Embed Size (px)
Citation preview
Epilepsyand Treatment
Anup Patel, M.D.Pediatric Neurologist
Capitol Neurology

Epilepsy
Epilepsy is a treatable condition and epileptic patients can live a normal and healthy life. Epilepsy should not be a social taboo

Definitions
• Epileptic seizure:• The clinical manifestations (symptoms and
signs) of excessive and hyper synchronous, usually self limited, activity of neurons in the cerebral cortex.
• Epilepsy:• A chronic disorder characterized by
recurrent (more than 2) unprovoked seizures.

The ILAE classification of seizures• I. Partial (focal, local) seizures
• A. Simple partial seizures (consciousness not impaired)
• B. Complex partial seizures (with impairment of consciousness)
• C.partial seizures evolving to generalized seizures

The ILAE classification of epileptic seizures • II. Generalized seizures
• A. Absence seizures
• 1. Absence seizures
• 2. Atypical absence seizures
• B. Myoclonic seizures
• C. Clonic seizures
• D. Tonic seizures
• E. Tonic-clonic seizures
• F. Atonic seizures (astatic seizures)
• III. Unclassified seizures

The International League Against Epilepsy classification of epilepsies and epileptic syndromes
I. Localization-related (focal, local, partial) epilepsies and syndromesA. Idiopathic (with age-related onset). At present, two
syndromes are established:1. Benign childhood epilepsy with centro temporal spikes
2. Childhood epilepsy with occipital paroxysms
B. Symptomatic. This category comprises syndromes of great individual variability.

The International League Against Epilepsy classification of epilepsies and epileptic syndromes
II. Generalized epilepsies and syndromes
A. Idiopathic (with age-related onset, in order of age appearance)
1. Benign neonatal familial convulsions2. Benign neonatal convulsions3. Benign myoclonic epilepsy in infancy4. Childhood absence epilepsy (pyknolepsy,
petit mal)5. Juvenile absence epilepsy6. Juvenile myoclonic epilepsy (impulsive petit
mal)7. Epilepsy with grand mal seizures on
awakening

The International League Against Epilepsy classification of epilepsies and epileptic syndromes
II. Generalized epilepsies and syndromes
B. Idiopathic, symptomatic, or both (in order of age of appearance)
1. Infantile Spasms
2. Lennox Gastaux
3. Epilepsy with myoclonic-astatic seizures
4. Epilepsy with myoclonic absences

The International League Against Epilepsy classification of epilepsies and epileptic syndromes
C. Symptomatic1. Nonspecific cause, early myoclonic encephalopathy
2. Specific syndromes. Epileptic seizures may complicate many disease states. Under this heading are included those diseases in which seizures are a presenting or predominant feature.

The International League Against Epilepsy classification of epilepsies and epileptic syndromes
III. Epilepsies and syndromes undetermined as to whether they are focal or generalizedA. With both generalized and focal seizures
1.Neonatal seizures2. Severe myoclonic epilepsy in infancy3. Epilepsy with continuous spikes and
waves during slow-wave sleep4. Acquired epileptic aphasia (Landau-
Kleffner syndrome)B. Without unequivocal generalized or focal features

The International League Against Epilepsy classification of epilepsies and epileptic syndromes
IV. Special syndromesA. Situation-related seizures
1. Febrile convulsions2. Seizures related to other identifiable
situations, such as stress, hormones, drugs, alcohol, or sleep deprivationB. Isolated, apparently unprovoked epileptic eventsC. Epilepsies characterized by the specific modes of seizures precipitatedD. Chronic progressive epilepsia partialis continua of childhood

