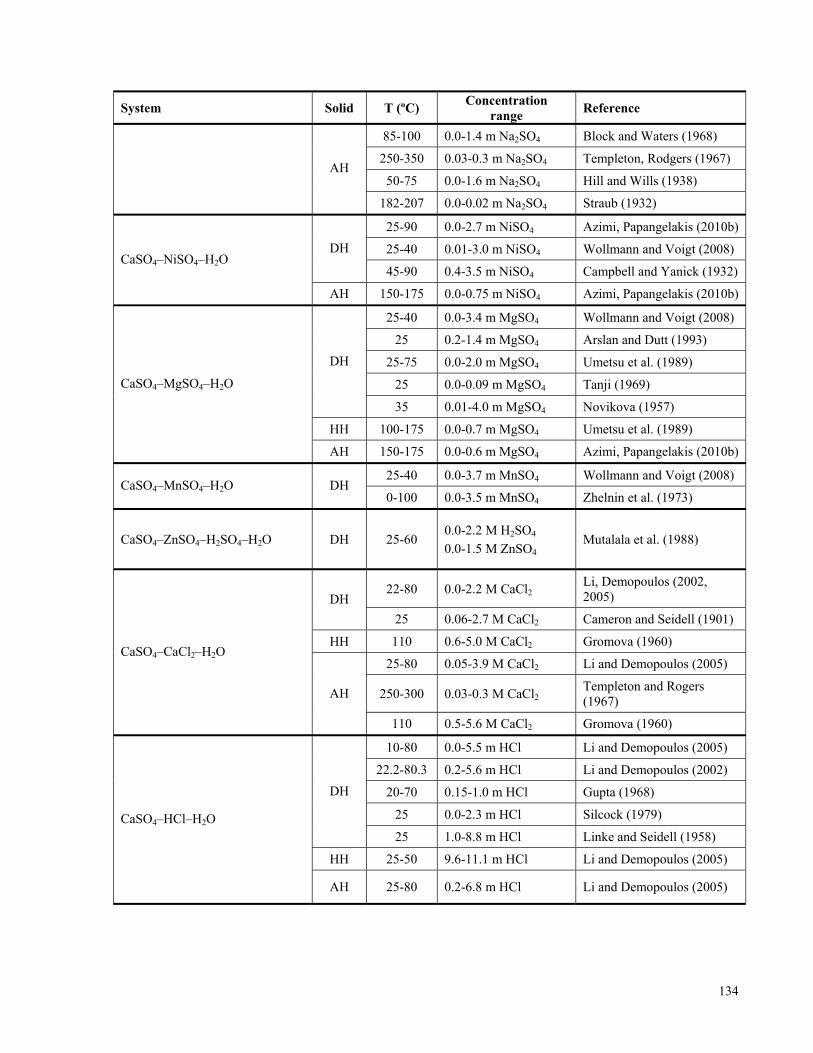
134
System Solid T (ºC) Concentration range Reference
85-100 0.0-1.4 m Na2SO4 Block and Waters (1968)
250-350 0.03-0.3 m Na2SO4 Templeton, Rodgers (1967)
50-75 0.0-1.6 m Na2SO4 Hill and Wills (1938) AH
182-207 0.0-0.02 m Na2SO4 Straub (1932)
25-90 0.0-2.7 m NiSO4 Azimi, Papangelakis (2010b)
25-40 0.01-3.0 m NiSO4 Wollmann and Voigt (2008) DH
45-90 0.4-3.5 m NiSO4 Campbell and Yanick (1932) CaSO4–NiSO4–H2O
AH 150-175 0.0-0.75 m NiSO4 Azimi, Papangelakis (2010b)
25-40 0.0-3.4 m MgSO4 Wollmann and Voigt (2008)
25 0.2-1.4 m MgSO4 Arslan and Dutt (1993)
25-75 0.0-2.0 m MgSO4 Umetsu et al. (1989)
25 0.0-0.09 m MgSO4 Tanji (1969)
DH
35 0.01-4.0 m MgSO4 Novikova (1957)
HH 100-175 0.0-0.7 m MgSO4 Umetsu et al. (1989)
CaSO4–MgSO4–H2O
AH 150-175 0.0-0.6 m MgSO4 Azimi, Papangelakis (2010b)
25-40 0.0-3.7 m MnSO4 Wollmann and Voigt (2008) CaSO4–MnSO4–H2O DH
0-100 0.0-3.5 m MnSO4 Zhelnin et al. (1973)
CaSO4–ZnSO4–H2SO4–H2O DH 25-60 0.0-2.2 M H2SO4
0.0-1.5 M ZnSO4 Mutalala et al. (1988)
22-80 0.0-2.2 M CaCl2 Li, Demopoulos (2002, 2005) DH
25 0.06-2.7 M CaCl2 Cameron and Seidell (1901)
HH 110 0.6-5.0 M CaCl2 Gromova (1960)
25-80 0.05-3.9 M CaCl2 Li and Demopoulos (2005)
250-300 0.03-0.3 M CaCl2 Templeton and Rogers (1967)
CaSO4–CaCl2–H2O
AH
110 0.5-5.6 M CaCl2 Gromova (1960)
10-80 0.0-5.5 m HCl Li and Demopoulos (2005)
22.2-80.3 0.2-5.6 m HCl Li and Demopoulos (2002)
20-70 0.15-1.0 m HCl Gupta (1968)
25 0.0-2.3 m HCl Silcock (1979)
DH
25 1.0-8.8 m HCl Linke and Seidell (1958)
HH 25-50 9.6-11.1 m HCl Li and Demopoulos (2005)
CaSO4–HCl–H2O
AH 25-80 0.2-6.8 m HCl Li and Demopoulos (2005)

