Embed Size (px)
Citation preview

of December 2, 2018.This information is current as
Expression of Homing Chemokinesand Marginal Zone Formation and Proper Essential Role of RelB in Germinal Center
Debra S. Weih, Z. Buket Yilmaz and Falk Weih
http://www.jimmunol.org/content/167/4/1909doi: 10.4049/jimmunol.167.4.1909
2001; 167:1909-1919; ;J Immunol
Referenceshttp://www.jimmunol.org/content/167/4/1909.full#ref-list-1
, 29 of which you can access for free at: cites 77 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2001 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on Decem
ber 2, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on D
ecember 2, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

Essential Role of RelB in Germinal Center and Marginal ZoneFormation and Proper Expression of Homing Chemokines1
Debra S. Weih, Z. Buket Yilmaz, and Falk Weih2
High levels of the Rel/NF-�B family member RelB are restricted to specific regions of thymus, lymph nodes, and Peyer’s patches.In spleen, RelB is expressed in periarteriolar lymphatic sheaths, germinal centers (GCs), and the marginal zone (MZ). In thisstudy, we report that RelB-deficient (relB�/�) mice, in contrast to nfkb1�/�, but similar to nfkb2�/� mice, are unable to form GCsand follicular dendritic cell networks upon Ag challenge in the spleen. RelB is also required for normal organization of the MZand its population by macrophages and B cells. Reciprocal bone marrow transfers demonstrate that RelB expression in radiation-resistant stromal cells, but not in bone marrow-derived hemopoietic cells, is required for proper formation of GCs, folliculardendritic cell networks, and MZ structures. However, the generation of MZ B cells requires RelB in hemopoietic cells. Expressionof TNF ligand/receptor family members is only moderately altered in relB�/� splenocytes. In contrast, expression of homingchemokines is strongly reduced in relB�/� spleen with particularly low mRNA levels of the chemokine B lymphocyte chemoat-tractant. Our data indicate that activation of p52-RelB heterodimers in stromal cells downstream of TNF/lymphotoxin is requiredfor normal expression of homing chemokines and proper development of spleen microarchitecture. The Journal of Immunology,2001, 167: 1909–1919.
T he adaptive immune system is characterized by the spe-cific recognition of Ags, central and peripheral tolerancefor self Ags, clonal expansion of Ag-specific lympho-
cytes, and immunological memory. In contrast to innate immuneresponses, the adaptive immune system requires highly organizedand specialized lymphoid organs to exert its function. Develop-ment and maturation of B and T lymphocytes take place in primarylymphoid organs, such as bone marrow (BM)3 and thymus,whereas adaptive immune responses to pathogens are initiated insecondary lymphoid organs, such as the spleen and lymph nodes.
NF-�B plays an important role in immune, inflammatory, andstress responses (1). Five members of this transcription factor fam-ily have been identified in vertebrates: NF-�B1 (encoding the pre-cursor molecule p105 and the processed form p50), NF-�B2 (en-coding the precursor p100 and the processed form p52), RelA(p65), RelB, and c-Rel. The DNA-binding activity of Rel/NF-�Bcomplexes is regulated by members of the I�B family, and severaldistinct I�B molecules with homologies to ankyrin repeats havebeen described. In most cell types, Rel/NF-�B proteins are trappedin the cytoplasm by the I�B inhibitors. A wide range of stimuliactivates the I�B kinase complex, resulting in the phosphorylation,
ubiquitination, and degradation of I�Bs. Consequently, the Rel/NF-�B proteins translocate to the nucleus and bind to so-called �Bsequence motifs (2–4).
The classical NF-�B activity is a p50-RelA heterodimer, butmost other possible homo- and heterodimeric complexes can occurdepending on cell type and activation status. One exception isRelB, which only dimerizes with p50 or p52 forming potent tran-scriptional activators. In the mouse, high levels of RelB expressionare restricted to specific regions of lymphoid organs, such as thethymic medulla, periarteriolar lymphatic sheaths (PALS) of thespleen, and the paracortex of lymph nodes. The basal �B-bindingactivity in thymus and spleen largely consists of p50-RelB andp52-RelB heterodimers, suggesting a role of RelB in the constitu-tive expression of �B-regulated genes in these tissues, whereasRelA and c-Rel complexes appear to be involved in the inducible�B-binding activity and gene activation (5).
The analysis of Rel/NF-�B knockout mice revealed that theseproteins have essential, but distinct roles in development and func-tion of the immune system (6, 7). Mice with a targeted disruptionof RelB display a complex phenotype, including multiorgan in-flammation and multifocal defects in immune responses. RelB-deficient mice have thymic atrophy due to a reduced population ofdendritic and medullary epithelial cells, lack clearly developedlymph nodes, and develop splenomegaly due to extramedullaryhemopoiesis in the red pulp (8–11). Humoral responses in RelB-deficient mice may be impaired due to an abnormal microarchi-tecture of the spleen, which does not support proper germinal cen-ter (GC) and marginal zone (MZ) formation. GCs are sites ofintense B cell proliferation, selection, maturation, and death duringAb responses. Follicular dendritic cells (FDCs) are restricted to thelight zones of GCs, and their ability to trap and retain immunecomplexes (ICs) on their surfaces for long periods of time may beimportant for the maintenance of immunological memory (12).The splenic MZ is the major route of entry of Ags, APCs, andlymphocytes into the white pulp. The flow of blood from termi-nating arterioles filters past macrophages, B cells, and dendriticcells (DCs) before reaching the red pulp and rejoining circulationvia venous sinuses. Because asplenic people and animals are
Forschungszentrum Karlsruhe, Institute of Toxicology and Genetics, Karlsruhe, Ger-many
Received for publication December 14, 2000. Accepted for publication June 5, 2001.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by the Deutsche Forschungsgemeinschaft (GrantsWe2224/1-1 and We2224/2-1).2 Address correspondence and reprint requests to Dr. Falk Weih, ForschungszentrumKarlsruhe, Institute of Toxicology and Genetics, P.O. Box 3640, 76021 Karlsruhe,Germany. E-mail address: [email protected] Abbreviations used in this paper: BM, bone marrow; BLC, B lymphocyte chemoat-tractant; DAB, diaminobenzidine; DC, dendritic cell; ELC, EBV-induced molecule 1ligand chemokine; FDC, follicular DC; GC, germinal center; IC, immune complex;LT, lymphotoxin; MAdCAM-1, mucosal addressin cellular adhesion molecule-1;MMM, metallophilic marginal macrophage; MZ, marginal zone; MZM, MZ macro-phage; NIK, NF-�B-inducing kinase; PALS, periarteriolar lymphatic sheath; PNA,peanut agglutinin; SLC, secondary lymphoid organ chemokine; TD, T cell-dependent;Tg, transgenic; TI, T cell-independent; wt, wild type.
Copyright © 2001 by The American Association of Immunologists 0022-1767/01/$02.00
by guest on Decem
ber 2, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

highly susceptible to encapsulated bacteria, it is thought that theMZ may have a critical role in alerting the immune system to thesepathogens (13, 14).
In the present study, we focus our analysis on the phenotypicalchanges in the spleen microarchitecture in mice lacking p50/NF-�B1 or RelB. Several cell types could be identified that are affectedby the lack of RelB, but that appear normal in nfkb1�/� mice.Adoptive transfer experiments revealed that RelB expression inradiation-resistant stromal cells, but not in BM-derived hemopoi-etic cells, is required for the establishment of GCs, FDC networks,and MZ structures. However, the generation of MZ B cells re-quires RelB in hemopoietic cells. We also demonstrate that ex-pression of chemokines that play an important role in the organi-zation of lymphoid organs is reduced in RelB-deficient mice.
Materials and MethodsMice
Generation of nfkb1�/�, nfkb2�/�, and relB�/� mice has been describedpreviously (10, 15, 16). Analyses were performed on mice with a mixedB6 � 129 genetic background (for adoptive BM transfer experiments, seebelow). All animals were housed and bred under standardized conditionswith water and food ad libitum in a specific pathogen-free mouse facilityat the Forschungszentrum Karlsruhe, Institute of Toxicology and Genetics.
Immunizations and IC trapping
SRBCs (ACILA GMN, Walldorf, Germany) were used to elicit T cell-dependent (TD) immune responses in mice. SRBCs in Alsevers werewashed three times in PBS, and mice were injected i.p. with 100 �l 10%SRBC suspension in PBS and sacrificed 10 days later. For IC trapping,mice were immunized with SRBCs, and 6 days later injected i.v. with 200�g preformed peroxidase antiperoxidase ICs (Dako Diagnostika, Ham-burg, Germany) (17). Spleens were removed 24 h later, embedded in Poly-freeze (Polysciences, Warrington, PA), and stored at �80°C. Frozen sec-tions were cut at 10 �m and acetone fixed, and the HRP Ag was detectedby diaminobenzidine (DAB), followed by hematoxylin counterstaining andcoverslipping.
Immunohistochemical analyses
Frozen blocks were cut at 8–10 �m, and after air drying, sections werefixed in cold acetone before immunohistochemical staining. Staining forGCs was performed with biotinylated peanut agglutinin (PNA; 1/100) andvisualized with glucose oxidase reagents (Vector Laboratories, Burlin-game, CA). All other immunohistochemical staining procedures were per-formed with standard avidin/biotin peroxidase complex procedures usingproducts from Vector Laboratories. Endogenous peroxidase activity wasquenched in 0.3% hydrogen peroxide, and sections were blocked with avi-din D/biotin reagents, followed by 0.5% casein in PBS with 1.5% rat orrabbit serum, as required for appropriate blocking of nonspecific Ab bind-ing. Primary Ab incubation was either at 4°C overnight or at room tem-perature for 2 h. Appropriate biotinylated secondary anti-rat or anti-rabbitAb was diluted 1/100 and applied for 30 min. Either DAB or 3-amino-9-ethylcarbazole reagents were used for visualization of the immunostaining,followed by hematoxylin counterstaining and coverslipping. Frozen sec-tions were stained for: IgD (clone 11-26, diluted 1/200; Southern Biotech-nology Associates, Birmingham, AL); FDC-M1 (diluted 1/200; gift fromM. Kosco-Vilbois, Sereno Pharmaceutical Research Institute, Geneva,Switzerland); CR1/CD35 (clone 8C12, diluted 1/100; PharMingen, SanDiego, CA); mucosal addressin cellular adhesion molecule-1 (MAd-CAM-1, clone MECA-367, diluted 1/25; PharMingen); ER-TR9 (diluted1/100; Bachem, Heidelberg, Germany); MOMA-1 (diluted 1/50; Bachem);and ER-TR7 (diluted 1/100; Bachem). Paraffin sections were cut andstained with hematoxylin and eosin (H&E) or for RelB immunohistochem-istry with polyclonal rabbit anti-RelB IgG (C-19, diluted 1/200; Santa CruzBiotechnology, Santa Cruz, CA). For negative control slides, the primaryAb was substituted with normal mixed serum. All negative control slideswere free of staining. Micrographs were taken with a Zeiss Axioskop anda Jenoptik ProgRes 3012 digital camera system.
