Embed Size (px)
Citation preview

Delivered by Ingenta to: Guest UserIP: 75.108.130.155 On: Mon, 30 May 2016 16:05:42
Copyright: American Scientific Publishers
Copyright © 2015 American Scientific PublishersAll rights reservedPrinted in the United States of America
ArticleJournal of
Pharmaceutical Sciencesand Pharmacology
Vol. 2, 259–276, 2015www.aspbs.com/jpsp
Evaluating the Effects of Different Molecular Weights ofPolymers in Stabilizing Supersaturated Drug Solutionsand Formulations Using Various Methodologies of theModel Drug: Fenofibrate
Smruti P. Chaudhari and Rutesh H. Dave∗
Division of Pharmaceutical Sciences, Arnold and Marie Schwartz College of Pharmacy and Health Sciences, Long Island University,Brooklyn, New York 11201, USA
Assessing the effect of the molecular weight of excipients like Hydroxypropyl methyl cellulose (HPMC) and Polyvinylpyrroli-done (PVP) on their ability to attain and maintain the supersaturation of drug-based solutions and solid dispersion willprovide useful information for the design of solid dosage formulations. In this study, three grades of HPMC (HPMC E5,HPMC E15 and HPMC E50) and PVP (PVP K12, PVP K29 and PVP K90) were chosen to study their effect as antiprecip-itants on the model drug Fenofibrate. Two approaches were used in this study: a non-formulated drug method (solventshift) and a formulated drug method (a miniaturized solvent casting method and spray-dried solid dispersion). The effectsof accelerated temperature and humidity were also studied in the spray dried solid dispersions and the solvent castingmethods. Miniaturized testing of the polymer using the solvent shift and the solvent casting method suggest that HPMCE5 is a favorable polymer that gives the best extend and stability for the formed supersaturated solutions in the screeningassay and also shows a better release profile in in-vitro assay. Out of all the grades of PVP studied, PVP K29 showsthe best extend and stability of a formed supersaturated solution; however, the spray-dried dispersions were vulnerableto temperature and humidity. The stability of Fenofibrate spray-dried dispersion is dependent on the molecular weight ofthe polymer and the amount of the polymer in the dispersion. A high molecular weight polymer shows good stability ascompared to a lower molecular grade polymer at higher drug loading; however, the polymer governs the dissolution ofhigh molecular weight of polymer dispersions.
KEYWORDS: Fenofibrate, Supersaturation, Spray-Dried Solid Dispersion, HMPC, PVP.
INTRODUCTIONMost of the drugs discovered in the past decade arehydrophobic in nature. They belong to the Biopharmaceu-tics Classification System (BCS) of Class II compounds,which are characterized by high permeability and lowaqueous solubility (Friesen et al., 2008). Even though theymay not fit the “rule of five” (Lipinski et al., 1997), thesedrugs are safe and efficacious, and hence, their devel-opment is critical. Unfortunately, it is difficult to retainthe potency and efficacy of these drug candidates whileimproving their solubility (Ruben et al., 2006). Two types
∗Author to whom correspondence should be addressed.Email: [email protected]: 31 December 2015Accepted: 2 March 2016
of approaches are used to address the low solubility chal-lenges, which include chemical modification, such as saltformation (Serajuddin, 2007), prodrug (Stella and Nti-Addae, 2007), or formulation methods, such as lipid for-mulations (Akhlaquer Rahman et al., 2011), cocrystalsformations (Thakuria et al., 2013), particle size reduc-tion (Brittain, 2002), inclusion complexes with cyclodex-trins (Moya-Ortega et al., 2011), amorphous dispersionsof drug and polymer (Serajuddin, 1999), nanocrystals(Müller et al., 2011; Murdande et al., 2015) and nanopar-ticles (Al-Nemrawi and Dave, 2014). Of these methodsamorphous solid dispersion is gaining momentum sinceit increases the dissolution of these drugs and therebyincreases bioavailability (Singh et al., 2011), althoughadditional experiments are needed for each drugs. Addi-tionally, solid dispersion offers the possibility of presenting
J. Pharm. Sci. Pharmacol. 2015, Vol. 2, No. 3 2333-3715/2015/2/259/018 doi:10.1166/jpsp.2015.1066 259

Delivered by Ingenta to: Guest UserIP: 75.108.130.155 On: Mon, 30 May 2016 16:05:42
Copyright: American Scientific Publishers
Evaluating the Effects of Different Molecular Weights of Polymers Chaudhari and Dave
the drugs in solid dosage forms, which have high patientcompliance (Hancock and Zografi, 1997). Various meth-ods are available to convert crystalline drugs into an amor-phous form, such as mechanical milling (Peltonen andHirvonen, 2010), fusion (DiNunzio et al., 2010), hot meltextrusion (Lakshman et al., 2008), spray-drying (Patelet al., 2013; Patel et al., 2014; Alhalaweh et al., 2009),freeze drying (Yang et al., 2008), and supercritical fluidprecipitation (Bouchard et al., 2008). Of these, hot meltextrusion and spray drying are the most widely used meth-ods for manufacturing of solid dispersions, since thesetechnologies are scalable. The spray drying method offerstwo advantages over hot melt extrusion. First, the materi-als are not exposed to extreme temperatures, and second,it is possible to granulate material using the same equip-ment (Hugo et al., 2013). In this research we have usedthe spray drying method to convert crystalline drug intoan amorphous form.In spite of several advantages of solid dispersion, sta-
bility and reproducibility have limited the commercial useof solid dispersion. To address this issue, the BCS ClassII compound fenofibrate (FENO) was selected. FENO ispractically insoluble in water, has very low tg and is highlylipophilic (logP = 5�24 (Vogt et al., 2008)) in nature.In light of Fick’s first law, we know that if the permeabilityis good, dissolution is the only rate limiting step. Severalreports in the literature have indicated that FENO has avery low tg of −21.3 �C (de Waard et al., 2008; Górniaket al., 2011; Sanganwar and Gupta, 2008, Zhou et al.,2002) and very high molecular mobility. As a result, itshows a tendency for spontaneous recrystallization, whichin turn, affects the dissolution characteristics which meansit, has an unpredictable drug release profile. Differentpolymers have been shown to be beneficial in inhibitingdrug crystallization, including polyvinylpyrrolidone (PVP)(Khougaz and Clas, 2000), hydroxypropylmethyl cellulose(HPMC) (Raghavan et al., 2001) and many others as well.This polymer follows what is called a spring and parachuteapproach. A spring is a high energy form of drug or self-emulsifying system, which allows for the rapid dissolutionof poorly soluble drugs at a supersaturated concentration.A formulation component which stabilizes the metastablesupersaturated system acts as a parachute, hindering nucle-ation, or crystal growth (Guzmán et al., 2007; Guzman,2004; Gao et al., 2004; Gao and Morozowich, 2006).Sporanox (itraconazole) is an example of this stratagem(Peeters et al., 2002). Itraconazole is a poorly water sol-uble drug (∼1 ng/ml at ph7) with a logP > 5 and melt-ing point of 167 �C. The marketed formulation was basedon the development of the solid solution of the drug in apolymeric matrix. HPMC was used as polymeric matrix.The drug and HPMC were dissolved in a common sol-vent and coated on sugar spheres. In this formulation,HPMC acts as an inhibitor of drug nucleation and crystalgrowth for a sufficient period of time, which leads to sig-nificant absorptions and bioavailability. The optimization
of the system features will allow for sustained supersat-uration, which in turn, leads to improved bioavailability.The use of supersaturation approaches has been widelyreported in topical and transdermal therapies (Raghavanet al., 2001; Raghavan et al., 2003; Davis and Hadgraft,1991; Hadgraft, 1999) and has led to marketed products(Strickley, 2004). Another approach used by scientists isto increase the saturation solubility of drugs. In all ofthese methods the selection of the excipient i.e., polymericmatrix is the crucial step, which in turn, stabilizes thehigh energy form of the drug. Some of the reports in theliterature have reported multiple in-vitro assays to eval-uate supersaturation, precipitation or precipitation inhibi-tion by use of the excipients, i.e., polymers. In this paper,the solvent shift and solvent casting methods are usedto evaluate the supersaturation potential of the polymer.These two techniques are miniaturized testing methodsinvolving �g quantity of drug; hence spray dried soliddispersions (SD) are prepared in order to evaluate theireffect of FENO on a large scale. Two polymer fami-lies are used in this study: PVP (PVP K12, PVP K29and PVP K90) and HPMC (HPMC E5, HPMC E15 andHPMC E50).
MATERIAL AND METHODSMaterialsFENO was obtained from Sigma Aldrich. PVP K12(Plasdone® k12), PVP K29 (Plasdone® K29); PVPK90 (Plasdone® K90) was received from ISP tech-nologies and HPMC E5 (Methocel® E5), HPMCE15 (Methocel® E15) and HPMC E50 (Methocel®
E50) were procured from Dow Chemicals. Sodiumtaurocholate (Sigma Aldrich) and lecithin (SpectrumChemicals) were used for simulated intestinal fluid prepa-ration. Chemicals such as Sodium dihydrogen phosphate(NaH2PO4�, sodium chloride (NaCl), sodium hydrox-ide (NaOH), hydrochloric acid (HCl) and HPLC gradeacetonitrile and water were obtained from SpectrumChemicals.
Quantification of FENOHigh performance liquid chromatography (HPLC) wasused for quantification of FENO. HPLC analysis was per-formed using an Agilent 1100 series HPLC system fittedwith a binary pump, with a plate auto sampler, a thermo-stat in the column compartment and a diode array detec-tor controlled by Chemstation, software version A.10 .02.A Supleco C18 Column of 4.6 mm×150 mm with particlesize of 5 um was also used. The mobile phase was com-posed of 90% acetonitrile and 10% water. The flow ratewas 1 ml/min. The column temperature was maintained at25 �C and UV detection was carried out at 291 nm andthe injection volume used was 10 uL with 6 min of runtime and retention time of 4.1 min.
260 J. Pharm. Sci. Pharmacol. 2, 259–276, 2015

