Embed Size (px)
Citation preview

Cell Reports, Volume 19
Supplemental Information
Enhanced Rate of Acquisition of Point
Mutations in Mouse Intestinal Adenomas
Compared to Normal Tissue
Natalia Lugli, Vasilis S. Dionellis, Paloma Ordóñez-Morán, Irene Kamileri, Sotirios K.Sotiriou, Joerg Huelsken, and Thanos D. Halazonetis

1
This PDF file contains:
Figure S1
Supplemental Figure Legend
Tables S1-S4
Experimental Procedures
Supplemental References
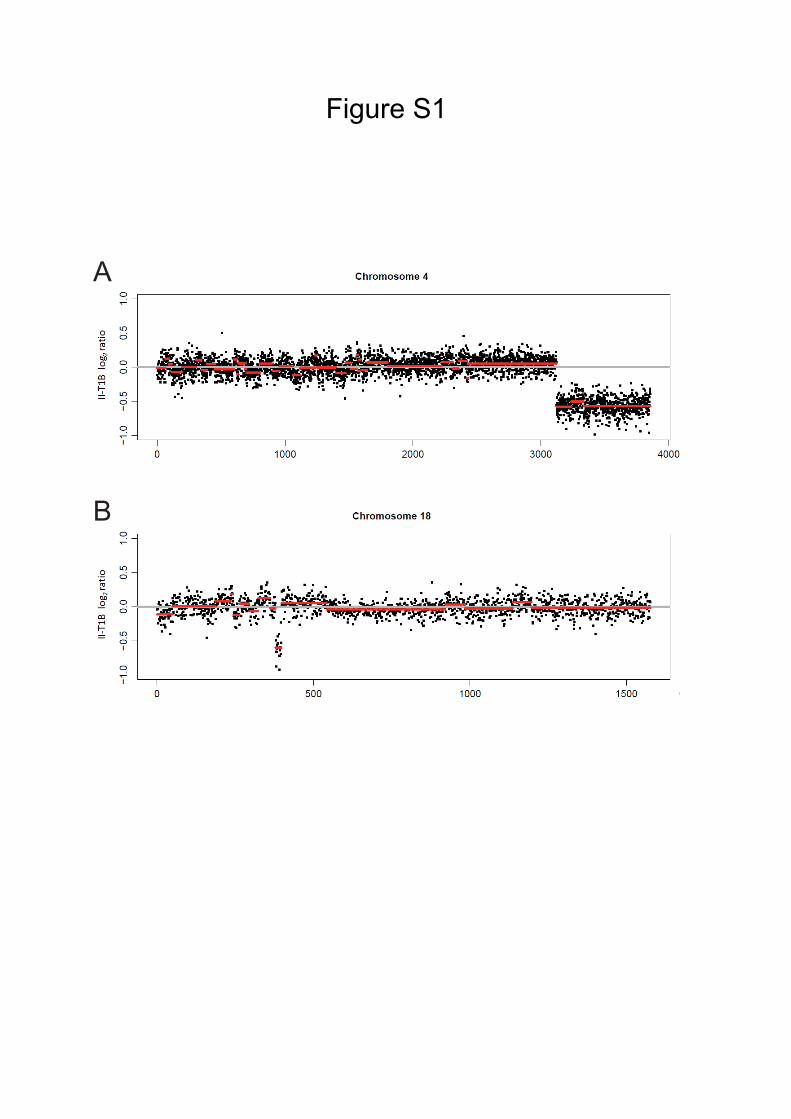
Figure S1
A
B

3
SUPPLEMENTAL FIGURE LEGEND
Figure S1. Copy Number Alterations (CNAs), Related to Figure 4
(A) CNA in tumor-derived organoid II-T1B resulting in deletion of the telomeric 13
Mb of the q arm of chromosome 4.
(B) CNA in tumor-derived organoid II-T1B resulting in a focal deletion of 0.23 Mb
within the q arm of chromosome 18, involving the Apc locus.

4
Table S1. Sequencing coverage of normal and tumor-derived organoids, Related
to Figure 2
See separate file.

5
Table S2. Single nucleotide substitutions in the coding regions of the sequenced
normal and tumor-derived organoids, Related to Figure 3
See separate file.

6
Table S3. Indels in the coding regions of the sequenced normal and tumor-
derived organoids, Related to Figure 4
See separate file.

