Embed Size (px)
Citation preview

At the Intersection of Health, Health Care and Policy
doi: 10.1377/hlthaff.2014.0122
, 33, no.8 (2014):1453-1459Health AffairsMarket Withdrawals
Era Of Faster FDA Drug Approval Has Also Seen Increased Black-Box Warnings AndWolfe, Orlaith Heymann, Leah Zallman and Karen E. Lasser
Cassie Frank, David U. Himmelstein, Steffie Woolhandler, David H. Bor, Sidney M.Cite this article as:
http://content.healthaffairs.org/content/33/8/1453.full.html
available at: The online version of this article, along with updated information and services, is
For Reprints, Links & Permissions: http://healthaffairs.org/1340_reprints.php
http://content.healthaffairs.org/subscriptions/etoc.dtlE-mail Alerts : http://content.healthaffairs.org/subscriptions/online.shtmlTo Subscribe:
written permission from the Publisher. All rights reserved.mechanical, including photocopying or by information storage or retrieval systems, without prior
may be reproduced, displayed, or transmitted in any form or by any means, electronic orAffairs HealthFoundation. As provided by United States copyright law (Title 17, U.S. Code), no part of
by Project HOPE - The People-to-People Health2014Bethesda, MD 20814-6133. Copyright © is published monthly by Project HOPE at 7500 Old Georgetown Road, Suite 600,Health Affairs
Not for commercial use or unauthorized distribution
at PENN STATE UNIV on August 12, 2014Health Affairs by content.healthaffairs.orgDownloaded from
at PENN STATE UNIV on August 12, 2014Health Affairs by content.healthaffairs.orgDownloaded from

By Cassie Frank, David U. Himmelstein, Steffie Woolhandler, David H. Bor, Sidney M. Wolfe,Orlaith Heymann, Leah Zallman, and Karen E. Lasser
Era Of Faster FDA Drug ApprovalHas Also Seen Increased Black-Box Warnings And MarketWithdrawals
ABSTRACT After approval, many prescription medications that patientsrely on subsequently receive new black-box warnings or are withdrawnfrom the market because of safety concerns. We examined whether thefrequency of these safety problems has increased since 1992, when thePrescription Drug User Fee Act, legislation designed to accelerate thedrug approval process at the Food and Drug Administration, was passed.We found that drugs approved after the act’s passage were more likely toreceive a new black-box warning or be withdrawn than drugs approvedbefore its passage (26.7 per 100.0 drugs versus 21.2 per 100.0 drugs at upto sixteen years of follow-up). We could not establish causality, however.Our findings suggest the need for reforms to reduce patients’ exposure tounsafe drugs, such as a statement or symbol in the labeling, medicationguides for patients, and marketing materials indicating that a drug wasapproved only recently.
Adverse reactions tomedications arecommon and sometimes fatal.1 Theprimary mission of the Food andDrug Administration (FDA) is toprotect the public’s health by en-
suring the safety and efficacy of drugs, medicaldevices, and biological products. Many adversedrug reactions are not detected until after a drughas been approved by the FDA and is in use. Thepremarketing review of drugs is a critical stepintended to identify safety problems beforedrugs enter widespread public use.ThePrescriptionDrugUserFeeAct (PDUFA)—
first enacted in 1992 and renewed in 1997, 2002,2007, and 2012—authorizes the FDA to collectfees from drug companies to expedite the drugapproval process. Congress enacted the PDUFAin response to widespread concerns that theprocess was taking too long.2,3 The act estab-lishes performance goals for various steps inthe FDA review process that the agency shouldmeet to ensure that new drugs make it to market
in a timely fashion.Since PDUFA was enacted, median drug ap-
proval times for new molecular entities(NMEs)—active ingredients that have never be-fore been marketed in the United States in anyform—have decreased from 33.6 months in1979–86 to 16.1months in 1997–02 (a 52 percentreduction).4 However, some critics have raisedconcerns that PDUFA may have expedited drugapproval at the expense of the public’s safety.5
Notably, between 1999 and 2009 outpatient pre-scribers in the United States wrote more than30 million prescriptions for each of nine drugs(for a total of more than 270 million prescrip-tions) that subsequently were withdrawn fromthe market for safety reasons or that receivedblack-box warnings for potentially lethal sideeffects.6 Other researchers, however, have ar-gued that such concerns about faster drug ap-provals are not justified.4,7,8
Previous studies4,7,9 have examined changes inthe rate of new black-box warnings or safety
doi: 10.1377/hlthaff.2014.0122HEALTH AFFAIRS 33,NO. 8 (2014): 1453–1459©2014 Project HOPE—The People-to-People HealthFoundation, Inc.