Delivered by Ingenta to: Guest UserIP: 75.108.130.155 On: Mon, 30 May 2016 16:05:42
Copyright: American Scientific Publishers
Chaudhari and Dave Evaluating the Effects of Different Molecular Weights of Polymers
Preparation of MediaIn this study, the fasted state simulated intestinal fluid(FaSSIF) was used as the test medium for the solventshift method and the solvent casting method. FaSSIF wasprepared according to the formula described in Vertzoniet al. (Vertzoni et al., 2004). Drug release experimentswere also carried out in blank FaSSIF with 1% w/vsodium lauryl sulfate (SLS) without sodium taurocholateand lecithin. The composition of FaSSIF was as follows:pH 6.5, 3 mM sodium Taurocholate, 0.75 mM Lecithin,29 mM NaH2PO4, 13.8 mM NaOH, 106 mM NaCl. pHwas adjusted to 6.5 by 1 N NaOH or 1 N HCl.
Solubility MeasurementsThe solubility of FENO was assessed in an aqueousmedium by the shaking flask method: approximately10 mg of FENO was dispersed in 5 ml of medium andshaken for 24 hrs at room temperature (RT) and 37 �C. Theundissolved materials were separated from the solution bycentrifugation (14000 rpm×10 mins). The supernatant wasdiluted with mobile phase and analyzed by HPLC.
STABILIZING EFFECT OF POLYMERSolvent Shift MethodPolymers were dissolved in FaSSIF at concentrations of0.01%, 0.1 %, 1%, 2% w/v and FENO was dissolvedin dimethyl sulfoxide (DMSO) at a concentration of50 mg/ml. Then 30 ml of polymer solution was placed ina beaker and stirred continuously with the help of a mag-netic stirrer. Organic solution was then added drop-wise tothe polymer solution until a visible precipitate was notice-able. 1 ml samples were withdrawn after 5, 30, 60, 90, 120minutes post drug addition. Samples were filtered using a0.45 um GHP filter and diluted immediately with mobilephase and analyzed in the HPLC. In every case, only 2%of organic phase was added.
Viscosity MeasurementViscosity measurements were also performed on AP solu-tion using Gilmont Falling Ball viscometer, size 1. 5–10 mlsolution was filled in the viscometer and capped. The timerequired by stainless steel ball to pass between two set offiduciary line is measured with a stop watch and viscositywas calculated by
� = K��1−��t (1)
Where, K = 0�3 (size 1), �1 = density of stainlesssteel ball, � = Density of the liquid, t = time of descent(minutes).
Density of solution was measured with the help of glasspycnometer.
Solvent Casting MethodThe formulations were prepared in 96 well plates. Ethanolwas used as the solvent to prepare the excipient-drug stock
solution. Ethanol was chosen as the solvent since it dis-solves all the excipients and drugs used in this study.A stock solution with the drug excipient was prepared and200 uL was dispensed in each plate. The drug concentra-tion in each plate was kept at 0.1 mg/ml and the polymervaried according to the drug load in each well. After dis-pensing the liquid, the solvent was evaporated in a vacuumoven at 50 �C for 2.5 hrs. After drying, each well containeda film or pellet, which was sealed with a paraffin film andkept at room temperature overnight. This step was done togive unstable formulation the opportunity to recrystallize.Replicates of the plates were prepared and stored in roomtemperature and humidity for three months and dissolu-tion testing was performed again after 15 days, 1 monthand 3 months. Plates were also stored at accelerated sta-bility conditions at 40 �C and 75% relative humidity (RH)(described below) and analyzed after 2 weeks, 4 weeksand 6 weeks.
Drug Release Testing from Solvent CastThe drug release testing from the solvent casts was per-formed by adding 200 uL of FaSSIF (pH 6.5) in eachwell and then the plates were shaken in the shaker for0.5, 1 and 4 hrs at 95 rpm. After shaking, the contentsof the plates were transferred to the 0.45 um GHP mem-brane filter plate (Pall Life Sciences). The samples werepulled through the filter with the help of the vacuum.100 uL of sample was diluted with the mobile phaseimmediately to avoid precipitation. The drug release stud-ies were carried out on the stability samples as well; theplates were shaken for 4 hrs only to determine the drugcontent.
Preparation of Spray Dried Solid DispersionThe spray dried solid dispersion was prepared usinga Buchi spray dryer B-290 (Buchi Laboretechnik AG,Flawil, Switzerland) equipped with an inert loop B-295.The spray drying solution was prepared by dissolving theexcipient and the drug in ethanol. An 8% w/v feed con-centration was used for PVP K12 and K29, a 3.2% w/vfeed concentration was used for HPMC E5, HPMC E15and a 2% w/v feed concentration was used with HPMCE50. The inlet temperature was set as 110 �C and the solu-tion was sprayed at a 10 ml/min flow rate. The aspiratorrate was set as 100% with a nozzle size of 0.7 mm anddrying air flow (473 L/Hr) was kept constant throughoutthe experiment. The resulting spray dried powder was col-lected and stored in tightly closed vials and stored overdesiccators at room temperature.
Stability StudiesStability studies of the formulation were performed at40 �C in sealed glass chambers. A saturated NaCl saltsolution was prepared by using chemically pure NaCland distilled water. This solution was placed in glass
J. Pharm. Sci. Pharmacol. 2, 259–276, 2015 261

Delivered by Ingenta to: Guest UserIP: 75.108.130.155 On: Mon, 30 May 2016 16:05:42
Copyright: American Scientific Publishers
Evaluating the Effects of Different Molecular Weights of Polymers Chaudhari and Dave
chambers and allowed to equilibrate at 40 �C to main-tain the accelerated stability condition of 40 �C and 75%RH. SD was stored in these glass chambers in open glassvials. The samples were pulled after 1, 2 and 6 weeks.The solid dispersion collected was gently separated andscreened through USP mesh no 30. All stability sampleswere then thoroughly dried in a silica gel (0% RH) des-iccator for 48 hrs and then stored in sealed glass vials in0% RH desiccator until further analysis.
Drug Load QuantificationThe FENO content in SD and stability samples were deter-mined by suspending the amount of powder equivalent to5 mg of FENO in 10 ml of DMSO and stirred with thehelp of a magnetic stirrer for 4 hrs. After 4 hours, the sam-ples were filtered through a 0.45 �m PTFE syringe filterand diluted appropriately and analyzed in HPLC.
CHARACTERIZATION OF SOLIDDISPERSIONPowder X-ray DiffractionPowder X-ray diffraction (PXRD) was done using scan-ning diffractometer (Advanced Diffraction System, ScintagInc., Model XI Cupertino, CA) controlled by a computerwith diffraction management system software for WindowsNT. The X-ray radiation used was generated by a copperK� filter with a wavelength of 1.54 A0 at 45 KV and40 mA. Solid samples were placed on a sample holder withdimensions of 2 cm× 2 cm× 2 mm. Powder was placedon the holder using a spatula and then flattened. Sampleswere scanned over a range of 5� to 50� 2� degree using ascan rate 2�/min and scan step of 0.05.
Modulated Differential Scanning CalorimetryModulated differential scanning calorimetry (MDSC) wasapplied as an additional method to detect crystallinity inthe sample apart from X-ray. It has been reported in the lit-erature that small crystals might not be detected by PXRD,even if it is above the limit of detection (Munson, 2009).MDSC analysis was performed using Q200 (TA Instru-ments, USA) equipped with a cooling system. Nitrogenwas used as a purge gas with a flow rate of 50 ml/min.5–10 mg samples were weighed into aluminum pans witha pinhole on the lid and then hermetically sealed. Sam-ples were heated from 20–200 �C at a heating rate of5 �C/min with modulation of 1.59 �C every 60 s. All thedata handling was performed using a Universal Analysis2000 software package (TA Instruments).
Fourier Transform Infrared SpectroscopyFourier Transform Infrared Spectroscopy (FTIR) spectrafor all solid dispersion were obtained using MAGNA-IR-60 Spectrophotometer (Nicolet Instrument Corp., Madi-son, WI). A small quantity of each sample was triturated
with pure potassium bromide in a mortar and pestle andcompressed to form a semitransparent film. Each film wasscanned in the region of 400 to 4000 cm−1. 64 scans werecollected for each sample.
Dissolution TestingIn-vitro release experiments were carried out in USPApparatus 2 (Distek Dissolution Systems 2100A, EastBrunswick, NJ). The release behavior of the SD, the physi-cal mixture and the pure drug were assessed in two media:FaSSIF and blank FaSSIF (without sodium taurocholateand lecithin) with 1% SLS: to ensure sufficient wetting,media was conditioned at 37 �C with a stirring speed of50 rpm. A sample amount equivalent to 25 mg of FENOwas used throughout the studies in 250 ml of media.A sample volume of 3 ml was drawn through a stainlesssteel cannula at 15, 30, 45, 60, 90 and 120 min. All sam-ples were filtered through a 0.45 �m PTFE syringe filter.The first milliliter of the sample was discarded and rest ofthe sample was diluted with mobile phase and analyzed inHPLC.
Particle Size AnalysisParticle size analysis was performed using Flowcam(Scarborough, ME) and data was analysed using Flowcamsoftware version 3.4.5. Drug particle and dispersions weresuspended in mineral oil and particle size analysis was per-formed at a flow rate of 0.5 ml/min for 10 minutes usinga 4× optical lens.
RESULTS AND DISCUSSIONSolubility StudiesSolubility of FENO in water was found to be 0.3 �g/ml±0.01 �g/ml at 25 �C and 37 �C, respectively. It was seenthat temperature did not affect the solubility of FENOin water. Media plays an important role in solubility ofFENO. The solubility of FENO in FaSSIF (0.5 �g/ml±0.02 �g/ml at 25 �C, 13.5 �g/ml± 0.3 �g/ml at 37 �C)was significantly enhanced, as compared to blank FaSSIF(0.3 �g/ml at 25 �C and 37 �C), which indicates FENOis solubilized in the micelles of the simulated intestinalmedia. The significant increase in solubility upon additionof taurocholate and lecithin corresponds with the docu-mented positive food effect of FENO (Guichard, 2000;Guivarc’h et al., 2004; Sauron, 2006; Yun et al., 2006).The solubility of FENO is greatly enhanced with the addi-tion of 1% w/v SLS to Blank FaSSIF (337 �g/ml±14 �g/ml at 37 �C). The critical micelle concentration(CMC) of SLS is 8 mM (Cheng et al., 2006; Dave et al.,2012). We have used SLS above its CMC concentration,and hence, the solubility of FENO is more in blank FaSSIFwith 1% w/v SLS. The excipient also has an effect on sol-ubility (Rodríguez-hornedo and Murphy, 1999; Strickley,2004). However, when the solubility of FENO in FaSSIF
262 J. Pharm. Sci. Pharmacol. 2, 259–276, 2015