RNA analyses
RNA was extracted from spleen using peqGOLD TriFast reagent accordingto the manufacturer’s specifications (Peqlab Biotechnologie, Erlangen,Germany). For semiquantitative RT-PCR, 2 �g total RNA was oligo(dT)
primed and reverse transcribed using SuperScript II from Life Technolo-gies (Rockville, MD). The following PCR primers were used: TNF (5�-ATG AGC ACA GAA AGC ATG ATC-3� and 5�-TAC AGG CTT GTCACT CGA ATT-3�); lymphotoxin (LT) � (5�-ATG ACA CTG CTC GGCCGT CT-3� and 5�-CTA CAG TGC AAA GGC TCC AAA-3�); LT� (5�-TTG TTG GCA GTG CCT ATC ACT GTC C-3� and 5�-CTC GTG TACCAT AAC GAC CCG TAC-3�); LIGHT (5�-AGA CTG CTG ACC TGCTTT G-3� and 5�-CCC TTC TTT CCT CCC TTT CC-3�); TNFR-I (5�-GAA CCT ACT TGG TGA GTG AC-3� and 5�-CAC AAC TTC ATACAC TCC TC-3�); LT�R (5�-TTA TCG CAT AGA AAA CCA GAC TTGC-3� and 5�-TCA AAG CCC AGC ACA ATG TC-3�); B lymphocyte che-moattractant (BLC) (5�-ATG AGG CTC AGC ACA GCA AC-3� and 5�-CCA TTT GGC ACG AGG ATT CAC-3�); EBV-induced molecule 1 li-gand chemokine (ELC) (5�-GCC TCA GAT TAT CTG CCA T-3� and5�-AGA CAC AGG GCT CCT TCT GGT-3�); secondary lymphoid organchemokine (SLC) (5�-ATG ATG ACT CTG AGC CTC C-3� and 5�-GAGCCC TTT CCT TTC TTT CC-3�); CXCR5 (5�-ACT ACC CAC TAA CCCTGG AC-3� and 5�-AGG TGA TGT GGA TGG AGA GGA G-3�); CCR7(5�-GAG AGA CAA GAA CCA AAA GCA C-3� and 5�-GGG AAG AATTAG GAG GAA AAG G-3�); and �-actin (5�-AGA GGT ATC CTG ACCCTG AAG TAC C-3� and 5�-CCA CCA GAC AAC ACT GTG TTG GCAT-3�). Amplification conditions using an MJ Research (Cambridge, MA)PTC-225 thermal cycler were 94°C for 1 min, 60°C for 1 min, and 72°Cfor 1 min for 25 cycles in the presence of 1 �Ci [�-32P]dCTP. For negativecontrols, reverse transcriptase was omitted. Amplified products were sep-arated in 6% polyacrylamide gels. Northern analysis using 15 �g totalspleen RNA was performed as described previously (18). Equal loadingwas controlled by methylene blue staining of the membrane. Purifiedprobes were labeled with [�-32P]dCTP using a random priming kit fromAmersham Pharmacia Biotech (Piscataway, NJ). Quantifications were per-formed with a Fuji film FLA-3000 fluorescent image analyzer.
Flow cytometric analyses
Flow cytometry was performed using a BD Biosciences (Mountain View,CA) FACStarPlus flow cytometer and cell sorter. Splenocytes were isolatedand RBCs were lysed according to standard procedures (19). For analysisof MZ B cells, splenocytes were labeled with anti-CD23 PE (clone B3B4,1/200 dilution; PharMingen) and anti-CD21/CD35 FITC (clone 7G6, 1/100dilution; PharMingen) mAbs in FACS buffer (Ca2�/Mg2�-free PBS, 0.5%BSA). For analysis of surface expression of LT�R ligands, splenocyteswere cultured overnight (2 � 106/ml) in RPMI 1640 supplemented with10% heat-inactivated FCS, penicillin (100 U/ml), streptomycin (100 �g/ml), L-glutamine (2 mM), and 2-ME (50 �M), and either induced with 80nM PMA and 0.5 �M ionomycin or treated with DMSO as a solventcontrol. For FACS staining, cells were treated with Fc Block (clone 2.4G2,1/200 dilution; PharMingen) and then incubated with anti-CD4 FITC(clone RM4-5, 1/100 dilution; PharMingen) or anti-IgD FITC (clone 11-26c.2a, 1/100 dilution; PharMingen) mAbs in FACS buffer. Ligand bindingto the LT�R was detected with a rLT�R human IgG1 fusion protein (20)(1/200 dilution), followed by biotinylated mouse-absorbed goat F(ab�)2
anti-human IgG (1/100 dilution; Southern Biotechnology Associates) andstreptavidin-PE (1/200 dilution; PharMingen). All incubations were for 30min on ice, followed by two washes with FACS buffer. Analysis was re-stricted to small cells with a low sideward scatter. An average of 104 cellswas recorded in each case.
Adoptive BM transfers and analysis of chimerism
BM cells were isolated from femora of 2- to 3-mo-old wild-type (wt) orrelB�/� mice and injected (4–6 � 106 cells i.v. per mouse) into eitherrelB�/� or wt controls (2–3 months old). Before injection, recipient micehad been irradiated with 2 � 550 rad (3-h interval) and rested for 4–6 hafter the second irradiation. The following transfers were performed:wt3wt (B63B6), relB�/�3wt (B63B6), and wt3relB�/� (B6 �1293B6 � 129). Six to 8 wk later, recipient mice were injected i.p. withSRBCs. For immunohistochemical analysis, spleens were collected 10days after immunization. For the analysis of chimerism, peripheral bloodwas collected by cardiac puncture at necropsy, leukocytes were preparedusing ACK lysis buffer, and DNA was extracted for PCR genotyping, aspreviously described (21). Only mice in which the genotype of PBLs wascompletely of donor origin were further analyzed.
ResultsImmunohistochemical analysis of RelB expression in the spleen
In situ hybridization and immunohistochemistry experimentsshowed that RelB expression in spleen of naive wt mice isrestricted to the white pulp, with high levels in DCs (22, 23). To
1910 RelB IS REQUIRED FOR GC AND MZ FORMATION
by guest on Decem
ber 2, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

examine whether RelB is expressed in GCs, we immunized wtmice with SRBCs and stained spleen sections with RelB-specificAbs (Fig. 1). White pulp, red pulp, PALS, GCs, and MZ couldclearly be identified by H&E staining (Fig. 1A). Anti-RelB immu-nohistochemistry demonstrated very strong expression in PALS ofthe T cell area (Fig. 1, B and C). Lower levels of RelB expressioncould also be detected in GCs (Fig. 1, B and D) and in the MZ (Fig.1, B and E). The specificity of the Ab was demonstrated by the lackof labeling in sections from RelB-deficient mice (data not shown).
RelB-deficient mice lack GCs
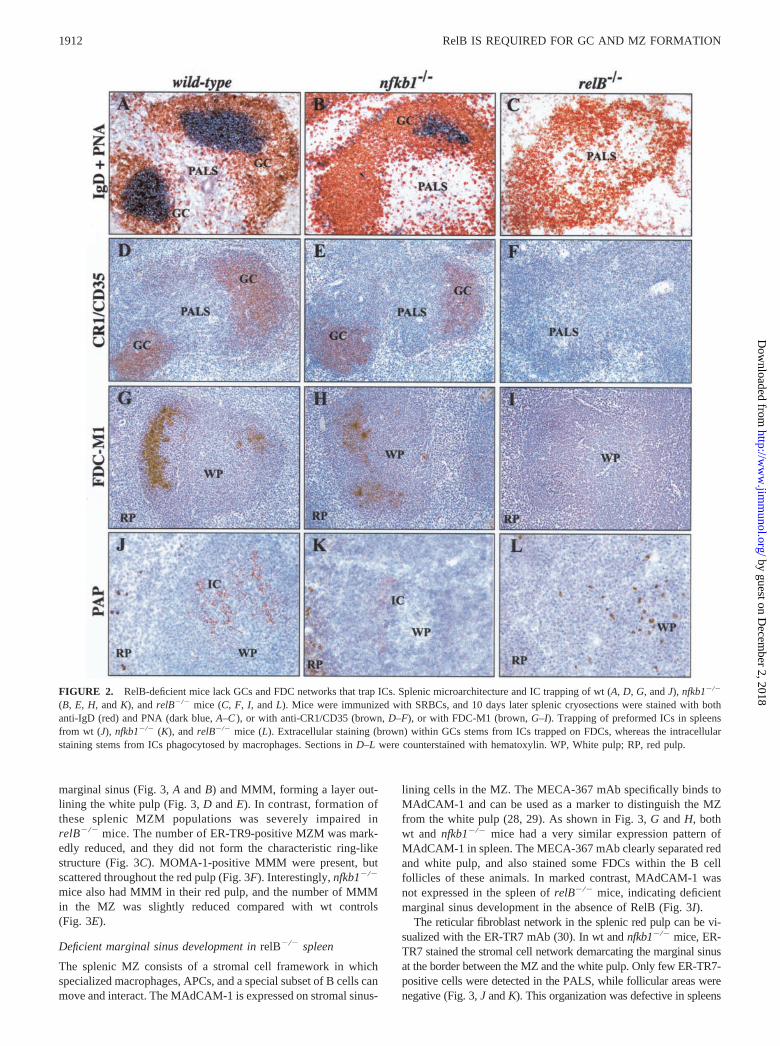
Previous studies revealed that RelB is required for normal Ab re-sponses after immunization with TD Ags. In particular, RelB-de-ficient mice show reduced isotype switching, whereas IgM re-sponses are higher than in control animals (11). To analyzewhether the impaired Ab response to TD Ags relates to an im-paired microarchitecture in secondary lymphoid organs that cannotsupport proper GC formation, we immunized mice with SRBCs,isolated the spleens 10 days after challenge, and prepared cryo-sections for immunohistochemical evaluation. Control mice devel-oped typical GCs with PNA� clusters surrounded by IgD� B cells(Fig. 2A). The plant lectin PNA binds to centroblasts/centrocytes,whereas IgD is highly expressed on mature follicular B cells anddown-regulated on most GC B cells. Confirming previous reports(24, 25), nfkb1�/� mice also developed GCs, although PNA stain-ing was overall decreased compared with wt mice (Fig. 2B). Sim-ilar staining in spleens of RelB-deficient mice failed to detect anyGCs and PNA� clusters. In addition, primary B cell follicles weredisorganized and IgD� B cells were scattered in the T cell area ofthe splenic white pulp (Fig. 2C). Lack of B and T cell segregationin relB�/� spleen was also observed in sections stained with B220and CD4/CD8 mAbs (data not shown).
