Embed Size (px)
Citation preview

R
Ahoovgfascsimiar
KP
I
cDaasoalaa
tb[m
FI
©d
eview Article
Epilepsy Syndromes in InfancyChristian M. Korff, MD and Douglas R. Nordli, Jr., MD
odeHoscwrgtmt2emp
ES
siWtetas[mrdst[cm
CDB
n increasing number of infantile epilepsy syndromesave been recognized. However, a significant numberf infants (children aged 1-24 months) do not fit in anyf the currently used subcategories. This article re-iews the clinical presentation, electroencephalo-raphic findings, evolution, and management of theollowing entities: early infantile epileptic encephalop-thy, early myoclonic epilepsy, infantile spasms/Westyndrome, severe myoclonic epilepsy of infancy, myo-lonic-astatic epilepsy, generalized epilepsy with febrileeizures plus, malignant migrating partial seizures ofnfancy, hemiconvulsions-hemiplegia-epilepsy, benign
yoclonic epilepsy, and benign familial/nonfamilialnfantile seizures. Issues related to their classificationre addressed. © 2006 by Elsevier Inc. All rightseserved.
orff CM, Nordli Jr DR. Epilepsy syndromes in infancy.ediatr Neurol 2006;34:253-263.
ntroduction
An accurate diagnosis is considered the first and mostritical step in the management of pediatric epilepsy [1,2].elineating epilepsy syndromes serves that purpose. In
ddition to improving communication between caregiversnd establishing clear criteria for research studies, epilepsyyndromes can suggest underlying disease pathophysiol-gy, provide prognostic information to the families, andid in the selection of appropriate diagnostic tests. Epi-epsy syndromes are the rational basis for selection ofppropriate antiepileptic drugs, surgical procedures, orlternative treatment options [1,3,4].
The epilepsy syndrome classification system adopted byhe International League Against Epilepsy in 1989 [5] haseen demonstrated to be reliable and applicable to children6,7]. Moreover, an increasing number of infantile (1-24onths) epileptic syndromes are being isolated and rec-
rom the Epilepsy Center, Children’s Memorial Hospital, Chicago,llinois.
dR
2006 by Elsevier Inc. All rights reserved.oi:10.1016/j.pediatrneurol.2005.08.005 ● 0887-8994/06/$—see front matter
gnized as such by the majority, thanks to the recentevelopments in diagnostic techniques, such as video-lectroencephalography, brain imaging, and cytogenetics.owever, despite this remarkable progress, up to a quarterf infants with epilepsy do not fit in any of the proposedyndromes, or are classified into broad and unspecificategories [8]. It is likely that additional entities areaiting to be defined in this age group [1]. This article
eviews the clinical characteristics, electroencephalo-raphic findings, evolution, and management of the infan-ile epileptic syndromes identified in infancy so far. Theirain features are summarized in Table 1, and specific
reatment options for some of them are presented in Table. Issues related to their classification are addressed at thend of the review, and suggestions for further improve-ents in the classification of infantile epilepsies are
roposed.
arly Infantile Epileptic Encephalopathy Withuppression-Burst (Ohtahara Syndrome)
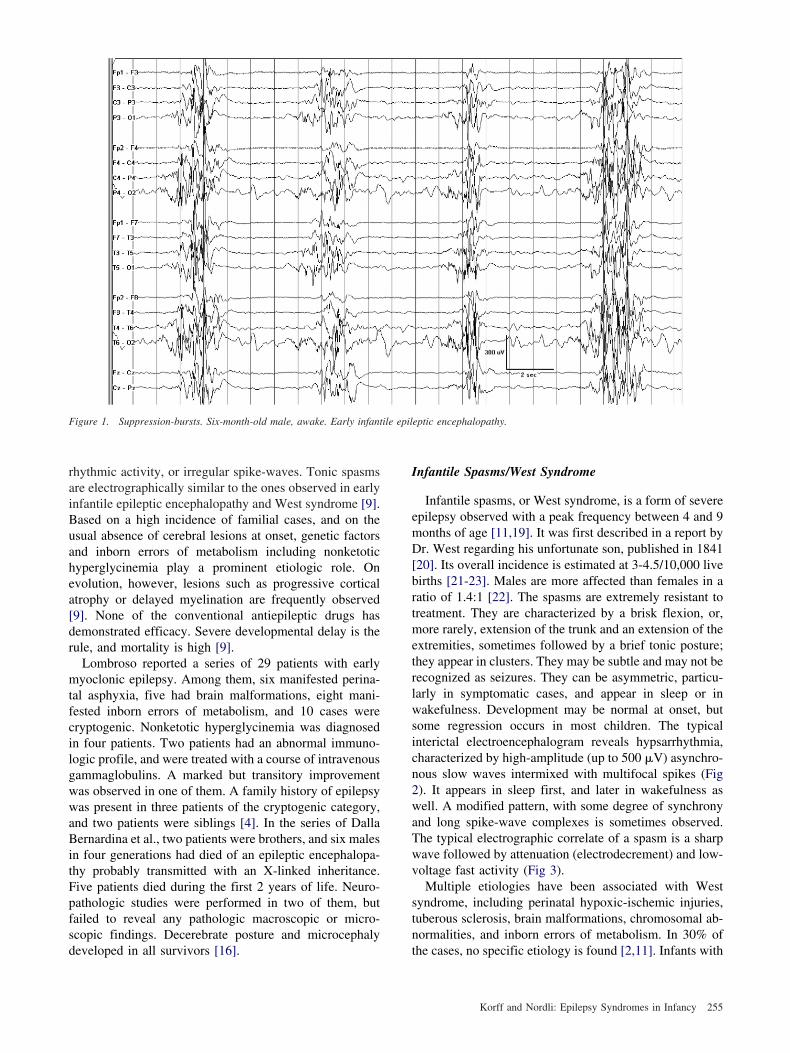
Early infantile epileptic encephalopathy, or Ohtaharayndrome, is a severe neurologic condition first describedn 1976 [9]. It is a rare entity, with a relative prevalence to
est syndrome estimated at 1/40 or less [9]. It is charac-erized by epileptic tonic spasms and a suppression-burstlectroencephalogram (Fig 1). The suppression-burst pat-ern is present in both waking and sleeping states, andppears in the first 3 months of life [9]. Partial toniceizures can be observed, but myoclonic seizures are rare9]. Etiology is heterogeneous, and includes brain malfor-ations, migration disorders, mitochondrial diseases, and,
arely, inborn errors of metabolism, such as Leigh’sisease or cytochrome oxidase deficiency [9,10]. Theeizures are usually intractable to antiepileptic medica-ions, and the developmental prognosis is uniformly poor9]. Surgery can be considered in those patients withortical malformations, such as cortical dysplasia or hemi-egalencephaly. Ohtahara syndrome frequently evolves
ommunications should be addressed to:r. Nordli; Director, Epilepsy Center; Children’s Memorial Hospital;ox #29; 2300 Children’s Plaza; Chicago, IL 60614-3394. E-mail:
[email protected] May 5, 2005; accepted July 27, 2005.253Korff and Nordli: Epilepsy Syndromes in Infancy

iGwnttc
ifsefoii3a2chtpsooAcoa
hiapOieldewg[
E
cdc“eboaeeississcacsamc
T
I
D
M
B
T
EEID
M
MG
HBB r; parti
2
nto West syndrome, and for some, later, into Lennox-astaut syndrome [4,11,12]. Controversies exist as tohether Ohtahara syndrome, West syndrome, and Len-ox-Gastaut syndrome are part of the same spectrum ofhe age-dependent epileptic encephalopathies, or whetherhey each represent isolated syndromes with specificharacteristics [12,13].