Delivered by Ingenta to: Guest UserIP: 75.108.130.155 On: Mon, 30 May 2016 16:05:42
Copyright: American Scientific Publishers
Chaudhari and Dave Evaluating the Effects of Different Molecular Weights of Polymers
Table I. Physiochemical properties of polymers.
Viscosity in mpa ·sPolymer Tg (�C) Molecular weight (Da) 0.1 mg/ml (n = 3) 1 mg/ml (n = 3) 10 mg/ml (n = 3) 20 mg/ml (n = 3)
PVP K12 117 4�000 0.91 0.91 1�1 1�42PVP K29 176 58�000 1.03 1.13 1�24 1�87PVP K90 181 1�300�000 1.08 1.33 3�07 7�37HPMC E5 150 28�700a 0.97 1.11 2�06 5�02HPMC E15 154 60�000a 1.06 1.26 4�02 15�00a
HPMC E50 164 86�700a 1.03 1.38 ND 50�00a
Notes: avalues are taken from published literature (Keary, 2001), ND: not determined.
in the presence of 2% w/v excipient was tested, it wasfound that the excipient does not have any effect on thesolubility of FENO (data not shown). The physiochemicalproperties of the polymer are listed in Table I. Molecularweight increases as we go from PVP K12 to PVP K90 forPVP, and HPMC E5 to HPMC E15, which is associatedwith the increase in glass transition temperature. The vis-cosity of the solution is concentration dependent. Highermolecular weight HPMC and PVP has the higher viscos-ity. The viscosity of 10 mg/ml solution of HPMC E50 and20 mg/ml solution of HPMC E15 and HPMC E50 wasnot determined due to limitation of gilmont falling ballviscometer, size 1.
STABILIZING EFFECT OF POLYMERSolvent Shift MethodIn the blank experiment, the supersaturation degree andstability was determined for the supersaturated FENOsolution without any antiprecipitant. The supersaturationdegree, expressed as a concentration of FENO in solu-tion after 5 min post drug addition, was 8.48 �g/ml±0.03 �g/ml. After 2 hrs, there was 3.19 �g/ml ±0.01 �g/ml of FENO in the solution. These data sug-gests that we can achieve a significant degree of super-saturation (DS) even without the addition of the excipientcompared to thermodynamic solubility. DS is expressed
Figure 1. Spring and parachute approach.
as shown in Eq. (2)
S = C
Ceq
(2)
Where C is the drug concentration and Ceq is the equi-librium drug concentration.It also represents the state of drug
DS= 0, Drug is in a state of saturation,DS< 1, Drug is in an unsaturated state,DS> 1, Drug is in a supersaturated state.To evaluate the extent of supersaturation produced and thestability of the supersaturated solution, the excipient gainfactor (EGF) was calculated from Figure 1, Eq. (3)
Excipient Gain Factor= Area A+Area B+Area C
Area A+Area B(3)
For most of the polymers studied, the stability of supersat-uration was superior compared to blank. Table II shows thedata for the degree of supersaturation achieved after 5 minpost drug addition. It is seen that K29 shows the highestdegree of supersaturation after 5 min post drug addition inthe PVP family, whereas HPMC E5 shows superior resultsin the HPMC family. In general, increasing the polymerconcentration does not increase the supersaturation pro-duced. However, it only increases the stability of supersat-urated FENO in solution for PVP K12 and K29 polymers,which is shown by an increase in the excipient gain fac-tor until a polymer concentration of 1 mg/ml. Increasingthe concentration further has a reverse effect and stabil-ity decreases, since EGF decreases, as shown in Table III.Increasing the concentration of the HPMC polymer also
Table II. Supersaturation of drug created in the presence ofexcipients.
Supersaturation produced after 5 mins post drug addition
0.1 mg/ml 1 mg/ml 10 mg/ml 20 mg/ml(n = 3) (n = 3) (n = 3) (n = 3)
PVP K12 1.8 1.7 1.6 1.5PVP K29 1.9 1.9 1.9 1.3PVP K90 1.3 1.6 1.6 1.4HPMC E5 3.1 2.7 2.2 1.9HPMC E15 2.9 2.2 1.6 1.3HPMC E50 2.0 1.5 1.5 0.9
J. Pharm. Sci. Pharmacol. 2, 259–276, 2015 263
Delivered by Ingenta to: Guest UserIP: 75.108.130.155 On: Mon, 30 May 2016 16:05:42
Copyright: American Scientific Publishers
Evaluating the Effects of Different Molecular Weights of Polymers Chaudhari and Dave
Table III. Stability of supersaturated drug solution at the endof 2 hours.
Excipient gain factor
0.1 mg/ml 1 mg/ml 10 mg/ml 20 mg/ml(n = 3) (n = 3) (n = 3) (n = 3)
PVP K12 1.45 1.47 1.44 1.43PVP K29 1.45 1.46 1.45 1.42PVP K90 1.49 1.44 1.45 1.42HPMC E5 1.59 1.58 1.56 1.53HPMC E15 1.56 1.51 1.49 1.40HPMC E50 1.50 1.51 1.36 1.31
has a negative effect on the degree of supersaturation gen-erated characterized by a decrease in EGF.
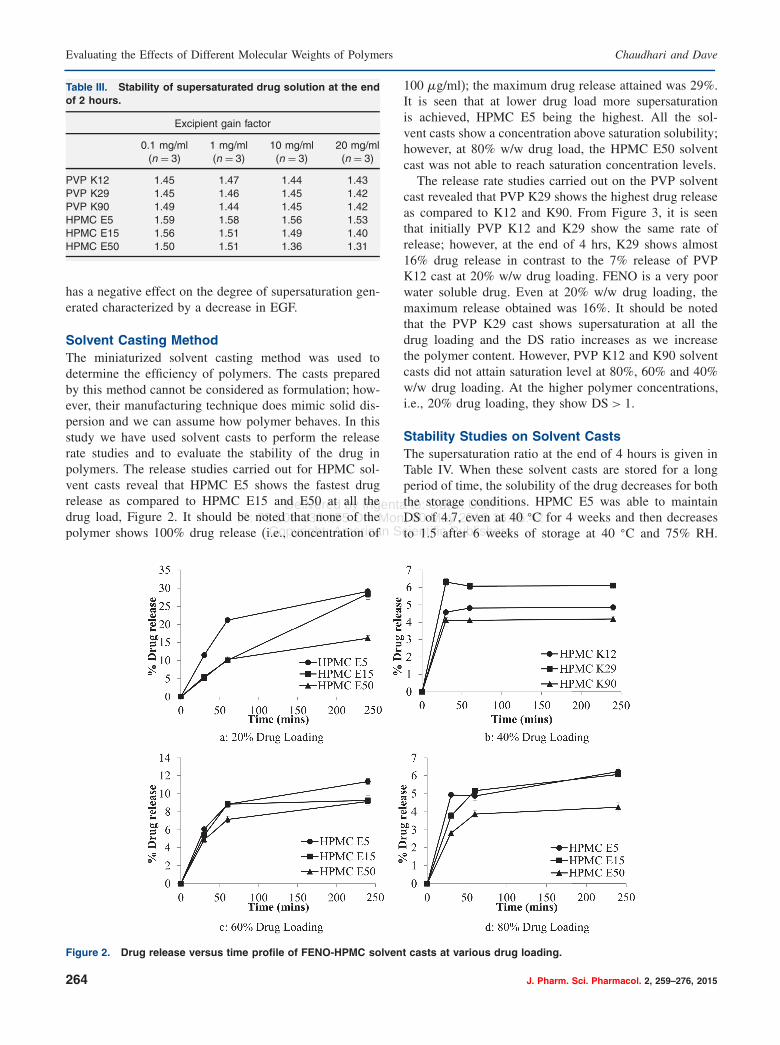
Solvent Casting MethodThe miniaturized solvent casting method was used todetermine the efficiency of polymers. The casts preparedby this method cannot be considered as formulation; how-ever, their manufacturing technique does mimic solid dis-persion and we can assume how polymer behaves. In thisstudy we have used solvent casts to perform the releaserate studies and to evaluate the stability of the drug inpolymers. The release studies carried out for HPMC sol-vent casts reveal that HPMC E5 shows the fastest drugrelease as compared to HPMC E15 and E50 at all thedrug load, Figure 2. It should be noted that none of thepolymer shows 100% drug release (i.e., concentration of
Figure 2. Drug release versus time profile of FENO-HPMC solvent casts at various drug loading.
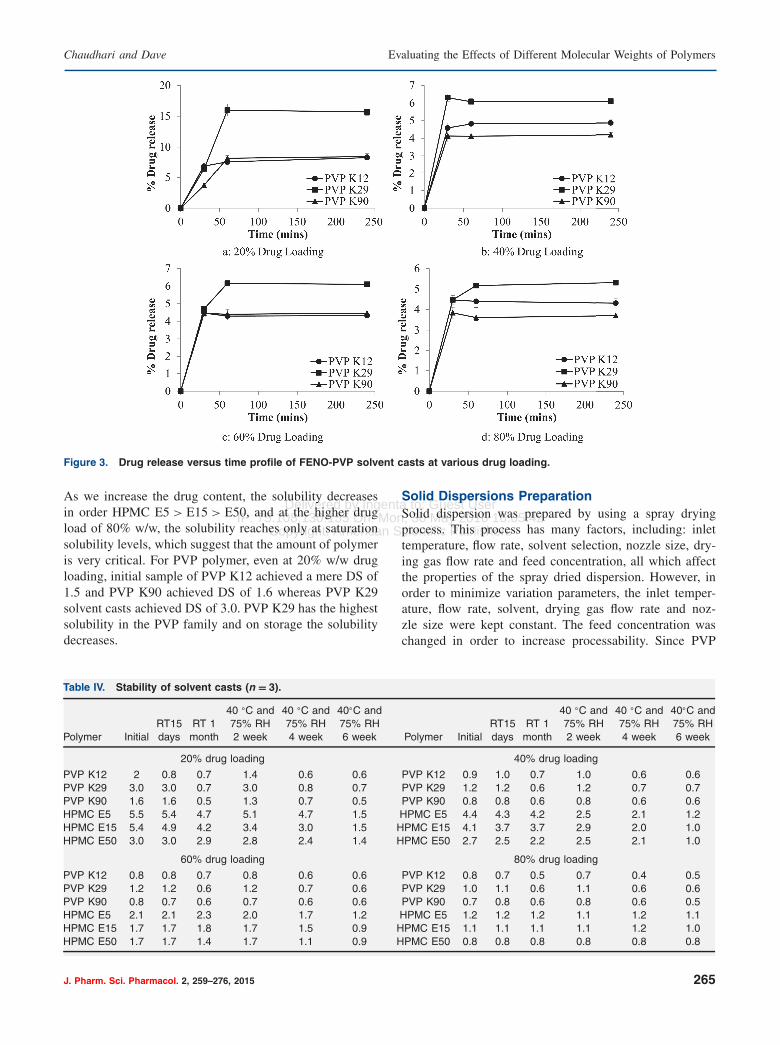
100 �g/ml); the maximum drug release attained was 29%.It is seen that at lower drug load more supersaturationis achieved, HPMC E5 being the highest. All the sol-vent casts show a concentration above saturation solubility;however, at 80% w/w drug load, the HPMC E50 solventcast was not able to reach saturation concentration levels.The release rate studies carried out on the PVP solvent
cast revealed that PVP K29 shows the highest drug releaseas compared to K12 and K90. From Figure 3, it is seenthat initially PVP K12 and K29 show the same rate ofrelease; however, at the end of 4 hrs, K29 shows almost16% drug release in contrast to the 7% release of PVPK12 cast at 20% w/w drug loading. FENO is a very poorwater soluble drug. Even at 20% w/w drug loading, themaximum release obtained was 16%. It should be notedthat the PVP K29 cast shows supersaturation at all thedrug loading and the DS ratio increases as we increasethe polymer content. However, PVP K12 and K90 solventcasts did not attain saturation level at 80%, 60% and 40%w/w drug loading. At the higher polymer concentrations,i.e., 20% drug loading, they show DS> 1.
Stability Studies on Solvent CastsThe supersaturation ratio at the end of 4 hours is given inTable IV. When these solvent casts are stored for a longperiod of time, the solubility of the drug decreases for boththe storage conditions. HPMC E5 was able to maintainDS of 4.7, even at 40 �C for 4 weeks and then decreasesto 1.5 after 6 weeks of storage at 40 �C and 75% RH.
264 J. Pharm. Sci. Pharmacol. 2, 259–276, 2015
Delivered by Ingenta to: Guest UserIP: 75.108.130.155 On: Mon, 30 May 2016 16:05:42
Copyright: American Scientific Publishers
Chaudhari and Dave Evaluating the Effects of Different Molecular Weights of Polymers
Figure 3. Drug release versus time profile of FENO-PVP solvent casts at various drug loading.
As we increase the drug content, the solubility decreasesin order HPMC E5 > E15 > E50, and at the higher drugload of 80% w/w, the solubility reaches only at saturationsolubility levels, which suggest that the amount of polymeris very critical. For PVP polymer, even at 20% w/w drugloading, initial sample of PVP K12 achieved a mere DS of1.5 and PVP K90 achieved DS of 1.6 whereas PVP K29solvent casts achieved DS of 3.0. PVP K29 has the highestsolubility in the PVP family and on storage the solubilitydecreases.
Table IV. Stability of solvent casts (n = 3).
40 �C and 40 �C and 40�C and 40 �C and 40 �C and 40�C andRT15 RT 1 75% RH 75% RH 75% RH RT15 RT 1 75% RH 75% RH 75% RH
Polymer Initial days month 2 week 4 week 6 week Polymer Initial days month 2 week 4 week 6 week
20% drug loading 40% drug loading
PVP K12 2 0.8 0.7 1.4 0.6 0.6 PVP K12 0.9 1.0 0.7 1.0 0.6 0.6PVP K29 3.0 3.0 0.7 3.0 0.8 0.7 PVP K29 1.2 1.2 0.6 1.2 0.7 0.7PVP K90 1.6 1.6 0.5 1.3 0.7 0.5 PVP K90 0.8 0.8 0.6 0.8 0.6 0.6HPMC E5 5.5 5.4 4.7 5.1 4.7 1.5 HPMC E5 4.4 4.3 4.2 2.5 2.1 1.2HPMC E15 5.4 4.9 4.2 3.4 3.0 1.5 HPMC E15 4.1 3.7 3.7 2.9 2.0 1.0HPMC E50 3.0 3.0 2.9 2.8 2.4 1.4 HPMC E50 2.7 2.5 2.2 2.5 2.1 1.0
60% drug loading 80% drug loading
PVP K12 0.8 0.8 0.7 0.8 0.6 0.6 PVP K12 0.8 0.7 0.5 0.7 0.4 0.5PVP K29 1.2 1.2 0.6 1.2 0.7 0.6 PVP K29 1.0 1.1 0.6 1.1 0.6 0.6PVP K90 0.8 0.7 0.6 0.7 0.6 0.6 PVP K90 0.7 0.8 0.6 0.8 0.6 0.5HPMC E5 2.1 2.1 2.3 2.0 1.7 1.2 HPMC E5 1.2 1.2 1.2 1.1 1.2 1.1HPMC E15 1.7 1.7 1.8 1.7 1.5 0.9 HPMC E15 1.1 1.1 1.1 1.1 1.2 1.0HPMC E50 1.7 1.7 1.4 1.7 1.1 0.9 HPMC E50 0.8 0.8 0.8 0.8 0.8 0.8
Solid Dispersions PreparationSolid dispersion was prepared by using a spray dryingprocess. This process has many factors, including: inlettemperature, flow rate, solvent selection, nozzle size, dry-ing gas flow rate and feed concentration, all which affectthe properties of the spray dried dispersion. However, inorder to minimize variation parameters, the inlet temper-ature, flow rate, solvent, drying gas flow rate and noz-zle size were kept constant. The feed concentration waschanged in order to increase processability. Since PVP
J. Pharm. Sci. Pharmacol. 2, 259–276, 2015 265