135
System Solid T (ºC) Concentration range Reference
10-110 0.0-4.7 m NaCl Marshall and Slusher (1966)
28-90 0.0-5.5 m NaCl Ostroff and Metler (1966)
25-100 0.0-4.0 m NaCl Marshall et al. (1964) DH
15-50 0.0-4.4 m NaCl Silcock (1979)
HH 125 0.0-4.0 m NaCl Marshall et al. (1964)
100-300 0.0-6.0 m NaCl Blount and Dickson (1969)
250-325 0.0-5.7 m NaCl Templeton and Rogers (1967)
125-200 0.0-4.0 m NaCl Marshall et al. (1964)
25-50 0.0-5.0 m NaCl Bock (1961)
CaSO4–NaCl–H2O
AH
125-200 0.9-5.5 m NaCl Silcock (1979)
25 0.0-5.3 m MgCl2 Zdanovskii, Chernova (1976)
38-70 0.0-0.35 m MgCl2 Ostroff and Metler (1966)
25 0.0-5.0 m MgCl2 Linke and Seidell (1958) DH
25 0.0-5.3 m MgCl2 Cameron and Seidell (1901)
CaSO4–MgCl2–H2O
AH 250-300 0.03-0.3 m MgCl2 Templeton, Rogers (1967)
CaSO4–AlCl3–H2O DH 25-80 0.0-1.5 m AlCl3 Li and Demopoulos (2006a)
CaSO4–FeCl3–HCl–H2O DH 25-80 0.0-2.2 m FeCl3
0.5, 3.0 m HCl Li and Demopoulos (2006a)
22-80 0.0-2.2 m CaCl2
1.0, 3.0, 5.0 m HCl Li and Demopoulos (2005)
22.2-80.3 0.01-2.1 m CaCl2
1.0 m HCl Li and Demopoulos (2002) DH
20 0.0-3.0 m CaCl2
0.0-6.0 m HCl Silcock (1979)
HH 60-80 0.05-4.7 m CaCl2
3.0, 6.0 m HCl Li and Demopoulos (2005)
CaSO4–CaCl2–HCl–H2O
AH 25-80 0.05-4.3 m CaCl2
3.0, 6.0 m HCl Li and Demopoulos (2005)