A recent report on 16 patients diagnosed with earlynfantile epileptic encephalopathy indicated that the age atirst seizure ranged from 1 day to 3 months. There was noex predominance. All of the patients had tonic spasms,ither in clusters or isolated. Hemiconvulsions, erraticocal motor seizures, and asymmetric tonic seizures werebserved in one third to one half of the patients. General-zed clonic seizures were observed in one patient. Thenterictal electroencephalogram was characterized by 1- to-second bursts of high-voltage (150-350 �V) slow wavesnd multifocal spikes, separated by suppression phases of-5 seconds duration. This pattern was independent of theircadian rhythm. The spasms were accompanied by aigh-voltage slow wave followed by attenuation, some-imes with low-voltage fast activity. The interictal sup-ression-burst pattern often disappeared during a cluster ofpasms. Partial seizures could precede or follow a clusterf spasms, but were also observed independently. Etiologyr associated conditions included two suspected cases oficardi syndrome, one case of hemimegalencephaly, one
ase of porencephaly, one case of hydrocephalus, one casef lissencephaly, and four cases of nonspecific cerebraltrophy or dysgenesis. Adrenocorticotrophic hormone
able 2. Specific treatment options
Epileptic Syndrome Treatment Options
nfantile spasms (West) Adrenocorticotrophic hormone, vigabatrin,ketogenic diet
ravet Stiripentol–valproic acid–clobazam,topiramate
yoclonic astatic epilepsy Valproic acid, levetiracetam, ketogenicdiet
enign myoclonic Valproic acid
able 1. Main characteristics of the epileptic syndromes that pres
Syndrome
arly infantile epileptic encephalopathy First moarly myoclonic epilepsy First mo
nfantile spasms First yearavet syndrome First yea
generayoclonic astatic epilepsy First yea
and balignant migrating partial seizures of infancy First yeaeneralized epilepsy with febrile seizures (�) Variable
afebriemiconvulsion-hemiplegia-epilepsy Prolongeenign myoclonic epilepsy First 3 yenign familial/nonfamilial seizures First yea
Pepilepsy
54 PEDIATRIC NEUROLOGY Vol. 34 No. 4
elped two patients with cryptogenic etiology, but wasneffective in all others. Three patients had their seizurectivity controlled on zonisamide. This publication wasarticularly important because of the prognostic findings.n evolution, two groups with different prognoses were
dentified: one of them included seven patients, whovolved from Ohtahara syndrome to West syndrome andater to Lennox-Gastaut syndrome, and all of these patientsied; the second included patients with spike foci onlectroencephalogram: only one patient died, and sevenere seizure-free. Nevertheless, all patients in the secondroup were physically and mentally severely handicapped14].
arly Myoclonic Epilepsy/Encephalopathy
This rare entity was termed “neonatal myoclonic en-ephalopathy” in its initial description [15]. It was furtherelineated and renamed “early myoclonic epileptic en-ephalopathy” by Dalla Bernardina et al. in 1983 [16], andearly myoclonic epilepsy” by Aicardi in 1985 [17]. Likearly infantile epileptic encephalopathy, it is characterizedy intractable seizures with onset during the first 3 monthsf life, suppression-burst interictal electroencephalogram,nd poor developmental prognosis [9]. Early myoclonicpilepsy is, however, distinguished from early infantilepileptic encephalopathy by its predominant clinical man-festations, erratic myoclonias, and by the presence of auppression-burst pattern mainly during sleep. Partialeizures are also observed, and include tonic spasms,solated or in clusters, versive seizures, migrating cloniceizures, and asymmetric tonic seizures with occasionalecondary generalization [9]. Patients with early myo-lonic epilepsy share most electroencephalographic char-cteristics with patients with early infantile epileptic en-ephalopathy, although the duration of bursts anduppression epochs, and the length of burst-burst intervalsre more variable in early myoclonic epilepsy [9,18]. Mostyoclonias have no associated electroencephalographic
hanges, and are therefore considered nonepileptic [4,9].
nfancy
Main Characteristics
nic seizures, spasms; suppression-burst; severe outcomeyoclonias, spasms; suppression-burst (sleep); severe outcomes; hypsarrhythmia; developmental delay
al and generalized seizures, myoclonias; normal EEG at onset; later:pike-waves and multifocal spikes; severe outcomeralized myoclonic and astatic seizures; interictal parietal theta activityspike-waves; variable outcomenuous electrographic seizures, multiple areas of onset; severe outcomeand developmental phenotype in addition to febrile seizures and
ralized convulsionsteral febrile seizures; subsequent hemiparesis and partial epilepsyyoclonic seizures; normal interictal EEG; normal developmental seizures; normal development
ent in i
nths; tonths; mr; spasmr; partilized sr; gene
ilateralr; contiseizure
le gened unilaears; m
artial seizures manifest various electrographic patterns of
raiBuahea[dr
mtfcilgwwaBitFpfsd
I
emD[brtmetrlwsicn2waTwv
stn
F ile epil
hythmic activity, or irregular spike-waves. Tonic spasmsre electrographically similar to the ones observed in earlynfantile epileptic encephalopathy and West syndrome [9].ased on a high incidence of familial cases, and on thesual absence of cerebral lesions at onset, genetic factorsnd inborn errors of metabolism including nonketoticyperglycinemia play a prominent etiologic role. Onvolution, however, lesions such as progressive corticaltrophy or delayed myelination are frequently observed9]. None of the conventional antiepileptic drugs hasemonstrated efficacy. Severe developmental delay is theule, and mortality is high [9].