Lack of FDC networks and IC trapping in RelB-deficient mice
FDC networks trap and retain ICs on their surfaces and play acrucial role in selecting Ag-specific B cells during Ab responses.To analyze the FDC network in detail, spleen sections from im-munized mice were stained with anti-mouse CR1/CD35 (clone8C12) and FDC-M1 mAbs, both recognizing FDCs. Immunohis-tochemical analysis revealed a normal pattern of CR1/CD35 stain-ing within splenic B cell follicles in both wt and nfkb1�/� mice
(Fig. 2, D and E). In contrast, CR1/CD35� cells were absent fromthe spleen of relB�/� mice (Fig. 2F). A similar result was ob-served in spleen sections stained with the FDC-M1 mAb. TypicalFDC networks were detected within B cell follicles of wt mice,whereas relB�/� animals did not show any FDC-M1 staining.FDC-M1 staining could also be detected in spleens from nfkb1�/�
mice, although fewer cells were stained compared with wt controls(Fig. 2G–I).
FDCs in GCs retain Ag in an unprocessed form on their surfacefor the selection of B cells (12). To address to which extent Agretention occurred in spleens from control and mutant mice, weperformed IC trapping experiments. Animals were immunizedwith SRBCs, and 6 days later injected with preformed peroxidaseantiperoxidase ICs. Spleens were removed next day, and the HRPAg was detected by DAB histochemistry on cryosections. At thistime point, FDCs in the GC region of the spleen are the only celltypes with extracellular enzyme (17). ICs were readily trapped onFDCs in control mice (Fig. 2J). Whereas reduced IC trapping wasobserved in nfkb1�/� mice (Fig. 2K), relB�/� mice showed no ICtrapping in the splenic white pulp (Fig. 2L). The strong intracel-lular staining stems from ICs phagocytosed by macrophages,which in wt and nfkb1�/� mice were largely restricted to the redpulp and the MZ, but which were scattered within the white pulpof relB�/� mice. In summary, RelB is essential for the formationof GCs and FDC networks and for the retention of native Ags inthe spleen, whereas the p50 subunit of NF-�B contributes onlyminimally to these phenomena.
Impaired formation of splenic MZ macrophage (MZM)populations in RelB-deficient mice
To investigate the role of p50/NF-�B1 and RelB in developmentand organization of the splenic MZ, we performed a comparativeimmunohistochemical analysis of spleen sections from wt,nfkb1�/�, and relB�/� mice. The MZ is characterized by the pres-ence of two distinct macrophage populations, MZM and metallo-philic marginal macrophages (MMM), separating red and whitepulp in the spleen. These specialized macrophages can be discrim-inated by the ER-TR9 and MOMA-1 mAbs, respectively (26, 27).Staining of spleen sections from wt and nfkb1�/� mice revealedthe two distinct macrophage populations with MZM outside of the
FIGURE 1. Immunohistochemical detection ofRelB in spleen from SRBC-stimulated wt mice. A,Staining of spleen section with H&E showing whitepulp (WP), red pulp (RP), PALS, GC, and MZ. B,Staining with anti-RelB Ab reveals expression inPALS, GCs, and the MZ of the white pulp. C–E,High power views of RelB-positive cells in the whitepulp shown in B. C, High expression of RelB inPALS of the T cell area. Lower levels of RelB ex-pression can also be detected in GCs (D) and the MZ(E). Sections in B–E were counterstained withhematoxylin.
1911The Journal of Immunology
by guest on Decem
ber 2, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
marginal sinus (Fig. 3, A and B) and MMM, forming a layer out-lining the white pulp (Fig. 3, D and E). In contrast, formation ofthese splenic MZM populations was severely impaired inrelB�/� mice. The number of ER-TR9-positive MZM was mark-edly reduced, and they did not form the characteristic ring-likestructure (Fig. 3C). MOMA-1-positive MMM were present, butscattered throughout the red pulp (Fig. 3F). Interestingly, nfkb1�/�
mice also had MMM in their red pulp, and the number of MMMin the MZ was slightly reduced compared with wt controls(Fig. 3E).
Deficient marginal sinus development in relB�/� spleen
The splenic MZ consists of a stromal cell framework in whichspecialized macrophages, APCs, and a special subset of B cells canmove and interact. The MAdCAM-1 is expressed on stromal sinus-
lining cells in the MZ. The MECA-367 mAb specifically binds toMAdCAM-1 and can be used as a marker to distinguish the MZfrom the white pulp (28, 29). As shown in Fig. 3, G and H, bothwt and nfkb1�/� mice had a very similar expression pattern ofMAdCAM-1 in spleen. The MECA-367 mAb clearly separated redand white pulp, and also stained some FDCs within the B cellfollicles of these animals. In marked contrast, MAdCAM-1 wasnot expressed in the spleen of relB�/� mice, indicating deficientmarginal sinus development in the absence of RelB (Fig. 3I).
The reticular fibroblast network in the splenic red pulp can be vi-sualized with the ER-TR7 mAb (30). In wt and nfkb1�/� mice, ER-TR7 stained the stromal cell network demarcating the marginal sinusat the border between the MZ and the white pulp. Only few ER-TR7-positive cells were detected in the PALS, while follicular areas werenegative (Fig. 3, J and K). This organization was defective in spleens
FIGURE 2. RelB-deficient mice lack GCs and FDC networks that trap ICs. Splenic microarchitecture and IC trapping of wt (A, D, G, and J), nfkb1�/�
(B, E, H, and K), and relB�/� mice (C, F, I, and L). Mice were immunized with SRBCs, and 10 days later splenic cryosections were stained with bothanti-IgD (red) and PNA (dark blue, A–C ), or with anti-CR1/CD35 (brown, D–F), or with FDC-M1 (brown, G–I). Trapping of preformed ICs in spleensfrom wt (J), nfkb1�/� (K), and relB�/� mice (L). Extracellular staining (brown) within GCs stems from ICs trapped on FDCs, whereas the intracellularstaining stems from ICs phagocytosed by macrophages. Sections in D–L were counterstained with hematoxylin. WP, White pulp; RP, red pulp.
1912 RelB IS REQUIRED FOR GC AND MZ FORMATION
by guest on Decem
ber 2, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
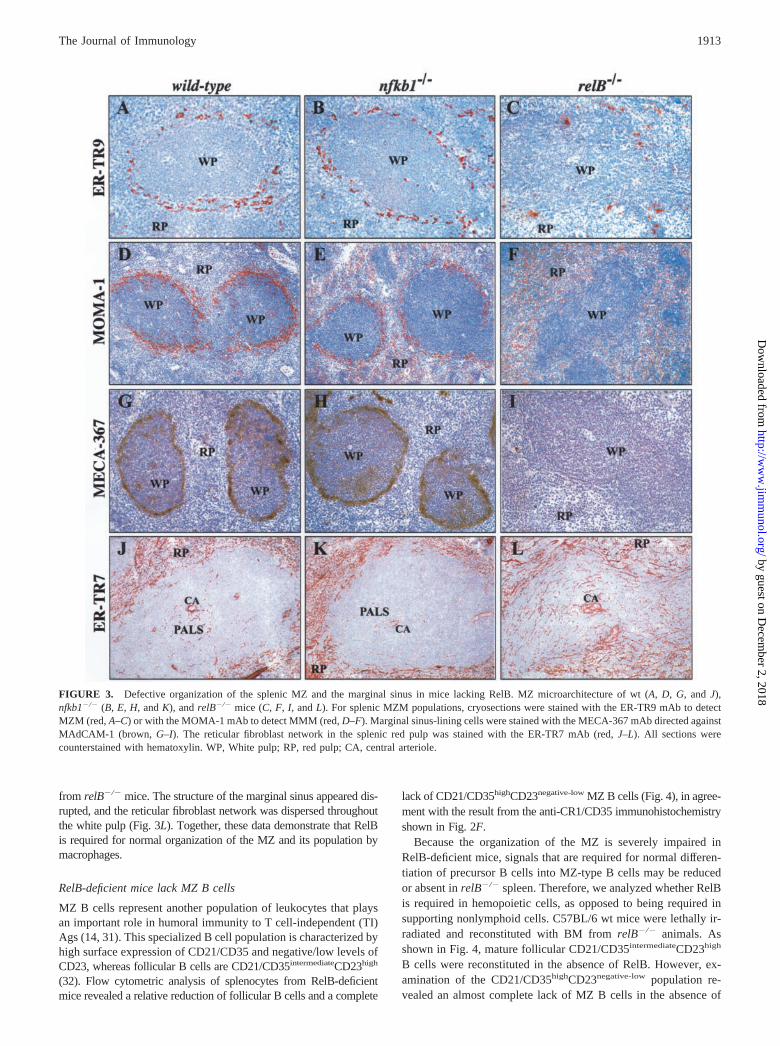
from relB�/� mice. The structure of the marginal sinus appeared dis-rupted, and the reticular fibroblast network was dispersed throughoutthe white pulp (Fig. 3L). Together, these data demonstrate that RelBis required for normal organization of the MZ and its population bymacrophages.
RelB-deficient mice lack MZ B cells
MZ B cells represent another population of leukocytes that playsan important role in humoral immunity to T cell-independent (TI)Ags (14, 31). This specialized B cell population is characterized byhigh surface expression of CD21/CD35 and negative/low levels ofCD23, whereas follicular B cells are CD21/CD35intermediateCD23high
(32). Flow cytometric analysis of splenocytes from RelB-deficientmice revealed a relative reduction of follicular B cells and a complete
lack of CD21/CD35highCD23negative-low MZ B cells (Fig. 4), in agree-ment with the result from the anti-CR1/CD35 immunohistochemistryshown in Fig. 2F.
