Embed Size (px)
Citation preview

Emilie Taymor and Panos Kanavos
A Comparative Analysis of the Regulatory Requirements of Orphan Medicinal Products in the United States and European Union
Working Paper No: 45/2015 January 2015 LSE Health

Comparative Analysis of the Regulatory Requirements of Orphan Medicinal Products in the United States and European Union Emilie Taymor1 and Panos Kanavos1
1 LSE Health, London School of Economics and Political Science
Working Paper No. 45/2015 First published in January 2015 by: LSE Health The London School of Economics and Political Science Houghton Street London WC2A 2AE © Emilie Taymor, Panos Kanavos All rights reserved. No part of this paper may be reprinted or reproduced or utilised in any form or by any electronic, mechanical or other means, now known or hereafter invented, including photocopying and recording, or in any information storage or retrieve system, without permission in writing from the publishers. British Library Cataloguing in Publication Data. A catalogue record for this publication is available from the British Library. ISSN 2047-8879Corresponding Author: Panos Kanavos Department of Social Policy Old Building London School of Economics and Political Science Houghton Street London WC2A 2AE [email protected]

1
Abstract
Introduction: Both the Food and Drug Administration (FDA) and European Medicines Agency
(EMA) have regulatory incentives for manufacturers of orphan medicines used to treat rare
diseases. This study compares the similarities and differences of orphan drugs and their
regulations in the US and Europe.
Methods: The study includes a literature review, a comparison between FDA and EMA
regulatory requirements and incentives; an analysis of orphan designation and market
authorization (MA) applications for orphan drugs between the two regulatory bodies; a
comparative analysis on a sample of 14 applications; and finally, a breakdown of the coverage
decisions of the drug samples across 4 European HTA bodies.
Results: As of 01/07/2014 from the start of the EMA’s regulations in 2000, the FDA has granted
275 orphan designations and market authorized 275 of these. In the EU there are 1025 orphan
designations, 76 of which are currently market authorized. Some products the EMA denied
orphan designation or MA to have been designated and approved in the US. The HTA bodies
analyzed have not accepted many of the drugs approved for MA in Europe.
Discussion and Conclusion: The EMA lags behind the FDA in how many orphan designations
and MAA the agency has approved. Based on the results from the study there are a number of
possible explanations for this including differences in: application designation procedure,
incentives given to orphan drugs, MA submission and acceptance dates, and criteria for MA.
Because the small number of HTA bodies granted coverage for these orphan drugs, many
patients are denied access to crucial medicines. Manufacturers of orphan drugs must work with
both the regulatory and HTA bodies to ensure drugs for rare diseases continue to be produced,
marketed, and covered by insurance.

2
Contents 1. Introduction ............................................................................................................................... 5
1.1. Background and Policy Context..............................................................................................................5
1.1.1. Orphan Diseases...................................................................................................................................5
1.1.2. Orphan Drug Designation..................................................................................................................5
1.1.3. Marketing Authorization....................................................................................................................6
1.2. Study Objectives...........................................................................................................................................7
2. Methodology .............................................................................................................................. 8
2.1. Literature Review.........................................................................................................................................8
2.2. Regulatory Requirements and Incentives for Orphan Designations............................................9
2.3. Comparison of Evidentiary Requirements for Orphan Designations..........................................9
2.4. Breakdown of Orphan Designation.....................................................................................................10
2.4.1. Positive Opinions..............................................................................................................................10
2.4.2. Negative Opinions.............................................................................................................................10
2.5. Breakdown and Comparison of European Public Assessment Reports (EPARs) Compared to Corresponding FDA Reports.....................................................................................................................10
2.5.1. Approved MA.....................................................................................................................................11
2.5.2. Refused MA........................................................................................................................................11
2.5.3. Withdrawn MA..................................................................................................................................11
2.5.4 MA Sample Comparison..................................................................................................................11
2.6. Breakdown and Comparison of HTA Appraisals...........................................................................13
2.7. Study Limitations.......................................................................................................................................13
2.8. Ethics Approval.............................................................................Error!Bookmarknotdefined.
3. Results ...................................................................................................................................... 14
3.1. Orphan Drug Applications and Designation.....................................................................................14
3.1.1. Application Requirements..............................................................................................................14
3.1.2. Joint Application................................................................................................................................15
3.2. Incentives......................................................................................................................................................16
3.2.1. Marketing Exclusivity......................................................................................................................16
3.2.2. Scientific Advice...............................................................................................................................16
3.2.3. Fee Reductions...................................................................................................................................17
3.2.4. Grant Funding.....................................................................................................................................17
3.2.5. Additional Benefits...........................................................................................................................17

3
3.3. Orphan Drug Designations and Approvals.......................................................................................18
3.3.1. EMA Orphan Designations............................................................................................................18
3.3.1.1. Positive Opinions...........................................................................................................................19
3.3.1.2. Refused Designations...................................................................................................................19
3.3.2. EPAR Breakdown.............................................................................................................................20
3.3.2.1. Approved MA.................................................................................................................................20
3.3.2.2. Refused MA.....................................................................................................................................21
3.3.2.3. Withdrawn MA...............................................................................................................................21
3.4. Breakdown of Sample MAA..................................................................................................................22
3.4.1. Designation and Approval Dates.................................................................................................22
3.4.2. Approval Decisions..........................................................................................................................22
3.4.4. Comparator Used in Pivotal Trial................................................................................................25
3.4.5. Endpoints Used in Pivotal Trial....................................................................................................26
3.5. From MA to Coverage Decisions: The Role of HTA....................................................................26
4. Discussion................................................................................................................................. 27
5. CONCLUSION ....................................................................................................................... 31
References .................................................................................................................................... 33
Published EPAR References:.........................................................................................................................37
Published FDA Report References:.............................................................................................................39
Published HTA References:............................................................................................................................40
Appendix ...................................................................................................................................... 44

4
Terms BLA – biologic license application CA – conditional approval CDER – Center for Drug Evaluation and Research CHMP – Committee for Medicinal Products for Human Use COMP – Committee for Orphan Medicinal Products EC – European Commission EEA – European Economic Area EMA – European Medicines Agency EPAR – European Public Assessment report FDA – Food and Drug Administration GMP – Good manufacturing practice IOM – Institute of Medicine IRDiRC – International Rare Diseases Research Consortium MA – marketing authorization MAA – marketing authorization application NORD – National Organization for Rare Disorders OD – orphan drug ODA – Orphan Drug Act OOPD – Office of Orphan Products Development RDRD - The Rare Disease Repurposing Database TL – total

5
1. Introduction
1.1. Background and Policy Context
Orphan diseases and orphan drugs are an increasingly common topic in the health care literature
because of the vast amount of people affected by orphan diseases and the lack of necessary
treatment options for these diseases. Although policy makers are making strides towards
increasing incentives for pharmaceutical companies to develop such technologies for orphan
diseases, there is still a long way to go. It is crucial to understand the barriers that pharmaceutical
companies face in R&D of orphan medicines and how to increase access worldwide to these
treatments.
1.1.1. Orphan Diseases
No single definition for an orphan disease exists worldwide, but it is generally a disease that
affects a small portion of the population. Between 6,000 and 7,000 rare diseases have been
discovered, and new diseases are frequently recorded in scientific literature (Orphanet 2012).
This number varies depending on the specific definition used for what constitutes a rare disease.
In Europe, it is a condition with a prevalence of no more than 5 in 10,000 in Europe, and in the
United States (US), it is a disease that affects no more than 200,000 patients (EMA 2014a). For
the majority of these diseases, no treatment is available. Although each disease only affects a
small number of a given population, it is estimated that over 55 million people suffer from an
orphan disease in the US or Europe (Stolk 2006). Approximately 80 percent of these orphan
diseases are due to genetics (IOM 2010).
1.1.2. Orphan Drug Designation
As with any drug, the research and development (R&D) of orphan drugs is extremely costly, and
because each orphan disease impacts so few patients, pharmaceutical companies may be
discouraged from developing drugs for such diseases due to the lack of return on investment.
Because of this, the US, EU and other countries have implemented policies to encourage the
R&D of drugs for orphan diseases. In 1983, the US was the first country to pass legislation
regarding orphan drugs, with the Food and Drug Administration (FDA) signing into place the
Orphan Drug Act (ODA). In 2000, the European Commission (EC) adopted its own set of rules
for designated orphan medicines, EC Regulation Number 141/2000 of the European Parliament
and the Council, commonly known as the Orphan Regulation (European Parliament 1999). Since