Lombroso reported a series of 29 patients with earlyyoclonic epilepsy. Among them, six manifested perina-
al asphyxia, five had brain malformations, eight mani-ested inborn errors of metabolism, and 10 cases wereryptogenic. Nonketotic hyperglycinemia was diagnosedn four patients. Two patients had an abnormal immuno-ogic profile, and were treated with a course of intravenousammaglobulins. A marked but transitory improvementas observed in one of them. A family history of epilepsyas present in three patients of the cryptogenic category,
nd two patients were siblings [4]. In the series of Dallaernardina et al., two patients were brothers, and six males
n four generations had died of an epileptic encephalopa-hy probably transmitted with an X-linked inheritance.ive patients died during the first 2 years of life. Neuro-athologic studies were performed in two of them, butailed to reveal any pathologic macroscopic or micro-copic findings. Decerebrate posture and microcephaly
igure 1. Suppression-bursts. Six-month-old male, awake. Early infant
eveloped in all survivors [16]. t
nfantile Spasms/West Syndrome
Infantile spasms, or West syndrome, is a form of severepilepsy observed with a peak frequency between 4 and 9onths of age [11,19]. It was first described in a report byr. West regarding his unfortunate son, published in 1841
20]. Its overall incidence is estimated at 3-4.5/10,000 liveirths [21-23]. Males are more affected than females in aatio of 1.4:1 [22]. The spasms are extremely resistant toreatment. They are characterized by a brisk flexion, or,ore rarely, extension of the trunk and an extension of the
xtremities, sometimes followed by a brief tonic posture;hey appear in clusters. They may be subtle and may not beecognized as seizures. They can be asymmetric, particu-arly in symptomatic cases, and appear in sleep or inakefulness. Development may be normal at onset, but
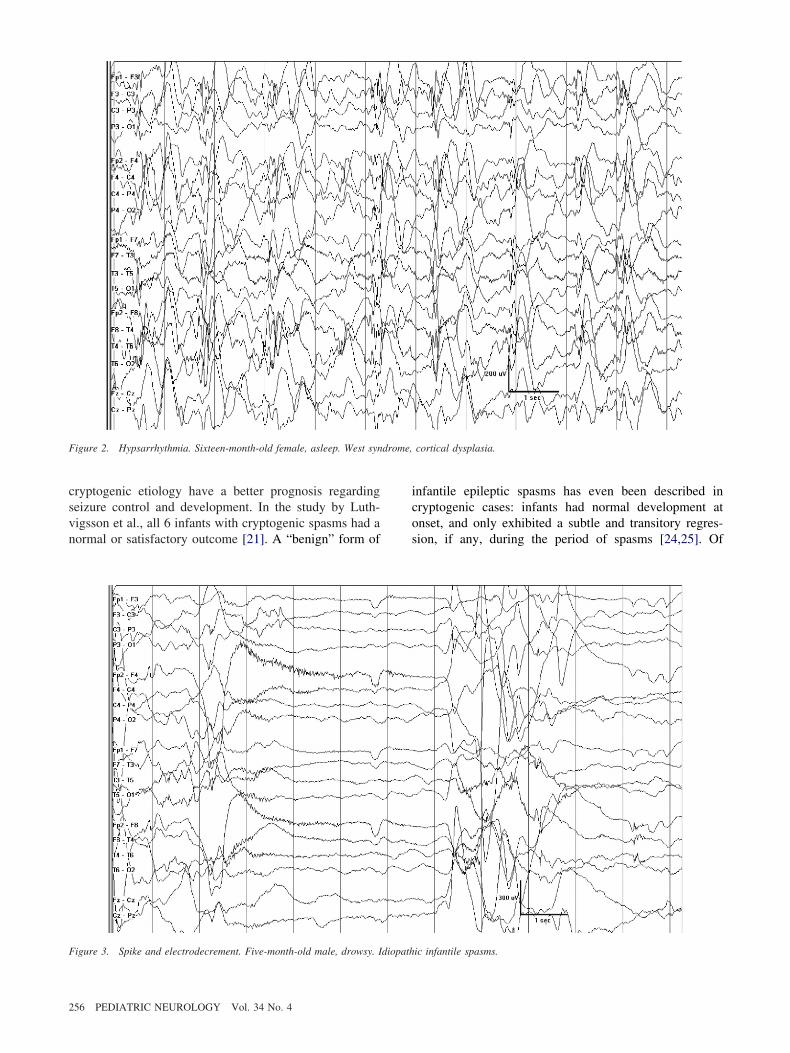
ome regression occurs in most children. The typicalnterictal electroencephalogram reveals hypsarrhythmia,haracterized by high-amplitude (up to 500 �V) asynchro-ous slow waves intermixed with multifocal spikes (Fig). It appears in sleep first, and later in wakefulness asell. A modified pattern, with some degree of synchrony
nd long spike-wave complexes is sometimes observed.he typical electrographic correlate of a spasm is a sharpave followed by attenuation (electrodecrement) and low-oltage fast activity (Fig 3).Multiple etiologies have been associated with West
yndrome, including perinatal hypoxic-ischemic injuries,uberous sclerosis, brain malformations, chromosomal ab-ormalities, and inborn errors of metabolism. In 30% of
eptic encephalopathy.
he cases, no specific etiology is found [2,11]. Infants with
255Korff and Nordli: Epilepsy Syndromes in Infancy
csvn
icos
F drome,
F
2
ryptogenic etiology have a better prognosis regardingeizure control and development. In the study by Luth-igsson et al., all 6 infants with cryptogenic spasms had aormal or satisfactory outcome [21]. A “benign” form of
igure 2. Hypsarrhythmia. Sixteen-month-old female, asleep. West syn
igure 3. Spike and electrodecrement. Five-month-old male, drowsy. Idiopath
56 PEDIATRIC NEUROLOGY Vol. 34 No. 4
nfantile epileptic spasms has even been described inryptogenic cases: infants had normal development atnset, and only exhibited a subtle and transitory regres-ion, if any, during the period of spasms [24,25]. Of
cortical dysplasia.
ic infantile spasms.

p[bbpaciCd
sdoArf
tsnsiatVmctriqessl
s(mhdrrlhgewReccm
cp
dppiw
DE
sclmzdueBtitatDplp
o[[masoaioaoe
taaftctNatssm
articular note was that their visual function was preserved25]. In these infants, the interictal electroencephalogrametween spasms and during a cluster returned to theaseline hypsarrhythmia, and no focal anomalies wereresent [24,25]. However, extensive neuropsychologicalssessment in cryptogenic cases revealed that the risk ofognitive deficits in preschool children was significantlyncreased, even in those with rapid control of spasms.ognitive dysfunction could vary from mild learningisability to specific deficits with normal intelligence [26].Epilepsy with other types of seizures follows West
yndrome in 50-60% of the cases, Lennox-Gastaut syn-rome being the most common form observed [13]. Theptimal treatment of infantile spasms remains uncertain.drenocorticotrophic hormone, prednisolone and, more
ecently, vigabatrin have been traditionally consideredirst choices.
An evidence-based review of the literature on thereatment of infantile spasms revealed the following ob-ervations that summarize the current situation: “1) Adre-ocorticotropic hormone is probably effective for thehort-term treatment of infantile spasms, but there isnsufficient evidence to recommend the optimum dosagend duration of treatment; 2) There is insufficient evidenceo determine whether oral corticosteroids are effective; 3)igabatrin is possibly effective for the short-term treat-ent of infantile spasm and is possibly also effective for
hildren with tuberous sclerosis; 4) Concerns about retinaloxicity suggest that serial ophthalmologic screening isequired in patients on vigabatrin; however, the data arensufficient to make recommendations regarding the fre-uency or type of screening; 5) There is insufficientvidence to recommend any other treatment of infantilepasms; 6) There is insufficient evidence to conclude thatuccessful treatment of infantile spasms improves theong-term prognosis” [27].