Delivered by Ingenta to: Guest UserIP: 75.108.130.155 On: Mon, 30 May 2016 16:05:42
Copyright: American Scientific Publishers
Evaluating the Effects of Different Molecular Weights of Polymers Chaudhari and Dave
Table V. % yield of FENO-SD.
25% w/w 10% w/w 5% w/wdrug loading drug loading drug loading
% yield % yield % yieldPolymer (n = 3) Stdev (n = 3) Stdev (n = 3) Stdev
PVP K12 50 1.58 75 3.02 73 1.97PVP K29 70 1.47 71 1.20 72 2.57HPMC E5 58 1.59 59 0.51 57 0.43HPMC E15 42 1.57 51 0.69 43 0.21HPMC E50 28 0.85 36 0.69 35 0.55
K90 and HPMC E50 have very high viscosity, we reducedthe feed concentration to 1% w/v for PVP K90 and to 2%w/v for HPMC E50. Despite reducing the feed concentra-tion, the processing parameters were inadequate for PVPK90 and we did not get any product; thus the PVP K90dispersion was not prepared. The % yield for FENO isgiven in Table V. It is seen from Table V that the yieldof HPMC E50 is less, as compared to HPMC E15 and E5in the HPMC family and the yield of PVP K12 is less,as compared to K29. The viscosity of PVP K12 is lessas compared to PVP K29, and hence, in a dilute solu-tion very small particles formed and which escaped thecyclone and got deposited on the filter (Patel et al., 2013;Patel et al., 2014). This explains the reason for the lowyield for PVP K12. The yield for HPMC E5 is higher ascompared to HPMC E15. HPMC E15 has a higher vis-cosity as compared to HPMC E5 and hence, the viscosityof the feed solution is higher for E15. Due to the higherviscosity, most of the dispersion was lost due to stick-ing in the drying chamber of the spray dryer and hencewe get lower yield. HPMC E50 shows very low yield ascompared to other HPMC E5, HPMC E50, the feed con-centration of HPMC E50 was reduced to 1% w/v for better
Figure 4. Overlay of MDSC thermogram of FENO-SD (a) HPMC-SD (b) PVP-SD.
Figure 5. The effect of increase in drug loading on the glasstransition temperature of drug: polymer SD.
processability. However, owing to the high viscosity of theHPMC E50, most of the dispersion was lost on dryingchamber of the spray dryer.
CHARACTERIZATION OF SOLIDDISPERSIONSModulated Differential Scanning Calorimetry andPowder X-ray DiffractionMany factors, such as glass transition temperature, plas-ticization, storage temperature and humidity, as wellas viscosity play an important role in determining thekinetic stability of high energy amorphous solid dosageforms. Since FENO is prone to spontaneous recrystalliza-tion, efforts were employed to physically stabilize FENOthrough spray dried solid dispersion. In this study var-ious polymers were evaluated as potential inhibitors ofrecrystallization.Three different grades of HPMC and PVP were investi-
gated as stabilizing agents based on their molecular weight
266 J. Pharm. Sci. Pharmacol. 2, 259–276, 2015