6
then, both the ODA and Orphan Regulation have been revised and clarified.
Both acts have been successful in increasing the number of orphan drugs available on the market.
In the US, since the inception of the ODA, there have been more than 2,800 orphan drug
designations (USFDA 2014). In 2013 alone, the FDA granted 260, which is a 38 percent increase
from 2012 (Reardon 2014). In the Orphan Regulation’s first decade, 1,235 applications were
reviewed by the Committee for Orphan Medicinal Products (COMP), more than 850 of which
were granted a positive opinion for orphan designation in the EU (COMP 2011).
Although implemented for the same purposes, the FDA and European Medicines Agency (EMA)
have different qualifications for orphan designation, and orphan medicines receive different
benefits in the US versus EU. This study seeks to analyze the differences in regulatory
requirements and benefits and how these differences affect which medicines are granted orphan
designation and marketing authorization (MA) in the two regions.
1.1.3. Marketing Authorization
Even if a drug receives orphan designation, this does not mean that the drug will receive MA. In
2012 for instance, less than 10 percent of orphan designated drugs had received MA in the EU
(Michel 2012). Although orphan designation allows for certain incentives, drugs must still
undergo similar procedures for MA to that of non-orphan drugs. In the US, sponsors must submit
either a New Drug Application (NDA) or a Biologic License Application (BLA) to the Center
for Drug Evaluation and Research or the Center for Biologics Evaluation and Research. The
application must provide data to demonstrate that (1) the drug is safe and effective for its
proposed use with a positive benefit-risk ratio; (2) the proposed labeling is appropriate; and (3)
that the sponsor performed good manufacturing practice (FDA 2013).
In the EU, a marketing authorization application (MAA) must be submitted to the Committee of
Medicinal Products for Human Use (CHMP), which evaluates the data and makes its
recommendation to the European Commission (EC) who is ultimately responsible for refusing or
granting MA.
There have been many studies examining the discrepancies in numbers between orphan drug
designations and MA for these drugs in either the EMA or the FDA (Kesselheim 2011; Joppi

7
2006; Heemstra 2008; Heemstra 2011), but few studies compare the decisions and regulations
between the two for the same drug-indication pair.
According to one study, there are differences in clinical trials to support approval of orphan
versus non-orphan drugs for cancer (Kesselheim 2011). It found that orphan trials were less
likely to be randomized, blinded and were more likely to assess disease response rather than
overall survival (OS) than non-orphan drug trials. Orphan trials on average were smaller than
non-orphan trials. The study also showed more treated patients in orphan drug trials having
serious adverse events than those patients in non-orphan cancer trials (Kesselheim 2011). Others
give the possible explanation that it is the poor quality of the applications submitted to the EMA
resulting in the lack of MA approvals. Lack of reliable methods for evaluating the effect of drugs
on small numbers of patients may be why orphan drug applications are of poor quality (Joppi
2006). Another study found that prior experience of a company in developing orphan drugs is a
key predictor for future authorization of orphan drugs in EU; it also found that orphan drug
approval was strongly associated with the sponsor’s previous experience in obtaining approval
for an orphan drug (Heemstra 2008). Yet another study identified the interaction and relationship
between the FDA and sponsor, or lack thereof, as a key indicator between orphan designated
drugs that were and were not MA in the USA (Heemstra 2011).
1.2. Study Objectives
The aim of this study is to study the similarities and differences that may exist between the FDA
and the EMA with regards to orphan status designation, MA, and the incentives provided to
orphan drugs developers for this purpose. Additionally, the study investigates the impact that the
existing regulatory frameworks have played on new drugs applying for orphan drug status. The
study analyzes the potential benefits these special regulations for orphan drug indications have
allowed for the advancement of medicine for rare diseases in the two jurisdictions and will
compare the differences between the FDA and the EMA’s orphan designation application
procedure, incentives given, MAAs, and numbers of approved designations and marketed orphan
drugs, with specific objectives for each. In doing so, the study uses a sample of orphan drug-
indication pairs to:

8
Compare applications and incentives: To understand how differences between FDA and
EMA may impact pharmaceutical companies from applying for orphan designation in one
region but not the other;
Understand the evidentiary requirements: To understand whether or not MA is obtained from
similar supporting evidence by both agencies and to highlight any differences in
requirements by the two agencies; and
Research the impact on access post-MA: To understand the differences in coverage decisions
across key HTA bodies in Europe and investigate whether EMA MA leads to full access
across selected European countries.
2. Methodology Multiple research methods were used to perform this study. First a literature review was
conducted to obtain proper background and policy information on orphan drug regulation and the
current thoughts on these regulations. Next, a comparison between FDA and EMA regulatory
requirements and incentives was performed, followed by a general comparison of orphan
designation and MA applications for orphan drugs between the two regulatory bodies.
Subsequently, a comparative analysis was performed on a sample of 14 applications to highlight
key differences between the EMA and FDA acceptances and requirements. Finally, the coverage
decisions of the drug samples were compared across 4 European HTA bodies.
2.1. Literature Review
The literature review was conducted according to guidelines from the Centre for Reviews and
Dissemination’s Guidance for Undertaking Reviews in Health Care (CRD 2008). A keyword
search on Medline, PubMed, CINAHL, and EMBASE was carried out using the search terms
‘orphan drug,’ ‘orphan drug regulation,’ ‘orphan drug designation,’ ‘orphan drug legislation,’
‘orphan drug indication’, ‘Orphan Drug Act’, and ‘EU orphan drug.’ Health Affairs and Nature
were searched additionally using the same key terms, and legislative documents and websites of
pertinent organizations (FDA, EMA, EURORDIS, and Orphanet) were reviewed.
The objective of the literature review was to identify previous studies analyzing orphan drug
regulations in the US, Europe, or a comparison of the two. In addition it seeks to provide the

9
essential regulatory documents for evaluation of the different regulatory requirements of both
regions.
2.2. Regulatory Requirements and Incentives for Orphan Designations
This study seeks to compare the dossiers submitted in the US and EU for the same orphan drug-
indication pairs to highlight potential differences the two regulatory bodies may have in what
they consider adequate data to grant MA.
Requirements: A comparison of the different requirements for a drug to obtain orphan
designation status between the EMA and FDA was performed. The EMA’s online application for
orphan-medicinal-product designation form was downloaded and compared to the FDA’s
Electronic Code of Federal Regulations, Part 316 – orphan drugs, Title 21: Food and Drugs
(EMA n.d.; eCFR 2014). In addition, the joint application that allows a company to apply for
designation in both jurisdictions was downloaded and analyzed, along with the additional
requirements needed specifically for the EMA or FDA.
Incentives: The benefits that sponsors are granted for orphan designation were compared using
information from the EMA’s webpage on orphan incentives and a FDA PowerPoint presentation
outlining FDA orphan drug designation incentives (EMA 2014c, Needleman 2011).
2.3. Comparison of Evidentiary Requirements for Orphan Designations
The number of drug-disease pairs each year that receive orphan designation and MA by the
EMA and the FDA was compared using the cutoff date of 01 July 2014. For the FDA
information, a search by each year from the first to the last day was performed first for all
designations and then for only approved products on the “FDA Search Orphan Designations and
Approvals” website (USFDA 2014a). Individual numbers per year were collected starting from
2000; the designations and approvals from 1983-2000 were grouped together because the EMA
did not put in place its orphan drug regulation until 2000.
For the EMA, the list of positive opinions on EMA’s “Rare disease (orphan) designations”
website was downloaded and the drug-indication pairs were sorted by year and then counted
(EMA 2014d). The list of approved orphan medicines was downloaded from EMA’s database on
“European public assessment reports” and sorted by date to obtain how many orphan drugs were
approved per year (EMA 2014b).