Because the organization of the MZ is severely impaired inRelB-deficient mice, signals that are required for normal differen-tiation of precursor B cells into MZ-type B cells may be reducedor absent in relB�/� spleen. Therefore, we analyzed whether RelBis required in hemopoietic cells, as opposed to being required insupporting nonlymphoid cells. C57BL/6 wt mice were lethally ir-radiated and reconstituted with BM from relB�/� animals. Asshown in Fig. 4, mature follicular CD21/CD35intermediateCD23high
B cells were reconstituted in the absence of RelB. However, ex-amination of the CD21/CD35highCD23negative-low population re-vealed an almost complete lack of MZ B cells in the absence of
FIGURE 3. Defective organization of the splenic MZ and the marginal sinus in mice lacking RelB. MZ microarchitecture of wt (A, D, G, and J),nfkb1�/� (B, E, H, and K), and relB�/� mice (C, F, I, and L). For splenic MZM populations, cryosections were stained with the ER-TR9 mAb to detectMZM (red, A–C) or with the MOMA-1 mAb to detect MMM (red, D–F). Marginal sinus-lining cells were stained with the MECA-367 mAb directed againstMAdCAM-1 (brown, G–I). The reticular fibroblast network in the splenic red pulp was stained with the ER-TR7 mAb (red, J–L). All sections werecounterstained with hematoxylin. WP, White pulp; RP, red pulp; CA, central arteriole.
1913The Journal of Immunology
by guest on Decem
ber 2, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
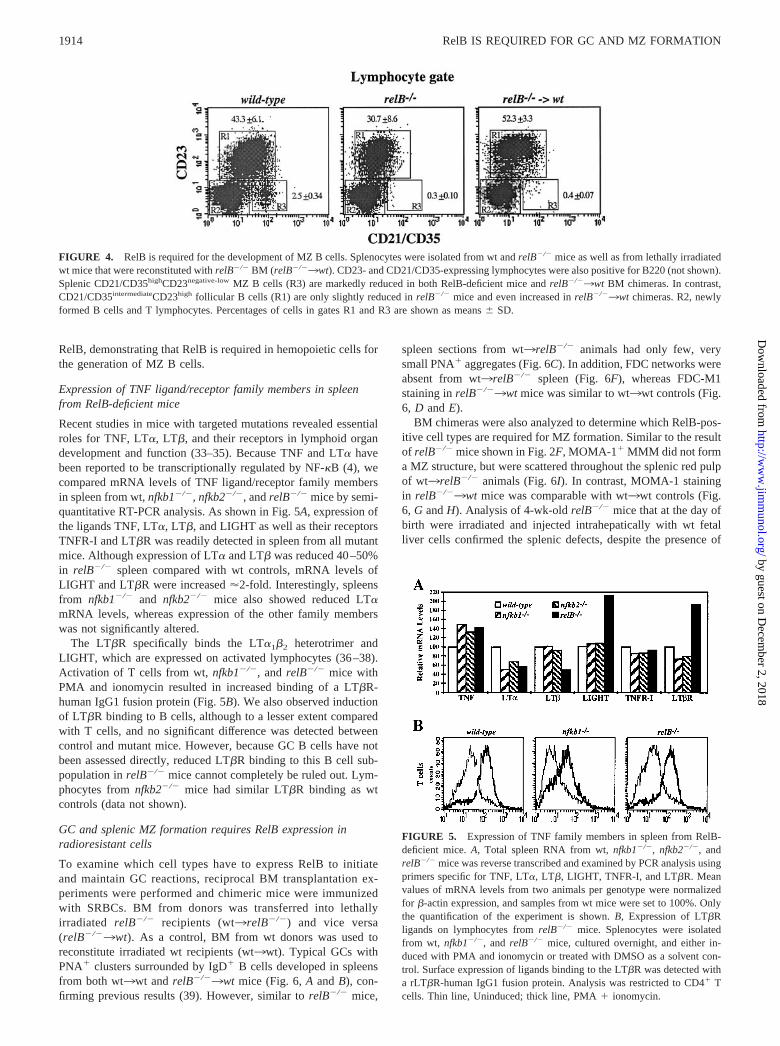
RelB, demonstrating that RelB is required in hemopoietic cells forthe generation of MZ B cells.
Expression of TNF ligand/receptor family members in spleenfrom RelB-deficient mice
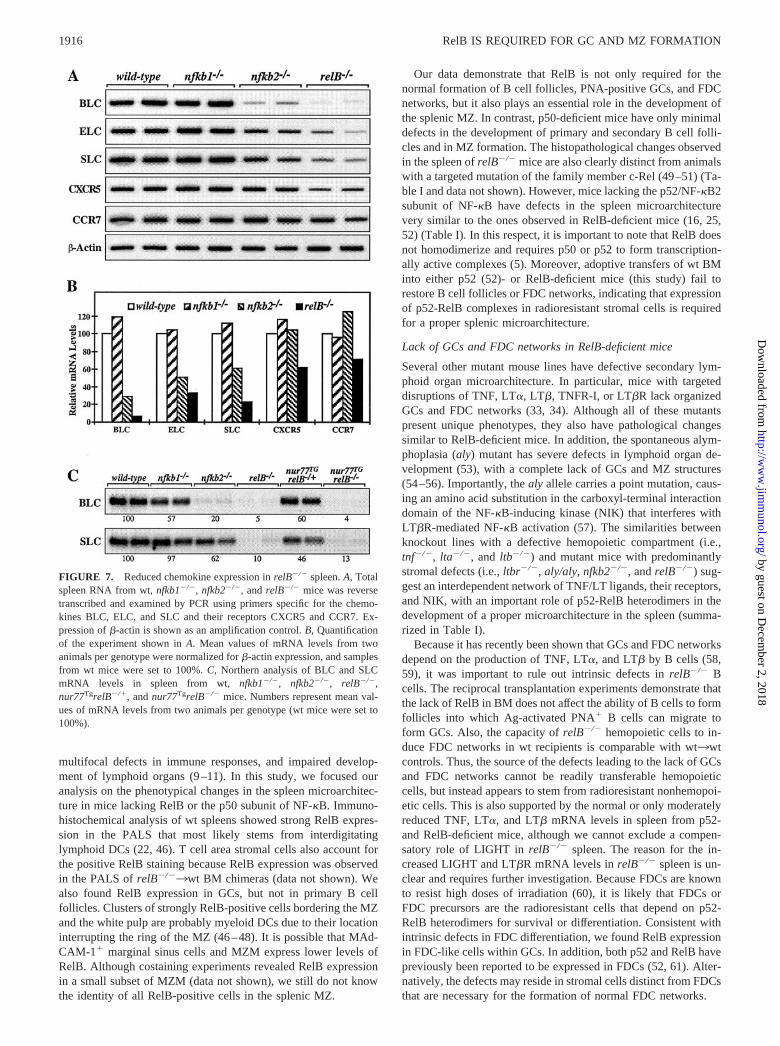
Recent studies in mice with targeted mutations revealed essentialroles for TNF, LT�, LT�, and their receptors in lymphoid organdevelopment and function (33–35). Because TNF and LT� havebeen reported to be transcriptionally regulated by NF-�B (4), wecompared mRNA levels of TNF ligand/receptor family membersin spleen from wt, nfkb1�/�, nfkb2�/�, and relB�/� mice by semi-quantitative RT-PCR analysis. As shown in Fig. 5A, expression ofthe ligands TNF, LT�, LT�, and LIGHT as well as their receptorsTNFR-I and LT�R was readily detected in spleen from all mutantmice. Although expression of LT� and LT� was reduced 40–50%in relB�/� spleen compared with wt controls, mRNA levels ofLIGHT and LT�R were increased �2-fold. Interestingly, spleensfrom nfkb1�/� and nfkb2�/� mice also showed reduced LT�mRNA levels, whereas expression of the other family memberswas not significantly altered.
The LT�R specifically binds the LT�1�2 heterotrimer andLIGHT, which are expressed on activated lymphocytes (36–38).Activation of T cells from wt, nfkb1�/�, and relB�/� mice withPMA and ionomycin resulted in increased binding of a LT�R-human IgG1 fusion protein (Fig. 5B). We also observed inductionof LT�R binding to B cells, although to a lesser extent comparedwith T cells, and no significant difference was detected betweencontrol and mutant mice. However, because GC B cells have notbeen assessed directly, reduced LT�R binding to this B cell sub-population in relB�/� mice cannot completely be ruled out. Lym-phocytes from nfkb2�/� mice had similar LT�R binding as wtcontrols (data not shown).
GC and splenic MZ formation requires RelB expression inradioresistant cells
To examine which cell types have to express RelB to initiateand maintain GC reactions, reciprocal BM transplantation ex-periments were performed and chimeric mice were immunizedwith SRBCs. BM from donors was transferred into lethallyirradiated relB�/� recipients (wt3relB�/�) and vice versa(relB�/�3wt). As a control, BM from wt donors was used toreconstitute irradiated wt recipients (wt3wt). Typical GCs withPNA� clusters surrounded by IgD� B cells developed in spleensfrom both wt3wt and relB�/�3wt mice (Fig. 6, A and B), con-firming previous results (39). However, similar to relB�/� mice,
spleen sections from wt3relB�/� animals had only few, verysmall PNA� aggregates (Fig. 6C). In addition, FDC networks wereabsent from wt3relB�/� spleen (Fig. 6F), whereas FDC-M1staining in relB�/�3wt mice was similar to wt3wt controls (Fig.6, D and E).
BM chimeras were also analyzed to determine which RelB-pos-itive cell types are required for MZ formation. Similar to the resultof relB�/� mice shown in Fig. 2F, MOMA-1� MMM did not forma MZ structure, but were scattered throughout the splenic red pulpof wt3relB�/� animals (Fig. 6I). In contrast, MOMA-1 stainingin relB�/�3wt mice was comparable with wt3wt controls (Fig.6, G and H). Analysis of 4-wk-old relB�/� mice that at the day ofbirth were irradiated and injected intrahepatically with wt fetalliver cells confirmed the splenic defects, despite the presence of
FIGURE 5. Expression of TNF family members in spleen from RelB-deficient mice. A, Total spleen RNA from wt, nfkb1�/�, nfkb2�/�, andrelB�/� mice was reverse transcribed and examined by PCR analysis usingprimers specific for TNF, LT�, LT�, LIGHT, TNFR-I, and LT�R. Meanvalues of mRNA levels from two animals per genotype were normalizedfor �-actin expression, and samples from wt mice were set to 100%. Onlythe quantification of the experiment is shown. B, Expression of LT�Rligands on lymphocytes from relB�/� mice. Splenocytes were isolatedfrom wt, nfkb1�/�, and relB�/� mice, cultured overnight, and either in-duced with PMA and ionomycin or treated with DMSO as a solvent con-trol. Surface expression of ligands binding to the LT�R was detected witha rLT�R-human IgG1 fusion protein. Analysis was restricted to CD4� Tcells. Thin line, Uninduced; thick line, PMA � ionomycin.