Delivered by Ingenta to: Guest UserIP: 75.108.130.155 On: Mon, 30 May 2016 16:05:42
Copyright: American Scientific Publishers
Chaudhari and Dave Evaluating the Effects of Different Molecular Weights of Polymers
Figure 6. The effect of temperature and humidity on FENO-HPMC SD at 5% drug loading.
and three different drug loading i.e., 25%, 10% and 5%w/w was used to test these polymers. All the polymersused were able to convert crystalline FENO into an amor-phous form, which is evident from the MDSC thermo-grams, as noted in Figure 4. The PXRD was performed
Figure 7. DSC thermogram of FENO-HPMC solid dispersions at different drug loading and its stability samples stored in 40 �C% 75% RH for 1 week, 2 weeks, and 6 weeks.
on solid dispersion to check for presence of crystallinefenofibrate. The PXRD diffraction pattern did not showthe presence of crystalline FENO in the solid dispersionand stability samples (data not shown).It is well known that as the polymer content increases,
the tg increases, as seen in Figure 5. HPMC E5 showslower tg as compared to E15 and E50, which can beexplained due to the molecular weight difference betweenHPMC polymers. It was demonstrated that the storage ofindomethacin at 40–50 �C below tg prevents crystalliza-tion for long periods of time (Yoshioka, 1995); hence, thehigher tg of dispersion is desired. At 5% of drug load-ing, all HPMC polymers retain FENO in the amorphousstate even after storage at an elevated temperature andhumidity (40 �C and 75% RH); however, a decrease intg was observed, see Figure 6. The tg of HPMC E5 dis-persion with 5% FENO shows a decrease in tg to 109.55after 6 weeks of storage; however, E50 doesn’t showsmuch change in tg. Figures 7 and 8, show an overlayof DSC thermogram of FENO-polymer SD at differentdrug loading and their stability samples for HPMC and
J. Pharm. Sci. Pharmacol. 2, 259–276, 2015 267

Delivered by Ingenta to: Guest UserIP: 75.108.130.155 On: Mon, 30 May 2016 16:05:42
Copyright: American Scientific Publishers
Evaluating the Effects of Different Molecular Weights of Polymers Chaudhari and Dave
PVP, respectively. In Figure 7, it can be seen that at 10%w/w drug loading, HPMC E5 shows a decrease in tg after1 week of storage samples; after that, 2 tg are observed,which indicates immiscibility and we can see crystallineFENO peak in 6 week samples. All the polymers wereable to form an amorphous form of FENO at 10% w/wand below drug loading. At 25% w/w, drug loading all theSD forms were crystalline characterized by peak near thedrug melting point as seen in Figures 7 and 8.PVP polymer shows similar results as HPMC; an
increase in tg is observed as we increase the polymer con-tent for both grades of PVP, as seen in Figure 5. FENO-PVP K29 SD with 5% w/w drug loading shows tg of155 �C, which is the highest tg obtained for both HPMCand the PVP series. However, the drug crystallizes out after1 week of storage at 40 �C and 75% RH. From Figure 8,it is seen that PVP SD is more susceptible to temperatureand humidity than HPMC SD.The percent crystallinity of the SD and its stability sam-
ples were calculated by Eq. (3) by pan (Pan et al., 2006;Pan et al., 2008)
Crystallinity of drug substance
= �A/Wt�/H×100% (4)
Where, A = area under the melting endotherm, Wt =amount of FENO in the solid dispersion as measured byHPLC and H = heat of fusion of pure crystalline FENO.The % crystallinity was calculated for all the SD pre-
pared and shown in Figure 9. The SD prepared with25% w/w drug loading shows 6% of crystallinity initially
Figure 8. DSC thermogram of FENO-PVP solid dispersions at different drug loading and its stability samples stored in 40 �C %75% RH for 1 week, 2 weeks, and 6 weeks.
Figure 9. FENO crystallinity in spray dried dispersions dur-ing 6 weeks stability.
for HPMC E5 and 7% crystallinity for HPMC E15 andE50. After storage of the samples, crystallinity increasesin all HPMC grades used; however, E15 and E50 weremost affected with 15% crystallinity in the 6-week storage
268 J. Pharm. Sci. Pharmacol. 2, 259–276, 2015

Delivered by Ingenta to: Guest UserIP: 75.108.130.155 On: Mon, 30 May 2016 16:05:42
Copyright: American Scientific Publishers
Chaudhari and Dave Evaluating the Effects of Different Molecular Weights of Polymers
samples. HPMC dispersions with 10% w/w were amor-phous in nature initially after storage while FENO crys-tallizes out and shows 9% crystallinity in E50, and 7%crystallinity in E15. E5 dispersion were amorphous at10% w/w of drug loading even after storage. Increas-ing the HPMC content further in dispersion increases thestability with no crystallinity detected after 6 weeks ofstorage.
PVP K12 forms amorphous dispersion at all the drugloading studied; however, they were susceptible to elevatedtemperature and humidity and show 18% crystallinity in6 weeks of storage at 25% w/w drug loading and 11%crystallinity at 10% and 5% w/w drug loading. In PVPpolymer PVP K12, dispersion was most affected. This maybe due to lower viscosity of PVP K12 as compared toPVP K29.
Figure 10. FTIR spectra of FENO-HPMC physical mixtures and solid dispersions at different drug loading.
Fourier Transform Infrared SpectroscopyTo investigate the mechanism of forming amorphous soliddispersion, FTIR was used to investigate potential inter-actions between FENO and polymeric excipients. In highenergy form of drug i.e., SD, the interactions between thedrug and polymers may be relevant to stabilization of theSD (Wang et al., 2009). FENO has four functional groupsthat can act as proton acceptors: they have two hydroxylgroups ((O–H) groups) and two oxygen atoms of carbonyl(C O) but they lack a proton donor. Some scientist havereported that FENO forms hydrogen bonds with polymerstypically used in the pharmaceutical industry (Yun et al.,2006). The less resolved peaks and broader band shapesin the FTIR spectra of FENO SD suggest the presence ofamorphous FENO (Heinz et al., 2009). Crystalline FENOshows that carbonyl stretching peaks at 1729 cm−1 and
J. Pharm. Sci. Pharmacol. 2, 259–276, 2015 269