Cassie Frank ([email protected]) is an instructorof medicine at CambridgeHealth Alliance and HarvardMedical School, in Boston,Massachusetts.
David U. Himmelstein is aprofessor at the School ofPublic Health, City Universityof New York (CUNY).
Steffie Woolhandler is aprofessor at the School ofPublic Health, CUNY.
David H. Bor is an associateprofessor of medicine atCambridge Health Alliance andHarvard Medical School.
Sidney M. Wolfe is a senioradviser at the HealthResearch Group, PublicCitizen, in Washington, D.C.
Orlaith Heymann is a researchassistant at Boston MedicalCenter and the School ofMedicine, Boston University,in Massachusetts.
Leah Zallman is an instructorof medicine at CambridgeHealth Alliance and HarvardMedical School.
Karen E. Lasser is anassociate professor ofmedicine at Boston MedicalCenter and the School ofMedicine, Boston University.
August 2014 33:8 Health Affairs 1453
Prescription Drugs
at PENN STATE UNIV on August 12, 2014Health Affairs by content.healthaffairs.orgDownloaded from

withdrawals (two indicators of drug safety prob-lems) before and after the enactment of PDUFA.These studies found no difference in rates of newblack-box warnings or safety withdrawals afterthe act was passed. However, the data covered arelatively short time span after PDUFA’s enact-ment, and none of the studies examined bothnew black-box warnings and safety withdrawalsin the aggregate. This limited the number ofsafety events examined and hence the studies’statistical power to detect possible differences. Afourth study8 noted no association between theFDA’s review time for a drug and the number ofadverse drug event reports voluntarily submittedto the FDA in the three years following drugapproval.In contrast, other studies have found evidence
of increased drug safety problems following theenactment of PDUFA. Mary Olson10 found thatdrugs receiving faster reviews (as has been thecase since PDUFA’s enactment) are associatedwith increased counts of serious adverse drugreactions. Thomas Moore and Curt Furberg11 re-cently reported that in 2008 clinical trials forexpedited-review drugs were smaller, comparedto trials for other drugs, and that many postmar-keting studies that the FDA requested or re-quired as a condition of approval (among bothgroups of drugs) to address safety concerns hadnot been completed by as late as 2013.Another study12 found that many years may
elapse between drug approval and the recogni-tion of problems that eventually require the cre-ation of a risk evaluation and mitigation strate-gy. Such a strategy may involve elements such asamedicationguide, a package insert forpatients,and a communication plan to manage a knownorpotential serious risk associatedwithadrugorbiological product.13 Given the conflicting find-ings of the studies mentioned above, the ques-tion of whether drug risks have increased underthe PDUFA merits further exploration.As in our earlier study,14 in the study reported
on here, we examined the aggregate outcome ofnew black-box warnings and safety withdrawalsover time. Both of these widely accepted mea-sures of drug safety problems indicate that im-portant new hazards were recognized only afterdrug approval. This combinedmeasure has a keyadvantage over the use of safety-related with-drawals alone, since withdrawals are rare (lessthan one per year) and hence analyses of thisoutcome alone have low statistical power. Wehypothesized that new black-box warnings andsafety withdrawals have increased followingPDUFA’s enactment, perhaps as a result of anexpedited review process thatmay not adequate-ly detect serious drug safety problems in thepreapproval period.
Study Data And MethodsData Sources And Definitions We obtained alist from the Tufts Center for Drug Developmentof all NMEs that the FDA approved in the period2000–09,15 updating the list used in our previousstudy that covered the years 1975–99.14 We ex-cluded over-the-countermedications, diagnosticagents, and biologics, to ensure comparability tothe previous study.The previous study relied exclusively on the
Physicians’ Desk Reference (PDR) to identifynew black-box warnings. However, in recentyears the print version of this publication hasomitted label information formanydrugs,whichmakes it difficult to identify black-box warningsthat are embedded in the drug package labeling.Many drugs (including those with black-boxwarnings) are only briefly listed in the PDR,withno information available other than the drug’sname and manufacturer.Hence, in addition to the PDR,we searched the
PDR website (PDR.net), which includes morelabel information for many drugs than is inthe print version, and the FDA’s Internet-basedresource of approved drug products, drugs@FDA,16 to identify new black-box warnings. Un-fortunately, drug label information on the FDAwebsite is also incomplete; in particular, lesslabel information is available for generic drugsthan for brand-name drugs.17
We identified drugs withdrawn for safety rea-sons from the FDA website and from a 2005report by the FDA’s Center for Drug Evaluationand Research (CDER).18 The report outlines thecenter’s initiatives and documents performanceacross program areas, including drug safety,during 2005. It includes lists of drug recallsand safety-based drug withdrawals.We defined withdrawn drugs according to the
definition used by the CDER, which includesdrug recalls. The center defines recalled drugsas those withdrawn from the market becauseofproblems in themanufacturingordistributionphase that may pose a significant publichealth risk.Our definition also included safety-based drug
withdrawals, in which an intrinsic property of adrug prompts its withdrawal for safety reasons.In some cases, the FDA initiated the drug remov-al. In other cases, the manufacturer voluntarilywithdrew the drug from the market after theidentification of serious adverse drug reactions.This was the case, for example, with some of thefluoroquinolones, a class of antibiotics used in avariety of infections.Our definition was consistent with those used
in previous studies.14,19 Because some with-drawals were initiated by the company and werenot counted by the FDA as safety related, our
Prescription Drugs
1454 Health Affairs August 2014 33:8
at PENN STATE UNIV on August 12, 2014Health Affairs by content.healthaffairs.orgDownloaded from

study may underestimate the number of safety-related withdrawals.Other studies20,21 have noted that safety con-
cerns are infrequently the primary reason forvoluntary drug withdrawal. We did not includedrugs withdrawn for other reasons, such as poorsales or the introduction of similar drugs with abetter risk-benefit profile.