136
System Solid T (ºC) Concentration range Reference
25-80 0.7-3.3 m MgCl2
0.5 m HCl Li and Demopoulos (2006a)
50-80 0.0-2.2 m MgCl2
0.5 m HCl Li and Demopoulos (2002) DH
25-50 0.3-3.5 m MgCl2
3.0 m HCl Li and Demopoulos (2006a)
CaSO4–MgCl2–HCl–H2O
AH 80 0.3-3.5 m MgCl2
3.0 m HCl Li and Demopoulos (2006a)
CaSO4–NaCl–HCl–H2O DH 50-80 0.5-3.0 m NaCl
0.5 m HCl Li and Demopoulos (2006a)
DH 50 0.2-1.5 m CaCl2
1.0 m MgCl2
0.5 m HCl Li and Demopoulos (2006a)
CaSO4–CaCl2–MgCl2–HCl–H2O
HH 60 0.3-1.9 m CaCl2
1.0 m MgCl2
6.0 m HCl Li and Demopoulos (2006a)
25 0.0-5.9 m Na2SO4
0.0-1.9 m NaCl Yeatts and Marshall (1972)
25-70 0.0-2.0 m Na2SO4
0.0-4.0 m NaCl Block and Waters (1968) DH
25 0.4-1.8 m Na2SO4
0.5-5.6 m NaCl Cameron et al. (1907)
85-100 0.0-1.1 m Na2SO4
0.5-2.0 m NaCl Block and Waters (1968)
100 0.01-0.03 m Na2SO4
2.0 m NaCl Furby et al. (1968)
CaSO4–Na2SO4–NaCl–H2O
AH
250-300 0.0-0.3 m Na2SO4
0.0-0.9 m NaCl Templeton, Rodgers (1967)
CaSO4–Na2SO4–MgCl2–H2O DH 40 0.0-2.0 m Na2SO4
0.0-0.6 m MgCl2 Barba et al. (1984)
DH 28-250 0.0-5.5 m NaCl
0.0-4.4 m MgCl2 Ostroff and Metler (1966)
CaSO4–NaCl–MgCl2–H2O AH 250-300
0.0-0.5 m NaCl
0.0-0.16 m MgCl2 Templeton, Rodgers (1967)
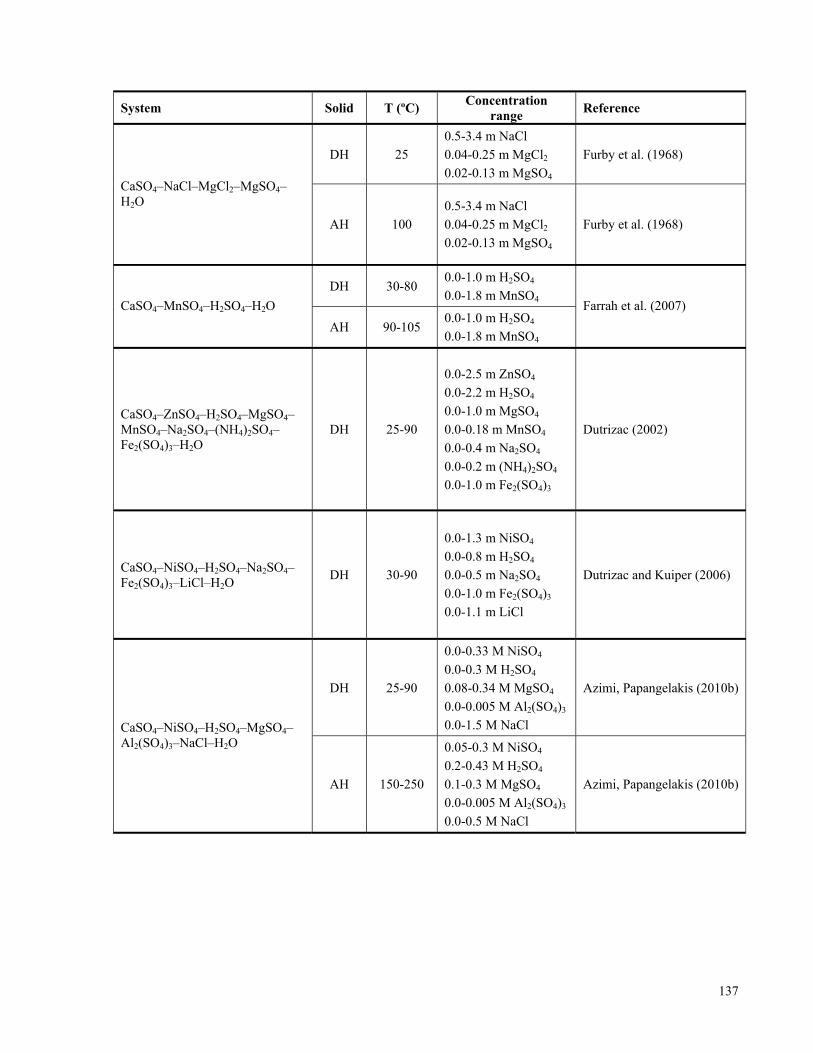
137
System Solid T (ºC) Concentration range Reference
DH 25 0.5-3.4 m NaCl
0.04-0.25 m MgCl2
0.02-0.13 m MgSO4
Furby et al. (1968)
CaSO4–NaCl–MgCl2–MgSO4–H2O
AH 100 0.5-3.4 m NaCl
0.04-0.25 m MgCl2
0.02-0.13 m MgSO4 Furby et al. (1968)
DH 30-80 0.0-1.0 m H2SO4
0.0-1.8 m MnSO4 CaSO4–MnSO4–H2SO4–H2O AH 90-105
0.0-1.0 m H2SO4
0.0-1.8 m MnSO4
Farrah et al. (2007)
CaSO4–ZnSO4–H2SO4–MgSO4–MnSO4–Na2SO4–(NH4)2SO4–Fe2(SO4)3–H2O
DH 25-90
0.0-2.5 m ZnSO4
0.0-2.2 m H2SO4
0.0-1.0 m MgSO4
0.0-0.18 m MnSO4
0.0-0.4 m Na2SO4
0.0-0.2 m (NH4)2SO4
0.0-1.0 m Fe2(SO4)3
Dutrizac (2002)
CaSO4–NiSO4–H2SO4–Na2SO4–Fe2(SO4)3–LiCl–H2O DH 30-90
0.0-1.3 m NiSO4
0.0-0.8 m H2SO4
0.0-0.5 m Na2SO4
0.0-1.0 m Fe2(SO4)3
0.0-1.1 m LiCl
Dutrizac and Kuiper (2006)
DH 25-90
0.0-0.33 M NiSO4
0.0-0.3 M H2SO4
0.08-0.34 M MgSO4
0.0-0.005 M Al2(SO4)3
0.0-1.5 M NaCl
Azimi, Papangelakis (2010b)
CaSO4–NiSO4–H2SO4–MgSO4–Al2(SO4)3–NaCl–H2O
AH 150-250
0.05-0.3 M NiSO4
0.2-0.43 M H2SO4