Delivered by Ingenta to: Guest UserIP: 75.108.130.155 On: Mon, 30 May 2016 16:05:42
Copyright: American Scientific Publishers
Evaluating the Effects of Different Molecular Weights of Polymers Chaudhari and Dave
1652 cm−1. A shift in peak position indicates the strengthof the FENO-polymer hydrogen bonding (Lynne S. Taylor,1997). Physical mixtures (PM) of FENO-HPMC do notshow any change in peak positions at all drug loading,i.e., no interaction between FENO and HPMC, as shownin Figure 10. SD formed at 25% w/w drug loading in allHPMC polymers show peak at 1730 cm−1 and 1652 cm−1,which is almost the same as crystalline FENO. Higherpolymer content shows a larger shift. At 10% w/w drugloading, there is peak at 1733 cm−1 and 1657 cm−1 forall HPMC polymer, as shown in Figure 10. SD containing5% w/w FENO shows a similar peak shift to SD con-taining 10% w/w FENO. From this data we can concludethat FENO shows hydrogen bonding with the polymer atthe higher polymer content, i.e., 10% and 5% w/w drugloading, owing to the larger spectral shifts. However, itshould be noted that all grades of HPMC show similarresults. These dispersions, when subjected to elevated tem-perature and humidity, show a decrease in spectral shiftas compared to the initial shifts indicating a breakageof the hydrogen bonds. From Figure 11, it is seen thatafter 6 weeks at 25% w/w drug loading, the spectral shiftdecreases to 1729 cm−1 and 1652 cm−1, which is the sameas that of crystalline FENO. HPMC E15 and E50 SDcontaining 10% w/w FENO show a decrease in spectralshift to 1731 cm−1 and 1653 cm−1 after 6 weeks, andHPMC E5 SD shows peaks at 1720 cm−1 and 1656 cm−1.
Figure 11. FTIR spectra of FENO-HPMC solid dispersions at different drug loading and their stability at 1 week, 2 weeks, and6 weeks of storage in 40 �C % 75% RH.
The spectral shift is less in stability samples of E5 ascompared to E15 and E50. This data supports the fact thatE15 and E50 show greater crystallinity as compared to E5.IR spectra of physical mixtures of FENO-PVP are
shown in Figures 12(a and b). PVP shows peak at1678 cm−1. It is seen that at 25% w/w drug loading, thephysical mixture of FENO-PVP shows peak same as thatof FENO. However, when we increase the polymer contentpeak at 1729 cm−1 and 1652 cm−1 is seen as mere shoulderin IR spectra of both the grades of PVP. SD of FENO-PVPshows the larger spectral shift to 1679 cm−1 at 5% w/wdrug loading. The IR spectra of FENO-PVP, looks similarto that of the PVP polymer, as seen in Figure 12. The shiftin FENO-PVP SD shows a similar trend as that of HPMC.As we increase the drug content in SD, the spectral shiftdecreases; peak at 1729 cm−1 in FENO is seen as shoulderat 5% w/w and at 10% w/w drug loading. A decrease inthe spectral shift is observed when these dispersions aresubjected to elevated temperature and humidity, as notedin Figure 13. The 1652 cm−1 is seen as shoulder after1 week of storage in FENO-PVP at 25% w/w drug load-ing; however, no such peak is seen at 10% or 5% w/wdrug loading.Some reports have demonstrated that drug-polymer
interactions are important in order to stabilize solid disper-sion (Lynne S. Taylor, 1997). It was found that the crys-tallization tendency of a series of benzodiazepines with
270 J. Pharm. Sci. Pharmacol. 2, 259–276, 2015

Delivered by Ingenta to: Guest UserIP: 75.108.130.155 On: Mon, 30 May 2016 16:05:42
Copyright: American Scientific Publishers
Chaudhari and Dave Evaluating the Effects of Different Molecular Weights of Polymers
Figure 12. FTIR spectra of FENO-PVP physical mixtures and solid dispersions at different drug loading.
different functional groups was prevented only when thecompound was able to form a hydrogen bond with the car-rier (phospholipid) (Konno and Taylor, 2006). It has alsobeen argued that the anti-plasticizing effect of the polymerplays an important role in the stabilization of amorphousdrugs, in contrast to drug-polymer interactions. As the vis-cosity of the drug polymer system is increased, the dif-fusion of drug molecules is inhibited, which is necessaryfor recrystallization (Van den Mooter et al., 2001). In the
Figure 13. FTIR spectra of FENO-PVP solid dispersions at different drug loading and their stability at 1 week, 2 weeks, and6 weeks of storage in 40 �C% 75% RH.
present study, the type of polymer and drug/polymer ratio,rather than hydrogen bonding, affected the amorphouscharacter of FENO, since amorphous FENO in FENO-PVP dispersion was the least physically stable.
Dissolution StudiesThe solubility of FENO in blank FaSSIF with 1% w/v SLSis 337 �g/ml±14 �g/ml at 37 �C. The dose of FENO inboth the release media corresponds to a theoretical con-
J. Pharm. Sci. Pharmacol. 2, 259–276, 2015 271

Delivered by Ingenta to: Guest UserIP: 75.108.130.155 On: Mon, 30 May 2016 16:05:42
Copyright: American Scientific Publishers
Evaluating the Effects of Different Molecular Weights of Polymers Chaudhari and Dave
centration of 100 �g/ml. In FaSSIF, 100 �g/ml representsa supersaturated state. In blank FaSSIF with 1% SLS, sol-ubility of FENO is 337 �g/ml±14 �g/ml at 37 �C, with afinal concentration of 100 �g/ml, sink condition was thusmaintained throughout the experiment.Dissolution results could be well predicted with DSC
and FTIR analysis. The dissolution profile of SD of FENOwith HPMC and PVP, along with the stability sample inblank FaSSIF with 1% SLS and FASSIF, is shown inFigures 14–17. Comparing the dissolution profile of 25%FENO with HPMC polymer in sink conditions, shows thatas the viscosity increases there is a decrease in the rateof the release of the drug, Figure 14. The release fromHPMC E50 is the slowest in all the drug loading, fol-lowed by HPMC E15 and E5. An increase in the rate ofdrug release is observed from 25% to 10% w/w FENO forall the HPMC polymers. However, when drug loading isincreased further, the rate of drug release slows down ini-tially. A DSC thermogram of these SD shows that SD with10% w/w FENO is amorphous in nature; increasing thepolymer concentration increases the stability of the disper-sions. However, in sink conditions it is shown to slow thedissolution as compared to SD with 10% FENO. In non-sink conditions, Figure 15, it is seen that at 25% w/w drug
Figure 14. Comparison of dissolution profiles of FENO SD, PM and 6 weeks stability samples using various HPMC polymers atdifferent drug loading in blank FaSSIF with 1% SLS.
loading, the final concentration released in the media is amere 11.6 �g/ml for HPMC E5 SD, 11.4 �g/ml for E15,and 9 �g/ml for E50. The release from SD with 25% w/wFENO is expected to be less as the drug was in crystallineform. The release profile was improved in SD with 10%w/w FENO; the maximum concentration achieved was57 �g/ml for E5, 40 �g/ml for E15 and 33 �g/ml for E50.It is seen that E5 dispersion has a better release profile ascompared to E15 and E50. Further increasing the polymercontent in SD does not show slowing of the drug releaseas seen in non-sink conditions. Overall, after the samplesare kept in stability, the release profile is decreased in bothsink as well as non-sink conditions.FENO-PVP SD follows same trend as FENO-HPMC
SD. In blank FaSSIF with SLS, Figure 16 PVP K12 showsbetter release when compared to PVP K29. PVP K12shows 100% release in contrast to 90% release of PVPK29 at 10% drug loading. However, when these disper-sions were tested in FaSSIF, Figure 17, PVP K12, releaseis slow as compared to PVP K29. At 25% drug load-ing, K12 shows only 19% release, and at the same time,K29 shows 40% drug release. As we increase the polymercontent, the drug release increases up to 10% w/w drugloading; increasing the polymer content further does not
272 J. Pharm. Sci. Pharmacol. 2, 259–276, 2015

Delivered by Ingenta to: Guest UserIP: 75.108.130.155 On: Mon, 30 May 2016 16:05:42
Copyright: American Scientific Publishers
Chaudhari and Dave Evaluating the Effects of Different Molecular Weights of Polymers
Figure 15. Comparison of dissolution profiles of FENO SD, PM and 6 weeks stability samples using various HPMC polymers atdifferent drug loading in FaSSIF.
show much change in the release profile. With regard tostability, PVP SD shows a slowing of the drug releasedue to crystallization of the FENO, which was also shownby MDSC.
Figure 16. Comparison of dissolution profiles of FENO SD, PM and 6 weeks stability samples using various PVP polymers atdifferent drug loading in blank FaSSIF with 1% SLS.
Particle SizeThe particle size of FENO-SD is given in Table VI. FENOhas a mean particle size of 16.69 �g± 0.03 �g. In gen-eral the particle size was reduced to a certain extent in
J. Pharm. Sci. Pharmacol. 2, 259–276, 2015 273