FIGURE 4. RelB is required for the development of MZ B cells. Splenocytes were isolated from wt and relB�/� mice as well as from lethally irradiatedwt mice that were reconstituted with relB�/� BM (relB�/�3wt). CD23- and CD21/CD35-expressing lymphocytes were also positive for B220 (not shown).Splenic CD21/CD35highCD23negative-low MZ B cells (R3) are markedly reduced in both RelB-deficient mice and relB�/�3wt BM chimeras. In contrast,CD21/CD35intermediateCD23high follicular B cells (R1) are only slightly reduced in relB�/� mice and even increased in relB�/�3wt chimeras. R2, newlyformed B cells and T lymphocytes. Percentages of cells in gates R1 and R3 are shown as means � SD.
1914 RelB IS REQUIRED FOR GC AND MZ FORMATION
by guest on Decem
ber 2, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

RelB-positive transferred cells in the white pulp of the recipients(data not shown). Together, these data indicate an essential role ofRelB in nonhemopoietic cells for the development of proper B cellfollicles, GCs, and FDC networks. Moreover, RelB expression inboth radioresistant and hemopoietic cells is required for normalMZ formation and its population by macrophages and B cells.
Reduced expression of homing chemokines in relB�/� spleen
One possible explanation for the observed defects in the organi-zation and microarchitecture of the spleen in relB�/� mice is thatradioresistant stromal cells require RelB to generate signals thatare needed for proper lymphocyte migration. Recently, several re-ports have shown an essential role for chemokines and their re-ceptors in cell migration and organization of secondary lymphoidorgans (40–42). We focused our analysis on three chemokines thatare constitutively expressed in lymphoid organs. Both ELC andSLC bind to the chemokine receptor CCR7, whereas BLC interactswith CXCR5, also termed BLR-1 (40–42).
Semiquantitative RT-PCR analysis from total spleen RNA re-vealed that expression of all chemokines and chemokine receptorswas comparable between nfkb1�/� mice and wt controls. In con-trast, a marked reduction in BLC mRNA levels was observed inspleens from nfkb2�/� (3.5-fold) and even more pronounced inrelB�/� mice (15-fold). ELC and SLC expression was also re-duced in spleen from nfkb2�/� and relB�/� mice, but to a lesserextent. Expression of the chemokine receptors CXCR5 and CCR7
was only slightly affected in relB�/� mice and normal in nfkb2�/�
animals (Fig. 7, A and B). Interestingly, BLC and SLC mRNAlevels were reduced to a similar level in both relB�/� mice andwt3relB�/� chimeras, whereas the reduced expression of ELC inrelB�/� spleen was partially rescued by adoptive transfer of wtBM (data not shown).
To rule out that the reduced BLC and SLC expression inrelB�/� spleen results from a dilution effect due to a relative de-crease of lymphoid areas in enlarged spleens, we also analyzednur77-transgenic (Tg) relB�/� mice (nur77TgrelB�/�). The con-stitutive overexpression of the orphan nuclear receptor Nur77 inthymocytes results in massive apoptosis and a dramatic reductionof peripheral T cells (43). We have previously shown thatnur77TgrelB�/� mice do not develop multiorgan inflammation orsplenomegaly (44, 45). Northern analysis confirmed the reducedmRNA levels of BLC (20-fold) and SLC (10-fold) in relB�/�
spleen (Fig. 7C). Moreover, BLC and SLC expression was alsoclearly reduced in nur77TgrelB�/� spleen (15- and 3.5-fold, re-spectively) compared with Tg controls. These results indicate thatdecreased expression of the homing chemokines BLC and SLC bystromal cells contributes to the defective splenic microarchitecturein RelB-deficient mice.
DiscussionRelB-deficient mice develop a complex phenotype, including anautoimmune-like inflammatory syndrome, myeloid hyperplasia,
FIGURE 6. GC and splenic MZ formation requires RelB expression in stromal cells. Irradiated recipients were reconstituted with BM from donors, asindicated. After 6–8 wk, chimeras were immunized i.p. with SRBCs. Ten days later, animals were sacrificed and analyzed for their degree of chimerism,and spleen cryosections were stained with both anti-IgD (red) and PNA (dark blue, A–C ), or with FDC-M1 (brown, D–F), or with MOMA-1 (red, G–I).Sections in D–I were counterstained with hematoxylin. WP, White pulp; RP, red pulp.
1915The Journal of Immunology
by guest on Decem
ber 2, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
multifocal defects in immune responses, and impaired develop-ment of lymphoid organs (9–11). In this study, we focused ouranalysis on the phenotypical changes in the spleen microarchitec-ture in mice lacking RelB or the p50 subunit of NF-�B. Immuno-histochemical analysis of wt spleens showed strong RelB expres-sion in the PALS that most likely stems from interdigitatinglymphoid DCs (22, 46). T cell area stromal cells also account forthe positive RelB staining because RelB expression was observedin the PALS of relB�/�3wt BM chimeras (data not shown). Wealso found RelB expression in GCs, but not in primary B cellfollicles. Clusters of strongly RelB-positive cells bordering the MZand the white pulp are probably myeloid DCs due to their locationinterrupting the ring of the MZ (46–48). It is possible that MAd-CAM-1� marginal sinus cells and MZM express lower levels ofRelB. Although costaining experiments revealed RelB expressionin a small subset of MZM (data not shown), we still do not knowthe identity of all RelB-positive cells in the splenic MZ.
Our data demonstrate that RelB is not only required for thenormal formation of B cell follicles, PNA-positive GCs, and FDCnetworks, but it also plays an essential role in the development ofthe splenic MZ. In contrast, p50-deficient mice have only minimaldefects in the development of primary and secondary B cell folli-cles and in MZ formation. The histopathological changes observedin the spleen of relB�/� mice are also clearly distinct from animalswith a targeted mutation of the family member c-Rel (49–51) (Ta-ble I and data not shown). However, mice lacking the p52/NF-�B2subunit of NF-�B have defects in the spleen microarchitecturevery similar to the ones observed in RelB-deficient mice (16, 25,52) (Table I). In this respect, it is important to note that RelB doesnot homodimerize and requires p50 or p52 to form transcription-ally active complexes (5). Moreover, adoptive transfers of wt BMinto either p52 (52)- or RelB-deficient mice (this study) fail torestore B cell follicles or FDC networks, indicating that expressionof p52-RelB complexes in radioresistant stromal cells is requiredfor a proper splenic microarchitecture.
Lack of GCs and FDC networks in RelB-deficient mice
Several other mutant mouse lines have defective secondary lym-phoid organ microarchitecture. In particular, mice with targeteddisruptions of TNF, LT�, LT�, TNFR-I, or LT�R lack organizedGCs and FDC networks (33, 34). Although all of these mutantspresent unique phenotypes, they also have pathological changessimilar to RelB-deficient mice. In addition, the spontaneous alym-phoplasia (aly) mutant has severe defects in lymphoid organ de-velopment (53), with a complete lack of GCs and MZ structures(54–56). Importantly, the aly allele carries a point mutation, caus-ing an amino acid substitution in the carboxyl-terminal interactiondomain of the NF-�B-inducing kinase (NIK) that interferes withLT�R-mediated NF-�B activation (57). The similarities betweenknockout lines with a defective hemopoietic compartment (i.e.,tnf�/�, lta�/�, and ltb�/�) and mutant mice with predominantlystromal defects (i.e., ltbr�/�, aly/aly, nfkb2�/�, and relB�/�) sug-gest an interdependent network of TNF/LT ligands, their receptors,and NIK, with an important role of p52-RelB heterodimers in thedevelopment of a proper microarchitecture in the spleen (summa-rized in Table I).
Because it has recently been shown that GCs and FDC networksdepend on the production of TNF, LT�, and LT� by B cells (58,59), it was important to rule out intrinsic defects in relB�/� Bcells. The reciprocal transplantation experiments demonstrate thatthe lack of RelB in BM does not affect the ability of B cells to formfollicles into which Ag-activated PNA� B cells can migrate toform GCs. Also, the capacity of relB�/� hemopoietic cells to in-duce FDC networks in wt recipients is comparable with wt3wtcontrols. Thus, the source of the defects leading to the lack of GCsand FDC networks cannot be readily transferable hemopoieticcells, but instead appears to stem from radioresistant nonhemopoi-etic cells. This is also supported by the normal or only moderatelyreduced TNF, LT�, and LT� mRNA levels in spleen from p52-and RelB-deficient mice, although we cannot exclude a compen-satory role of LIGHT in relB�/� spleen. The reason for the in-creased LIGHT and LT�R mRNA levels in relB�/� spleen is un-clear and requires further investigation. Because FDCs are knownto resist high doses of irradiation (60), it is likely that FDCs orFDC precursors are the radioresistant cells that depend on p52-RelB heterodimers for survival or differentiation. Consistent withintrinsic defects in FDC differentiation, we found RelB expressionin FDC-like cells within GCs. In addition, both p52 and RelB havepreviously been reported to be expressed in FDCs (52, 61). Alter-natively, the defects may reside in stromal cells distinct from FDCsthat are necessary for the formation of normal FDC networks.
FIGURE 7. Reduced chemokine expression in relB�/� spleen. A, Totalspleen RNA from wt, nfkb1�/�, nfkb2�/�, and relB�/� mice was reversetranscribed and examined by PCR using primers specific for the chemo-kines BLC, ELC, and SLC and their receptors CXCR5 and CCR7. Ex-pression of �-actin is shown as an amplification control. B, Quantificationof the experiment shown in A. Mean values of mRNA levels from twoanimals per genotype were normalized for �-actin expression, and samplesfrom wt mice were set to 100%. C, Northern analysis of BLC and SLCmRNA levels in spleen from wt, nfkb1�/�, nfkb2�/�, relB�/�,nur77TgrelB�/�, and nur77TgrelB�/� mice. Numbers represent mean val-ues of mRNA levels from two animals per genotype (wt mice were set to100%).