0.1-0.3 M MgSO4
0.0-0.005 M Al2(SO4)3
0.0-0.5 M NaCl
Azimi, Papangelakis (2010b)
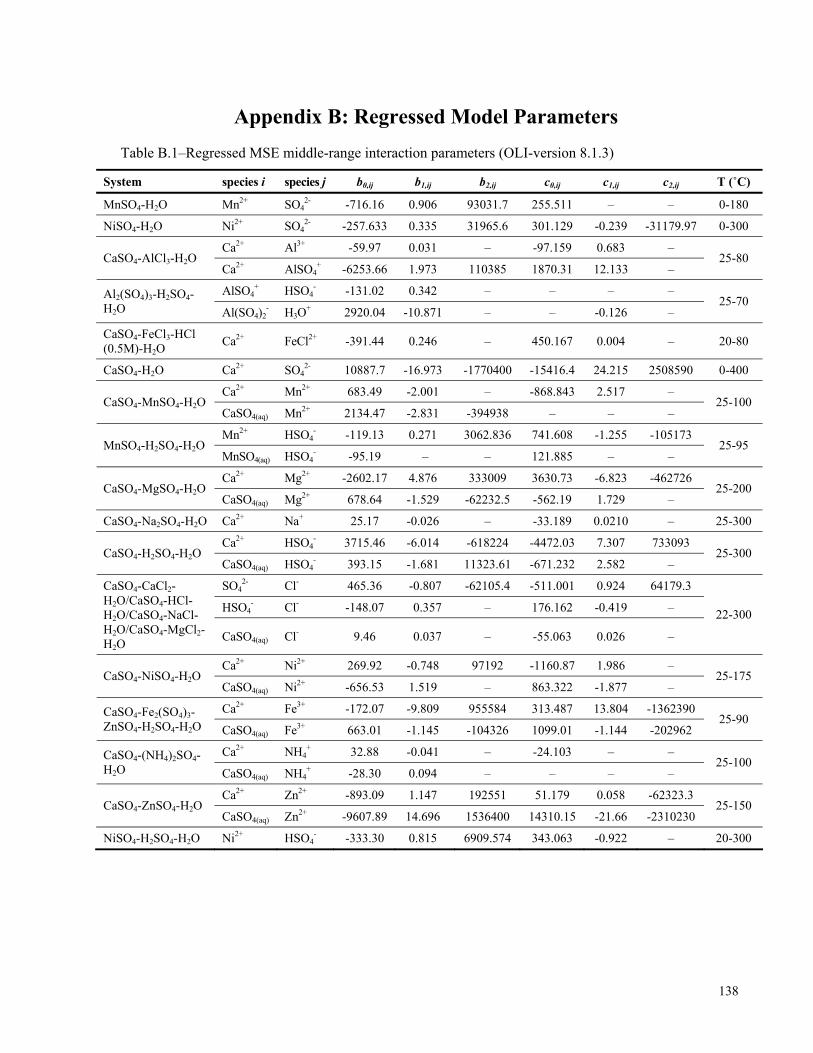
138
Appendix B: Regressed Model Parameters Table B.1–Regressed MSE middle-range interaction parameters (OLI-version 8.1.3)
System species i species j b0,ij b1,ij b2,ij c0,ij c1,ij c2,ij T (˚C)
MnSO4-H2O Mn2+ SO42- -716.16 0.906 93031.7 255.511 – – 0-180
NiSO4-H2O Ni2+ SO42- -257.633 0.335 31965.6 301.129 -0.239 -31179.97 0-300
Ca2+ Al3+ -59.97 0.031 – -97.159 0.683 – CaSO4-AlCl3-H2O
Ca2+ AlSO4+ -6253.66 1.973 110385 1870.31 12.133 –
25-80
AlSO4+ HSO4
- -131.02 0.342 – – – – Al2(SO4)3-H2SO4-H2O Al(SO4)2
- H3O+ 2920.04 -10.871 – – -0.126 – 25-70
CaSO4-FeCl3-HCl (0.5M)-H2O Ca2+ FeCl2+ -391.44 0.246 – 450.167 0.004 – 20-80
CaSO4-H2O Ca2+ SO42- 10887.7 -16.973 -1770400 -15416.4 24.215 2508590 0-400
Ca2+ Mn2+ 683.49 -2.001 – -868.843 2.517 – CaSO4-MnSO4-H2O
CaSO4(aq) Mn2+ 2134.47 -2.831 -394938 – – – 25-100
Mn2+ HSO4- -119.13 0.271 3062.836 741.608 -1.255 -105173
MnSO4-H2SO4-H2O MnSO4(aq) HSO4
- -95.19 – – 121.885 – – 25-95
Ca2+ Mg2+ -2602.17 4.876 333009 3630.73 -6.823 -462726 CaSO4-MgSO4-H2O
CaSO4(aq) Mg2+ 678.64 -1.529 -62232.5 -562.19 1.729 – 25-200
CaSO4-Na2SO4-H2O Ca2+ Na+ 25.17 -0.026 – -33.189 0.0210 – 25-300
Ca2+ HSO4- 3715.46 -6.014 -618224 -4472.03 7.307 733093
CaSO4-H2SO4-H2O CaSO4(aq) HSO4
- 393.15 -1.681 11323.61 -671.232 2.582 – 25-300
SO42- Cl- 465.36 -0.807 -62105.4 -511.001 0.924 64179.3
HSO4- Cl- -148.07 0.357 – 176.162 -0.419 –
CaSO4-CaCl2-H2O/CaSO4-HCl-H2O/CaSO4-NaCl-H2O/CaSO4-MgCl2-H2O CaSO4(aq) Cl- 9.46 0.037 – -55.063 0.026 –
22-300
Ca2+ Ni2+ 269.92 -0.748 97192 -1160.87 1.986 – CaSO4-NiSO4-H2O
CaSO4(aq) Ni2+ -656.53 1.519 – 863.322 -1.877 – 25-175
Ca2+ Fe3+ -172.07 -9.809 955584 313.487 13.804 -1362390 CaSO4-Fe2(SO4)3-ZnSO4-H2SO4-H2O CaSO4(aq) Fe3+ 663.01 -1.145 -104326 1099.01 -1.144 -202962
25-90
Ca2+ NH4+ 32.88 -0.041 – -24.103 – – CaSO4-(NH4)2SO4-
H2O CaSO4(aq) NH4+ -28.30 0.094 – – – –
25-100
Ca2+ Zn2+ -893.09 1.147 192551 51.179 0.058 -62323.3 CaSO4-ZnSO4-H2O
CaSO4(aq) Zn2+ -9607.89 14.696 1536400 14310.15 -21.66 -2310230 25-150
NiSO4-H2SO4-H2O Ni2+ HSO4- -333.30 0.815 6909.574 343.063 -0.922 – 20-300