10
2.4. Breakdown of Orphan Designation
2.4.1. Positive Opinions
The number of orphan designations was searched for on the FDA’s orphan designation and
approval website both for total number of designations from the start of the FDA’s regulations
until 01 July 2014 as well as the number of FDA orphan designations during the same time
period as the EMA’s published list – from 2000 until 2014.
2.4.2. Negative Opinions
The drugs that received a negative opinion for orphan designation by COMP were compared
against the response by the FDA for these same drug-indication pairs. The list of rare disease
designations was downloaded from the EMA’s website 9 July 2014. As of this date, the COMP
has given 18 negative opinions (EMA 2014d). These 18 drug-designation pairs were then
searched on the FDA orphan drug designation and approvals search to compare the FDA’s
response to these drug-designation pairs (USFDA 2014a). The active substance name was first
searched and if no response came up, it was then searched by indication. The drug-indication
pairs that have been given orphan designation were marked as well as those that have been given
both orphan designation and MA, along with the dates of these decisions.
To further understand why a negative decision was granted, the public summary reports for each
of COMP’s negative opinions were downloaded, and the reasons behind the opinion were
analyzed.
2.5. Breakdown and Comparison of European Public Assessment Reports (EPARs)
Compared to Corresponding FDA Reports
The basis for the comparison of MA was the EMA through its EPARs, while the FDA was used
as the comparator, given the lack of relevant data available from the FDA compared to EMA’s
EPARs that are available to the public.
The list of EPARs for orphan medicines was downloaded and the 85 published EPARs were
sorted to find how many received MA, how many were refused MA, and how many had their
MA withdrawn.
The FDA publishes summary reports of all drug approvals on their Drugs@FDA website
(USFDA 2014b). This website provides drug details as well as approval history, letters, reviews

11
and other documents in conjunction with the product’s approval. This database was used to
cross-compare matching orphan drug-indication pairs with the relevant EPARs.
2.5.1. Approved MA
The EPARs for MA orphan drugs were further sorted into those with conditional approval or
exceptional circumstances. Additionally, these drug-indication pairs were searched for on the
Drugs@FDA website to see the MA status in the US.
2.5.2. Refused MA
Of the orphan medicines published which had been refused MA by the EMA, the Drugs@FDA
website and “FDA orphan drug designations and approvals database” were searched to compare
which of these drugs have been approved and/or granted orphan designation by the FDA.
(USFDA 2014b; USFDA 2014a).
To understand specifically why the EMA refused MA, the EPARs were analyzed and broken
down by the reasoning behind the refusal based on: (a) Negative benefit-risk ratio; (b) Lack of
clinical evidence; (c) Inappropriate comparator; (d) Unacceptable endpoint; (e) Lack of good
manufacturing practice; (f) Poor study design; (g) lack of quality or (h) safety; (i) Dose choice;
(j) Inappropriate supporting scientific literature; and (k)Another drug already has marketing
exclusivity for orphan indication.
2.5.3. Withdrawn MA
A similar analysis was conducted on the applications withdrawn post-approval to identify the
reasons why they were withdrawn and which corresponding drug-indications have been
designated or approved in the US.
2.5.4 MA Sample Comparison
A sample of 14 drug-indication pairs granted MA in both the US and EU were compared for a
more in-depth analysis of the similarities and differences between what and why orphan drugs
receive MA based on their applications.
The table below provides the list of sample drug-pair indications selected and provides an
alphabetical key for the breakdown and comparison of the applications.

12
Table 2.1: Sample of Approved Orphan Drugs
ActiveIngredient Name Indication Sponsor
A macitentan OpsumitPulmonaryarterialhypertension ActelionPharmaceuticals
B lenalidomide Revlimid Multiplelymphoma Celgene
C cysteaminebitartrate Procysbi Cystinosis RaptorTherapeutics
D bedaquiline Sirturo Activetuberculosis Janssen
E teduglutide[rDNAorigin] Gattex/Revestive Shortbowelsyndrome NPSPharmaceuticals
F pasireotide Signifor Cushing'sdisease Novartis
G cabozantinib Cometriq Medullarythyroidcarcinoma Exelixis
H ivacaftor Kalydeco Cysticfibrosis VertexPharmaceuticals
I ruxolitinibphosphate Jakafi/Jakavi Myelofibrosis Incyte
J eculizumab SolirisAtypicalhemolyticuremicsyndrome AlexionPharmaceuticals
K icatibant Firazyr Angioedema ShireOrphanTherapies
L brentuximabvedotin AdcetrisAnaplasticlargecelllymphoma SeattleGenetics
M velaglucerasealfa Vpriv GaucherdiseaseShireHumanGeneticsTherapies
N aztreonam Cayston Cysticfibrosis GileadSciences
The dates for when each drug-indication pair received orphan designation and MA as well as
when the sponsor submitted the application for MA were obtained from the two databases to see
which regulatory was the pioneer and if there were substantial differences in time it takes to
receive MA after submission.
Data on the MA applications were obtained from the EPARs and the FDA published summary
reports. The documents published by both agencies were broken down by:
If there were post marketing requirements;
If accelerated approval or accelerated assessment was given;
If protocol assistance or scientific advice was given;
If conditional approval was given
How many trials were used to support efficacy, including phase I, phase II, and phase III
trials;
The comparator used in the pivotal trial; and
The pivotal trial’s primary endpoint.
13
2.6. Breakdown and Comparison of HTA Appraisals
To better understand the accessibility of the orphan products, it is necessary to understand which
of the orphan designated drugs have been apprised by various European agencies and the
outcomes of these appraisals. The same sample of drug-indications was used to compare HTA
appraisals in 4 European countries: England & Wales, France, Scotland, and Sweden, who all
have HTA bodies. The sample drug-indication pairs were searched for under the following HTA
agencies’ online databases:
Table2.2:HTAAgencyandWebsite
Country HTABody WebAddress
England & Wales National Institute for Health and Clinical Excellence (NICE)
http://www.nice.org.uk/guidance/published
France Haute Autorité de Santé (HAS) http://www.has-sante.fr/portail/jcms/fc_1249926/en/evaluation-des-technologies-de-sante-et-des-actes
Scotland Scottish Medicines Consortium (SMC)
http://www.scottishmedicines.org.uk/Home
Sweden Tandvårds- och läkemedelsförmånsverket (TLV)
http://www.tlv.se/in-english/reimbursement-review/avslutade-genomgangar/
Each HTA agency uses a different method for evaluation so to properly compare the agencies’
appraisals to one another, the decisions were lumped into either A, “accepted” (recommended,
list, or ASMR I-IV); AC, “accepted with criteria or stipulations”; R, “rejected” (not
recommended, reject, do not list, or ASMR V); P, “in progress or ongoing”; or NA, “not
appraised.” Examining the 4 countries’ decisions on the orphan drugs helps shed light on which
countries accept orphan products and therefore allow patients in these countries to access these
products.
2.7. Study Limitations
One limitation of the study is the lack of data the FDA makes available to the public compared to
the EMA. For this reason the study emphasizes the EMA’s data and uses the FDA as the
comparator. An email for additional information regarding application numbers and published
reports on refused applications was requested, but this request was refused, making it difficult to
conduct a complete comparison. In addition, the information made publicly available by the
FDA is neither as complete nor organized as the EMA’s. Additionally, compared to the EMA,
the FDA reviews have varied formats, making it difficult to compare to one another.

14
3. Results
3.1. Orphan Drug Applications and Designation
3.1.1. Application Requirements
To receive orphan designation, a drug must satisfy specific criteria. In the EU, it must be
intended to treat, prevent, or diagnose a disease that is life threatening or chronically debilitating;
the prevalence of the condition must be no more than 5 in 10,000 in the EU or it must be unlikely
that the medicine will ever generate sufficient returns to justify an investment for the drug’s
development; and there is no satisfactory alternative authorized, or the drug must be of
substantial additional value to those affected by the condition (EMA 2014d). In the US, a
sponsor must demonstrate a specific prevalence that shows that the disease occurs in less than
200,000 persons in the US. If a range exists for the prevalence, then the highest value in the
estimate is used. The request must also provide sufficient scientific rationale that demonstrates
‘promise’ that the product will treat, diagnose or prevent the orphan indication (Reese 2014).
Demonstration of quality, safety and efficacy is not necessary for a product to receive orphan
designation (EMA 2014d).
In the EU, prior to applying, a sponsor must notify the EMA of its intention to submit an
application at least two months prior to the planned submission date. Sponsors may request to
partake in a free pre-submission meeting, which according to the EMA, may increase the success
of the submitted application (EMA 2014e). In the US, a sponsor may submit an orphan
designation request during the pre-clinical development to clinical development stages (Reese
2014).
In Europe, the COMP is in charge of examining applications for orphan designation, gives either
a positive opinion or raises a list of questions to the sponsor, and then forwards the opinion to the
EC, who makes the final decision. If COMP gives a negative opinion, the sponsor has the ability
to appeal the decision. The EMA’s complete review process takes a maximum of 90 days to
complete, and on average, it also takes 90 days for the review cycle in the US. If orphan
designation is approved in the EU, the information is published under the rare disease
designations, and the orphan designation is entered into the Community register of designated
orphan-medicinal-products. In the US, the FDA Office of Orphan Products Development