Delivered by Ingenta to: Guest UserIP: 75.108.130.155 On: Mon, 30 May 2016 16:05:42
Copyright: American Scientific Publishers
Evaluating the Effects of Different Molecular Weights of Polymers Chaudhari and Dave
Figure 17. Comparison of dissolution profiles of FENO SD, PM and 6 weeks stability samples using various PVP polymers atdifferent drug loading in FaSSIF.
FENO SD. Since the parameters used for spray drying wassame, there is little difference in the particle size of differ-ent drug loaded dispersions. FENO is known for increasingits solubility on the reduction of the particle size to nano-crystal range (Zuo et al., 2013). However, spray dried dis-persion does not reduce particle size in nano range; thereis only a slight reduction of particle size as compared tothe pure drug, and hence, the difference in release patternand stability of solid dispersion in different solid disper-sion formed is due to the polymer itself.
SUMMARYIn this study, two types of experiments are performed onFENO: non-formulated drug and formulated drug. In nonformulated drug, two methods have been used: solventshift method and solvent casting method. In these methodability to attain and maintain supersaturation in presence ofdifferent molecular weight PVP and HPMC was studied.The solvent casts prepared were subjected to drug releasetesting and were also stored at elevated temperature and
Table VI. Mean particle size of FENO-SD.
25% 10% 5%
Particle Particle Particlesize size size(�g) (�g) (�g)
Polymer (n = 3) Stdev (n = 3) Stdev (n = 3) Stdev
PVP K12 18.41 0�17 15.17 0.12 15.09 0.09PVP K29 17.90 17�75 12.40 0.01 16.33 0.98HPMC E5 14.34 0�03 13.46 0.02 13.53 0.08HPMC E15 14.43 0�07 13.79 0.02 13.96 0.05HPMC E50 14.43 0�19 13.53 0.02 13.77 0.25
humidity. In formulated drug, solid dispersions were pre-pared using different molecular weight PVP and HPMC.Solid dispersions were prepared at 25%, 10% and 5% w/wdrug loading. These dispersions were also subjected to ele-vated temperature and humidity. Physical characterizationslike mDSC, IR, PXRD and dissolution studies were per-formed on these dispersions.
CONCLUSIONIn this study we examined the anti-precipitant effect ofpolymers using the solvent shift and the solvent castingmethod. PVP and HPMC were able to generate supersat-uration in simulated intestinal fluid by the solvent shiftmethod. However, the rapid crystallization tendency lim-its the stabilization of the supersaturated solution whenFENO concentration drops below saturation solubility atthe end of experiment. The polymers were able to main-tain a supersaturated state of FENO by the solvent castingmethod. PVP K29 and HPMC E5 casts showed immedi-ate drug release as compared to other grades used in theirrespective categories. HPMC E5 shows superior resultsas compared to PVP K29. However, these casts are vul-nerable to elevated temperature and humidity. HPMC E5casts show better stability than the other polymers used.Spray dried dispersions of these polymers were prepared.Although there are many factors, like inlet temperature,flow rate, solvent selection, drying gas flow rate, nozzlesize and feed concentration that affect the properties ofspray dried dispersions, we tried to minimize the variationsby adjusting the fixed inlet temperature, drying gas, flowrate constant and feed concentration to obtain product.Analysis revealed that FENO is converted to an amorphousform at 10% w/w and below drug loading. DSC analysis
274 J. Pharm. Sci. Pharmacol. 2, 259–276, 2015

Delivered by Ingenta to: Guest UserIP: 75.108.130.155 On: Mon, 30 May 2016 16:05:42
Copyright: American Scientific Publishers
Chaudhari and Dave Evaluating the Effects of Different Molecular Weights of Polymers
revealed that 5% w/w, drug loaded HPMC SD was stableeven at 40 �C and 75% RH. The SD prepared using PVPshows immiscibility in storage. FTIR analysis revealed thattypes of polymer and drug polymer ratios affect the amor-phous nature of FENO. High viscosity polymer inhibits thediffusion of FENO and increases stability. Non-sink mediaused for dissolution gives a better correlation than sinkmedia in FENO dispersions. This study will help scientistto understand the effect of the molecular weight of HPMCand PVP on neutral drugs with very low water solubility.
AbbreviationsHPMC—Hydroxypropyl methyl cellulosePVP—PolyvinylpyrrolidoneBCS—Biopharmaceutics classification systemFENO—FenofibrateNaH2PO4—Sodium dihydrogen phosphateNaCl—Sodium chlorideNaOH—Sodium hydroxideHCl—Hydrochloric acidFaSSIF—Fasted state simulated intestinal fluidDMSO—Dimethyl sulfoxideHPLC—High performance liquid chromatographySLS—Sodium lauryl sulfateRH—Relative humidityMDSC—Modulated differential scanning calorimetryPXRD—Powder X-ray diffractionFTIR—Fourier transform infrared spectroscopyRT—Room temperatureDS—Degree of supersaturationEGF—Excipient gain factorPM—Physical mixture.
REFERENCESAkhlaquer Rahman, M., Harwansh, R., Aamir Mirza, M., Hussain, S.,and Hussain, A. (2011). Oral lipid based drug delivery system (LBDDS):Formulation, characterization and application: A review. Current DrugDelivery 8, 330–345.
Al-Nemrawi, N. K. and Dave, R. H. (2014). Formulation and charac-terization of acetaminophen nanoparticles in orally disintegrating films.Drug Delivery 1–10.
Alhalaweh, A., Andersson, S., and Velaga, S. P. (2009). Preparation ofzolmitriptan–chitosan microparticles by spray drying for nasal delivery.European J. Pharm. Sci. 38, 206–214.
Bouchard, A., Jovanovic, N., Hofland, G. W., Jiskoot, W., Mendes, E.,Crommelin, D. J. A., and Witkamp, G.-J. (2008). Supercritical fluiddrying of carbohydrates: Selection of suitable excipients and processconditions. European Journal of Pharmaceutics and Biopharmaceutics68, 781–794.
Brittain, H. G. (2002). Effects of mechanical processing on phase com-position. J. Pharm. Sci. 91, 1573–1580.
Cheng, Y., Ye, X., Huang, X. D. and Ma, H. R. (2006). Reentrant wet-ting transition on surfactant solution surfaces. The Journal of ChemicalPhysics 125, 164709.
Dave, R. H., Patel, A. D., Donahue, E., and Patel, H. H. (2012). Toevaluate the effect of addition of an anionic surfactant on solid dispersionusing model drug indomethacin. Drug Dev. Ind. Pharm. 38, 930–939.
Davis, A. F. and Hadgraft, J. (1991). Effect of supersaturation on mem-brane transport: 1. Hydrocortisone acetate. Int. J. Pharm. 76, 1–8.
De Waard, H., Hinrichs, W. L. J., and Frijlink, H. W. (2008). A novelbottom–up process to produce drug nanocrystals: Controlled crystalliza-tion during freeze-drying. J. Controlled Release 128, 179–183.
Dinunzio, J. C., Brough, C., Hughey, J. R., Miller, D. A., Williams, III,R. O., and Mcginity, J. W. (2010). Fusion production of solid dispersionscontaining a heat-sensitive active ingredient by hot melt extrusion andKinetisol® dispersing. European Journal of Pharmaceutics and Biophar-maceutics 74, 340–351.
Friesen, D. T., Shanker, R., Crew, M., Smithey, D. T., Curatolo, W. J.,and Nightingale, J. A. S. (2008). Hydroxypropyl methylcellulose acetatesuccinate-based spray-dried dispersions: An overview. Molecular Phar-maceutics 5, 1003–1019.
Gao, P. and Morozowich, W. (2006). Development of supersaturatableself-emulsifying drug delivery system formulations for improving the oralabsorption of poorly soluble drugs. Expert Opinion on Drug Delivery3, 97–110.
Gao, P., Guyton, M. E., Huang, T., Bauer, J. M., Stefanski, K. J., andLu, Q. (2004). Enhanced oral bioavailability of a poorly water solu-ble drug PNU-91325 by supersaturatable formulations. Drug Dev. Ind.Pharm. 30, 221–229.
Górniak, A., Wojakowska, A., Karolewicz, B., and Pluta, J. (2011). Phasediagram and dissolution studies of the fenofibrate–acetylsalicylic acidsystem. J. Therm. Anal. Calorim. 104, 1195–1200.
Guichard, J. P., Blouquin, P., and Qing, Y. (2000). A new formulationof fenofibrate: Suprabioavailable tablets. Current Medical Research andOpinion 16, 134–138.
Guivarc’h, P.-H., Vachon, M. G., and Fordyce, D. (2004). A newfenofibrate formulation: Results of six single-dose, clinical studies ofbioavailability under fed and fasting conditions. Clinical Therapeutics26, 1456–1469.
Guzman, H., T. M., Zhang Z., Ratanabanangkoon, P., Shaw, P.,Gardner, C., Chen, H., Moreau, J., Almarsson, O., and Remenar, J.(2004). A “spring and parachute” approach to designing solid celecoxibformulations having enhanced oral absorption. AAPS J, T2189.
Guzmán, H. R., Tawa, M., Zhang, Z., Ratanabanangkoon, P., Shaw, P.,Gardner, C. R., Chen, H., Moreau, J.-P., Almarsson, Ö., and Remenar,J. F. (2007). Combined use of crystalline salt forms and precipitationinhibitors to improve oral absorption of celecoxib from solid oral formu-lations. J. Pharm. Sci. 96, 2686–2702.
Hadgraft, J. (1999). Passive enhancement strategies in topical and trans-dermal drug delivery. Int. J. Pharm. 184, 1–6.
Hancock, B. C. and Zografi, G. (1997). Characteristics and significance ofthe amorphous state in pharmaceutical systems. J. Pharm. Sci. 86, 1–12.
Heinz, A., Gordon, K. C., Mcgoverin, C. M., Rades, T., and Strachan,C. J. (2009). Understanding the solid-state forms of fenofibrate—A spec-troscopic and computational study. European Journal of Pharmaceuticsand Biopharmaceutics 71, 100–108.
Hugo, M., Kunath, K., and Dressman, J. (2013). Selection of excipient,solvent and packaging to optimize the performance of spray-dried formu-lations: Case example fenofibrate. Drug Dev. Ind. Pharm. 39, 402–412.
Keary, C. M. (2001). Characterization of METHOCEL cellulose ethersby aqueous SEC with multiple detectors. Carbohydr. Polym. 45, 293–303.
Khougaz, K. and Clas, S.-D. (2000). Crystallization inhibition in soliddispersions of MK-0591 and poly(vinylpyrrolidone) polymers. J. Pharm.Sci. 89, 1325–1334.
Konno, H. and Taylor, L. S. (2006). Influence of different polymers on thecrystallization tendency of molecularly dispersed amorphous felodipine.J. Pharm. Sci. 95, 2692–2705.
J. Pharm. Sci. Pharmacol. 2, 259–276, 2015 275