139
Table B.2–Regressed standard state Gibbs free energy and entropy of formation of various solids in comparison with the literature data
From literature Regressed values
Lange's Handbook / Golam Mostafa (1995) error%*
Solids ΔGf
º Sfº ΔGf
º Sfº ΔGf
º Sfº
T (°C)
CaSO4•0.5H2O -343903 31.479 -343732 / -322875 31.22 / 31.34 0.05/6.5 0.83/0.43 0–200 NiSO4•7H2O -588592 95.807 -589043 / -599260 90.66 / 94.18 0.08/1.78 5.68/1.72 0–32 NiSO4•6H2O -531852 80.678 -531000 / -540811 75.10 / 83.67 0.16/1.65 7.43/3.57 32–100 NiSO4•1H2O -244864 30.956 – / -248566 – / 31.11 – /1.49 – /0.49 100–220 MnSO4•7H2O -632452 114.66 – / -639346 – / 111.8 – /1.08 – /2.52 0–10 MnSO4•5H2O -518996 81.760 – / -522448 – / 81.02 – /0.66 – /0.91 10–25 MnSO4•1H2O -291986 22.136 – / -288652 – / 26.72 – /1.15 – /17.0 25–180
*100% ×
−=
valueliterature
valueliteraturevalueregressederror

140
Appendix C: Experimental Measurements in Laterite PAL Solutions
Table C.1 – Solubility of CaSO4 dihydrate (gypsum) in water at various NiSO4 concentrations
25°C 45°C 70°C 90°C NiSO4 CaSO4 density* CaSO4 density* CaSO4 density* CaSO4 density* mol/L mol/L g/mL mol/L g/mL mol/L g/mL mol/L g/mL
0.0 0.0154 0.998 0.0153 0.990 0.0146 0.974 0.0136 0.970 0.1 0.0110 1.014 0.0117 1.009 0.0117 1.001 0.0117 0.983 0.5 0.0128 1.075 0.0140 1.070 0.0160 1.060 0.0163 1.045 1.1 0.0145 1.148 0.0166 1.138 0.0183 1.130 0.0202 1.115 1.6 0.0143 1.216 0.0161 1.203 0.0188 1.192 0.0214 1.177 2.1 - - 0.0147 1.270 0.0184 1.253 0.0218 1.236 2.7 - - 0.0121 1.354 0.0154 1.321 0.0182 1.300
*density at temperature
Table C.2 – Solubility of CaSO4 anhydrite in water at various NiSO4 concentrations
150°C 175°C NiSO4 CaSO4 density* CaSO4 density* mol/L mol/L g/ml mol/L g/ml
0.0 0.0017 0.997 0.0010 0.997 0.1 0.0014 1.013 0.0009 1.013 0.3 0.0021 1.041 0.0012 1.039 0.6 0.0029 1.085 0.0019 1.084 0.8 0.0036 1.130 0.0026 1.127
*density at 25°C
Table C.3 – Solubility of CaSO4 anhydrite in water at various MgSO4 concentrations
150°C 175°C MgSO4 CaSO4 density* CaSO4 density* mol/L mol/L g/mL mol/L g/mL
0.00 0.0017 0.997 0.0010 0.997 0.10 0.0014 1.010 0.0008 1.010 0.20 0.0018 1.022 0.0010 1.022 0.40 0.0023 1.045 0.0014 1.045 0.60 0.0029 1.065 0.0020 1.065
*density at 25°C
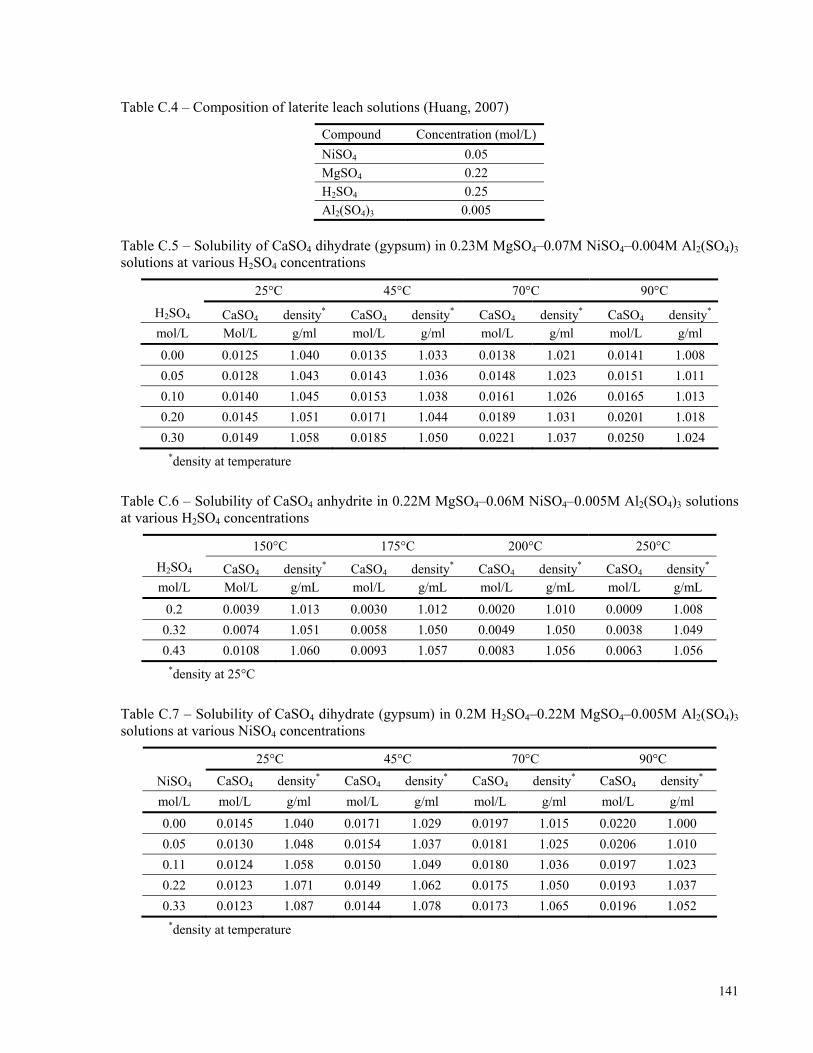
141
Table C.4 – Composition of laterite leach solutions (Huang, 2007)
Compound Concentration (mol/L) NiSO4 0.05 MgSO4 0.22 H2SO4 0.25 Al2(SO4)3 0.005
Table C.5 – Solubility of CaSO4 dihydrate (gypsum) in 0.23M MgSO4–0.07M NiSO4–0.004M Al2(SO4)3 solutions at various H2SO4 concentrations
25°C 45°C 70°C 90°C
H2SO4 CaSO4 density* CaSO4 density* CaSO4 density* CaSO4 density*
mol/L Mol/L g/ml mol/L g/ml mol/L g/ml mol/L g/ml
0.00 0.0125 1.040 0.0135 1.033 0.0138 1.021 0.0141 1.008 0.05 0.0128 1.043 0.0143 1.036 0.0148 1.023 0.0151 1.011 0.10 0.0140 1.045 0.0153 1.038 0.0161 1.026 0.0165 1.013 0.20 0.0145 1.051 0.0171 1.044 0.0189 1.031 0.0201 1.018 0.30 0.0149 1.058 0.0185 1.050 0.0221 1.037 0.0250 1.024
*density at temperature
Table C.6 – Solubility of CaSO4 anhydrite in 0.22M MgSO4–0.06M NiSO4–0.005M Al2(SO4)3 solutions at various H2SO4 concentrations
150°C 175°C 200°C 250°C H2SO4 CaSO4 density* CaSO4 density* CaSO4 density* CaSO4 density* mol/L Mol/L g/mL mol/L g/mL mol/L g/mL mol/L g/mL
0.2 0.0039 1.013 0.0030 1.012 0.0020 1.010 0.0009 1.008 0.32 0.0074 1.051 0.0058 1.050 0.0049 1.050 0.0038 1.049 0.43 0.0108 1.060 0.0093 1.057 0.0083 1.056 0.0063 1.056
*density at 25°C
Table C.7 – Solubility of CaSO4 dihydrate (gypsum) in 0.2M H2SO4–0.22M MgSO4–0.005M Al2(SO4)3 solutions at various NiSO4 concentrations
25°C 45°C 70°C 90°C
NiSO4 CaSO4 density* CaSO4 density* CaSO4 density* CaSO4 density* mol/L mol/L g/ml mol/L g/ml mol/L g/ml mol/L g/ml
0.00 0.0145 1.040 0.0171 1.029 0.0197 1.015 0.0220 1.000 0.05 0.0130 1.048 0.0154 1.037 0.0181 1.025 0.0206 1.010 0.11 0.0124 1.058 0.0150 1.049 0.0180 1.036 0.0197 1.023 0.22 0.0123 1.071 0.0149 1.062 0.0175 1.050 0.0193 1.037 0.33 0.0123 1.087 0.0144 1.078 0.0173 1.065 0.0196 1.052
*density at temperature

142
Table C.8 – Solubility of CaSO4 anhydrite in 0.3M H2SO4–0.22M MgSO4–0.005M Al2(SO4)3 solutions at various NiSO4 concentrations
150°C 175°C 200°C
NiSO4 CaSO4 density* CaSO4 density* CaSO4 density* mol/L mol/L g/ml mol/L g/ml mol/L g/ml
0.05 0.0072 1.049 0.0058 1.048 0.0048 1.048 0.10 0.0065 1.055 0.0052 1.055 0.0043 1.054 0.20 0.0056 1.075 0.0042 1.071 0.0032 1.071 0.30 0.0057 1.087 0.0041 1.086 0.0030 1.084 *density at 25°C
Table C.9 – Solubility of CaSO4 dihydrate (gypsum) in 0.2M H2SO4–0.05M NiSO4–0.005M Al2(SO4)3 solutions at various MgSO4 concentrations
25°C 45°C 70°C 90°C
MgSO4 CaSO4 density* CaSO4 density* CaSO4 density* CaSO4 density* mol/L Mol/L g/mL mol/L g/mL mol/L g/mL mol/L g/mL
0.08 0.0125 1.033 0.0149 1.022 0.0187 1.010 0.0226 1.000 0.16 0.0120 1.043 0.0144 1.033 0.0171 1.020 0.0190 1.006 0.25 0.0121 1.055 0.0140 1.043 0.0157 1.028 0.0175 1.015 0.34 0.0121 1.065 0.0136 1.054 0.0154 1.040 0.0169 1.025
*density at temperature
Table C.10 – Solubility of CaSO4 anhydrite in 0.3M H2SO4–0.06M NiSO4–0.005M Al2(SO4)3 solutions at various MgSO4 concentrations
150°C 175°C 200°C 250°C
MgSO4 CaSO4 density* CaSO4 density* CaSO4 density* CaSO4 density* mol/L Mol/L mg/L mol/L mg/L mol/L mg/L mol/L mg/L
0.1 0.0106 1.038 0.0093 1.038 0.0079 1.038 0.0059 1.039 0.2 0.0075 1.048 0.0059 1.048 0.0047 1.047 0.0030 1.050 0.3 0.0053 1.054 0.0042 1.055 0.0030 1.056 0.0020 1.058 *density at 25°C