A randomized, prospective, multicenter, controlledtudy on 208 infants with spasms compared vigabatrinminimum 100 mg/kg/day) with oral prednisolone (mini-um 40 mg/day) or intramuscular adrenocorticotrophic
ormone (minimum 40 international units on alternateays) using control of spasms at 13 and 14 days afterandomization as the principal outcome measure. Theesults indicated that spasms were significantly moreikely to cease in infants assigned adrenocorticotrophicormone or prednisolone (73% controlled) than in thoseiven vigabatrin (54% controlled). There were no differ-nces between the two hormonal treatments, in contrastith the previously mentioned evidence-based report.esponse to treatment was not affected by the underlyingtiology, but children with tuberous sclerosis were ex-luded on the assumption that vigabatrin was more effi-ient in such cases. No deaths were observed. Outcomeeasures at 14 months are expected to be reported [28].Both adrenocorticotrophic hormone and vigabatrin
arry potentially serious side effects, namely immunosup-
ression, gastric hemorrhage, cardiac insufficiency, and aeath for adrenocorticotrophic hormone; and irreversibleeripheral retinopathy for vigabatrin. These potential com-lications underline the need for detailed pretreatmentnvestigations and closely monitored follow-up visitshen these drugs are used.
ravet Syndrome (Severe Myoclonicpilepsy of Infancy)
Dravet syndrome was described in 1978 [29]. It is aevere epilepsy characterized by generalized and unilaterallonic or tonic-clonic seizures appearing in the first year ofife, later evolution to other types of seizures, includingyoclonias, atypical absences, alternating unilateral sei-
ures, or nonconvulsive status epilepticus. Progressiveevelopmental delay is invariably observed on evolution,sually from the second year on. Tonic seizures arexceptional. All seizures are resistant to treatment [29,30].ecause Dravet syndrome typically presents with a seizure
riggered by fever in an otherwise normal infant, it may bendistinguishable from febrile seizures at onset. However,he prolonged character of the first event and the rapidppearance of additional seizures usually allow differen-iation of Dravet syndrome from febrile seizures. The termravet syndrome was progressively preferred to otherroposed denominations, such as severe myoclonic epi-epsy in infancy, because myoclonias are not invariablyresent at onset.In a recent series, Dravet syndrome accounted for 8.2%
f patients who had their first seizure before the age of 329]. Males are more affected than females in a ratio of 2:129]. The interictal electroencephalogram is usually nor-al at presentation, and later manifests generalized spike-
nd-wave complexes, as well as focal and multifocalpikes. A theta rhythm in the central and vertex region isbserved early in the majority, but tends to disappear withge [13]. Photosensitivity is frequently demonstrated, buts inconstant during the course of the disease [29,30]. It isften a cause of self-stimulation. Massive myoclonias aressociated with bursts of irregular spike-and-waves; on thether hand, erratic myoclonias do not correlate withlectroencephalographic changes [13].
Convulsive seizures are thought to have a focal onset inhe majority of cases, with several foci firing successivelynd giving them a “falsely generalized” or “unstable”spect; truly generalized seizures could in fact be lessrequent than reported [29]. Atypical absences correspondo generalized 2-3.5 Hz irregular spike-waves [29]. Non-onvulsive status epilepticus is electrographically charac-erized by diffuse slow waves and intermixed spikes [29].euroimaging studies usually do not reveal brain lesions
t onset, but may reveal hippocampal sclerosis on evolu-ion [31]. Genetics plays an important role in Dravetyndrome, and a family history of epilepsy or febrileeizures is present in approximately 25% [29]. Affectedonozygotic pairs of twins have been reported. The
ssociation with a mutation in the sodium-channel gene
257Korff and Nordli: Epilepsy Syndromes in Infancy

Svtaid(dsesfgntdafsttctoa[bepw
M
TaanicsmoMecPtbiglldhtm
tedp7osrvcspesatdCd
M
sciidldip
tlfmssoewslobaoC1plici
2
CN1A has been recently discovered, and is present inarious proportions of patients. Recent abstracts suggesthat de novo truncation mutations are particularly associ-ted with Dravet syndrome. The presence of this mutations considered supportive, but not necessary to make theiagnosis of Dravet syndrome. �-aminobutyric acidGABA) receptor gene GABRG2 mutations have also beenescribed, but only rarely [29]. For some authors, Dravetyndrome is the most severe phenotype in the generalizedpilepsy with febrile seizures (�) spectrum [32]. Theignification of de novo mutations in families with otherorms of epilepsy remains unclear, but a heterogeneousenetic background is likely [29,33]. The long-term prog-osis is universally poor. Seizures are extremely difficulto control and remain sensitive to fever or hyperthermia;evelopmental delay is present in all infants; neurologicbnormalities, such as ataxia and pyramidal signs arerequent; and the mortality rate is high (15.9% in Dravet’series) [34]. Topiramate is effective [35,36], and stiripen-ol (not approved in the United States), as add-on therapyo valproic acid and clobazam, has demonstrated signifi-ant efficacy on convulsive seizures in a placebo-con-rolled study on 41 patients [37]. Additional treatmentptions, such as zonisamide and the ketogenic diet, havelso yielded some efficacy in controlling seizure activity29]. On the other hand, certain drugs, such as phenobar-ital, phenytoin, carbamazepine, and lamotrigine, mayxacerbate seizures [4,29]. Prophylaxis of fever and hy-erthermia should be specifically emphasized in patientsith Dravet syndrome.
yoclonic-Astatic Epilepsy (Doose Syndrome)
Myoclonic-astatic epilepsy was described in 1970 [38].he main characteristics of the epileptic syndrome werelready recognized in the initial description. The seizuresre primary generalized with myoclonic or astatic compo-ents that often lead to severe injuries. They are observedn combination with absences, generalized tonic-clonic orlonic-tonic-clonic seizures (personal observations), toniceizures, and nonconvulsive status epilepticus with subtleyoclonias. The onset is between 1 and 5 years, most
ften between 3 and 4 years, in an otherwise normal child.ales are more often affected than females. The interictal
lectroencephalogram is characterized by bilateral syn-hronous irregular 2-3 Hz spike-and-wave complexes.arietal rhythmic theta may be observed independent of
he state of vigilance. Myoclonic seizures are accompaniedy irregular spikes and polyspikes. The astatic components characterized by electrographic silence on electromyo-raphy, and can present as subtle head drops or massiveoss of muscle tone and fall to the ground. Organic brainesions are rarely encountered [39]. As with Dravet syn-rome, a genetic role is strongly suspected. A familyistory of seizures or electroencephalographic abnormali-ies was documented in up to 80% of the cases [38], and
yoclonic-astatic epilepsy was one of the clinical pheno- s
58 PEDIATRIC NEUROLOGY Vol. 34 No. 4
ypes encountered in the families in which generalizedpilepsy with febrile seizures (�) syndrome was initiallyescribed [40]. Outcome is variable. In a study on 52atients, 85% were developmentally normal at onset, and7% remained so at 7.5 years follow-up [41,42]. On thether hand, severe developmental delay and intractableeizures have also been described. The persistence of thetahythms until adolescence and adulthood, failure to de-elop a stable occipital alpha rhythm, generalized tonic-lonic convulsions during the first year of life, nonconvul-ive status epilepticus, and tonic seizures are consideredrognostically unfavorable [43]. In our experience, how-ver, patients with myoclonic-astatic epilepsy and toniceizures can have a normal development. Valproic acid isdvocated by many [39]. Ethosuximide is effective inreating absence seizures. Levetiracetam and the ketogeniciet have demonstrated promising results in our hands.omparative drug trials are definitely wanting in thisisorder.