Analysis We established whether or not adrug had a current black-box warning by search-ing the 2010 PDR and PDR.net starting Octo-ber 1, 2010, and concluding December 31,2010. We did not examine the years beyond2010 because the data for 2010 were the mostrecent available data at the time the study wasinitiated.We then manually searched previous editions
of the PDR. If a black-boxwarningwas present inthe PDRwhen the drug first appeared,wedidnotcount that as a new warning. In cases where wewere unable to establish from the PDRwhether ablack-box warning was included in the initiallabel (for example, in cases where no label forthe drug appeared in the PDR in the year after itsfirst approval),weused the first label available inthe online database drugs@FDA to establishwhether a black-box warning was present whenthe drug was introduced.For drugs with a recent label available on
drugs@FDA, we used that labeling informationto establish the date when a black-box warningfirst appeared. For other drugs, we manuallysearched the PDR from 2000 to 201022 to deter-mine the year in which the drug first received ablack-box warning and approximated the warn-ing date as January 1 of the year of the PDR inwhich the warning first appeared.For the ten drugs for which we could not pre-
cisely establish the date of the first appearance ofa new black-box warning using the PDR anddrugs@FDA, we imputed the black-box warningdate from the date of the first available label thatincluded a warning.We used the date when the
drug was first approved to approximate the datewhen it was firstmarketed.We counted each newwarning as a separate event.Nonsteroidal anti-inflammatory drugs and
antidepressants accounted for a large propor-tion of drugs with new black-box warnings andwithdrawals. Thus, we performed sensitivity an-alyses that excluded each of these drug classesfrom the main analysis, to determine whethereither drug class was driving the observed re-sults. We also performed a sensitivity analysisexcluding the ten drugs with new black-boxwarnings for which we had imputed the black-box warning acquisition date. For drugs ap-proved before 2000, we used data from the pre-vious study14 and searched for any newwarningsor withdrawals for these drugs.Statistical Methods We used the statistical
software SAS, version 9.3, to perform survivalanalyses using the proc lifetest procedure. Wegenerated Kaplan-Meier plots to determinehow much time elapsed between drug approvaland receipt of a black-box warning or with-drawal.We tested differences in the plots for NMEs
approved before and after the enactment ofPDUFA using the log-rank chi-square test. Weperformed this analysis before and after Janu-ary 1, 1993, because 1993 was the first calendaryear after PDUFA came into effect on Octo-ber 29, 1992.Limitations Our study had several limita-
tions. Our primarymeasures of drug safety prob-lems were a new black-box warning or a drugwithdrawal. Remarkably, no comprehensivesource of information on black-box warningsor withdrawals is available to clinicians, re-searchers, or the public. Our measures are im-perfect. However, they are the only measures ofsafety problems we know of that are availableacross all approved drugs in all years in our studyperiod.Importantly, our analysis did not establish
causality. In other words, we could not deter-mine with certainty whether the enactment ofPDUFA caused the increase in drug withdrawalsand acquisition of black-box warnings that weobserved. It is possible that other unmeasuredfactors caused the increased frequency of newblack-box warnings and withdrawals in the lateryears of the study period. For example, we didnot examine differences in the characteristics ofthe drugs approved before and after the enact-ment of PDUFA, such as a drug’s novelty or or-phan status, or whether it had undergone accel-erated approval.We could not control for other possible influ-
ences on the likelihood of a new black-box warn-ing, including changes in the FDA’s ability to
Danger signals are notbeing detected earlyenough to preventmillions of patientsfrom being exposed tounsafe drugs.