143
Table C.11 – Solubility of CaSO4 anhydrite in 0.25M H2SO4–0.2M MgSO4–0.005M Al2(SO4)3–0.05M NiSO4 solutions at 0.0 and 0.5M NaCl concentrations
T H2SO4 MgSO4 Al2(SO4)3 NiSO4 NaCl CaSO4 density* °C mol/L mol/L mol/L mol/L mol/L mol/L g/mL
150 0.22 0.2 0.005 0.06 0 0.0039 1.013 175 0.22 0.2 0.005 0.06 0 0.0030 1.012 200 0.22 0.2 0.005 0.06 0 0.0020 1.010 250 0.22 0.2 0.005 0.06 0 0.0009 1.008 150 0.25 0.2 0.004 0.05 0.5 0.0066 1.065 175 0.25 0.2 0.004 0.05 0.5 0.0055 1.064 200 0.25 0.2 0.004 0.05 0.5 0.0042 1.063 250 0.25 0.2 0.004 0.05 0.5 0.0028 1.063 *density at 25°C
Table C.12 – Solubility of CaSO4 dihydrate (gypsum) in 0.5M H2SO4 solutions at various NaCl concentrations
25°C 45°C 70°C 90°C NaCl CaSO4 density* CaSO4 density* CaSO4 density* CaSO4 density* mol/L mol/L g/mL mol/L g/mL mol/L g/mL mol/L g/mL
0.0 0.0193 1.030 0.0260 1.023 0.0403 1.015 0.0545 1.000 0.1 0.0200 1.034 0.0281 1.027 0.0409 1.016 0.0541 1.007 0.5 0.0235 1.050 0.0321 1.042 0.0444 1.033 0.0620 1.024 1.0 0.0248 1.068 0.0338 1.062 0.0506 1.056 0.0667 1.043 1.5 0.0263 1.090 0.0359 1.078 0.0537 1.071 0.0719 1.060
*density at temperature

144
Table C.13 – Solubility of CaSO4 dihydrate in 0.5M HCl solutions at various MgSO4 concentrations
25°C 45°C 70°C 90°C MgSO4 CaSO4 density* CaSO4 density* CaSO4 Density CaSO4 density* mol/L mol/L g/mL mol/L g/mL mol/L g/mL mol/L g/mL
0.0 0.0810 1.007 0.0940 1.005 0.1300 1.004 0.1560 0.997 0.1 0.0456 1.021 0.0594 1.013 0.0785 1.008 0.0987 0.999 0.5 0.0168 1.057 0.0205 1.047 0.0231 1.038 0.0292 1.030 1.0 0.0110 1.112 0.0139 1.100 0.0153 1.105 0.0188 1.080 1.5 0.0087 1.162 0.0105 1.158 0.0113 1.102 0.0135 1.128
*density at temperature
Table C.14 – Solubility of CaSO4 dihydrate in 0.5M H2SO4 solutions at various NiSO4 concentrations
25°C 45°C 70°C 90°C NiSO4 CaSO4 density* CaSO4 density* CaSO4 density* CaSO4 density* mol/L mol/L g/mL mol/L g/mL mol/L g/mL mol/L g/mL
0.00 0.0190 1.030 0.0258 1.022 0.0403 1.010 0.0545 1.002 0.10 0.0180 1.044 0.0234 1.037 0.0351 1.026 0.0454 1.015 0.25 0.0166 1.060 0.0210 1.056 0.0298 1.044 0.0383 1.029 0.50 0.0166 1.106 0.0196 1.095 0.0266 1.085 0.0329 1.071 1.00 0.0148 1.180 0.0172 1.170 0.0234 1.157 0.0314 1.141 1.50 0.0131 1.238 0.0158 1.230 0.0194 1.218 0.0245 1.202
*density at temperature

145
Appendix D: X-ray Diffraction Patterns
Figure D.1 X-ray diffraction pattern of the gypsum feed.
Figure D.2 X-ray diffraction pattern of the anhydrite feed (AII).

146
Figure D.3 X-ray diffraction pattern of hemihydrate*.
Figure D.4 X-ray diffraction pattern of soluble (AIII or γ) anhydrite (obtained from the ICDD, Bushuev et al., 1983). * Note: this XRD pattern is collected from a solid sample obtained in this work from gypsum dehydration
in pure water at 150°C inside an autoclave after 3 h. No hemihydrate was detected in the experiments carried out below 100°C.

147
XRD Patterns of the Solids in H2SO4 media at 70°C after 3 h
Figure D.5 X-ray diffraction pattern of the equilibrating solid phase in H2SO4 media at 70°C (retention time=3 h): (a) 2θ = 10–55° (b) 2θ = 37–44°. SEM image of this solid is presented in Fig. 5.14 (b).
no AII-AH characteristic peak
b
a