Seizure precipitants
• Stress, emotion• Sleep/sleep deprivation• Hyperventilation• Fever• Medications, metabolic disturbance• Reflex epilepsy
• Photic stimuli: TV, flashing lights, visual patterns
• Startle, music, reading, eating
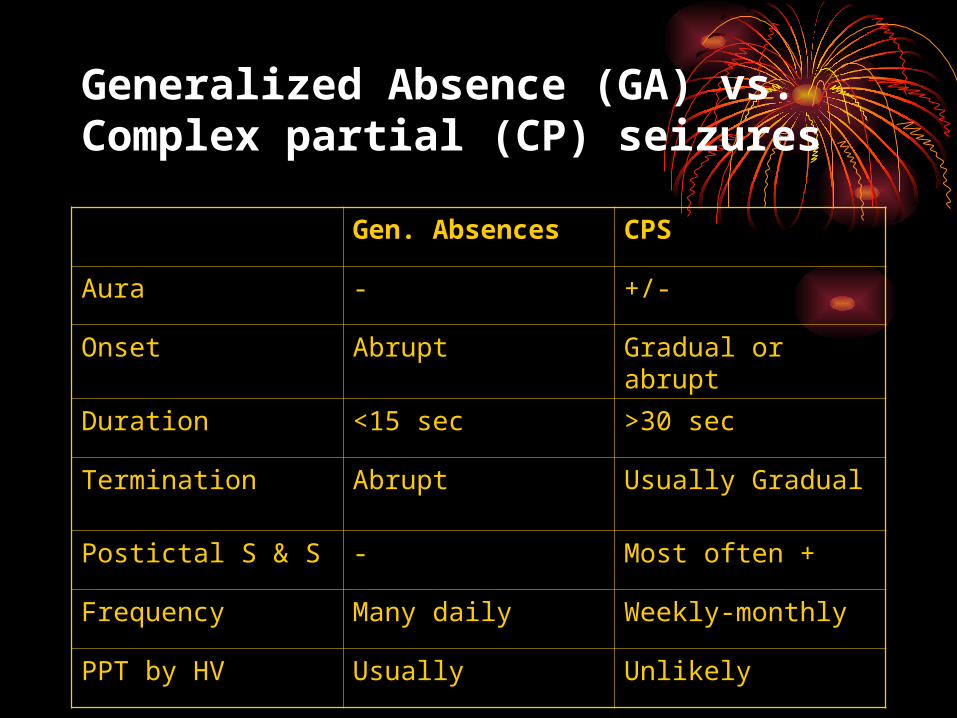
Generalized Absence (GA) vs. Complex partial (CP) seizures
Gen. Absences CPS
Aura - +/-
Onset Abrupt Gradual or abrupt
Duration <15 sec >30 sec
Termination Abrupt Usually Gradual
Postictal S & S - Most often +
Frequency Many daily Weekly-monthly
PPT by HV Usually Unlikely

Workup of a first unexplained seizure.
• EEG• MRI brain• 1 Unexplained seizure does not
necessitate AED treatment except:• Recognized epileptic syndrome with
high probability of recurrence.• Focal brain lesion.

EEG yield
• 1st EEG: 50%• With repeated EEG and
activation procedures the yield can go up to 90%
• No benefit after the fourth EEG, as it gives maximum yield
Treat or not to Treat
• The risk of recurrence of seizures is about 30-35% after the first unprovoked seizure
• The risk of recurrence is about 60% after second seizure

Drug Therapy basic principles• Use a single drug whenever possible.
• However, remember that roughly 60% of patients are controlled on monotherapy.
• Start low and go slow
• Increase the dose of that drug to either seizure control or toxicity (decreasing the dose if toxicity occurs).
• If a drug does not control seizures without toxicity, switch to another appropriate drug used alone, and again increase the dose until seizure control occurs or toxicity intervenes.

Drug therapy
Partial and Secondarily
Generalized Seizures • Carbamazepine, phenytoin, and valproic acid
are the first-line agents among most specialists for partial and secondarily generalized seizures
• Gabapentin, lamotrigine, oxcarbazepine, tiagabine, topiramate, and levetiracetam are new anticonvulsants that are recommended for treatment of partial seizures.