alignant Migrating Partial Seizures of Infancy
Malignant migrating partial seizures in infancy is aevere epileptic disorder described in 1995 [44]. It isharacterized by nearly continuous electrographic seizuresnvolving multiple independent areas of onset, beginningn the first 6 months of life in normal infants withevelopmental delay and intractable seizures. No under-ying etiology is evident, but a functional or metabolicysfunction is suspected. Its prevalence is unknown; seriesnvolving only small numbers of patients have beenublished [44-46].Coppola et al. reported a series of 14 patients fulfilling
hese criteria. The spectrum of clinical manifestations wasarge. All seizures had a motor component; most wereocal and accompanied by autonomic manifestations;any migrated from one side of the body to the other, and
ix of them secondarily generalized. Two patients pre-ented with status epilepticus. Mild clinical features werebserved during habitual seizures. The interictal electro-ncephalogram was normal in three patients at onset, butas always abnormal during evolution, with multifocal
pikes and sleep abnormalities. The ictal electroencepha-ogram was characterized by monomorphic rhythmic thetar alpha activity progressively involving adjacent areasefore decreasing. Additional seizures beginning in otherreas in either hemisphere could commence before the endf the first event, or immediately follow it (Fig 4).erebral magnetic resonance imaging was performed in0 patients and did not reveal any abnormalities. Elevenatients developed microcephaly during the first year ofife. Three patients died. Conventional treatments wereneffective, except for the combination of stiripentol andlonazepam in two children, in whom some neurologicmprovement was also observed [44].
We and others [47] suspect that there is a wider clinical
pectrum of this disorder. In particular, we have observed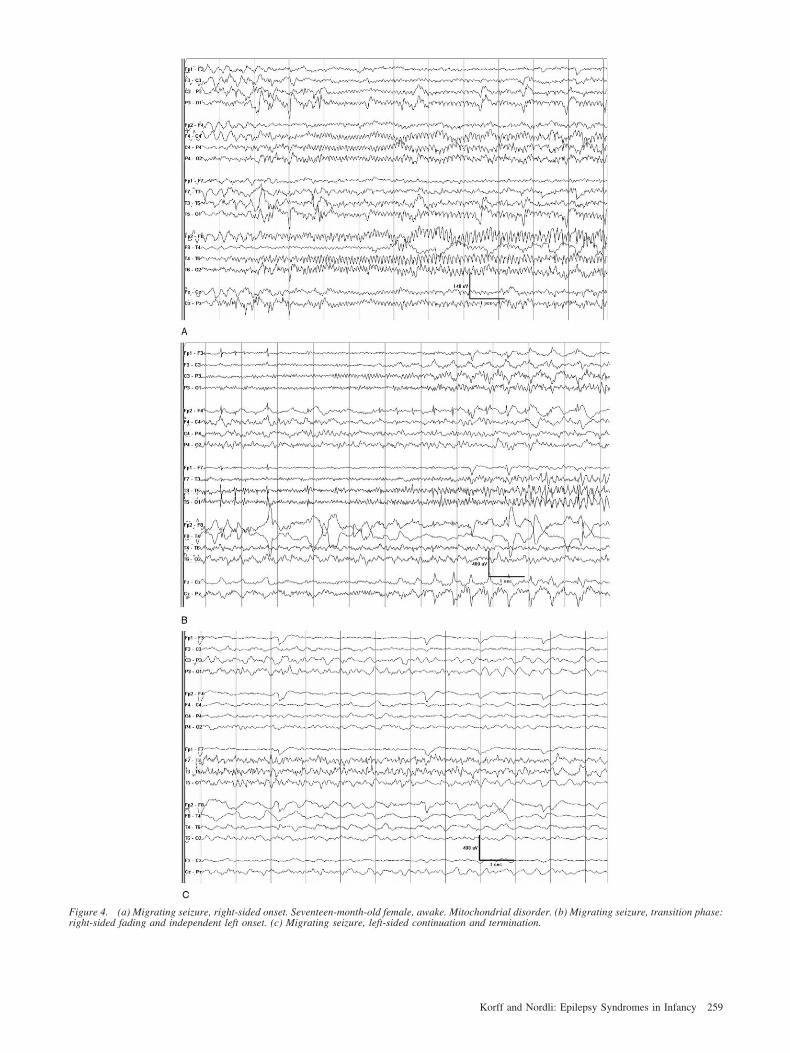
Fr
igure 4. (a) Migrating seizure, right-sided onset. Seventeen-month-old female, awake. Mitochondrial disorder. (b) Migrating seizure, transition phase:ight-sided fading and independent left onset. (c) Migrating seizure, left-sided continuation and termination.
259Korff and Nordli: Epilepsy Syndromes in Infancy

p[t
GP
sgaetrwsssataDdshavsac1oc�fwaa
H
1flrsaomvlecgew
mhdbaaboassdatfepiz
B
1olfafBteiicdsdsoiapoagtSemtnngs
2
atients with less-than-constant seizures, and Marsh et al.47] report patients with slightly better outcomes thanhose reported by Coppola et al.
eneralized Epilepsy With Febrile Seizureslus (GEFS�)
Generalized epilepsy with febrile seizures (�) was de-cribed in 1997 [40]. It is characterized by the association ofeneralized febrile seizures beyond the age of 6 years andfebrile generalized convulsions; a positive family history ofpilepsy with variable phenotypes; and a benign evolution inhe majority of cases. The first description in a large familyevealed that the clinical spectrum of generalized epilepsyith febrile seizures (�) comprised cases of simple febrile
eizures; febrile seizures beyond the age of 6 years [febrileeizures (�)]; febrile seizures (�) and absences; febrileeizures (�) and myoclonic seizures; febrile seizures (�) andtonic seizures; and myoclonic-astatic epilepsy. This spec-rum was further extended with other types of epilepsy, suchs febrile seizures (�) and temporal lobe epilepsy [48],ravet syndrome, and cryptogenic Lennox-Gastaut syn-rome [32]. The pattern of transmission in the first familyuggested an autosomal dominant inheritance. The authorsypothesized that a single major gene defect, with possibledditive gene effect, was responsible for the phenotypicariability on the entire spectrum of the generalized epilep-ies [40,48]. Extensive genetic analyses in these familiesllowed the identification of a mutation in the sodium-hannel �1 subunit gene (SCN�1) on chromosome region9q13.1 [49]. Additional mutations were later discovered inther genes, such as the sodium-channel �1 subunit gene onhromosome region 2q24 [50], or the GABAA receptor2-subunit gene on chromosome region 5q34 [51]. Theseindings underline the genetic complexity of this disorder,hich remains to be completely understood. Some have
rgued that this is not a specific epilepsy syndrome, but ratherdescription of genetic susceptibility.