148
XRD Patterns of the Solids in H2SO4 media at 70°C after 12 days
Figure D.6 X-ray diffraction pattern of the equilibrating solid phase in H2SO4 media at 70°C (retention time=12 days): (a) 2θ = 10–60° (b) 2θ = 31–45°. SEM image of this solid is presented in Fig. 5.14 (c).
Characteristic Peak of AII-AH
a
b
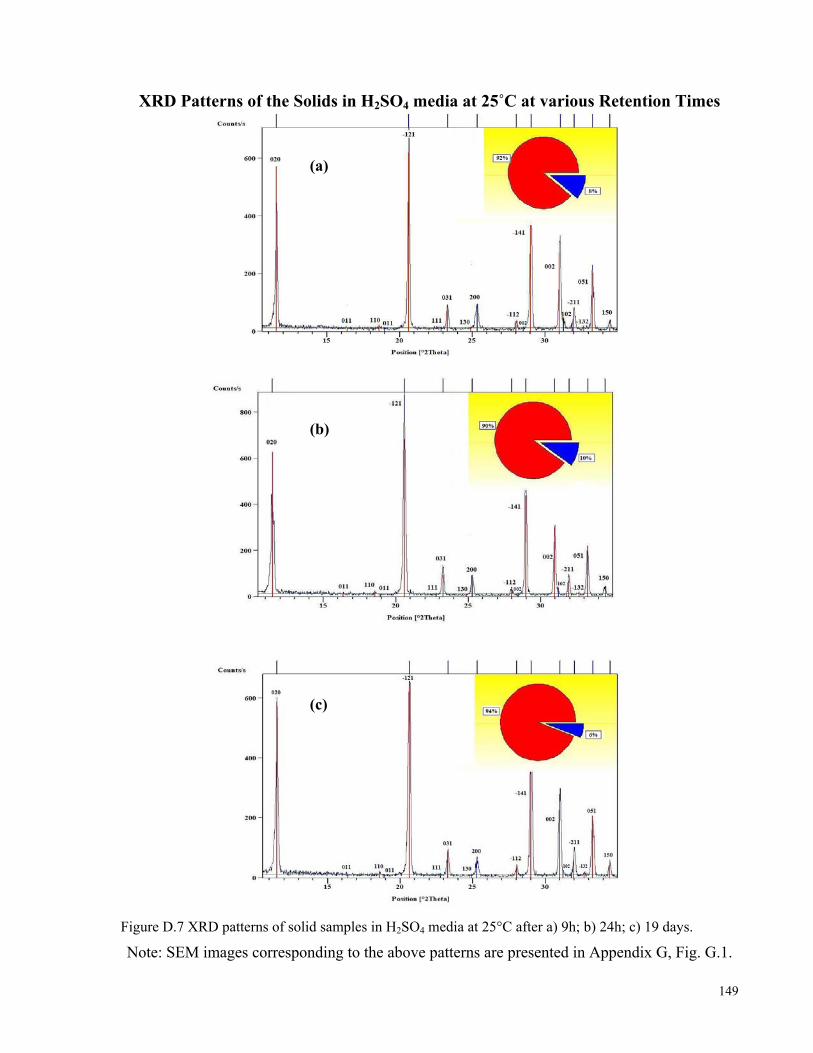
149
XRD Patterns of the Solids in H2SO4 media at 25˚C at various Retention Times
Figure D.7 XRD patterns of solid samples in H2SO4 media at 25°C after a) 9h; b) 24h; c) 19 days.
Note: SEM images corresponding to the above patterns are presented in Appendix G, Fig. G.1.
(a)
(b)
(c)
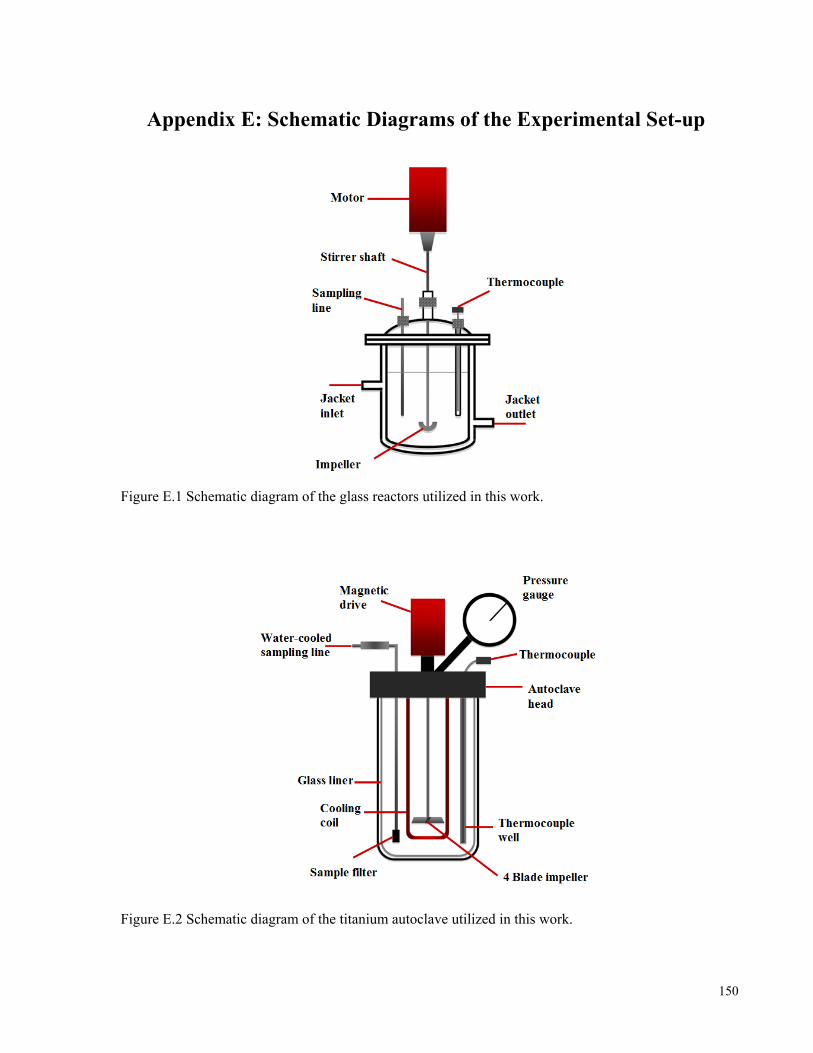
150
Appendix E: Schematic Diagrams of the Experimental Set-up
Figure E.1 Schematic diagram of the glass reactors utilized in this work.
Figure E.2 Schematic diagram of the titanium autoclave utilized in this work.

151
Appendix F: Experimental Measurements for DH-AH transformation
Table F.1– Gypsum–anhydrite transformation at 90°C in water (starting with 50 g/L gypsum+5 g/L anhydrite seeds as the saturating solid phase)
Solid% Sample ID Time (days) Gypsum Anhydrite #1 0.4 88 12 #2 1.0 90 10 #3 1.3 89 11 #4 1.9 90 10 #5 2.9 89 11 #6 5.0 60 40 #7 5.3 55 45 #8 7.3 27 73 #9 8.3 7 93 #10 9.8 0 100 #11 12.1 0 100 #12 13.1 0 100 #13 14.2 0 100
Table F.2– Concentration of CaSO4 and composition of saturating solid phases at various temperatures and residence times in 0.5 and 1.0 M H2SO4 solutions
[H2SO4]=0.5 M [H2SO4]=1.0 M Time
CaSO4 Solid (%) CaSO4 Solid (%) Sample ID
h Day T/°C Method
mol/L DH AH mol/L DH AH
#1 0 0 25 Heating – 100 0 – 100 0 #2 26 1 25 Heating 0.0187 95 5 0.0189 90 10 #3 50 2 45 Heating 0.0273 95 5 0.0288 90 10 #4 98 4 45 Heating 0.0271 95 5 0.0286 90 10 #5 122 5 60 Heating 0.0360 95 5 0.0391 90 10 #6 145 6 70 Heating 0.0417 95 5 0.0464 90 10 #7 169 7 70 Heating 0.0423 95 5 0.0471 90 10 #8 192 8 80 Heating 0.0506 95 5 0.0559 80 20 #9 264 11 90 Heating 0.0623 25 75 0.0581 0 100
#10 288 12 90 Heating 0.0455 0 100 0.0379 0 100 #11 315 13 70 Cooling 0.0422 0 100 0.0357 0 100 #12 344 14 45 Cooling 0.0295 15 85 0.0315 0 100 #13 390 16 25 Cooling 0.0193 100 0 0.0177 30 70