15
(OOPD) conducts the review. If an orphan designation is granted in the US, the sponsor must
submit annual reports to the FDA until MA (EMA 2014e; Reese 2014).
3.1.2. Joint Application
In November 2007 a joint application between the FDA and EMA was setup where sponsors can
apply using one common application. Even though a common application may be used, each
agency requires the information to be specific for either the US or EU and therefore different
data is required (HHS 2014a).
For both the EMA and the FDA, the applications require the sponsor to demonstrate that the
number of people affected by the disease in Europe or US is below the specific threshold and
must describe the methodology used. The sponsor must demonstrate that without incentives there
is no reasonable expectation that costs of R&D of the proposed orphan drug be recovered if the
medicinal product is authorized for marketing. Additionally for the EMA, the sponsor must show
that the marketing of the medicinal product in the EU would generate sufficient return in
necessary investment.
The applications must also provide information on (1) The name of the active substance specific
to Europe or US; (2) the proposed orphan designation, including the rationale for the use of the
drug for the indication; (3) the world-wide regulatory status and marketing history of the
product; (4) the sponsor’s information; (5) information on the manufacturer(s) and site(s) for
both the active substance and finished products; and (6) the sponsor’s declaration and signature.
There are additional requirements specific to the EMA with the joint application:
Specification of whether the active substance is authorized in EU centralized authorization,
mutual recognition, or national procedure.
Documentation that no other satisfactory methods of diagnosis, prevention, or treatment exist
in the EU; or that satisfactory methods exist, but this product will be of significant benefit.
Proof of establishment of the sponsor in the EEA.
Information on the sponsor whose main business is outside the EEA.
A copy of the scientific advice letter if advice was received.
Whether the sponsor intends to seek protocol assistance.
If known, the details for the planned submission of the MA application for the orphan
indication.
16
Whether the sponsor requests a fee reduction.
Information on the entity responsible for R&D of the medical product.
There are no additional requirements specific to the FDA (HHS 2014a).
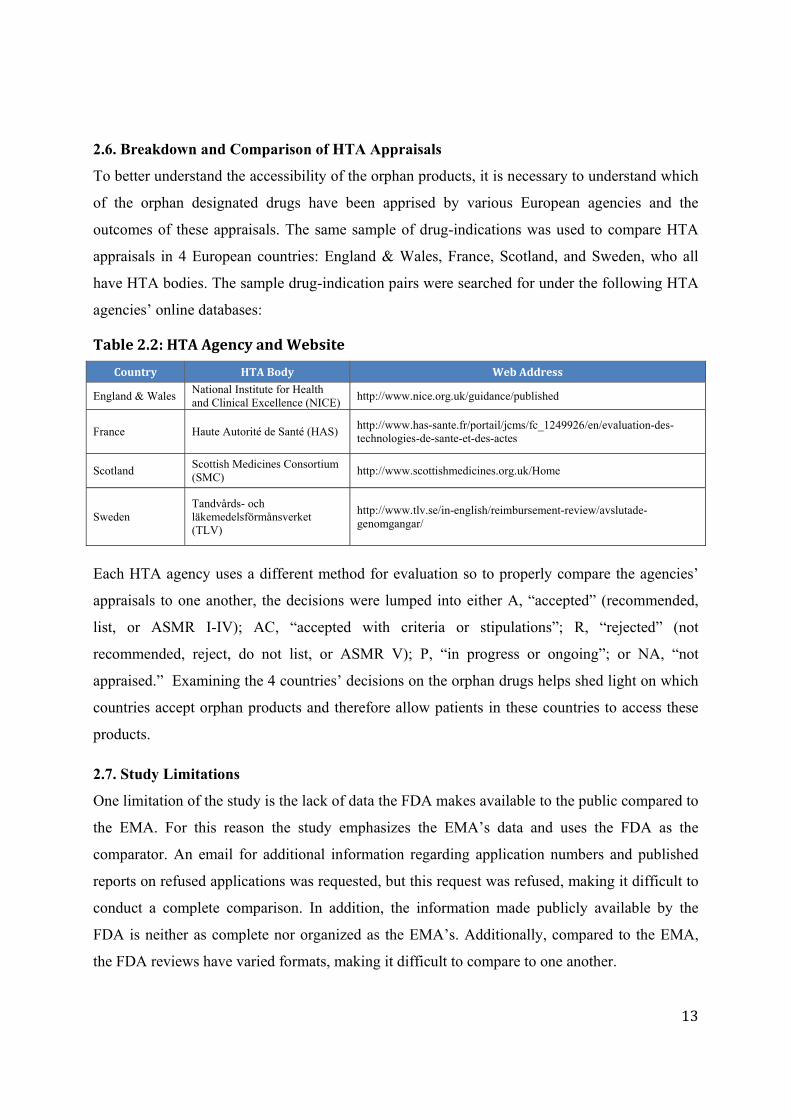
3.2. Incentives
The FDA and EMA offer different incentives to orphan drug developers. In the US, sponsors of
orphan designated drugs are offered marketing exclusivity, scientific advice, tax credits, and fee-
wavers to incentivize companies R&D of orphan drugs. The EMA offers market exclusivity,
scientific advice, fee reductions and conditional approval, but it does not offer any grant funding.
Table 3.1: Comparison of Incentives
Incentives FDA EMA
Marketexclusivity 7‐yearsfollowingFDAMA 10years;2yearextensionwithpaediatricinvestigationalplan
Scientificadvice Assistanceindrugdevelopmentprocessincludingindividualandparallelprotocolassistance.
Feereduction Taxcreditintheamountof50%ofclinicalinvestigationexpenses;exemptofpayingapplicationfees
Fullandpartialfeereductionsforprotocolassistance,initialandfollow‐uprequests;pre‐authorizationinspection;initialMA;andpost‐authorizationapplicationsandfees
Grantfunding Orphanproductgrantfunding No
Additional Priorityreview,acceleratedapproval,andafasttracksystem.
Conditionalapproval;approvalunderexceptionalcircumstances
(EMA2014c;Milto2014;EMA2013a)
3.2.1. Marketing Exclusivity
In the EU, sponsors have the ability to obtain 3 more years of marketing exclusivity than in the
US (10 versus 7 years) with the possibility of an additional 2 years for a medicine that complies
with the agreed pediatric investigation plan. Sponsors however must prove significant benefit at
the time of the application submission in the EU but not in the US, making for it harder to obtain
this exclusivity.
3.2.2. Scientific Advice
The FDA’s OOPD offers opportunities for both informal and formal meetings for sponsors to
receive answers to questions they have. The EMA provides specific scientific advice for orphan
drugs called protocol assistance. The EMA and FDA also offer parallel advice to sponsors,
which can be requested at any stage of the development process, and no advice is binding.

17
3.2.3. Fee Reductions
There are greater fee reductions available in the US compared to the EU. If successful in the US,
sponsors are exempt from paying application filing fees, which are over $2 million per
application, paid to the FDA regardless of whether the product is approved or not (Milto 2014).
The EMA recently added additional fee reductions as of 01/01/2014. Full and partial fee
reductions are available for protocol assistance, initial and follow-up requests; pre-authorization
inspection; MA and post-authorization applications and annual fees. There are additional fee
reductions available exclusive to small-medium enterprise (SME) sponsors and for pediatric-
related assistance in the EU.
3.2.4. Grant Funding
The perceived biggest benefit, and exclusive, to the USA, is the Orphan Product Grant Program.
As of 2014, the annual budget for grant funding is between $14 – 15 million. Clinical trials may
be awarded up to $200,000 per year for up to 3 years for phase 1 and up to $400,000 per year for
up to 4 years for phase 2 and 3 clinical trials. As of March 2014, the FDA has provided more
than $320 million for more than 530 grants for studies on orphan diseases. Over 50 approved
products partially funded by OOPD grants have been approved for marketing (McNeilly 2014).
Unlike the FDA, the EMA does not offer any grant funding for orphan designated products.
Sponsors of orphan drugs may be eligible for other grants from the EC and individual member
states (EMA 2013b).
3.2.5. Additional Benefits
In the US, many orphan drugs are able to apply to take part in expedited programs for serious
conditions, including priority review, accelerated approval, and a fast track system. With priority
review, MA takes 6 rather than the standard 10 months. Under an accelerated approval, a drug
may receive marketing approval based on adequate and well-controlled clinical trials using a
surrogate endpoint rather than needing to show irreversible morbidity or mortality. A sponsor
must complete post-marketing requirements if accelerated approval is given. Fast track
designation helps facilitate development and expedite the review process. If a drug receives fast
track, then the sponsor more frequently interacts with the FDA during the drug development
process and can submit sections of the application for guidance before the application is
complete (HHS 2014b).