emiconvulsions-Hemiplegia-Epilepsy
Hemiconvulsions-hemiplegia-epilepsy was described in960 [52]. It is characterized by prolonged unilateralebrile seizures followed by ipsilateral hemiparesis, and,ater, epilepsy. Pathologic studies in the acute stageevealed hemispheric swelling, hemorrhagic necrotic le-ions, and neuronal degeneration. In the chronic stage,trophy, cystic necrosis, and foci of demyelination werebserved. The temporal lobe was by far the most com-only involved region [52]. Vascular causes, such as
enous thromboses, were believed to be the most preva-ent. According to other authors, this may have beenxplained by infections involving the head and face,ommonly associated with cerebral thromboses. The sug-estion was advanced that these cases did not differ intiology from benign febrile seizures, and that the sequelae
ere caused by status epilepticus itself [53]. This state- q60 PEDIATRIC NEUROLOGY Vol. 34 No. 4
ent was supported by the fact that the frequency ofemiconvulsions-hemiplegia-epilepsy had considerablyecreased in parallel to the introduction of the early use ofenzodiazepines in the treatment of febrile convulsionsnd unilateral seizures [53]. Neuroimaging descriptionslso favor the idea that febrile status epilepticus can causerain injuries, with the demonstration of cytotoxic edeman diffusion-weighted magnetic resonance imaging in thecute phase [54-56], and of atrophy [54] and hypoperfu-ion on single-photon emission computed tomographytudies in later stages [57]. A recent report described theetection of human herpesvirus 6 and 7 deoxyribonucleiccid, and specific antibodies for human herpesvirus 7 inhe serum of an 18-month-old patient with generalizedebrile convulsions who exhibited early hemisphericdema and late atrophy [56], but the underlying patho-hysiologic mechanism remains unknown. Studies specif-cally addressing the influence of prolonged febrile sei-ures on temporal lobe epilepsy are currently under way.
enign Myoclonic Epilepsy of Infancy
Benign myoclonic epilepsy of infancy was described in981 [58]. It was initially characterized by the occurrencef easily treatable myoclonic seizures in the first 3 years ofife in infants who remained normal on evolution. Exceptor rare simple febrile seizures, no other types of seizuresre usually observed [59]. A family history of epilepsy orebrile convulsions is present in 30% of the cases [60].enign myoclonic epilepsy of infancy accounted for less
han 1% of all the epilepsies, and 2% of all generalizedpilepsies, in unpublished data from the Centre Saint-Pauln Marseilles. Males are more often affected than femalesn a ratio of approximately 2:1 [59]. The seizures areharacterized as brief myoclonic jerks, particularly duringrowsiness and, in some cases, are activated by photictimulation or sudden external stimuli [59]. They usuallyisappear during sleep. Although generalized tonic-cloniceizures and absence seizures have been described [59],thers regard these seizures as exclusionary criteria. Thenterictal electroencephalogram is normal. The myocloniasre accompanied by generalized spike-waves orolyspike-waves. In contrast, in benign nonepileptic my-clonus of infancy, no electroencephalographic changesre observed during myoclonias. Neuroradiologic investi-ations do not reveal abnormalities. Seizures are sensitiveo valproic acid, which is considered the drug of choice.ixty-seven of 76 patients (88%) with benign myoclonicpilepsy of infancy became seizure-free on valproateonotherapy [59]. In most cases they disappear in less
han 1 year of evolution. Recent data suggest that theeurodevelopmental outcome depends on an early recog-ition and on appropriate treatment. It is favorable in thereat majority, but mild mental retardation can be ob-erved and the real benignity of the syndrome has been
uestioned [59].
B
iAfimdbeomTTp[insptcphmeif
I
tretbiw
cdamsac
wfe““oei
csweedtg
pafdsdwmetaad
CL
R
J
s4
h
eep
ne
e5
Bd2
DD
i
WD
N
t
enign Familial/Nonfamilial Infantile Seizures
This entity is characterized by partial seizures appearingn the first year of life in otherwise normal infants [61,62].
family history of similar seizures at the same age isrequent, and an autosomal dominant inheritance withncomplete penetrance is suspected [63]. Linkage to chro-osome regions 19q, 2q24, and 16p12-q12 has been
emonstrated [63,64]. The seizures are characterized byehavioral arrest with versive components, cyanosis, gen-ralized hypertonia, and unilateral myoclonic jerks. Sec-ndary generalization can occur. They resumed at aedian age of 14-15 months in a series on 64 cases [62].he interictal electroencephalogram is normal [61,62].he ictal electroencephalographic recordings indicate aarieto-occipital onset, with occasional generalization61,63]. Seizures are well controlled on valproic acid [63];n mild cases, treatment might not even be necessary. Theeurodevelopmental prognosis is always favorable. Itseparation from the benign nonfamilial form of infantileartial seizures is controversial. In a study comparing thewo entities, Caraballo et al. did not find any electroclini-al differences. The only parameter that varied was theresence, in benign familial infantile seizures, of a familyistory of seizures. Paroxysmal choreoathetosis, a move-ent disorder occasionally associated with this entity on
volution, was also solely observed in familial cases, butn too small numbers to be considered as a significantinding [62].
ssues and Needs
Some major issues characterize the current classifica-ion of epileptic syndromes in infants. First, most of theeported epileptic syndromes are rare and may not beasily recognized. The available data suggest that whilehese syndromes often carry severe prognoses, they mayenefit from specific forms of treatment. Patients withnfantile epilepsies should therefore be referred to centersith experience in this domain.Second, there is a conspicuous absence of precise
orrelation between metabolic diseases and epilepsy syn-romes. It is likely that more detailed reports of seizuresnd electroencephalographic findings in patients withetabolic defects will help identify additional epilepsy
yndromes. Exceptions include early myoclonic epilepsynd West syndrome where metabolic etiologies havelearly been identified.
Third, there is a small, but important group of infantsith devastating encephalopathies that cannot be classi-
ied into specific syndromes. These unclear cases oftennd up in broad, ill-defined seizure categories, such asunclassified seizures”, and epilepsy subtypes, such asepilepsies with both generalized and focal seizures nottherwise specified” (3.1.0), or “epilepsies without un-quivocal generalized or focal seizures” (3.2). These cases
llustrate that there is a need for more precise seizure rlassification and for further work in classifying epilepsyyndromes. We suspect that there are new syndromesaiting to be discovered within this ill-defined group of
pilepsies. We furthermore speculate that severe inbornrrors of metabolism, severe channelopathies, and neuro-egenerative disorders may be responsible for some ofhese infantile epilepsies with mixtures of focal andeneralized features.Finally, the International League Against Epilepsy has
roposed a diagnostic scheme that is useful for thenalysis of patients with epilepsy. This scheme consists ofive axes that would allow a variety of approaches depen-ent on the particular purpose. These axes include ictalemiology, seizure type, epileptic syndrome, etiology, andegree of impairment [65]. Whatever the adopted scheme,e agree that, implicit in this ordering of diagnoses, oneust arrive at an accurate seizure classification before an
pilepsy syndrome can be established. We believe infan-ile seizures require an electroclinical diagnosis in order tochieve appropriate diagnostic precision. Current effortsre under way to thoroughly establish electroclinicalefinitions of infantile seizures.
hristian Korff, MD is the recipient of a scholarship from the Eugenioitta Foundation, Vaduz, Liechtenstein.
eferences
[1] Nordli DR Jr. Diagnostic difficulty in infants and children.Child Neurol 2002;17(Suppl. 1):S28-35.[2] Shields WD. Diagnosis of infantile spasms, Lennox-Gastaut
yndrome, and progressive myoclonic epilepsy. Epilepsia 2004;5(Suppl. 5):2-4.[3] Aicardi J. Syndromic classification in the management of child-
ood epilepsy. J Child Neurol 1994;9(Suppl. 2):14-8.[4] Lombroso CT. Early myoclonic encephalopathy, early infantile
pileptic encephalopathy, and benign and severe infantile myoclonicpilepsies: A critical review and personal contributions. J Clin Neuro-hysiol 1990;7:380-408.[5] Commission on Classification and Terminology of the Inter-
ational League Against Epilepsy. Proposal for revised classification ofpilepsies and epileptic syndromes. Epilepsia 1989;30:389-99.