1916 RelB IS REQUIRED FOR GC AND MZ FORMATION
by guest on Decem
ber 2, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
Despite the complete lack of GCs or FDC networks, RelB-de-ficient mice show Ig isotype switching in response to TD Ags,although at reduced levels and with delayed kinetics (11). Thisfinding correlates with normal in vitro maturation of relB�/� Bcells to Ig secretion and Ig class switching in response to distinctactivators in combination with various cytokines (62). Some af-finity maturation and isotype switching in the absence of properGCs and FDC networks have also been reported in other mutantlines, such as mice deficient for p52, LT��, TNFR-I, or LT�R, inparticular after repeated immunization with high doses of Ag (52,63–66). Thus, one role of GCs and FDC networks may be toprovide an optimal environment for effective Ab responses to lim-iting, and presumably more physiological, levels of Ags.
RelB-deficient mice lack splenic MZ
The spleen plays a major role in the protection against bacterialinfections, and the MZ in particular participates in immune re-sponses against TI polysaccharide Ags (13, 67). Using a panel ofmAbs against MZM, MMM, and marginal sinus-lining stromalcells, we found that MZ organization in relB�/� spleen is severelyimpaired, correlating with high susceptibility to bacterial infec-tions and markedly reduced isotype switching in response to TIAgs (11). In addition, RelB-deficient mice lack MZ B cells, aspecialized cell type that is required for normal TI type II humoralresponses to polysaccharide Ags (31). A marked reduction of MZB cells was also observed in relB�/�3wt chimeras, indicating anintrinsic defect in MZ B cell development. Similarly, it has beenshown that p50/NF-�B1 is required for MZ B cell generation, andthat RelA and c-Rel play significant, but less critical roles in thisprocess (68). Together these findings demonstrate that NF-�B isrequired for MZ B cell development and suggest a particular roleof p50-RelB complexes.
RelB-deficient mice also completely lack MECA-367� mar-ginal sinus-lining cells and have markedly reduced numbers ofER-TR9� MZM. Transfer of wt BM into RelB-deficient recipientsdoes not restore the disrupted marginal sinus structures, suggestingthat stromal cells are responsible for the MZ defects in RelB-deficient mice. The MECA-367 mAb recognizes an epitope ofthe addressin MAdCAM-1 on sinus-lining cells in the MZ.MAdCAM-1 has been shown to be important for extravasation ofblood lymphocytes into Peyer’s patches (69), however, not forhoming of lymphocytes into the white pulp of adult spleen (28). Itremains to be investigated whether MAdCAM-1 expression on
sinus-lining cells is required for proper MZ formation during earlystages of spleen development.
MOMA-1� MMM were found scattered throughout the red pulpin relB�/� spleen, indicating that RelB is not essential for thedevelopment of MMM, but rather for their proper localizationwithin the splenic MZ. This is supported by adoptive transfer ex-periments in which relB�/�3wt chimeras showed MOMA-1staining in the MZ similar to wt3wt controls. In contrast, p52-deficient mice lack MMM and have normal or only slightly re-duced numbers of ER-TR9� MZM (25). BM transfers restoreMMM to the MZ in p52-deficient recipients, indicating intrinsicdefects in this hemopoietic cell (52). Both p50 and p52 can alsointeract with the I�B family member Bcl-3 to form transcription-ally active complexes (70, 71). Interestingly, Bcl-3-deficient micehave dramatically reduced numbers of MMM, and ER-TR9�
MZM are virtually absent in spleen (72). These results suggestdifferent requirements for the development of these two distinctMZM populations with a particular role of p52 and Bcl-3 forMMM, whereas MZM depend on Bcl-3 and RelB.
Chemokine expression in RelB-deficient mice
Chemokines provide important signals for the proper localization oflymphocytes in specialized compartments within lymphoid organs(33, 40). In particular, BLC expressed by follicular stromal cells se-lectively attracts B cells via CXCR5, whereas expression of SLC byT zone stromal cells attracts T cells and DCs via CCR7 (73). BLC-deficient mice fail to organize B cells into polarized follicular clustersand lack follicle FDCs (74). In addition, mice lacking SLC expressionhave defects in T cell homing and DC localization in secondary lym-phoid organs (75). Thus, BLC and SLC play an essential role in theformation of organized lymphoid tissue.
BLC and SLC expression is markedly reduced in both relB�/�
and nfkb2�/� spleen, whereas expression of the corresponding re-ceptors CXCR5 and CCR7 is only slightly affected in relB�/� andnormal in nfkb2�/� mice. Reduced SLC levels have also beenobserved in thymus of RelB-deficient animals (76). Interestingly,the reduction in BLC and SLC mRNA levels in spleen is compa-rable between relB�/� mice and wt3relB�/� chimeras, indicatingthat RelB complexes in stromal cells have a role upstream of BLCand SLC. This is consistent with the observation that RelB andBLC are coexpressed in some GC cells, although not all BLC-positive cells also show RelB staining (data not shown). The che-mokine ELC is expressed by stromal cells and DCs in lymphoid
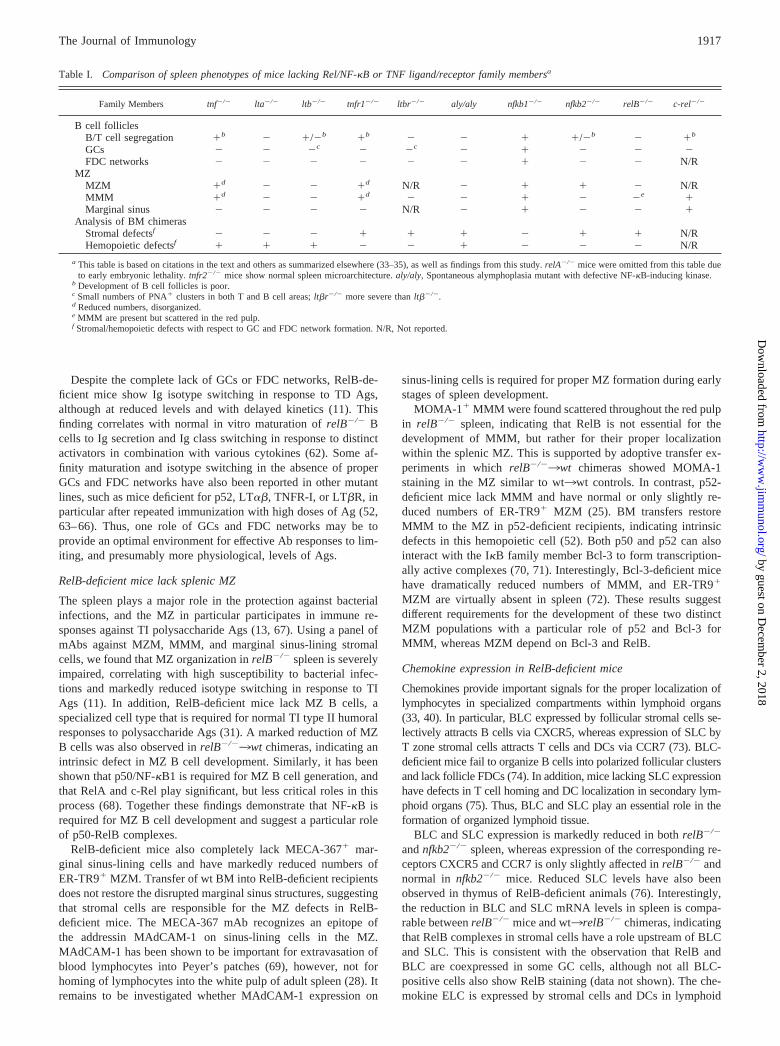
Table I. Comparison of spleen phenotypes of mice lacking Rel/NF-�B or TNF ligand/receptor family membersa
Family Members tnf�/� lta�/� ltb�/� tnfr1�/� ltbr�/� aly/aly nfkb1�/� nfkb2�/� relB�/� c-rel�/�
B cell folliclesB/T cell segregation �b � �/�b �b � � � �/�b � �b
GCs � � �c � �c � � � � �FDC networks � � � � � � � � � N/R
MZMZM �d � � �d N/R � � � � N/RMMM �d � � �d � � � � �e �Marginal sinus � � � � N/R � � � � �
Analysis of BM chimerasStromal defectsf � � � � � � � � � N/RHemopoietic defectsf � � � � � � � � � N/R
a This table is based on citations in the text and others as summarized elsewhere (33–35), as well as findings from this study. relA�/� mice were omitted from this table dueto early embryonic lethality. tnfr2�/� mice show normal spleen microarchitecture. aly/aly, Spontaneous alymphoplasia mutant with defective NF-�B-inducing kinase.
b Development of B cell follicles is poor.c Small numbers of PNA� clusters in both T and B cell areas; lt�r�/� more severe than lt��/�.d Reduced numbers, disorganized.e MMM are present but scattered in the red pulp.f Stromal/hemopoietic defects with respect to GC and FDC network formation. N/R, Not reported.
1917The Journal of Immunology
by guest on Decem
ber 2, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

tissues and strongly attracts naive T cells and activated B cells(77). The reduced expression of ELC correlates with reduced num-bers and abnormal distribution of DCs in spleen of RelB-deficientmice (9, 10, 47, 78). Although the reduced ELC levels may con-tribute to the disrupted microarchitecture in relB�/� spleen, adop-tive transfer of wt BM into RelB-deficient recipients partially re-stored ELC levels, but not GC and FDC network formation.
Recently, it has been shown that LT�� and TNF on hemopoieticcells are required for stromal cell expression of BLC, ELC, andSLC homing chemokines in B and T cell areas of the spleen (73).Lower ELC and SLC mRNA levels and drastically reduced BLCmRNA expression have also been reported in spleen from aly/alymice (79). Thus, our data indicate that activation of p52-RelB het-erodimers in stromal cells downstream of TNF and LT��, prob-ably mediated by TNFR-I/LT�R and NIK, is required directly orindirectly for the expression of factors, such as BLC and SLC, thatplay an essential role in B cell follicle and FDC network devel-opment and lymphocyte compartmentalization in the spleen.