Generalized seizures
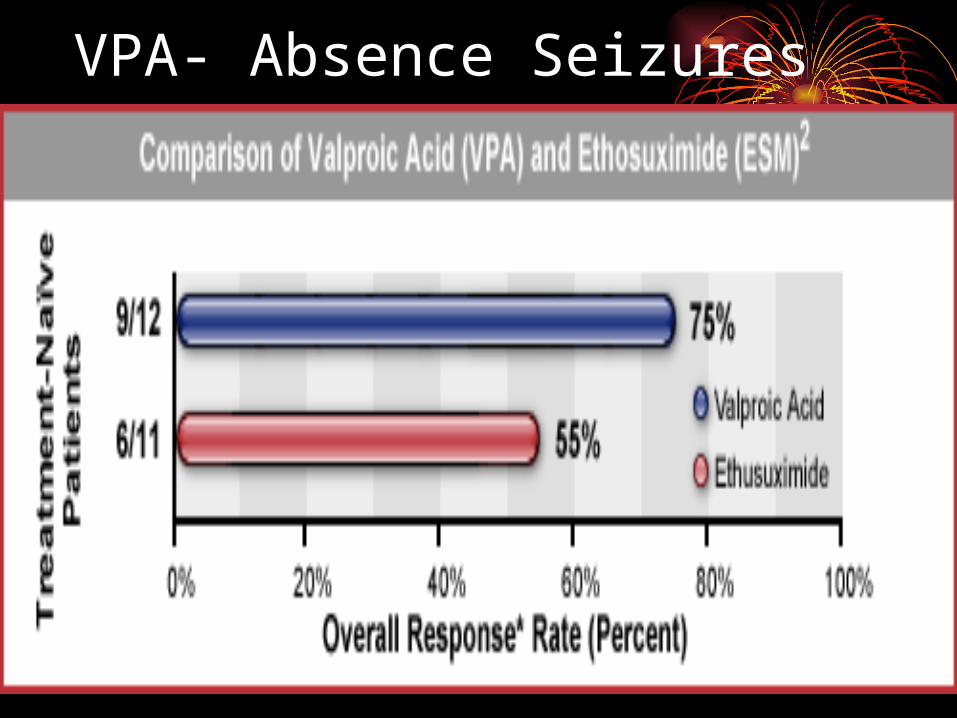
Primary Generalized Seizures• Ethosuximide and valproic acid are effective
for treating absence seizures, but ethosuximide is not effective for treatment of primary generalized tonic-clonic seizures.
• lamotrigine
• felbamate
• zonisamide
• topiramate
• levetiracetam

Ketogenic diet
• Ketosis improves seizure control
• The basic protocol calls for a diet with a fat-to–carbohydrate-plus-protein ratio of 4 to 1 on a caloric basis. A modification of the diet uses medium-chain triglyceride (MCT) oil and allows for a greater amount of carbohydrate. The MCT oil diet is not clearly more beneficial, nor
is it better tolerated. • beneficial in a subset of patients who have not
responded to antiepileptic drugs.

Common Pediatric Epilepsy Syndromes
• Absence Epilepsy• Juvenile Myoclonic Epilepsy• Benign Rolandic Epilepsy• Infantile Spasms• Lennox Gastaux