August 2014 33:8 Health Affairs 1455
at PENN STATE UNIV on August 12, 2014Health Affairs by content.healthaffairs.orgDownloaded from
detect postmarketing risks; changes in industrybehavior, such as direct-to-consumer advertis-ing; or changes in regulatory authority (whichincreased after 2007, following the publicationof the Institute of Medicine report23 on the USdrug safety system) to address postmarketingrisks. Markedly improved postmarketing sur-veillance could increase the detection of adversereactions. However, there is little evidence tosuggest that this has occurred.24
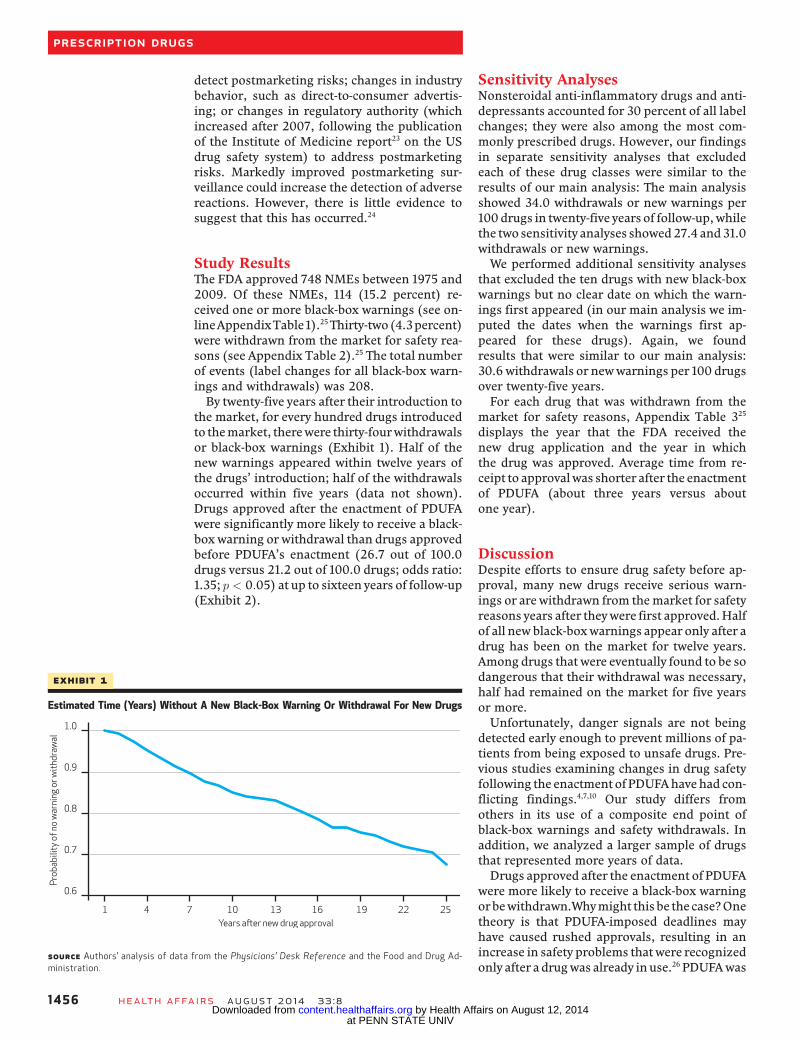
Study ResultsThe FDA approved 748 NMEs between 1975 and2009. Of these NMEs, 114 (15.2 percent) re-ceived one or more black-box warnings (see on-lineAppendixTable1).25Thirty-two(4.3percent)were withdrawn from the market for safety rea-sons (see Appendix Table 2).25 The total numberof events (label changes for all black-box warn-ings and withdrawals) was 208.By twenty-five years after their introduction to
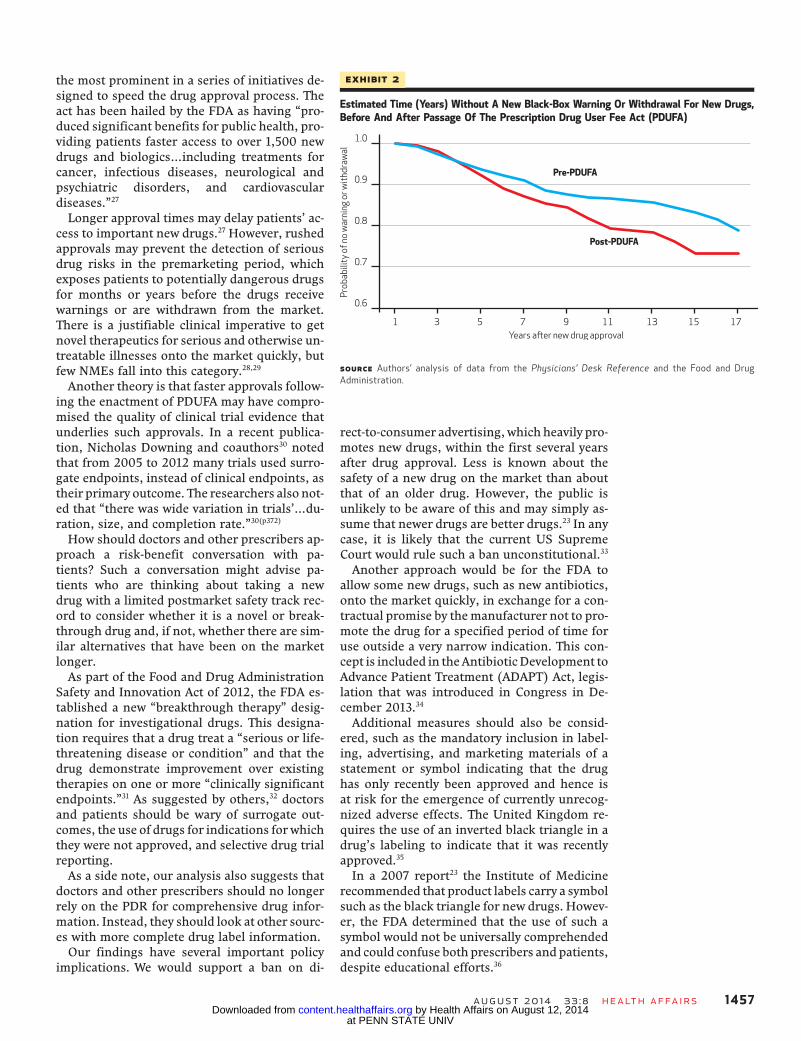
the market, for every hundred drugs introducedto themarket, therewere thirty-fourwithdrawalsor black-box warnings (Exhibit 1). Half of thenew warnings appeared within twelve years ofthe drugs’ introduction; half of the withdrawalsoccurred within five years (data not shown).Drugs approved after the enactment of PDUFAwere significantly more likely to receive a black-box warning or withdrawal than drugs approvedbefore PDUFA’s enactment (26.7 out of 100.0drugs versus 21.2 out of 100.0 drugs; odds ratio:1.35; p < 0:05) at up to sixteen years of follow-up(Exhibit 2).
Sensitivity AnalysesNonsteroidal anti-inflammatory drugs and anti-depressants accounted for 30 percent of all labelchanges; they were also among the most com-monly prescribed drugs. However, our findingsin separate sensitivity analyses that excludedeach of these drug classes were similar to theresults of our main analysis: The main analysisshowed 34.0 withdrawals or new warnings per100drugs in twenty-five years of follow-up,whilethe two sensitivity analyses showed27.4 and31.0withdrawals or new warnings.We performed additional sensitivity analyses