AcknowledgmentsWe gratefully acknowledge Claudia Stoll, Monika Pech, Norma Howells,and Reina Mebius for excellent technical assistance and advice. We areindebted to Marie Kosco-Vilbois for providing FDC-M1 mAb; JasonG. Cyster for BLC, ELC, and SLC cDNAs and advice; and JeffreyL. Browning and Paul D. Rennert for anti-LT� and anti-LT� mAbs as wellas LT�R-human IgG fusion proteins. We also thank Harm HogenEsch forvaluable comments on this manuscript, Peter Herrlich for continuing sup-port, and all the staff in the animal facility at the Institute of Toxicology andGenetics.
References1. Baeuerle, P. A., and T. Henkel. 1994. Function and activation of NF-�B in the
immune system. Annu. Rev. Immunol. 12:141.2. Ghosh, S., M. J. May, and E. B. Kopp. 1998. NF-�B and Rel proteins: evolu-
tionarily conserved mediators of immune responses. Annu. Rev. Immunol. 16:225.
3. Karin, M. 1999. How NF-�B is activated: the role of the I�B kinase (IKK)complex. Oncogene 18:6867.
4. Pahl, H. L. 1999. Activators and target genes of Rel/NF-�B transcription factors.Oncogene 18:6853.
5. Ryseck, R.-P., F. Weih, D. Carrasco, and R. Bravo. 1996. RelB, a member of theRel/NF-�B family of transcription factors. Braz. J. Med. Biol. Res. 29:895.
6. Attar, R. M., J. Caamano, D. Carrasco, V. Iotsova, H. Ishikawa, R.-P. Ryseck,F. Weih, and R. Bravo. 1997. Genetic approaches to study Rel/NF-�B/I�B func-tion in mice. Semin. Cancer Biol. 8:93.
7. Gerondakis, S., M. Grossmann, Y. Nakamura, T. Pohl, and R. Grumont. 1999.Genetic approaches in mice to understand Rel/NF-�B and I�B function: trans-genics and knockouts. Oncogene 18:6888.
8. Lo, D., H. Quill, L. Burkly, B. Scott, R. D. Palmiter, and R. L. Brinster. 1992. Arecessive defect in lymphocyte or granulocyte function caused by an integratedtransgene. Am. J. Pathol. 141:1237.
9. Burkly, L., C. Hession, L. Ogata, C. Reilly, L. A. Marconi, D. Olson, R. Tizard,R. Cate, and D. Lo. 1995. Expression of relB is required for the development ofthymic medulla and dendritic cells. Nature 373:531.
10. Weih, F., D. Carrasco, S. K. Durham, D. S. Barton, C. A. Rizzo, R.-P. Ryseck,S. A. Lira, and R. Bravo. 1995. Multiorgan inflammation and hematopoieticabnormalities in mice with a targeted disruption of RelB, a member of the NF-�B/Rel family. Cell 80:331.
11. Weih, F., G. Warr, H. Yang, and R. Bravo. 1997. Multifocal defects in immuneresponses in RelB-deficient mice. J. Immunol. 158:5211.
12. Kosco-Vilbois, M. H., J. Y. Bonnefoy, and Y. Chvatchko. 1997. The physiologyof murine germinal center reactions. Immunol. Rev. 156:127.
13. Kraal, G. 1992. Cells in the marginal zone of the spleen. Int. Rev. Cytol. 132:31.14. Cyster, J. G. 2000. B cells on the front line. Nat. Immunol. 1:9.15. Sha, W. C., H.-C. Liou, E. I. Tuomanen, and D. Baltimore. 1995. Targeted dis-
ruption of the p50 subunit of NF-�B leads to multifocal defects in immune re-sponses. Cell 80:321.
16. Caamano, J. H., C. A. Rizzo, S. K. Durham, D. S. Barton, C. Raventos-Suarez,C. M. Snapper, and R. Bravo. 1998. Nuclear factor (NF)-�B2 (p100/p52) isrequired for normal splenic microarchitecture and B cell-mediated immune re-sponses. J. Exp. Med. 187:185.
17. Chen, L. L., A. M. Frank, J. C. Adams, and R. M. Steinman. 1978. Distributionof horseradish peroxidase (HRP)-anti-HRP immune complexes in mouse spleenwith special reference to follicular dendritic cells. J. Cell Biol. 79:184.
18. Ryseck, R.-P., H. Macdonald-Bravo, M.-G. Mattei, S. Ruppert, and R. Bravo.1989. Structure, mapping and expression of a growth factor inducible gene en-coding a putative nuclear hormonal binding receptor. EMBO J. 8:3327.
19. Coligan, J. E., A. M. Kruisbeek, D. H. Margulies, E. M. Shevach, and W. Strober.1992. Isolation and fractionation of mononuclear cell populations. In CurrentProtocols in Immunology. J. Wiley and Sons, eds. Greene Publishing Associates& Wiley-Interscience, New York.
20. Force, W. R., B. N. Walter, C. Hession, R. Tizard, C. A. Kozak, J. L. Browning,and C. F. Ware. 1995. Mouse lymphotoxin-� receptor: molecular genetics, ligandbinding, and expression. J. Immunol. 155:5280.
21. Weih, F., S. K. Durham, D. S. Barton, W. C. Sha, D. Baltimore, and R. Bravo.1997. p50-NF-�B complexes partially compensate for the absence of RelB: se-verely increased pathology in p50�/�relB�/� double-knockout mice. J. Exp.Med. 185:1359.
22. Carrasco, D., R.-P. Ryseck, and R. Bravo. 1993. Expression of relB transcriptsduring lymphoid organ development: specific expression in dendritic antigen-presenting cells. Development 118:1221.
23. Weih, F., D. Carrasco, and R. Bravo. 1994. Constitutive and inducible Rel/NF-�Bactivities in mouse thymus and spleen. Oncogene 9:3289.
24. Schwarz, E. M., P. Krimpenfort, A. Berns, and I. M. Verma. 1997. Immunolog-ical defects in mice with a targeted disruption in Bcl-3. Genes Dev. 11:187.
25. Franzoso, G., L. Carlson, L. Poljak, E. W. Shores, S. Epstein, A. Leonardi,A. Grinberg, T. Tran, T. Scharton-Kersten, M. Anver, et al. 1998. Mice deficientin nuclear factor (NF)-�B/p52 present with defects in humoral responses, ger-minal center reactions, and splenic microarchitecture. J. Exp. Med. 187:147.
26. van Vliet, E., M. Melis, and W. van Ewijk. 1985. Marginal zone macrophages inthe mouse spleen identified by a monoclonal antibody: anatomical correlationwith a B cell subpopulation. J. Histochem. Cytochem. 33:40.
27. Kraal, G., and M. Janse. 1986. Marginal metallophilic cells of the mouse spleenidentified by a monoclonal antibody. Immunology 58:665.
28. Kraal, G., K. Schornagel, P. R. Streeter, B. Holzmann, and E. C. Butcher. 1995.Expression of the mucosal vascular addressin, MAdCAM-1, on sinus-lining cellsin the spleen. Am. J. Pathol. 147:763.
29. Szabo, M. C., E. C. Butcher, and L. M. McEvoy. 1997. Specialization of mucosalfollicular dendritic cells revealed by mucosal addressin-cell adhesion molecule-1display. J. Immunol. 158:5584.
30. van Vliet, E., M. Melis, J. M. Foidart, and W. Van Ewijk. 1986. Reticular fibro-blasts in peripheral lymphoid organs identified by a monoclonal antibody. J. His-tochem. Cytochem. 34:883.
31. Guinamard, R., M. Okigaki, J. Schlessinger, and J. V. Ravetch. 2000. Absence ofmarginal zone B cells in Pyk-2-deficient mice defines their role in the humoralresponse. Nat. Immunol. 1:31.
32. Oliver, A. M., F. Martin, G. L. Gartland, R. H. Carter, and J. F. Kearney. 1997.Marginal zone B cells exhibit unique activation, proliferative and immunoglob-ulin secretory responses. Eur. J. Immunol. 27:2366.
33. Fu, Y. X., and D. D. Chaplin. 1999. Development and maturation of secondarylymphoid tissues. Annu. Rev. Immunol. 17:399.
34. Matsumoto, M. 1999. Role of TNF ligand and receptor family in the lymphoidorganogenesis defined by gene targeting. J. Med. Invest. 46:141.
35. Pasparakis, M., S. Kousteni, J. Peschon, and G. Kollias. 2000. Tumor necrosisfactor and the p55TNF receptor are required for optimal development of themarginal sinus and for migration of follicular dendritic cell precursors intosplenic follicles. Cell. Immunol. 201:33.
36. Crowe, P. D., T. L. VanArsdale, B. N. Walter, C. F. Ware, C. Hession,B. Ehrenfels, J. L. Browning, W. S. Din, R. G. Goodwin, and C. A. Smith. 1994.A lymphotoxin-�-specific receptor. Science 264:707.
37. Mauri, D. N., R. Ebner, R. I. Montgomery, K. D. Kochel, T. C. Cheung, G. L. Yu,S. Ruben, M. Murphy, R. J. Eisenberg, G. H. Cohen, et al. 1998. LIGHT, a newmember of the TNF superfamily, and lymphotoxin � are ligands for herpesvirusentry mediator. Immunity 8:21.
38. Zhai, Y., R. Guo, T. L. Hsu, G. L. Yu, J. Ni, B. S. Kwon, G. W. Jiang, J. Lu,J. Tan, M. Ugustus, et al. 1998. LIGHT, a novel ligand for lymphotoxin � re-ceptor and TR2/HVEM induces apoptosis and suppresses in vivo tumor forma-tion via gene transfer. J. Clin. Invest. 102:1142.
39. Gerloni, M., D. Lo, and M. Zanetti. 1998. DNA immunization in relB-deficientmice discloses a role for dendritic cells in IgM3 IgG1 switch in vivo. Eur.J. Immunol. 28:516.
40. Cyster, J. G. 1999. Chemokines and cell migration in secondary lymphoid organs.Science 286:2098.
41. Zlotnik, A., and O. Yoshie. 2000. Chemokines: a new classification system andtheir role in immunity. Immunity 12:121.
42. Cyster, J. G. 2000. Leukocyte migration: scent of the T zone. Curr. Biol. 10:R30.43. Weih, F., R.-P. Ryseck, L. Chen, and R. Bravo. 1996. Apoptosis of nur77/N10-
transgenic thymocytes involves the Fas/Fas ligand pathway. Proc. Natl. Acad.Sci. USA 93:5533.
44. Weih, F., S. K. Durham, D. S. Barton, W. C. Sha, D. Baltimore, and R. Bravo.1996. Both multiorgan inflammation and myeloid hyperplasia in RelB-deficientmice are T cell dependent. J. Immunol. 157:3974.