Absence Seizures
• age of onset 3-8 years• abrupt cessation of activity with change of
facial expression and blank gaze• duration short usually < 15 seconds• child returns to normal and no postictal period• automatisms sometimes• activated by hyperventilation • characteristic EEG 3 Hz spike & wave• treat with AEDs (Ethosuxsimide, Valproate,
Topamax, and Lamictal)• patients usually grow out of seizures by teen
years
VPA- Absence Seizures
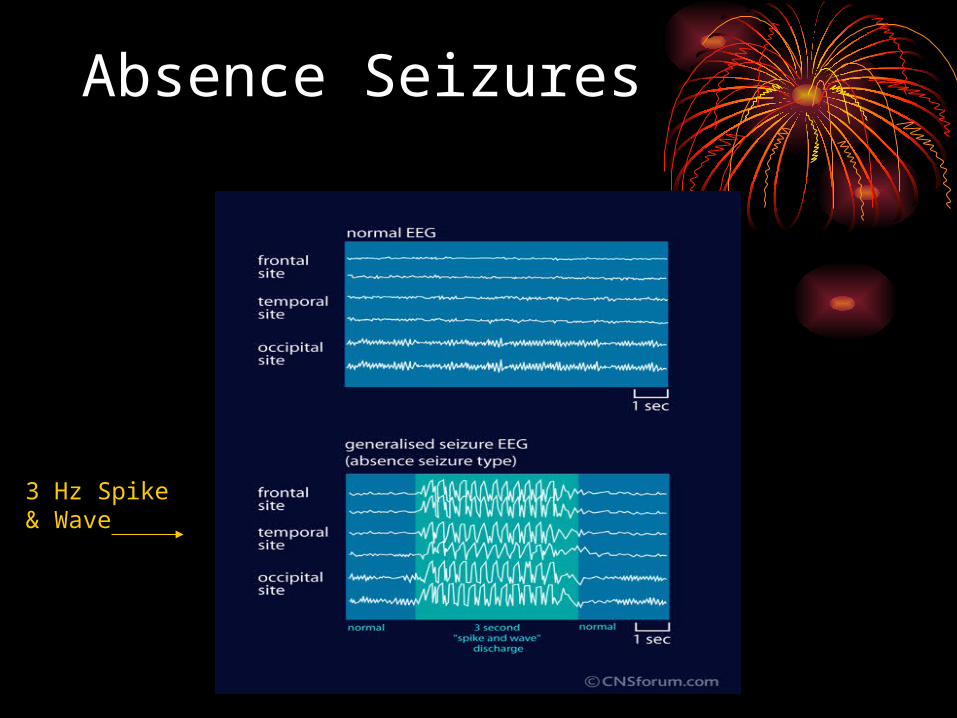
Absence Seizures
3 Hz Spike & Wave

What is JME?
• Also called Janz syndrome • First described in 1867• Triad includes myoclonic jerks,
absence, & tonic clonic seizures• Normal development• Normal imaging

What is JME?
• A common epilepsy syndrome: 10-15% of all epilepsies
• Age of onset 12-18 years• F=M• Accounts for 25% of patients with
idiopathic generalized epilepsies. • Most have myoclonic jerks, 85% have
GTC’s, and 15-38% have absence

Juvenile Myoclonic Epilepsy
• EEG with 3-6 Hz multispike and wave• Photosensitivity in 27%-41%• Focal EEG abnormalities in up to 55%• Triggers: AM wakening, lack of sleep,
fatigue, ETOH, and fasting• Requires life-long treatment• Little data on effective treatment

Treating JME
• Depakote• Keppra• Zonegran• Topamax• Lamictal
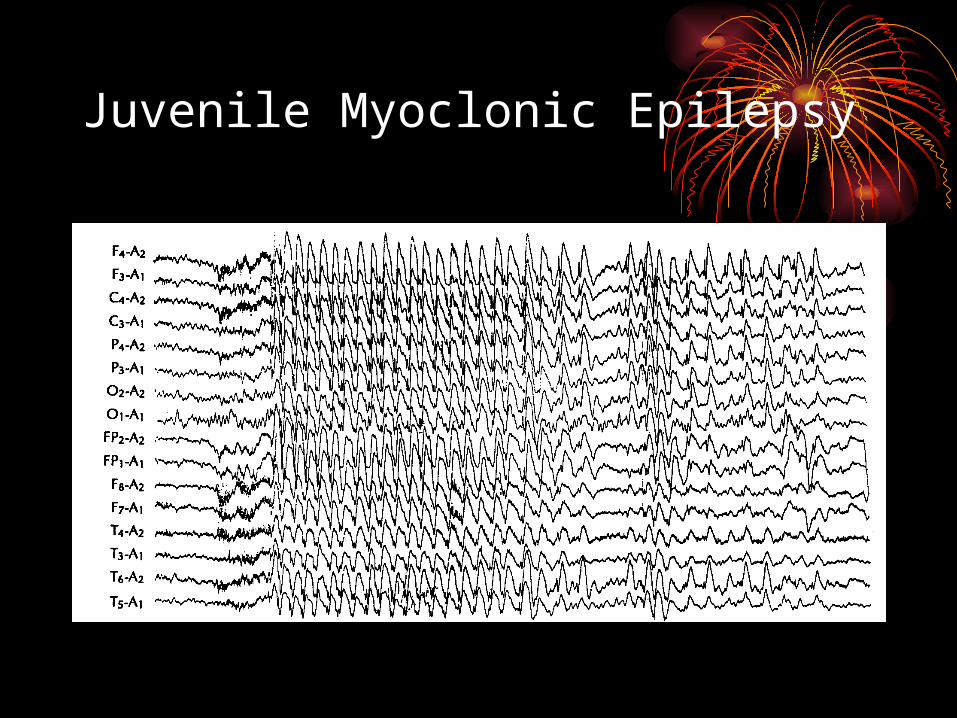
Juvenile Myoclonic Epilepsy