that excluded the ten drugs with new black-boxwarnings but no clear date on which the warn-ings first appeared (in our main analysis we im-puted the dates when the warnings first ap-peared for these drugs). Again, we foundresults that were similar to our main analysis:30.6withdrawals or newwarnings per 100 drugsover twenty-five years.For each drug that was withdrawn from the
market for safety reasons, Appendix Table 325
displays the year that the FDA received thenew drug application and the year in whichthe drug was approved. Average time from re-ceipt to approvalwas shorter after the enactmentof PDUFA (about three years versus aboutone year).
DiscussionDespite efforts to ensure drug safety before ap-proval, many new drugs receive serious warn-ings or arewithdrawn from themarket for safetyreasons years after theywere first approved.Halfof all new black-boxwarnings appear only after adrug has been on the market for twelve years.Among drugs that were eventually found to be sodangerous that their withdrawal was necessary,half had remained on the market for five yearsor more.Unfortunately, danger signals are not being
detected early enough to prevent millions of pa-tients from being exposed to unsafe drugs. Pre-vious studies examining changes in drug safetyfollowing the enactment of PDUFAhavehad con-flicting findings.4,7,10 Our study differs fromothers in its use of a composite end point ofblack-box warnings and safety withdrawals. Inaddition, we analyzed a larger sample of drugsthat represented more years of data.Drugs approved after the enactment of PDUFA
were more likely to receive a black-box warningorbewithdrawn.Whymight thisbe the case?Onetheory is that PDUFA-imposed deadlines mayhave caused rushed approvals, resulting in anincrease in safety problems thatwere recognizedonly after a drugwas already inuse.26 PDUFAwas
Exhibit 1
Estimated Time (Years) Without A New Black-Box Warning Or Withdrawal For New Drugs
SOURCE Authors’ analysis of data from the Physicians’ Desk Reference and the Food and Drug Ad-ministration.