18
There are similar special circumstance benefits in the EMA. Conditional approval (CA) is
obtained with less complete data than normally required and is only valid for 1 year. Exceptional
circumstances is granted to a drug without the data normally required for MA but is given
because the condition is rare or because it is not possible to collect substantial data given specific
circumstances such as small population size. Neither are exclusive to orphan-medicinal-products
(EMA 2014c).
3.3. Orphan Drug Designations and Approvals
A per year comparison between the FDA and EMA was made for both orphan designations and
MA between 2000 and 2014. As of 01/07/2014 from the start of the EMA’s regulations, the FDA
granted 275 orphan designations and market authorized 275 of these. The EMA granted 1025
orphan designations, 76 of which currently have MA. In total the FDA has granted 3122 orphan
designations and 473 of these are market authorized.
It is important to note that in the US, there is a difference between the indication for orphan
designation and approval. Often the approved indication is narrower than the designation
because designation is for a disease, while approval is for a specific indication. For instance,
Bosutinib was designated for the treatment of chronic myelogenous leukemia (CML) while
approved for the treatment of Philadelphia chromosome-positive (Ph+) CML with resistance, or
intolerance to prior therapy (Reese 2014).
3.3.1. EMA Orphan Designations
The COMP has published 1306 orphan designation applications on its website; 1025 have
positive opinions, 18 received negative opinions, 244 were withdrawn post designation, and 9
designations expired (EMA 2014d).
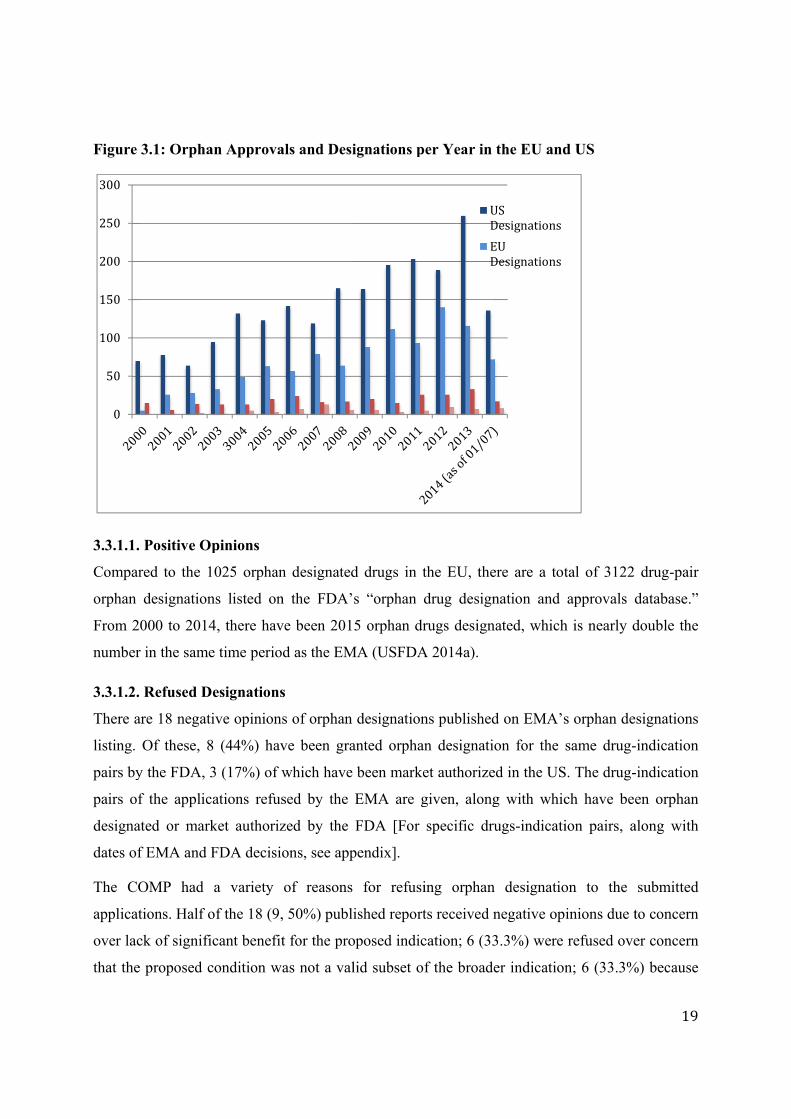
Figure 3
3.3.1.1.
Compar
orphan
From 20
number
3.3.1.2.
There ar
listing.
pairs by
pairs of
designat
dates of
The CO
applicat
over lac
that the
0
50
100
150
200
250
300
3.1: Orpha
Positive Op
red to the 1
designation
000 to 2014
in the same
Refused D
re 18 negati
Of these, 8
y the FDA, 3
f the applic
ted or mark
f EMA and F
OMP had
tions. Half o
ck of signifi
proposed c
an Approva
pinions
1025 orphan
ns listed on
4, there hav
e time perio
esignations
ive opinions
8 (44%) hav
3 (17%) of w
ations refus
ket authoriz
FDA decisio
a variety
of the 18 (9,
cant benefit
condition wa
als and Desi
n designated
n the FDA’
ve been 201
d as the EM
s
s of orphan
ve been gra
which have
sed by the
zed by the
ons, see app
of reasons
, 50%) publ
t for the pro
as not a val
ignations p
d drugs in t
s “orphan
5 orphan dr
MA (USFDA
designation
anted orpha
been marke
EMA are g
FDA [For
pendix].
for refusi
ished report
oposed indic
lid subset o
per Year in
the EU, the
drug design
rugs design
A 2014a).
ns published
an designatio
et authorize
given, along
specific dru
ing orphan
ts received n
cation; 6 (33
f the broade
UD
ED
the EU and
ere are a tot
nation and
nated, which
d on EMA’s
on for the
ed in the US
g with whic
ugs-indicati
n designatio
negative op
3.3%) were
er indication
USDesignations
EUDesignations
d US
tal of 3122
approvals d
h is nearly d
s orphan de
same drug-
S. The drug-
ch have bee
ion pairs, a
on to the
inions due t
refused ove
n; 6 (33.3%
19
drug-pair
database.”
double the
signations
-indication
-indication
en orphan
along with
submitted
to concern
er concern
%) because

20
the proposed condition is not considered an orphan disease; and 5 (27.8%) were refused because
insufficient data was provided to support the application. The majority of the applications were
refused due to multiple concerns by the COMP. The reasons of concern for each application,
many of which are refused for on multiple grounds, are outlined below:
Table 3.2: Reason’s for COMP’s Refusal
Significant benefit
Proposed Condition
Orphan disease criteria
Lack of data FDA designation
FDA MA
A B C D E F G
H I
J K L M N O
P Q R TL 9 6 6 5 8 3
3.3.2. EPAR Breakdown
There are 85 published EPARs for orphan medicines. Of these, 76 have been authorized, 7 were
refused MA, and 2 have had their MA withdrawn post-approval.
3.3.2.1. Approved MA
Of the orphan drugs approved in Europe, 7 have conditional approval (CA) and 14 have
exceptional circumstances (EC). Of these 76 drug-indication pairs, 52 are orphan designated and
approved for marketing for the same indication in the USA; an additional 18 are orphan
designated for the same designation but not approved for marketing [see appendix for complete
list].