7
Table S4. Estimated mutation rates per cell division per base pair in normal and
precancerous/cancer cells, calculated from data derived from high throughput
sequencing studies, Related to Figure 2
Study Model Type Mutation rate per cell division per bp (for exomes)
Current study Mouse Normal small intestine 0.37×10!!"
Adenoma small intestine 4×10!!"
Alexandrov et al., 2015 Human Colon cancer 19×10!!"
Nikolaev et al, 2012 Human
Hyperplastic polyps (normal)
Dysplastic polyps
(adenomas)
0.1×10!!"
20×10!!"

8
EXPERIMENTAL PROCEDURES
Mice
The C57BL/6J-ApcMin/J mice were purchased from the Jackson Laboratory (Moser
et al., 1990). All mice were kept on a 12-hour light/dark cycle in an SPF room. The
mice, all of which were males, were sacrificed at 4 months of age; all experiments
were authorized by the Canton of Lausanne and were performed according to
accepted guidelines for animal handling.
Organoid preparation and culture
Small intestine tissue was isolated from C57BL/6J-ApcMin/J mice. The tissue was
washed in cold PBS; healthy looking regions were separated from regions where
tumors were present; each tumor was treated separately. Each healthy part and each
tumor was cut into 2-3 mm pieces and incubated in PBS-EDTA for 30 minutes at 4°C.
Subsequently, the tissue slices were washed in PBS-FBS and dissociated crypts were
filtered through 70 µm cell strainers (BD Bioscience). After centrifugation, the pellets
were resuspended in media, and single crypts were picked both from the healthy part
and from each of the tumors. Each single healthy crypt or each single tumor crypt was
mixed with cold Matrigel® (Corning) and plated in 96-well plates. After the Matrigel®
formed a gel, tissue culture media was added. The tissue culture media was based on
AdDMEM/F12 (Life Technologies) supplemented with B27 and N2 (Life
Technologies) and 1.25 µM N-Acetylcysteine (Sigma-Aldrich). The following growth
factors were also added: 50 ng/mL murine recombinant EGF (Life Technologies) and
R-spondin 1 Fc fusion and noggin-6xHis (Ordóñez-Morán et al., 2015). The medium
was changed every 3 days and organoids were split every 4-5 days by mechanical
dissociation.
Immunofluorescence
Organoids were removed from Matrigel® using Cell Recovery Solution (Corning),
then embedded in OCT (Tissue-Tek); slices 10 µm thick were cut using a cryostat
(Leica CM 1850). Organoid sections were fixed for 1h in 4% paraformaldehyde at
room temperature and stained using standard immunofluorescence techniques and
commercially available antibody for γH2AX (Upstate). The nuclei were
counterstained with DAPI and images were acquired using a Zeiss 700 confocal

9
microscope.
DNA extraction and exome sequencing
DNA was extracted from both healthy and tumor organoids and from the liver of each
mouse using the Qiagen DNeasy Blood & Tissue Kit (Qiagen), according to the
manufacturer’s instructions, and quantified using a Qubit Fluorometer (Thermo
Fisher). The fragmentation of the extracted DNA was conducted using a Covaris
instrument. The resultant DNA fragments (~200bp) were subjected to exome capture
using the SureSelect Mouse All Exon kit (Agilent), followed by preparation of paired-
end libraries and sequencing on an Illumina Hiseq2000 platform.
Mapping and somatic SNS calling
The Burrows-Wheeler Alignment tool (v. 0.7.12) was used for the alignment of
sequenced reads on the mouse reference genome NCBI build GRCm38/mm10 and the
resultant sam files were processed by SAMtools v1.3:sam, in order to perform bam
transformation, sorting, removal of PCR duplicates and indexing. Base quality
recalibration and INDELs realignment was performed using the Genome Analysis
Tool Kit (GATK v.3.5.0). Somatic SNS calling was conducted using the default
parameters of MuTect2 algorithm, which is integrated in the GATK toolkit. In
addition to the embedded filtering process of MuTect (Cibulskis et al., 2013), we
applied some extra criteria regarding the somatic SNS calling: discard variants located
in snp loci, according to NCBI mouse dbsnp142; discard variants with depth less than
14x; shortlist variants with allele fraction of at least 20%; apply 10% tolerance to
strand bias, with the variant present in at least two times in forward (read 1) and
reverse (read 2) read respectively. Moreover, a panel of normal samples, containing
variants from all liver and normal intestine tissues of our mice, was used in order to
discard germline mutations with lower allele frequencies in the general population. In
order to test the sensitivity and the specificity of the algorithm and avoid possible
false positive calls, the aforementioned pipeline was repeated, switching the labels of
normal and tumor samples. Mutation rates were calculated using the following
formula:

10
Identification of LOH events
To detect LOH events, we used the HaplotypeCaller algorithm of the GATK toolkit in
order to call all the heterozygous SNSs variants in liver and all the homozygous SNSs
variants in normal and tumor organoids, and then look for possible overlaps. Raw
variants were first called using GATK v.3.5.0 Haplotypecaller algorithm with default
settings and additional parameters: stand_emit_conf 10 and stand_call_conf 30. Low
quality variants were eliminated using GATK VariantFiltration v.3.5.0 with the
options --filterExpression "QD < 2.0 || FS > 60.0 || MQ < 40.0 || MQRankSum < -12.5
|| ReadPosRankSum < -8.0" --filterName "snv_filter" and --Description="Low
quality". The shortlist of variants was further filtered by applying extra criteria:
discard variants with depth less than 14x; shortlist variants with allele fraction of at
least 20%; apply 10% tolerance to strand bias, with the variant present in at least two
times in forward (read 1) and reverse (read 2) read respectively.
Somatic small indels calling
Raw variants were called using GATK Haplotypecaller v.3.5.0 as previously
described. The quality was assessed by discarding variants using GATK
VariantFiltration v.3.5.0 with the following parameters: --filterExpression "QD < 2.0
|| FS > 200.0 || MQRankSum < -20", --filterName "indel_filter" and --
Description="Low quality". Since this algorithm is not designed for the direct
detection of somatic mutations, we first called all the small indels in the exome of
liver samples and then excluded the same variants detected in both normal and tumor
organoids. Additional filter processes with the same extra criteria as in SNSs calling,
as well as the usage of a panel of normal samples, secured the elimination of false
positives calls.
Somatic mutation signatures
The mutational spectra of detected somatic SNSs were examined using the
SomaticSignature v.2.10.0 R package for the analysis of all the 96 possible
trinucleotide changes.
!"#$#%&' !"#$ =!!"#$%& !" !"#$%&' !"!#!"#$%& !" !"#$%!&'( ! /!"#$%& !"#$%% !"#"$"%&$
!"#$%&' !"#$%& !"#!"# !"#$%ℎ × 2

11
Copy number alteration calling
For the detection of CNAs, bam files were analyzed by VarScan2 v.2.2.4 using the
recommended workflow (Koboldt et al., 2013). VarScan2 copy number mode run
with the parameters --min-coverage 50 --min-segment-size 400 --max-segment-size
500. The parameter --min-coverage 50 was also used during VarScan copyCaller step.
Circular binary segmentation (CBS) and plotting were handled by DNAcopy library
v.1.48.0 using the underneath script:
library(DNAcopy)
cn <- read.table("output.copynumber.called.center",header=T)
CNA.object <-CNA( genomdat = cn[,7], chrom = cn[,1], maploc = cn[,2], data.type
= "logratio", sampleid = "sample_name")
CNA.smoothed <- smooth.CNA(CNA.object)
segment <- segment(CNA.smoothed, verbose=0, min.width=2, undo.SD=3)
seg.pvalue <- segments.p(segment, ngrid=100, tol=1e-6, alpha=0.05,
search.range=100, nperm=1000)
write.table (seg.pvalue, file="DNAcopy.out.file", sep="\t")
plot(segment, plot.type = "p")
plot(segment, plot.type = "c")
In order to detect somatic CNA events, we excluded CNAs that were present in the
liver tissue of the mouse from which the organoids were prepared.

12
SUPPLEMENTAL REFERENCES
Cibulskis, K., Lawrence, M.S., Carter, S.L., Sivachenko, A., Jaffe, D., Sougnez, C.,
Gabriel, S., Meyerson, M., Lander, E.S., and Getz, G. (2013). Sensitive detection of
somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol.
31, 213-219.
Koboldt, D.C., Larson, D.E., and Wilson, R.K. (2013). Using VarScan 2 for Germline
Variant Calling and Somatic Mutation Detection. Curr. Protoc. Bioinformatics 44,
15.4.1-17.
Ordóñez-Morán, P., Dafflon, C., Imajo, M., Nishida, E., and Huelsken, J. (2015).
HOXA5 Counteracts Stem Cell Traits by Inhibiting Wnt Signaling in Colorectal
Cancer. Cancer Cell 28, 815-829.