152
Table F.3– Concentration of CaSO4 and composition of saturating solid phases at various temperatures and residence times in 1.5 and 2.0 M H2SO4 solutions
[H2SO4]=1.5 M [H2SO4]=2.0 M Time
CaSO4 Solid (%) CaSO4 Solid (%) Sample ID
h day T/°C Method
mol/L DH AH mol/L DH AH
#1 0 0 25 – – 100 0 – 100 0 #2 24 1 25 Heating 0.0179 85 15 0.0154 85 15 #3 50 2 45 Heating 0.0284 85 15 0.0246 82 18 #4 70 3 45 Heating 0.0285 85 15 0.0251 85 15 #5 94 4 60 Heating 0.0392 85 15 0.0357 85 15 #6 151 6 60 Heating 0.0391 85 15 0.0347 82 18 #7 171 7 70 Heating 0.0495 85 15 0.0466 82 18 #8 192 8 70 Heating 0.0502 85 15 0.0449 82 18 #9 216 9 70 Heating 0.0476 80 20 0.0456 70 30
#10 245 10 80 Heating 0.0600 80 20 0.0451 0 100 #11 266 11 80 Heating 0.0617 60 40 0.0321 0 100 #12 293 12 80 Heating 0.0461 0 100 0.0317 0 100 #13 359 15 90 Heating 0.0353 0 100 0.0274 0 100 #14 383 16 70 Cooling 0.0338 0 100 0.0245 0 100 #15 407 17 45 Cooling 0.0319 0 100 0.0232 0 100 #16 435 18 25 Cooling 0.0173 15 85 0.0131 5 95
* DH: gypsum, AH: anhydrite

153
Appendix G: Additional SEM Images
Solid samples in H2SO4 media at 25˚C at various retention times
Figure G.1 SEM images of solid samples in 1.5 M H2SO4 media at 25°C after a) 9h; b) 24h; c) and d) 19 days.
Note: XRD patterns corresponding to the above SEM images are presented in Appendix D, Fig. D.7.
a
b
c d

154
SEM images of solid samples in pure water at 90˚C at various retention times
Following SEM images present solid samples in pure water at 90°C, in the absence of anhydrite
seeds, at various retention times. X-ray diffraction analysis showed 100% gypsum for all the
solids.
t=6 h t=46 h
t=121 h t=175 h Figure G.2 SEM images of saturating solid samples in pure water at 90°C after various retention times (in the absence of anhydrite seeds).
(a) (b)
(c) (d)

155
Appendix H: The Rietveld Method (Full-Pattern Analysis)
In the present work, the quantification of XRD patterns were performed utilizing the Rietveld
refinement (or Rietveld method), which is embedded in the XRD HighScore Software used to
analyze the diffractograms. Rietveld refinement is a computer-based analytical procedure,
pioneered by Hugo Rietveld (1969), for the characterization and quantification of crystalline
materials, utilizing the full information (such as intensities height and width as well as the
position of the reflections) over the powder pattern.
The Rietveld method was originally developed as a method of refining crystal structures using
neutron powder diffraction data. The method requires knowledge of the approximate crystal
structure of all phases of interest in the pattern. In this method, a full-pattern fit is done until a
theoretical profile matches the measured profile. In the Rietveld method, the basic approach is
to, first, obtain the X-ray pattern of the sample, and then to identify all phases present (using
available databases such as ICDD) and to input basic structural data for all the phases. After
that, the data are processed until the best fit to the experimental pattern is obtained. The quantity
minimized in Rietveld refinement is the conventional least squares residual:
2
)()(∑ −=j
cjojj IIwR
where Ij(o) and Ij(c) are the intensities observed and calculated, respectively, at the jth step in the
data, and wj is the weight at the jth step. Detailed discussion of the Rietveld method is beyond
the scope of this work, however, it is important to mention that this method is more accurate
compared to other available quantification methods such as peak intensity-based methods,
because of the whole-pattern fitting approach.
The Variables of a Rietveld Refinement:
The following variables are refined in the Rietveld method for the characterization of the
measured profiles:
• Peak shape function (which describes the shape of the diffraction peaks, e.g., Gaussian
shape);

156
• Peak width function (starts with optimal full width at half maximum (FWHM) values);
• Preferred orientation function (introduces a factor based on deviation from randomness);
• The structure factor (calculated from the crystal structure data and includes site
occupancy information, cell dimensions, inter-atomic distances, temperature and
magnetic factors. Crystal structure data is usually obtained from the ICDD database);
• The scale factor (relating the intensity of the experimental data with that of the model
data).
The least-squares parameters are those varied in the model to achieve the best fit to the
experimental data. In the Rietveld method, these parameters are divided into two groups. The
first group includes the profile parameters, defining the positions, half-widths, possible
asymmetry of the diffraction peaks, and preferred orientation. The second group contains the
structure parameters, defining the contents of the asymmetric unit cell, such as overall scale
factor, overall isotropic temperature parameter, and coordinates of all atomic units. More details
regarding the Rietveld refinements are available in the literature (Rietveld, 1969).
In the present work, the accuracy of the Rietveld method was validated against mixtures of
gypsum and anhydrite feed with known composition. The estimated compositions, from the
Rietveld refinement, and the measured compositions were in good agreement (AARD%= 7.3 for
gypsum and 10.4 for anhydrite), as shown in the following table:
Table H.1– Estimated vs. measured composition of the mixtures of gypsum/anhydrite solid samples
Measured composition, wt%
Rietveld estimated composition, wt% Relative Error, % Sample
ID gypsum anhydrite gypsum anhydrite gypsum anhydrite
#1 100 0 100 0 0.0 0.0 #2 90 10 85 15 5.6 50.0 #3 80 20 77 23 3.8 15.0 #4 70 30 69 31 1.4 3.3 #5 50 50 46 54 8.0 8.0 #6 35 65 35 65 0.0 0.0 #7 15 85 9 91 40.0 7.1 #8 0 100 0 100 0.0 0.0
AARD% – – – – 7.3 10.4
∑−
=NP
i valueExp
valueCalculatedvalueExp
NPAARD
.
.100(%)





