21
3.3.2.2. Refused MA
Seven (8.3%) of the 85 published EPARs for orphan designated drugs were refused MA. 3 have
MA in the US, 3 have orphan designation, and only one has both MA and orphan designation.
[For specific drug-indication pairs along with FDA designation and MA status, see appendix].
There were a variety of reasons the CHMP refused to grant MA, the most common that benefits
do not outweigh risks of the orphan drug for the given indication. Reasons for a negative benefit-
risk ratio include insufficient overall evidence, lack of or choice of comparator used, choice of
effectiveness endpoint, failure of good manufacturing practice (GMP), poor study design, issues
pertaining to quality or safety of medicine, choice of dose, and poor supporting scientific
literature. All but one of the refusals were based on the previous criterion to support the
conclusion that the benefits do not outweigh the risk; elelyso for type-1 Gaucher disease
demonstrated a positive benefit-risk ratio but could not be granted MA because Vpriv has
marketing exclusivity for the same indication.
Table 3.3: EMA Reason Refused
3.3.2.3. Withdrawn MA
Two (2.4%) of the 85 the published EPARs for orphan designated drugs have had their MA
withdrawn post-approval. Both are MA in the US and one of the two is orphan designated.
Negativebenefit‐risk
Lackofevidence
ComparatorEnd‐point
GMPStudydesign
Quality Safety DoseScientificliterature
PriordrugMA
A
B
C
D
E
F
G
TL 6 3 3 2 1 4 1 3 2 1 1

22
Table 3.4: FDA Status of EMA Withdrawn MA
ActiveSubstance Indication ReasonsforEMARefusal
FDAOrphanDesignated FDAMA
celecoxib
Familialadenomatouspolyposis
Sponsor(Pfizer)didnotmeetthespecificrequirementstoprovidefurtherinformationregardingdataonitsefficacyandsafety. No 23.12.1998
rilonacept
Cryopirin‐associatedperiodicsyndromes MAHvoluntarilywithdrewMA 20.12.2004 27.02.2008
(HMDE2011;USFDA2014b;PressOffice2012;USFDA2014a)
3.4. Breakdown of Sample MAA
3.4.1. Designation and Approval Dates
Out of the sample of 14 drugs, only 2 (14.3%) were orphan designated first in Europe, while the
other 12 had already received orphan designation in the US. Despite the low rate of pioneer
designations, 5 (35.7%) were approved for marketing in Europe prior to the US, and of the drugs
that were first approved in the US, 7 of these were approved within 1 year following Europe.
Four (28.6%) of the 14 MA applications were submitted in the same month to both the EMA and
FDA and 4 were submitted first to the EMA (Soliris, non-comparable due to lack of FDA
submission data). Only 5 (35.7%) of these first obtained MA in Europe, 2 of which were more
than one year in advance. The most significant difference between EU and US application
timelines is in regards to the time it takes between the sponsor submitting the application to the
agency’s decision. On average it took 455 days in the EU to receive MA, while it took 238 days
in the US; it took nearly twice as long in the EU as in the US.
3.4.2. Approval Decisions
In the sample, 13 (92.9%) of the FDA decisions had post-marketing requirements, while the
COMP had requirements for 10 (71.4%). The FDA granted accelerated approval to 2 (14.3%) of
the sample drugs and the EMA granted 3 (21.4%) accelerated assessment, but denied Procysbi;
the drugs granted accelerated processes differed between regions. While the FDA gave scientific
advice to only 2 (14.3%), the EMA gave advice to 9 (64.3%) sponsors – 7 in the form of protocol
assistance (PA). The EMA conditionally approved 4 (28.6%) drugs. Cayston no longer is marked
with CA because its sponsor completed the necessary requirements to receive full MA.

23
Table 3.5: Orphan Designation and Approvals Dates
Designation EMAMA FDAMA
EMA FDA Dif.(days) Submit Approval lag
time Submit Approvallagtime
A 27.09.2011 03.09.2009 754 25.10.2012 20.12.2013 421 19.10.2012 18.10.2013 364B 12.12.2003 20.09.2001 813 28.02.2006 14.06.2007 471 30.12.2005 29.09.2006 273C 20.09.2010 24.10.2006 1427 01.03.2012 06.09.2013 554 30.03.2012 30.04.2013 396D 26.08.2005 10.01.2005 228 28.08.2012 05.03.2014 554 29.06.2012 28.12.2012 182E 11.12.2001 29.06.2000 530 03.03.2011 30.08.2012 546 30.11.2011 21.12.2012 387F 08.10.2009 24.07.2009 76 30.09.2010 24.04.2012 572 17.02.2012 14.12.2012 301G 06.02.2009 29.11.2010 ‐661 29.09.2012 21.03.2014 538 21.05.2012 29.11.2012 192H 08.07.2008 20.12.2006 566 27.10.2011 23.07.2012 270 18.10.2011 31.01.2012 105I 07.11.2008 05.09.2008 63 01.06.2011 23.08.2012 449 03.06.2011 16.11.2011 166J 24.07.2009 29.04.2009 86 25.09.2006 20.06.2007 268 N/A 23.09.2011 N/AK 17.02.2003 25.11.2003 ‐281 27.07.2007 11.07.2008 350 25.02.2011 25.08.2011 181L 15.01.2009 23.10.2008 84 31.05.2011 25.10.2012 513 28.02.2011 19.08.2011 172M 09.06.2010 08.06.2009 366 30.10.2009 26.08.2010 300 31.08.2009 26.02.2010 179N 21.06.2004 12.03.2002 832 07.03.2008 21.09.2009 563 13.08.2009 22.02.2010 193AVG. 349 455 238
Table3.6:ApprovalDecisions
Post‐MarketingRequirements
AcceleratedApproval/Assessment
ScientificAdvice ConditionalApproval
(EMASpecific) FDA EMA FDA EMA FDA EMA
A B (PA)1 C R2 (PA) D E (PA) F G H (PA) I J (PA) K (PA) L M N (PA) *TL 13 10 2 3 2 9 4*initially,nowswitchedtoregular
1PA:ProtocolAssistance2R:Requested,butnotgranted

24
Table 3.7: Number of Efficacy Trials Used for Clinical Evidence
EfficacyTrials PhaseI PhaseII PhaseIII ExtensionTrial
FDA EMA FDA EMA FDA EMA FDA EMA FDA EMAA 1 2 1 1 1 B 2 3 1* 1* 2 2 C 1 1 4 5 1 1 D 2 3 1 2 2 0 0 E 3 2 2 1 1 1F 2 3 1 1 1 1 1G 1 2 3 1 1 H 4 3 7 2 2 2 1I 2 2 2 2 J 4 6 1 1* 1 2 3 3K 4 3 1 1 3 2 L 3 2 4 4 2 2 0 0 M 3 4 2* 2* 2 2 N 5 4 2 2 1 1 2 2 AVG 2.6 2.9 4.0 2.1 1.5 1.3 1.5 1.4 2.0 1.5*1trialconsideredbothphase1andphase2
Table 3.8: Comparator Used in Pivotal Trial
Placebo Alternative None
A
B
C
D
E
F
G
H
I
J
K
L
M
N
Total 10 3 2

25
Table3.9:PrimaryEndpointUsedinPivotalTrial
FDA EMA
A
Timetofirstoccurrenceuptoendoftherapyofthefollowing:death,atrialseptostomy,lungtransplantation,initiationofIV,orotherworseningofcondition’ssymptoms
Reductionintheriskofmorbidity/mortalityeventsfromstartoftreatmenttofirsteventuptoendoftrial
B TimefromrandomizationtofirstdocumentationofPD3basedonmyelomaresponsecriteria
CDemonstrationofcomparabledepletionofsteady‐statecysteamine‐troughwhite‐blood‐cellcystinelevels
Whitebloodcellcystinelevelsatspecifictimepointsduringadministrationofprocysbi/comparatorinacross‐overdesign
D Timetosputumcultureconversionduringtreatmentwithbedaquiline/placebo
E ReductionfrombaselineinPN4volumeatvarioussubsequentstudytimepoints
Agradedcriterionaccountingfordurationandintensityofresponsebasedoff%reductioninPNvolume
F
Proportionofresponders,summarizedasapointestimatewithacalculated2‐sided95%CI.Sincenoplacebocontrolarm,determinationofwhatconstitutedasignificantresponsetotreatmentwasarbitrarilydefinedasalowerboundinthe95%CIexceeding15%
Theproportionofresponderstopasireotide600μgs.c.b.i.d.and900μgs.c.b.i.d.independentlyinpatientswithCushing’sdiseaseasmeasuredbymeanUFC≤upperlimitofnormalafter6monthstreatment
G Progressionfreesurvivalfromrandomizationtodocumentedprogressivedisease
H Absolutechangefrombaselinein%predictedFEV15through24weeksoftreatment
I
Statisticallysignificantdifferencebetweenthetwotreatmentarmsinthe%ofpatientswith≥35%spleenvolumereductionbyweek24ofthetreatment
Spleenvolumeresponserateatweeks24and48ofstudy
J Plateletcountchange,hematologicnormalization,orcompletethromboticmicroangiopathy
Haemoglobin stabilizationandunitsofpackedredbloodcellstransfusedduringthetreatmentphase
K Timetoonsetofsymptomrelief
L Overallresponserate(ORR)definedasthesumofthecompleteandpartialremissionrates
ORRdefinedasclinicalresponseofprogressiveorstablediseaseorpartialorcompleteremission
M Changeinhemoglobinconcentrationfrombaselinetoendofstudy
N
Initially:TimetoneedantibioticsotherthanCayston/placebo withdocumentedsymptom(s)predictiveofpulmonaryexacerbationfollowingstartofstudy
Changedto:changeinpulmonaryfunctionfrombaselineatday14
3.4.3. Trials Used for Clinical Evidence The number of trials used as support for clinical evidence in the applications differed between
the two agencies. From the sample, on average, however, similar amounts were used to prove
safety and efficacy, although more phase I trials were noted in the FDA applications than the
EMA’s.
3.4.4. Comparator Used in Pivotal Trial
In all 14 drug-indication pairs studied, the same comparator was used in the pivotal trials for
both the EMA and FDA applications. Nine (64.3%) samples used only a placebo, 2 (14.2%)
samples used only the current standard of care; 1 (7.1%), Firazyr used both a placebo and
3PD:Progressivedisease4PN:parenteralinfusionoffluid,energyand/orelectrolytes5FEV1:Forcedexpiratoryvolumein1second