45. Barton, D., H. HogenEsch, and F. Weih. 2000. Mice lacking the transcriptionfactor RelB develop T cell-dependent skin lesions similar to human atopic der-matitis. Eur. J. Immunol. 30:2323.
46. Steinman, R. M., M. Pack, and K. Inaba. 1997. Dendritic cells in the T-cell areasof lymphoid organs. Immunol. Rev. 156:25.
47. Wu, L., A. D’Amico, K. D. Winkel, M. Suter, D. Lo, and K. Shortman. 1998.RelB is essential for the development of myeloid-related CD8�� dendritic cellsbut not of lymphoid-related CD8�� dendritic cells. Immunity 9:839.
48. Crowley, M. T., C. R. Reilly, and D. Lo. 1999. Influence of lymphocytes on thepresence and organization of dendritic cell subsets in the spleen. J. Immunol.163:4894.
1918 RelB IS REQUIRED FOR GC AND MZ FORMATION
by guest on Decem
ber 2, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

49. Kontgen, F., R. J. Grumont, A. Strasser, D. Metcalf, R. Li, D. Tarlinton, andS. Gerondakis. 1995. Mice lacking the c-rel proto-oncogene exhibit defects inlymphocyte proliferation, humoral immunity, and interleukin-2 expression.Genes Dev. 9:1965.
50. Carrasco, D., J. Cheng, A. Lewin, G. Warr, H. Yang, C. Rizzo, F. Rosas,C. Snapper, and R. Bravo. 1998. Multiple hemopoietic defects and lymphoidhyperplasia in mice lacking the transcriptional activation domain of the c-Relprotein. J. Exp. Med. 187:973.
51. Liou, H.-C., Z. Jin, J. Tumang, S. Andjelic, K. A. Smith, and M.-L. Liou. 1999.c-Rel is crucial for lymphocyte proliferation but dispensable for T cell effectorfunction. Int. Immunol. 11:361.
52. Poljak, L., L. Carlson, K. Cunningham, M. H. Kosco-Vilbois, and U. Siebenlist.1999. Distinct activities of p52/NF-�B required for proper secondary lymphoidorgan microarchitecture: functions enhanced by Bcl-3. J. Immunol. 163:6581.
53. Miyawaki, S., Y. Nakamura, H. Suzuka, M. Koba, R. Yasumizu, S. Ikehara, andY. Shibata. 1994. A new mutation, aly, that induces a generalized lack of lymphnodes accompanied by immunodeficiency in mice. Eur. J. Immunol. 24:429.
54. Koike, R., T. Nishimura, R. Yasumizu, H. Tanaka, Y. Hataba, Y. Hataba,T. Watanabe, S. Miyawaki, and M. Miyasaka. 1996. The splenic marginal zoneis absent in alymphoplastic aly mutant mice. Eur. J. Immunol. 26:669.
55. Shinkura, R., F. Matsuda, T. Sakiyama, T. Tsubata, H. Hiai, M. Paumen,S. Miyawaki, and T. Honjo. 1996. Defects of somatic hypermutation and classswitching in alymphoplasia (aly) mutant mice. Int. Immunol. 8:1067.
56. Karrer, U., A. Althage, B. Odermatt, C. W. Roberts, S. J. Korsmeyer,S. Miyawaki, H. Hengartner, and R. M. Zinkernagel. 1997. On the key role ofsecondary lymphoid organs in antiviral immune responses studied in alympho-plastic (aly/aly) and spleenless (Hox11�/�) mutant mice. J. Exp. Med. 185:2157.
57. Shinkura, R., K. Kitada, F. Matsuda, K. Tashiro, K. Ikuta, M. Suzuki, K. Kogishi,T. Serikawa, and T. Honjo. 1999. Alymphoplasia is caused by a point mutationin the mouse gene encoding NF-�B-inducing kinase. Nat. Genet. 22:74.
58. Fu, Y. X., G. Huang, Y. Wang, and D. D. Chaplin. 1998. B lymphocytes inducethe formation of follicular dendritic cell clusters in a lymphotoxin �-dependentfashion. J. Exp. Med. 187:1009.
59. Endres, R., M. B. Alimzhanov, T. Plitz, A. Futterer, M. H. Kosco-Vilbois,S. A. Nedospasov, K. Rajewsky, and K. Pfeffer. 1999. Mature follicular dendriticcell networks depend on expression of lymphotoxin � receptor by radioresistantstromal cells and of lymphotoxin � and tumor necrosis factor by B cells. J. Exp.Med. 189:159.
60. Humphrey, J. H., D. Grennan, and V. Sundaram. 1984. The origin of folliculardendritic cells in the mouse and the mechanism of trapping of immune complexeson them. Eur. J. Immunol. 14:859.
61. Feuillard, J., M. Korner, A. Israel, J. Vassy, and M. Raphael. 1996. Differentialnuclear localization of p50, p52, and RelB proteins in human accessory cells ofthe immune response in situ. Eur. J. Immunol. 26:2547.
62. Snapper, C. M., F. R. Rosas, P. Zelazowski, M. A. Moorman, M. R. Kehry,R. Bravo, and F. Weih. 1996. B cells lacking RelB are defective in proliferativeresponses, but undergo normal B cell maturation to Ig secretion and Ig classswitching. J. Exp. Med. 184:1537.
63. Matsumoto, M., S. F. Lo, C. J. L. Carruthers, J. Min, S. Mariathasan, G. Huang,D. R. Plas, S. M. Martin, R. S. Geha, M. H. Nahm, and D. D. Chaplin. 1996.Affinity maturation without germinal centres in lymphotoxin-�-deficient mice.Nature 381:462.
64. Koni, P. A., and R. A. Flavell. 1999. Lymph node germinal centers form in theabsence of follicular dendritic cell networks. J. Exp. Med. 189:855.
65. Futterer, A., K. Mink, A. Luz, M. H. Kosco-Vilbois, and K. Pfeffer. 1998. Thelymphotoxin � receptor controls organogenesis and affinity maturation in periph-eral lymphoid tissues. Immunity 9:59.
66. Wang, Y., G. Huang, J. Wang, H. Molina, D. D. Chaplin, and Y. X. Fu. 2000.Antigen persistence is required for somatic mutation and affinity maturation ofimmunoglobulin. Eur. J. Immunol. 30:2226.
67. Amlot, P. L., D. Grennan, and J. H. Humphrey. 1985. Splenic dependence of theantibody response to thymus-independent (TI-2) antigens. Eur. J. Immunol. 15:508.
68. Cariappa, A., H.-C. Liou, B. H. Horwitz, and S. Pillai. 2000. Nuclear factor �Bis required for the development of marginal zone B lymphocytes. J. Exp. Med.192:1175.
69. Berg, E. L., L. M. McEvoy, C. Berlin, R. F. Bargatze, and E. C. Butcher. 1993.L-selectin-mediated lymphocyte rolling on MAdCAM-1. Nature 366:695.
70. Fujita, T., G. P. Nolan, H.-C. Liou, M. L. Scott, and D. Baltimore. 1993. Thecandidate proto-oncogene bcl-3 encodes a transcriptional coactivator that acti-vates through NF-�B p50 homodimers. Genes Dev. 7:1354.
71. Bours, V., G. Franzoso, V. Azarenko, S. Park, T. Kannno, K. Brown, andU. Siebenlist. 1993. The oncoprotein Bcl-3 directly transactivates through �Bmotifs via association with DNA-binding p50B homodimers. Cell 72:729.
72. Franzoso, G., L. Carlson, T. Scharton-Kersten, E. W. Shores, S. Epstein,A. Grinberg, T. Tran, E. Shacter, A. Leonardi, M. Anver, et al. 1997. Criticalroles for the Bcl-3 oncoprotein in T cell-mediated immunity, splenic microarchi-tecture and germinal center reactions. Immunity 6:479.
73. Ngo, V. N., H. Korner, M. D. Gunn, K. N. Schmidt, D. S. Riminton,M. D. Cooper, J. L. Browning, J. D. Sedgwick, and J. G. Cyster. 1999. Lym-photoxin �/� and tumor necrosis factor are required for stromal cell expressionof homing chemokines in B and T cell areas of the spleen. J. Exp. Med. 189:403.
74. Ansel, K. M., V. N. Ngo, P. L. Hyman, S. A. Luther, R. Forster, J. D. Sedgwick,J. L. Browning, M. Lipp, and J. G. Cyster. 2000. A chemokine-driven positivefeedback loop organizes lymphoid follicles. Nature 406:309.
75. Gunn, M. D., S. Kyuwa, C. Tam, T. Kakiuchi, A. Matsuzawa, L. T. Williams, andH. Nakano. 1999. Mice lacking expression of secondary lymphoid organ che-mokine have defects in lymphocyte homing and dendritic cell localization.J. Exp. Med. 189:451.
76. Tanabe, S., Z. Lu, Y. Luo, E. J. Quackenbush, M. A. Berman,L. A. Collins-Racie, S. Mi, C. Reilly, D. Lo, K. A. Jacobs, and M. E. Dorf. 1997.Identification of a new mouse �-chemokine, thymus-derived chemotactic agent 4,with activity on T lymphocytes and mesangial cells. J. Immunol. 159:5671.
77. Ngo, V. N., H. L. Tang, and J. G. Cyster. 1998. Epstein-Barr virus-inducedmolecule 1 ligand chemokine is expressed by dendritic cells in lymphoid tissuesand strongly attracts naive T cells and activated B cells. J. Exp. Med. 188:181.
78. Crowley, M. T., and D. Lo. 1999. Targeted gene knockouts: insights into den-dritic cell biology. In Dendritic Cells. M. T. Lotze and A. W. Thomson, eds.Academic Press, San Diego, p. 579.
79. Fagarasan, S., R. Shinkura, T. Kamata, F. Nogaki, K. Ikuta, K. Tashiro, andT. Honjo. 2000. Alymphoplasia (aly)-type nuclear factor �B-inducing kinase(NIK) causes defects in secondary lymphoid tissue chemokine receptor signalingand homing of peritoneal cells to the gut-associated lymphatic tissue system.J. Exp. Med. 191:1477.
1919The Journal of Immunology
by guest on Decem
ber 2, 2018http://w
ww
.jimm
unol.org/D
ownloaded from





