[6] Berg AT, Shinnar S, Levy SR, Testa FM. Newly diagnosedpilepsy in children: Presentation at diagnosis. Epilepsia 1999;40:445-2.[7] Berg AT, Shinnar S, Levy SR, Testa FM, Smith-Rapaport S,
eckerman B. How well can epilepsy syndromes be identified atiagnosis? A reassessment 2 years after initial diagnosis. Epilepsia000;41:1269-75.[8] Sarisjulis N, Gamboni B, Plouin P, Kaminska A, Dulac O.
iagnosing idiopathic/cryptogenic epilepsy syndromes in infancy. Archis Child 2000;82:226-30.[9] Ohtahara S, Yamatogi Y. Epileptic encephalopathies in early
nfancy with suppression-burst. J Clin Neurophysiol 2003;20:398-407.[10] Williams AN, Gray RG, Poulton K, Ramani P, WhitehouseP. A case of Ohtahara syndrome with cytochrome oxidase deficiency.ev Med Child Neurol 1998;40:568-70.[11] Aicardi J. Myoclonic epilepsies of infancy and childhood. Adv
eurol 1986;43:11-31.[12] Kelley KR, Shinnar S, Moshe SL. A 5-month-old with intrac-
able epilepsy. Semin Pediatr Neurol 1999;6:138-44; discussion 144-5.[13] Arzimanoglou A, Guerrini R, Aicardi J. Infantile spasms and
elated syndromes. In: Arzimanoglou A, Guerrini R, Aicardi J, eds.
261Korff and Nordli: Epilepsy Syndromes in Infancy

A&
at
(1
m2
BiE
Cc
2
1
E3
o
s
i3
i
dD
AMo1
ItL
m7
s
sE
c2
cm
mBi1
am
Pi
gp
cN
ei
sB
cr1
cG
6
pa
R4
p
sE
ee
gs
SG
et
M1
ve3
sn
NK
hD
ScS
2
icardi’s epilepsy in children, 3rd ed. Philadelphia: Lippincott WilliamsWilkins, 2004:14-37.[14] Yamatogi Y, Ohtahara S. Early-infantile epileptic encephalop-
thy with suppression-bursts, Ohtahara syndrome; its overview referringo our 16 cases. Brain Dev 2002;24:13-23.
[15] Aicardi J, Goutières F. [Neonatal myoclonic encephalopathyauthor’s transl.)] Rev Electroencephalogr Neurophysiol Clin 1978;8:99-01.[16] Dalla Bernardina B, Dulac O, Fejerman N, et al. Earlyyoclonic epileptic encephalopathy (E.M.E.E.). Eur J Pediatr 1983;140:
48-52.[17] Aicardi J. Early myoclonic epilepsy. In: Roger J, Dravet C,
ureau M, Dreifuss FE, Perret A, Wolf P, eds. Epileptic syndromes innfancy, childhood and adolescence, 2nd ed. London: John Libbeyurotext, 1985:12-20.[18] Otani K, Abe J, Futagi Y, Yabuuchi H, Aotani H, Takeuchi T.
linical and electroencephalographical follow-up study of early myo-lonic encephalopathy. Brain Dev 1989;11:332-7.
[19] Zupanc ML. Infantile spasms. Expert Opin Pharmacother003;4:2039-48.[20] West WJ. On a peculiar form of infantile convulsions. Lancet
841;1:724-5.[21] Luthvigsson P, Olafsson E, Sigurthardottir S, Hauser WA.
pidemiologic features of infantile spasms in Iceland. Epilepsia 1994;5:802-5.[22] Brna PM, Gordon KE, Dooley JM, Wood EP. The epidemiol-
gy of infantile spasms. Can J Neurol Sci 2001;28:309-12.[23] Sidenvall R, Eeg-Olofsson O. Epidemiology of infantile
pasms in Sweden. Epilepsia 1995;36:572-4.[24] Dulac O, Plouin P, Jambaque I, Motte J. [Benign epileptic
nfantile spasms]. Rev Electroencephalogr Neurophysiol Clin 1986;16:71-82.[25] Dulac O, Plouin P, Jambaque I. Predicting favorable outcome in
diopathic West syndrome. Epilepsia 1993;34:747-56.[26] Gaily E, Appelqvist K, Kantola-Sorsa E, et al. Cognitive
eficits after cryptogenic infantile spasms with benign seizure evolution.ev Med Child Neurol 1999;41:660-4.[27] Mackay MT, Weiss SK, Adams-Webber T, et al. American
cademy of Neurology; Child Neurology Society. Practice parameter:edical treatment of infantile spasms: Report of the American Academy
f Neurology and the Child Neurology Society. Neurology 2004;62:668-81.[28] Lux AL, Edwards SW, Hancock E, et al. The United Kingdom
nfantile Spasms Study comparing vigabatrin with prednisolone oretracosactide at 14 days: A multicentre, randomised controlled trial.ancet 2004;364:1773-8.[29] Dravet C, Bureau M, Oguni H, Fukuyama Y, Cokar O. Severeyoclonic epilepsy in infancy: Dravet syndrome. Adv Neurol 2005;95:
1-102.[30] Dravet C. Severe myoclonic epilepsy in infants and its related
yndromes. Epilepsia 2000;41(Suppl. 9):7.[31] Siegler Z, Barsi P, Neuwirth M, et al. Hippocampal sclerosis in
evere myoclonic epilepsy in infancy: A retrospective MRI study.pilepsia 2005;46:704-8.[32] Singh R, Andermann E, Whitehouse WP, et al. Severe myo-
lonic epilepsy of infancy: Extended spectrum of GEFS�? Epilepsia001;42:837-44.[33] Guerrini R, Aicardi J. Epileptic encephalopathies with myo-
lonic seizures in infants and children (severe myoclonic epilepsy andyoclonic-astatic epilepsy). J Clin Neurophysiol 2003;20:449-61.[34] Dravet C, Bureau M, Guerrini R, Giraud N, Roger J. Severeyoclonic epilepsy in infancy (Dravet syndrome). In: Roger J, Dravet C,ureau M, Dreifuss FE, Perret A, Wolf P, eds. Epileptic syndromes in
nfancy, childhood and adolescence, 2nd ed. London: John Libbey,992:75-88.[35] Coppola G, Capovilla G, Montagnini A, et al. Topiramate as
dd-on drug in severe myoclonic epilepsy in infancy: An Italian
ulticenter open trial. Epilepsy Res 2002;49:45-8.62 PEDIATRIC NEUROLOGY Vol. 34 No. 4
[36] Nieto-Barrera M, Candau R, Nieto-Jimenez M, Correa A, delortal LR. Topiramate in the treatment of severe myoclonic epilepsy in
nfancy. Seizure 2000;9:590-4.[37] Chiron C, Marchand MC, Tran A, et al., and the STICLO study
roup. Stiripentol in severe myoclonic epilepsy in infancy: A randomisedlacebo-controlled syndrome-dedicated trial. Lancet 2000;356:1638-42.[38] Doose H, Gerken H, Leonhardt R, Volzke E, Volz C. Centren-
ephalic myoclonic-astatic petit mal: Clinical and genetic investigation.europädiatrie 1970;2:59-78.[39] Neubauer BA, Hahn A, Doose H, Tuxhorn I. Myoclonic-astatic
pilepsy of early childhood: Definition, course, nosography, and genet-cs. Adv Neurol 2005;95:147-55.