26
tranexamic acid as comparator, and 2 (14.2%), Gattex/Revestive and Adcetris, did not use any
comparator in their pivotal trials because they were single arm.
3.4.5. Endpoints Used in Pivotal Trial
There was little consistency among the endpoints used in the pivotal trials for clinical efficacy.
Six (42.9%) samples used the same endpoint description for pivotal clinical efficacy trials for the
FDA and EMA applications.
Both the FDA and EMA’s pivotal trials for Cayston (N) used the time to need IV or inhaled
antibiotics as the primary endpoint. However, the FDA review raised issues with this endpoint
and requested the sponsor make the secondary endpoint, change in pulmonary function from
baseline at day 14, to the primary endpoint. The sponsor altered its application to receive FDA
MA.
3.5. From MA to Coverage Decisions: The Role of HTA
Of the 4 HTA agencies, SMC reviewed the most submissions (9) while NICE reviewed the least
(3, 2 currently under review).
Out of the sample of 14 MA orphan drug-indication pairs, NICE recommended 2 (14.3% of
sample), rejected 1 (7.1%), and is in the review process of 2 (14.3%) others.
HAS accepted 7 (50%), and rejected 1 (7.1%). HAS accepted the most, however, this may partly
be due to the scaled (I-V) decision used instead of a stricter recommend/do not recommend that
the other HTA bodies use. Additionally HAS was the pioneer of the 4 HTA bodies for 5 of the
submissions.
SMC accepted 2 (14.3%), 1 only after a resubmission; accepted 2 (14.3%) with restrictions, 1
following a resubmission; and rejected 5 (35.1%), 1 of which had been resubmitted. SMC
rejected the most, but also reviewed more than any other agency. Finally, TLV accepted 5
(35.1%), accepted 1 (7.1%) with restrictions, and rejected 1 (7.1%).
Of the 14 drug-indication pairs, 2 (14.3%), Revlimid and Jakavi, were reviewed by all 4
agencies; 3 (21.4%), Firazyr, Vpriv, and Cayston, were reviewed by 3 agencies; 5 (35.1%) were
reviewed by half of the agencies; and 4 (28.6%) have not received a final decision from any of
the agencies.

27
Table 3.10: HTA Decisions
Name Indication NICE HAS SMC TLV Totalreviewed
A Opsumit pulmonaryarterialhypertension
N/A N/A AC:03/2014 AC:05/2014 2
B Revlimid multiplelymphomaA:(sponsor to
pay):06/2009
A(III):06/2012
AC:03/2014* A:06/2010 4
C Procysbi cystinosis N/A N/A N/A N/A 0
D Sirturo activetuberculosis N/A N/A N/A N/A 0
E Revestive shortbowelsyndrome.
N/A N/A N/A N/A 0
F Signifor Cushing'sdisease N/AA(IV):07/2012 R:10/2012 N/A 2
G Cometriq medullarythyroidcarcinoma N/A N/A N/A N/A 0
H Kalydeco cysticfibrosis N/A A(II):11/2012 R:06/2013* N/A 2
I Jakavi myelofibrosis R:06/2013 A(III):01/2013
R:04/2013 A:04/2014 4
J Solirisatypicalhemolyticuremicsyndrome
P A(II):09/2012 R:02/2012 N/A 2
K Firazyr angioedema N/AA(IV):11/2008 A:03/2012* A:03/2010 3
L Adcetris anaplasticlargecelllymphoma P A(III):
03/2013 N/A A:06/2013 2
M Vpriv Gaucherdisease N/A R(V):12/2010 A:10/2012 R:05/2011 3
N Cayston cysticfibrosis A;03/2013 N/A R:02/2012 A:03/2012 3
*decisionfollowingresubmission(Source:PublishedHTAreferences)
4. Discussion The number of orphan designations is consistently higher per year in the US than in Europe. The
study’s results support a number of potential explanations for the difference in numbers between
Europe and US orphan designations and approvals.
Application designation procedure: The comparison between orphan application requirements
and criteria might explain why there are more designations in the US than in Europe. The EU has
more stringent requirements for orphan designation than the US does. Even with the joint
application, the EMA has additional requirements both in terms of application procedures and
the criteria for orphan designation. Only the EMA has the requirement that the product must

28
show significant benefit over the current standard of care at the time of orphan designation.
Because of this, more applications may be submitted to the FDA than the EMA.
One limitation of the study is that only the numbers of applications submitted to the EMA for
orphan designation are available, as the FDA does not provide this information publicly.
According to the EMA, the COMP gave positive opinions to 850 of the1,235 applications
submitted applications between 2000 and 2010 (COMP 2011). To better understand if the
difference in orphan designations is due to the number of applications submitted to both
agencies, similar data for the FDA will be necessary.
Incentives: The results show there are substantial differences between incentives for sponsors
granted orphan designation in the US compared to Europe. Most notably, the FDA provides
substantially more financial incentives, through tax credits and grant funding, compared to the
EMA. Literature discussing factors influencing the development of orphan drugs in Europe
raises the issue that there are no tax credits given in Europe, which may be why there are fewer
orphan drugs in Europe compared to the US (Heemstra 2008; Joppi 2006; Dear 2006). While the
EMA provides a longer marketing exclusivity period than the FDA (10 years versus 7), this 3
year difference is most likely not a reason to deter sponsors from applying for orphan
designation in the US. Indeed, the longer marketing exclusivity period in the EU may actually
act as a barrier to entry for obtaining orphan designation where there is one already on the
market for the same indication. The two different incentive structures align with the discrepancy
in numbers of orphan designations between the EU and US.
Negative EMA Designations: Another possible explanation is that the EMA denies more
applications than the FDA. The results of the breakdown of COMP’s negative opinion for orphan
designation highlight this. Nearly half of the drug-indication pairs that were not granted orphan
designation in the EU were granted orphan designation by the FDA signifying that it is may be
easier to receive orphan designation in the US than in EU.
There are other potential reasons that were not analyzed in this study but could be looked at in
future research. One possibility is that because the US implemented its regulations before
Europe, sponsors may be more familiar with the FDA’s orphan designation regulations
compared to those of the EMA. However, after the 14 years that the EU regulations have been in

29
place, it seems that this would be unlikely. Another reason could be due to the make-up of the
EU versus the US and that the EU is a group of member states with different regulating bodies
while the US is a single entity. In the EU, there are drugs that have orphan status in a member
state, but not at the EU level. For example in Belgium the levodopa/carbidopa combination has
orphan status despite not having EU orphan designation (Denis 2010).
Market authorization submission and acceptance dates: It takes a substantial amount of time for
a drug to be approved after it has been submitted to the regulatory body and even an even longer
amount of time from orphan designation to approval. Because orphan designation can be
obtained at any point during the trial timeline, orphan designation may be received years prior to
completion of the studies needed to fulfill MA requirements. On average it takes years to make a
drug, and many drugs are never completed. Because of the small population size for an orphan
drug, it is even harder to study and therefore may take longer than the average drug due to
finding the appropriate population on which to study. Many drugs therefore that are currently
orphan designated, may either currently be in the trial process or may have been terminated due
to insufficient funds or study pools. A portion of the orphan designated drugs are also currently
in the review process post MAA/NDA submission, but because of the lengthy lag time in review,
the decision has not yet been made. Because it takes nearly twice as long in the EU as it does in
the US to receive authorization after submission, there will be a higher number of drugs MA in
the US than in the EU.
Criteria for MA: Another reason there may be more MA orphan drugs in the US than EU is
because more drugs receive a positive opinion in the US than EU. This is conveyed in the
number of EMA refused MAA. Of the 7 orphan designated drugs that received a negative
opinion in Europe, 3 are MA in the US. The breakdown of reasons for why the EMA rejected the
MAA shows multiple reasons of why these drugs were not deemed acceptable to be marketed in
the EU. The FDA’s criteria to obtain MA may not be as stringent as the EMA’s. Notably
however, the sample of accepted MA applications in the US and EU had similar supporting data.
The FDA to EMA’s drug approval process normally differs in that a placebo is often an
acceptable comparator in the US while but not in Europe (EMA 2004; HHS 2001). In the
samples studied however, the comparison did not differ between the US and EU, and so it would
be necessary to analyze all approved orphan products to draw further conclusions. It is
significant to point out that both withdrawn MAA in the EU are currently marketed in the US.