Prescription Drugs
1456 Health Affairs August 2014 33:8
at PENN STATE UNIV on August 12, 2014Health Affairs by content.healthaffairs.orgDownloaded from
the most prominent in a series of initiatives de-signed to speed the drug approval process. Theact has been hailed by the FDA as having “pro-duced significant benefits for public health, pro-viding patients faster access to over 1,500 newdrugs and biologics…including treatments forcancer, infectious diseases, neurological andpsychiatric disorders, and cardiovasculardiseases.”27
Longer approval times may delay patients’ ac-cess to important new drugs.27 However, rushedapprovals may prevent the detection of seriousdrug risks in the premarketing period, whichexposes patients to potentially dangerous drugsfor months or years before the drugs receivewarnings or are withdrawn from the market.There is a justifiable clinical imperative to getnovel therapeutics for serious and otherwise un-treatable illnesses onto the market quickly, butfew NMEs fall into this category.28,29
Another theory is that faster approvals follow-ing the enactment of PDUFA may have compro-mised the quality of clinical trial evidence thatunderlies such approvals. In a recent publica-tion, Nicholas Downing and coauthors30 notedthat from 2005 to 2012 many trials used surro-gate endpoints, instead of clinical endpoints, astheir primary outcome. The researchers also not-ed that “there was wide variation in trials’…du-ration, size, and completion rate.”30(p372)
How should doctors and other prescribers ap-proach a risk-benefit conversation with pa-tients? Such a conversation might advise pa-tients who are thinking about taking a newdrug with a limited postmarket safety track rec-ord to consider whether it is a novel or break-through drug and, if not, whether there are sim-ilar alternatives that have been on the marketlonger.As part of the Food and Drug Administration
Safety and Innovation Act of 2012, the FDA es-tablished a new “breakthrough therapy” desig-nation for investigational drugs. This designa-tion requires that a drug treat a “serious or life-threatening disease or condition” and that thedrug demonstrate improvement over existingtherapies on one or more “clinically significantendpoints.”31 As suggested by others,32 doctorsand patients should be wary of surrogate out-comes, the use of drugs for indications for whichthey were not approved, and selective drug trialreporting.As a side note, our analysis also suggests that
doctors and other prescribers should no longerrely on the PDR for comprehensive drug infor-mation. Instead, they should look at other sourc-es with more complete drug label information.Our findings have several important policy
implications. We would support a ban on di-
rect-to-consumer advertising,which heavily pro-motes new drugs, within the first several yearsafter drug approval. Less is known about thesafety of a new drug on the market than aboutthat of an older drug. However, the public isunlikely to be aware of this and may simply as-sume that newer drugs are better drugs.23 In anycase, it is likely that the current US SupremeCourt would rule such a ban unconstitutional.33
Another approach would be for the FDA toallow some new drugs, such as new antibiotics,onto the market quickly, in exchange for a con-tractual promise by themanufacturer not to pro-mote the drug for a specified period of time foruse outside a very narrow indication. This con-cept is included in theAntibioticDevelopment toAdvance Patient Treatment (ADAPT) Act, legis-lation that was introduced in Congress in De-cember 2013.34
Additional measures should also be consid-ered, such as the mandatory inclusion in label-ing, advertising, and marketing materials of astatement or symbol indicating that the drughas only recently been approved and hence isat risk for the emergence of currently unrecog-nized adverse effects. The United Kingdom re-quires the use of an inverted black triangle in adrug’s labeling to indicate that it was recentlyapproved.35
In a 2007 report23 the Institute of Medicinerecommended that product labels carry a symbolsuch as the black triangle for new drugs. Howev-er, the FDA determined that the use of such asymbol would not be universally comprehendedand could confuse both prescribers and patients,despite educational efforts.36
Exhibit 2
Estimated Time (Years) Without A New Black-Box Warning Or Withdrawal For New Drugs,Before And After Passage Of The Prescription Drug User Fee Act (PDUFA)
SOURCE Authors’ analysis of data from the Physicians’ Desk Reference and the Food and DrugAdministration.
August 2014 33:8 Health Affairs 1457
at PENN STATE UNIV on August 12, 2014Health Affairs by content.healthaffairs.orgDownloaded from

Finally, as others have suggested,8 policy mak-ers should direct increased agency attention andresources to postmarketing surveillance. Giventhe FDA’s limited resources, there may be a bud-getary trade-off between longer clinical trials todetect risks and stronger postmarketing surveil-lance. Based on the available evidence, it seemsprudent to require stronger postmarketing sur-veillance for breakthrough drugs that are ap-proved quickly.