Benign Rolandic Epilepsy
• autonomic dominant• onset 3-13yrs with peak 6-8 years• usually nocturnal or during sleep • infrequent episodes that awake the child with drooling,
speech arrest, ipsilateral facial twitching or twisted to one side that are only minutes in duration
• can sometimes generalize• development and exam are normal• characteristic EEG that shows Midtemporal (T3,T4) and
Central (C3,C4) spikes• treatment usually not indicated if infrequent but can
treat with AEDs• usually outgrown by 14 years
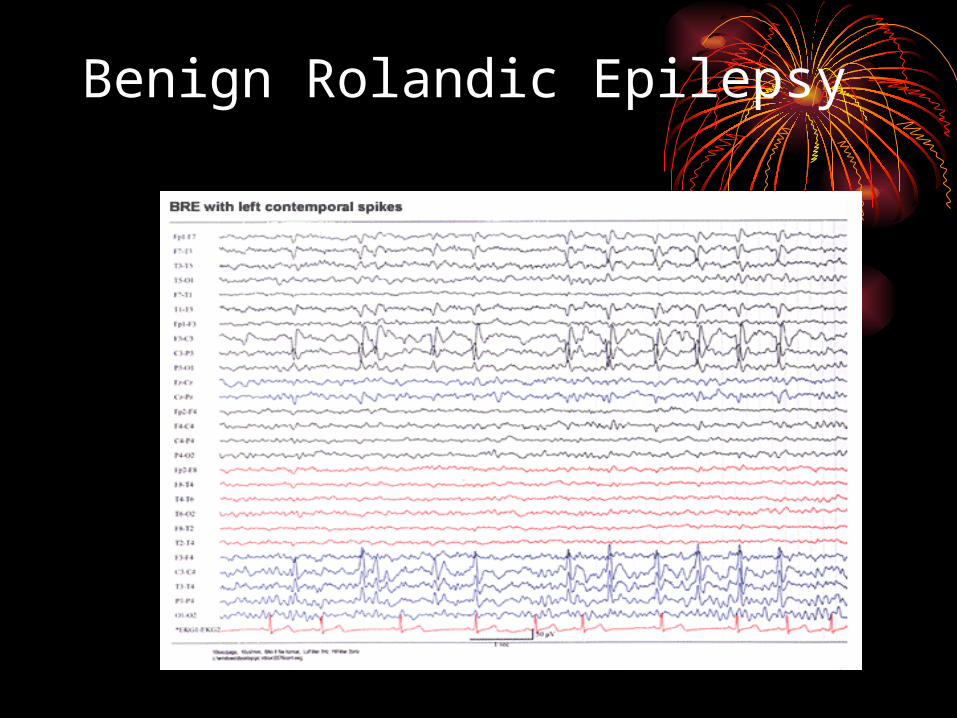
Benign Rolandic Epilepsy

Benign Occipital Epilepsy
• onset 15mos-15years, usually 4-8 years• initial seizure manifestations include visual
hallucinations (flashing lights), blindness, amaurosis, micropsia, metamorphopsia,
• loss of consciousness can occur• can have migraine and nausea afterward• different seizure types (GTC, CPS, unilateral
clonic) and occur mostly when transitioning from wakefulness to sleep
• EEG shows occipital spike & wave 1.5-2.5 Hz and eye opening enhances and sleep inhibits

Infantile Spasms (IS)
• specific type of seizure seen in infancy and early childhood
• onset is predominantly in the first year of life, typically < 1 year
• characteristic EEG called hypsarrhythmia• typical pattern is a sudden bending forward
and stiffening of the body, arms, and legs. Although there can also be arching of the torso.