[40] Scheffer IE, Berkovic SF. Generalized epilepsy with febrileeizures plus: A genetic disorder with heterogeneous clinical phenotypes.rain 1997;120:479-90.[41] Lagenstein I. [Epilepsies with myoclonic-astatic seizures. A
linical and electroencephalographical study in 95 patients. I. Clinicalesults (author’s transl.)] [Article in German]. Monatsschr Kinderheilkd980;128:711-6.[42] Lagenstein I. [Myoclonic-astatic petit mal and its course. A
linical and electroencephalographic study on 95 patients] [Article inerman]. Fortschr Med 1980;98:573-9.[43] Doose H. Myoclonic-astatic epilepsy. Epilepsy Res 1992(Suppl.
):163-8.[44] Coppola G, Plouin P, Chiron C, Robain O, Dulac O. Migrating
artial seizures in infancy: A malignant disorder with developmentalrrest. Epilepsia 1995;36:1017-24.
[45] Veneselli E, Perrone MV, Di Rocco M, Gaggero R, Biancheri. Malignant migrating partial seizures in infancy. Epilepsy Res 2001;6:27-32.[46] Gross-Tsur V, Ben-Zeev B, Shalev RS. Malignant migrating
artial seizures in infancy. Pediatr Neurol 2004;31:287-90.[47] Marsh E, Melamed SE, Barron T, Clancy RR. Migrating partial
eizures in infancy: Expanding the phenotype of a rare seizure syndrome.pilepsia 2005;46:568-72.[48] Singh R, Scheffer IE, Crossland K, Berkovic SF. Generalized
pilepsy with febrile seizures plus: A common childhood-onset geneticpilepsy syndrome. Ann Neurol 1999;45:75-81.
[49] Wallace RH, Wang DW, Singh R, et al. Febrile seizures andeneralized epilepsy associated with a mutation in the Na�-channel �1ubunit gene SCN1B. Nat Genet 1998;19:366-70.
[50] Escayg A, MacDonald BT, Meisler MH, et al. Mutations ofCN1A, encoding a neuronal sodium channel, in two families withEFS�2. Nat Genet 2000;24:343-5.[51] Baulac S, Huberfeld G, Gourfinkel-An I, et al. First genetic
vidence of GABA(A) receptor dysfunction in epilepsy: A mutation inhe gamma2-subunit gene. Nat Genet 2001;28:46-8.
[52] Gastaut H, Poirier F, Payan H, Salamon G, Toga M, Vigouroux. H.H.E. syndrome; hemiconvulsions, hemiplegia, epilepsy. Epilepsia
960;1:418-47.[53] Roger J, Dravet C, Bureau M. Unilateral seizures: Hemicon-
ulsions-hemiplegia syndrome (HH) and hemiconvulsions-hemiplegia-pilepsy syndrome (HHE). Electroencephalogr Clin Neurophysiol 1982;5(Suppl.):211-21.[54] Freeman JL, Coleman LT, Smith LJ, Shield LK. Hemiconvul-
ion-hemiplegia-epilepsy syndrome: Characteristic early magnetic reso-ance imaging findings. J Child Neurol 2002;17:10-6.[55] Herbst F, Heckmann M, Reiss I, Hugens-Penzel M, Gortner L,
eubauer B. [Hemiconvulsion-Hemiplegia-Epilepsy-Syndrome (HHE)].lin Padiatr 2002;214:126-7.[56] Kawada J, Kimura H, Yoshikawa T, et al. Hemiconvulsion-
emiplegia syndrome and primary human herpesvirus 7 infection. Brainev 2004;26:412-4.[57] Salih MA, Kabiraj M, Al-Jarallah AS, El Desouki M, Othman
, Palkar VA. Hemiconvulsion-hemiplegia-epilepsy syndrome: A clini-al, electroencephalographic and neuroradiological study. Childs Nervyst 1997;13:257-63.
[58] Dravet C, Bureau M. The benign myoclonic epilepsy of infancy
(N
A
it6
L1
fE
iP
Fs
pe
author’s transl.) [Article in French]. Rev Electroencephalogreurophysiol Clin 1981;11:438-44.[59] Dravet C, Bureau M. Benign myoclonic epilepsy in infancy.
dv Neurol 2005;95:127-37.[60] Dravet C, Bureau M, Genton P. Benign myoclonic epilepsy of
nfancy: Electroclinical symptomatology and differential diagnosis fromhe other types of generalized epilepsy of infancy. Epilepsy Res 1992;(Suppl.):131-5.[61] Vigevano F, Fusco L, Di Capua M, Ricci S, Sebastianelli R,
ucchini P. Benign infantile familial convulsions. Eur J Pediatr 1992;51:608-12.
[62] Caraballo RH, Cersosimo RO, Espeche A, Fejerman N. Benign namilial and non-familial infantile seizures: A study of 64 patients.pileptic Disord 2003;5:45-9.[63] Callenbach PM, de Coo RF, Vein AA, et al. Benign familial
nfantile convulsions: A clinical study of seven Dutch families. Eur Jaediatr Neurol 2002;6:269-83.[64] Caraballo RH, Cersosimo RO, Amartino H, Szepetowski P,
ejerman N. Benign familial infantile seizures: Further delineation of theyndrome. J Child Neurol 2002;17:696-9.
[65] Engel J Jr; International League Against Epilepsy (ILAE). Aroposed diagnostic scheme for people with epileptic seizures and withpilepsy: Report of the ILAE Task Force on Classification and Termi-
ology. Epilepsia 2001;42:796-803.263Korff and Nordli: Epilepsy Syndromes in Infancy













![Personalized translational epilepsy research - novel ... · focal (mostly lesional) epilepsy syndromes who are candidates for epilepsy surgery [6]. The ... characterized by hypo-,](https://img.dokumen.tips/doc/110x75/5f2c017e847cd27046085bd0/personalized-translational-epilepsy-research-novel-focal-mostly-lesional.jpg)











![EEG & Epilepsy syndromes report [Autosaved]](https://img.dokumen.tips/doc/110x75/55c907aebb61ebbb5b8b459b/eeg-epilepsy-syndromes-report-autosaved.jpg)