ConclusionNew drugs have a one-in-three chance of acquir-ing a newblack-boxwarning or beingwithdrawnfor safety reasons within twenty-five years of ap-proval. We believe that the ultimate solution isstronger US drug approval standards. In the in-terim, with the rare exception of truly break-through therapies, doctors should preferentiallyprescribe drugs that have been on the marketlonger and hence have a more established trackrecord of safety.32 ▪
Some of this research was presented atthe thirty-fifth annual meeting of theSociety of General Internal Medicine,Orlando, Florida, May 9–12, 2012.
NOTES
1 Lazarou J, Pomeranz BH, Corey PN.Incidence of adverse drug reactionsin hospitalized patients: a meta-analysis of prospective studies.JAMA. 1998;279(15):1200–5.
2 Food and Drug Administration. Forindustry: Prescription Drug User FeeAct (PDUFA) [Internet]. SilverSpring (MD): FDA; [last updated2014 May 6; cited 2014 Jun 10].Available from: http://www.fda.gov/ForIndustry/UserFees/PrescriptionDrugUserFee/default.htm
3 Kramer DB, Kesselheim AS. Userfees and beyond—the FDA Safety andInnovation Act of 2012. N Engl JMed. 2012;367(14):1277–9.
4 Berndt ER, Gottschalk AHB,Philipson TJ, StrobeckMW. Industryfunding of the FDA: effects of PDUFAon approval times and withdrawalrates. Nat Rev Drug Discov. 2005;4(7):545–54.
5 Moore TJ, Furberg CD. The safetyrisks of innovation: the FDA’s Ex-pedited Drug Development Pathway.JAMA. 2012;308(9):869–70.
6 Woolhandler S, Himmelstein DU.Unpublished analysis of data fromthe National Ambulatory MedicalCare Surveys and the National Hos-pital Ambulatory Medical Care Sur-veys, 1999–2010.
7 Begosh A, Goldsmith J, Hass E,Lutter RW, Nardinelli C, Vernon JA.Black box warnings and drug safety:examining the determinants andtiming of FDA warning labels [In-ternet]. Cambridge (MA): NationalBureau of Economic Research; 2006Dec [cited 2014 Jun 10]. (NBERWorking Paper No. 12803). Availablefrom: http://www.nber.org/papers/w12803.pdf
8 Grabowski H, Wang YR. Do fasterFood and Drug Administration drugreviews adversely affect patientsafety? An analysis of the 1992 Pre-scription Drug User Fee Act. J LawEcon. 2008;51:377–406.
9 Issa AM, Phillips KA, Van Bebber S,Nidamarthy HG, Lasser KE, Haas JS,et al. Drug withdrawals in the UnitedStates: a systematic review of theevidence and analysis of trends. CurrDrug Saf. 2007;2(3):177–85.
10 Olson MK. The risk we bear: the ef-fects of review speed and industryuser fees on new drug safety. JHealth Econ. 2008;27(2):175–200.
11 Moore TJ, Furberg CD. Developmenttimes, clinical testing, postmarketfollow-up, and safety risks for thenew drugs approved by the US Foodand Drug Administration: the classof 2008. JAMA Intern Med. 2014;174(1):90–5.
12 Rodriguez-Monguio R, SpielbergerK, Seoane-Vazquez E. Examinationof risk evaluation and mitigationstrategies and drug safety in the US.Res Social Admin Pharm. 2014;10(1):232–8.
13 Food and Drug Administration.Regulatory information: questionsand answers on the Federal Registernotice on drugs and biologicalproducts deemed to have risk eval-uation and mitigation strategies[Internet]. Silver Spring (MD): FDA;[last updated 2009 Jun 18; cited2014 Jun 10]. Available from: http://www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticActFDCAct/SignificantAmendmentstotheFDCAct/FoodandDrugAdministrationAmendmentsActof2007/ucm095439.htm
14 Lasser KE, Allen PD, WoolhandlerSJ, Himmelstein DU, Wolfe SM, BorDH. Timing of new black box warn-ings and withdrawals for prescrip-tion medications. JAMA. 2002;287(17):2215–20.
15 Tufts Center for the Study of DrugDevelopment. US approved drugsdata set. Boston (MA): The Center;2010.
16 Food and Drug Administration.
drugs@FDA [home page on the In-ternet]. Silver Spring (MD): FDA;[cited 2013 Feb 13]. Available from:http://www.accessdata.fda.gov/scripts/cder/drugsatfda/
17 Panagiotou OA, Contopoulos-Ioannidis DG, Papanikolaou PN,Ntzani EE, Ioannidis JP. Differentblack box warning labeling for same-class drugs. J Gen Intern Med.2011;26(6):603–10.