Delivered by Ingenta to: Guest UserIP: 75.108.130.155 On: Mon, 30 May 2016 16:05:42
Copyright: American Scientific Publishers
Evaluating the Effects of Different Molecular Weights of Polymers Chaudhari and Dave
Lakshman, J. P., Cao, Y., Kowalski, J., and Serajuddin, A. T. M. (2008).Application of melt extrusion in the development of a physically andchemically stable high-energy amorphous solid dispersion of a poorlywater-soluble drug. Molecular Pharmaceutics 5, 994–1002.
Lipinski, C. A., Lombardo, F., Dominy, B. W., and Feeney, P. J. (1997).Experimental and computational approaches to estimate solubility andpermeability in drug discovery and development settings. Advanced DrugDelivery Reviews 23, 3–25.
Lynne S. Taylor, G. Z. (1997). Spectroscopic characterization of inter-actions between PVP and indomethacin in amorphous molecular disper-sions. Pharm. Res. 14, 1691–1698.
Moya-Ortega, M., Messner, M., Jansook, P., Nielsen, T., Wintgens, V.,Larsen, K., Amiel, C., Sigurdsson, H., and Loftsson, T. (2011). Drugloading in cyclodextrin polymers: Dexamethasone model drug. Journalof Inclusion Phenomena and Macrocyclic Chemistry 69, 377–382.
Müller, R. H., Gohla, S., and Keck, C. M. (2011). State of the art ofnanocrystals—Special features, production, nanotoxicology aspects andintracellular delivery. European Journal of Pharmaceutics and Biophar-maceutics 78, 1–9.
Munson, E. J. (2009). Analytical techniques in solid-state characteri-zation. Developing Solid oral Dosage Forms, edited by Qiu, Y. Y. C.,Zhang, G. G. Z., Liu, L., Yu, L., Venkatramana Rao, and Porter, W.,Academic Press, San Diego.
Murdande, S. B., Shah, D. A., and Dave, R. H. (2015). Impact of nanosiz-ing on solubility and dissolution rate of poorly soluble pharmaceuticals.J. Pharm. Sci. 104, 2094–2102.
Pan, X., Julian, T., and Augsburger, L. (2006). Quantitative measurementof indomethacin crystallinity in indomethacin-silica gel binary systemusing differential scanning calorimetry and X-ray powder diffractometry.AAPS PharmSciTech. 7, E72–E78.
Pan, X., Julian, T., and Augsburger, L. (2008). Increasing the dissolutionrate of a low-solubility drug through a crystalline-amorphous transition:A case study with indomethicin. Drug Dev. Ind. Pharm. 34, 221–231.
Patel, A. D., Agrawal, A., and Dave, R. H. (2013). Development ofpolyvinylpyrrolidone-based spray-dried solid dispersions using responsesurface model and ensemble artificial neural network. J. Pharm. Sci.102, 1847–1858.
Patel, A. D., Agrawal, A., and Dave, R. H. (2014). Investigation of theeffects of process variables on derived properties of spray dried solid-dispersions using polymer based response surface model and ensembleartificial neural network models. European Journal of Pharmaceutics andBiopharmaceutics 86, 404–417.
Peeters, J., Neeskens, P., Tollenaere, J. P., Van Remoortere, P.,and Brewster, M. E. (2002). Characterization of the interaction of2-hydroxypropyl-�-cyclodextrin with itraconazole at pH 2, 4, and 7.J. Pharm. Sci. 91, 1414–1422.
Peltonen, L. and Hirvonen, J. (2010). Pharmaceutical nanocrystals bynanomilling: Critical process parameters, particle fracturing and stabi-lization methods. J. Pharm. Pharmacol. 62, 1569–1579.
Raghavan, S. L., Schuessel, K., Davis, A., and Hadgraft, J. (2003). For-mation and stabilisation of triclosan colloidal suspensions using super-saturated systems. Int. J. Pharm. 261, 153–158.
Raghavan, S. L., Trividic, A., Davis, A. F., and Hadgraft, J. (2001).Crystallization of hydrocortisone acetate: Influence of polymers. Int. J.Pharm. 212, 213–221.
Rodríguez-Hornedo, N. and Murphy, D. (1999). Significance of control-ling crystallization mechanisms and kinetics in pharmaceutical systems.J. Pharm. Sci. 88, 651–660.
Ruben, A. J., Kiso, Y., and Freire, E. (2006). Overcoming roadblocks inlead optimization: A thermodynamic perspective. Chemical Biology andDrug Design 67, 2–4.
Sanganwar, G. P. and Gupta, R. B. (2008). Dissolution-rate enhancementof fenofibrate by adsorption onto silica using supercritical carbon dioxide.Int. J. Pharm. 360, 213–218.
Sauron, R., Wilkins, M., Jessent, V., Dubois, A., Maillot, C., and Weil, A.(2006). Absence of a food effect with a 145 mg nanoparticle fenofibratetablet formulation. International Journal of Clinical Pharmacology andTherapeutics 44, 64–70.
Serajuddin, A. T. M. (1999). Solid dispersion of poorly water-solubledrugs: Early promises, subsequent problems, and recent breakthroughs.J. Pharm. Sci. 88, 1058–1066.
Serajuddin, A. T. M. (2007). Salt formation to improve drug solubility.Advanced Drug Delivery Reviews 59, 603–616.
Singh, A., Worku, Z. A., and Van Den Mooter, G. (2011). Oral formula-tion strategies to improve solubility of poorly water-soluble drugs. ExpertOpinion on Drug Delivery 8, 1361–1378.
Stella, V. J. and Nti-Addae, K. W. (2007). Prodrug strategies to overcomepoor water solubility. Advanced Drug Delivery Reviews 59, 677–694.
Strickley, R. (2004). Solubilizing excipients in oral and injectable formu-lations. Pharm. Res. 21, 201–230.
Thakuria, R., Delori, A., Jones, W., Lipert, M. P., Roy, L., and Rodríguez-Hornedo, N. (2013). Pharmaceutical cocrystals and poorly soluble drugs.Int. J. Pharm. 453, 101–125.
Van Den Mooter, G., Wuyts, M., Blaton, N., Busson, R., Grobet, P.,Augustijns, P., and Kinget, R. (2001). Physical stabilisation of amorphousketoconazole in solid dispersions with polyvinylpyrrolidone K25. Euro-pean J. Pharm. Sci. 12, 261–269.
Vertzoni, M., Fotaki, N., Nicolaides, E., Reppas, C., Kostewicz, E.,Stippler, E., Leuner, C., and Dressman, J. (2004). Dissolution media sim-ulating the intralumenal composition of the small intestine: Physiologicalissues and practical aspects. J. Pharm. Pharmacol. 56, 453–462.
Vogt, M., Kunath, K., and Dressman, J. B. (2008). Dissolutionenhancement of fenofibrate by micronization, cogrinding and spray-drying: Comparison with commercial preparations. European Journal ofPharmaceutics and Biopharmaceutics 68, 283–288.
Wang, R., Pellerin, C., and Lebel, O. (2009). Role of hydrogen bondingin the formation of glasses by small molecules: A triazine case study.J. Mater. Chem. 19, 2747–2753.
Yang, W., Tam, J., Miller, D. A., Zhou, J., Mcconville, J. T., Johnston,K. P., and Williams III, R. O. (2008). High bioavailability from nebu-lized itraconazole nanoparticle dispersions with biocompatible stabilizers.Int. J. Pharm. 361, 177–188.
Yoshioka, M., Hancock, B. C., and Zografi (1995). Inhibition ofindomethacin crystallization in poly(vinylpyrrolidone) coprecipitates.J. Pharm. Sci. 84, 983–986.
Yun, H.-Y., Lee, E., Chung, S., Choi, S.-O., Kim, H., Kwon, J.-T., kang,W., and Kwon, K.-I. (2006). The effects of food on the bioavailability offenofibrate administered orally in healthy volunteers via sustained-releasecapsule. Clinical Pharmacokinetics 45, 425–432.
Zhou, D., Zhang, G. G. Z., Law, D., Grant, D. J. W., and Schmitt,E. A. (2002). Physical stability of amorphous pharmaceuticals: Impor-tance of configurational thermodynamic quantities and molecular mobil-ity. J. Pharm. Sci. 91, 1863–1872.
Zuo, B., Sun, Y., Li, H., Liu, X., Zhai, Y., Sun, J., and He, Z. (2013).Preparation and in vitro/in vivo evaluation of fenofibrate nanocrystals.Int. J. Pharm. 455, 267–275.
276 J. Pharm. Sci. Pharmacol. 2, 259–276, 2015

















![Particle engineering of fenofibrate for advanced drug delivery … · 2019. 12. 30. · Carr’s index and Hausner ratio were calculated using Eqs. (2) and (3)[18]. Carr0s Index ¼](https://img.dokumen.tips/doc/110x75/60b2e27f0e2d5a3eba3b5d83/particle-engineering-of-fenofibrate-for-advanced-drug-delivery-2019-12-30-carras.jpg)