Infantile Spasms (IS)
• Spasms tend to begin soon after arousal from sleep. Individual spasms typically last for 1 to 5 seconds and occur in clusters, ranging from 2 to 100 spasms at a time.
• Infantile spasms usually stop by age 5, but are often replaced by other seizure types.
• West Syndrome is characterized by infantile spasms, hypsarrhythmia, and mental retardation.

Infantile Spasms (IS)
• Etiology• Cerebral malformations 35%, Perinatal
insult 15%, Metabolic 15%, • Tuberous Sclerosis 10%• Treatment usually starts with AEDs,
steroids, ACTH, Vigabatrin, B6, Surgery (if lesions)
• Prognosis depends on etiology. Worse prognosis with symptomatic as many, 50%, go on to have other types of seizures
• Many develop mental retardation or delayed development.

Infantile Spasms (IS)

Lennox Gastaut Syndrome
• Childhood epileptic encephalopathy (Lennox-Gastaut syndrome [LGS]) is a devastating pediatric epilepsy syndrome
• constituting 1-4% of childhood epilepsies• triad characterized by multiple types of
seizures, mental retardation or regression, and characteristic EEG
• abnormal EEG with generalized slow spike-and-wave discharges (1.5-2 Hz)

Lennox Gastaut Syndrome
• most common seizure types are tonic-axial, atonic, and absence seizures, but myoclonic, generalized tonic-clonic, and partial seizures can be observed. Seizures often are resistant to therapy.
• mean age at epilepsy onset is 3-5 years (range, 1 d to 14 y)
• 60% have underlying cause (TS, NF, perinatal insult) and 20% have history of Infantile Spasms
• diagnosis by history, PE, and EEG• treatment is difficult

Lennox Gastaut Syndrome

Acquired Epileptiform Aphasia
• Landau-Kleffner Syndrome• onset 2-12 years• acquired aphasia, verbal auditory agnosia,
decreased spontaneous speech• difficulty understanding speech and child
stops talking• several seizure types (GTC, Myoclonic,
Absence)• neuropsychological disturbances in >50%
but intelligence is not affected

Acquired Epileptiform Aphasia
• sometimes the child is diagnosed with Autism or being deaf
• EEG is normal during wakefulness but during sleep there is spike & wave mostly in parietal and temporal lobes, sometimes electrical status of sleep
• treatment with AEDs and steroids shows good control
• recovery of language is variable and if onset is before 6 years there is better outcome
• less than 50% live independent lives

Landau-Kleffner Syndrome
Landau-Kleffner EEG Shows S&W

Landau-Kleffner Syndrome

Epilepsy with Continuous Spike Waves During Slow Wave Sleep (CSWS)• also called electrical status epilepticus
of sleep• various seizure types occur during sleep• EEG shows continuous diffuse spike &
wave during slow wave sleep• prognosis guarded because of
neuropsychological disturbance and intellectual regression
• treated with AEDs and steroids

• chromosome 20 q• bizarre behavior and motor symptoms
during sleep • seizures begin in childhood and persists
in adulthood and can come in clusters• most attacks occur when dozing or
initiating sleep and can occur in clusters and a gasp or grunt will awake the child
Autosomal Dominant Nocturnal Frontal Epilepsy

Doose Syndrome
• Myoclonic-Astatic Epilepsy (MAE)• Is often resistant to medication• Is an idiopathic generalized epilepsy and the seizures
are generalized and different types • Onset of MAE occurs commonly between the first and
fifth year of life, with the mean age being three. • Statistics show that it usually affects children who have
previously developed normally, and boys are twice as likely as girls to develop MAE, other family members (immediate or extended) may also have epilepsy.
• Treatment with AEDs

Jeavons Syndrome (Eyelid Myoclonia with Absences)
• Eyelid myoclonia with absences has two components. The initial and more prominent is eyelid myoclonia. This may or may not progress to the second component, which is mild impairment of consciousness (absence). The seizure starts and ends abruptly with a duration of 3 to 5 seconds.
• The patient exhibits eyelid myoclonia with absences mainly on eye-closure and intermittent photic stimulation.
• These do not occur in the dark.

Thank you!





