18 Food and Drug Administration,Center for Drug Evaluation and Re-search. 2005 report to the nation:improving public health throughhuman drugs [Internet]. SilverSpring (MD): FDA; 2005 [cited 2014Jun 10]; Available from: http://www.fda.gov/downloads/AboutFDA/CentersOffices/CDER/WhatWeDo/ucm078935.pdf
19 Lexchin J. Drug withdrawals fromthe Canadian market for safety rea-sons, 1963–2004. CMAJ. 2005;172(6):756–67.
20 Qureshi ZP, Seoane-Vazquez E,Rodriguez-Monguio R, StevensonKB, Szeinbach SL. Market with-drawal of new molecular entitiesapproved in the United States from1980 to 2009. PharmacoepidemiolDrug Saf. 2011;20(7):772–7.
21 Wysowski DK, Swartz L. Adversedrug event surveillance and drugwithdrawals in the United States,1969–2002: the importance of re-porting suspected reactions. ArchIntern Med. 2005;165(12):1363–9.
22 Physicians’ desk reference. 54th–64th editions. Montvale (NJ): Med-ical Economics Co.; 2000–10.
23 Baciu A, Stratton K, Burke SP, edi-tors. The future of drug safety: pro-moting and protecting the health ofthe public. Washington (DC): Na-tional Academies Press; 2007.
24 Department of Health and HumanServices, Office of Inspector General.FDA lacks comprehensive data todetermine whether risk evaluation
Prescription Drugs
1458 Health Affairs August 2014 33:8
at PENN STATE UNIV on August 12, 2014Health Affairs by content.healthaffairs.orgDownloaded from

and mitigation strategies improvedrug safety [Internet]. Washington(DC): HHS; 2013 Feb [cited 2014Jun 10]. Available from: https://oig.hhs.gov/oei/reports/oei-04-11-00510.pdf
25 To access the Appendix, click on theAppendix link in the box to the rightof the article online.
26 Carpenter D, Zucker EJ, Avorn J.Drug-review deadlines and safetyproblems. N Engl J Med. 2008;358(13):1354–61.
27 Woodcock J. Statement on FDA userfee agreements: strengthening FDAand the medical products industryfor the benefit of patients [Internet].Silver Spring (MD): Food and DrugAdministration; 2012 Mar 29 [cited2014 Jun 10]. Available from: http://www.fda.gov/newsevents/testimony/ucm297390.htm
28 Gagne JJ, Choudhry NK. How many“me-too” drugs is too many? JAMA.2011;305(7):711–2.
29 Lurie P, Sasich LD. Safety of FDA-
approved drugs. JAMA. 1999;282(24):2297N8.
30 Downing NS, Aminawung JA, ShahND, Krumholz HM, Ross JS. Clinicaltrial evidence supporting FDA ap-proval of novel therapeutic agents,2005–2012. JAMA. 2014;311(4):368–77.
31 Sherman RE, Li J, Shapley S, RobbM, Woodcock J. Expediting drugdevelopment—the FDA’s new“breakthrough therapy” designa-tion. N Engl J Med. 2013;369(20):1878.
32 Schiff GD, Galanter WL, Duhig J,Lodolce AE, Koronkowski MJ,Lambert BL. Principles of conserva-tive prescribing. Arch Intern Med.2011;171(16):1433–40.
33 Shuchman M. Drug risks and freespeech—can Congress ban consumerdrug ads? N Engl J Med. 2007;356(22):2236–9.
34 Congressman Gene Green [Inter-net]. Washington (DC): Congress-man Gene Green. Press release,
Green, Gingrey introduce ADAPTActto safeguard public health; 2013 Dec12 [cited 2014 Jun 10]. Availablefrom: http://green.house.gov/press-release/green-gingrey-introduce-adapt-act-safeguard-public-health
35 Medicines and Healthcare ProductsRegulatory Agency. Black Trianglescheme—new medicines and vac-cines subject to EU-wide additionalmonitoring [Internet]. London:MHRA; [last modified 2014 Jun 5;cited 2014 Jun 10]. Available from:http://www.mhra.gov.uk/Safetyinformation/Howwemonitorthesafetyofproducts/Medicines/BlackTriangleproducts/index.htm#5
36 Shuren J. Statement on FDA userfees 2012: how innovation helpspatients and jobs [Internet]. SilverSpring (MD): Food and Drug Ad-ministration; 2012 Apr 18 [cited2014 Jun 10]. Available from: http://www.fda.gov/NewsEvents/Testimony/ucm300576.htm
August 2014 33:8 Health Affairs 1459
at PENN STATE UNIV on August 12, 2014Health Affairs by content.healthaffairs.orgDownloaded from





