30
The small number makes it difficult to make any definitive statements, but this discrepancy still
reflects the greater number of approved orphan products in the US than EU.
Based on the various data collected, it appears that there are multiple reasons for the difference
in numbers between the EU and US’s orphan designated and approved drug-indication pairs.
Although the EMA has attempted to simulate the FDA’s regulations to incentivize R&D of
orphan-medicinal-products, it still lags behind.
Regarding the sample of drugs authorized in both regions, there were little significant
discrepancies between the data submitted to one agency compared to the other and neither
agency seemed substantially more rigorous in their review. This may be in part with the
introduction of the joint application, meaning that sponsors would submit identical supporting
evidence to both agencies. The EMA’s process appears more organized than the FDA’s, as
shown in the standardized EPAR format versus the FDA’s various supporting review documents,
which had no cohesive style. This may be a reason why it took almost twice the time to grant
MA after application submission in the EMA versus the FDA – the more standardized process
may result in a lengthier review conducted in Europe than in the US.
The EMA is moving towards an adaptive licensing approach through the use of its CA. Four out
of the sample are approved conditionally and of the total number of published orphan EPARs, 7
were listed with CA. There is a difference in the EPARs between conditional approval and MA
with conditions. Some of the sample drugs have conditions under which they must first abide by,
such as further efficacy or safety studies, to receive MA, but these drugs are not labeled with CA.
The drugs in the sample labeled with CA were Sirturo, Cometriq, Adcetris, and Votubia. In
addition, however, the EMA requested Revestive, Signifor, Kalydeco, Jakavi, Soliris, Adcetris,
and Cayston to complete ongoing studies or perform additional studies by a given date to receive
complete MA. This discrepancy could be because conditional approvals only last for one year so
initially the EMA granted CA, or the equivalent before it was formally put in place in 2007 to
these drugs, and thereafter the sponsor remedied the conditions, switching from conditional to
full approval on the drug (CHMP 2006). Cayston, for example, was given conditional approval
initially but fulfilled post-marketing requirements and so it is no longer marked with CA. The
use of adaptive licensing such as CA by the EMA may help narrow the gap between MA orphan
medicines available in the EU compared to the US.

31
As seen in the comparison of HTA decisions, many MA drugs in Europe are not reimbursed in
some or any of the countries studied. This further limits access for patients to orphan medicines
in many countries in Europe compared to the US. Only 2 drugs in the sample were reviewed by
all 4 agencies, while twice as many have yet to be reviewed by any. Patients across France,
Scotland, England & Wales, and Sweden have limited, if any access, to these drugs, which may
be the only viable treatment for these rare diseases.
5. Conclusion
The orphan designation regulations setup in the US, EU, and elsewhere throughout the world
have stimulated R&D for orphan diseases, and the EMA-FDA common application and
continued dialogues have further allowed patients suffering from rare diseases to receive
treatment. This was seen in the study results of the continual increase of orphan designation and
MA from both agencies since 2000 along with the nearly identical data submitted to both
agencies seen in the sample drugs studied. Unfortunately however, the current fragmentation of
reimbursement policies across Europe has limited the access to these drugs. It is not enough to
grant drugs orphan designation and MA. A similar process is needed to promote the
reimbursement of these orphan designated products so that patients around the world are able to
afford such treatments.
In alignment with the study that found a positive association with communication between
sponsor and the FDA, drug manufacturers should engage with regulatory agencies to increase
chances of MA (Heemstra 2011). Additionally, sponsors should engage with HTA bodies to
increase their chance of HTA approval across Europe. Even if orphan drugs are granted MA,
they may not receive reimbursement, vastly limiting access to these medicines. Hughes et al
discuss special status for funding orphan medicines, which may not be deemed cost effective and
therefore will not be reimbursed in many European markets (Hughes 2005). Until there are more
regulations across HTA bodies for orphan medicines, sponsors should participate in early
engagement activities such as adaptive licensing or cost sharing.
In the past 14 years since the start of the EMA’s drug regulations, there has been a continued
increase in R&D and awareness of orphan medicines, but there still is a long way to go before

32
patients have access to the drugs they need. Regulatory and HTA bodies must continue to work
together and better align with one another to treat the millions of patients around the world
affected by rare diseases.

33
References
Centre for Reviews and Dissemination (CDR). 2008. CDR’s guidance for undertaking reviews in
health care. York: York Publishing Services. Committee for Medicinal Products for Human Use (CHMP). 2006. Guideline on the scientific
application and the practical arrangements necessary to implement commission regulation (EC) No 507/2006 on the conditional MA for medicinal products for human use falling within the scope of regulation (EC) No 726/2004.
The Committee for Orphan Medicinal Products (COMP) and the European Medicines Agency
Scientific Secretariat. 2011. European regulation on orphan medicinal products: 10 years of experience and future perspectives. Nature Reviews, 10, pp. 341—349.
Coté, T. R., Xu, K. & Pariser, A. R. 2010. Accelerating orphan drug development. Nature
Reviews Drug Discovery, 9 (12), pp. 901—902. Dear, J.W., Lilitkarntakul, P., & Webb, D.J. 2006. Are rare diseases still orphans or happily
adopted? The challenges of developing and using orphan medicinal products. Br J Clin Pharmacol, 62, pp. 264—271.
Denis, A., Mergaert, L., Fostier, C., et al. 2010. Issues surrounding orphan disease and orphan
drug policies in Europe. App Health Econ Health Policy, 8(5), pp. 343—50. Electronic Code of Federal Registrations (eCFR). 2014. Part 316 – Orphan Drugs. Available
from: http://www.ecfr.gov/cgi-bin/retrieveECFR?gp=&SID=91b7be5e87481538e33a4c0a76ba7183&n=21y5.0.1.1.6&r=PART&ty=HTML [Accessed 5 August 2014].
European Medicines Agency (EMA). 2014b. European Public Assessment Reports. Available
from: http://www.ema.europa.eu/ema/index.jsp?searchType=name&taxonomyPath=&genericsKeywordSearch=Submit&searchGenericType=orphan&keyword=Enter+keywords&alreadyLoaded=true&curl=pages%2Fmedicines%2Flanding%2Fepar_search.jsp&status=Authorised&status=Withdrawn&status=Suspended&status=Refused&mid=WC0b01ac058001d125&treeNumber=&murl=menus%2Fmedicines%2Fmedicines.jsp&searchTab=searchByAuthType&pageNo=2[Accessed 5 August 2014].
European Medicines Agency (EMA). 2014a. Medicines for rare diseases. Available from:
http://www.ema.europa.eu/ema/index.jsp?curl=pages/special_topics/general/general_content_000034.jsp&mid=WC0b01ac058002d4eb ; http://www.ecfr.gov/cgi-bin/text-idx?c=ecfr&SID=51cf70689d51f0ea4147c0a8ac649321&rgn=div5&view=text&node=21:5.0.1.1.6&idno=21 [Accessed 5 August 2014].
European Medicines Agency (EMA). 2014c. Orphan incentives. Available from:
http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_0

34
00393.jsp&mid=WC0b01ac058061f017[Accessed 5 August 2014]. European Medicines Agency (EMA). 2014d. Rare disease (orphan) designations. Available
from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/landing/orphan_search.jsp&mid=WC0b01ac058001d12b [Accessed 5 August 2014].
European Medicines Agency (EMA). 2014e. How to apply for orphan designation. Available
from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000519.jsp&mid=WC0b01ac05804ece5e [Accessed 21 August 2014].
European Medicines Agency (EMA). 2013a. Fee reductions for designated orphan medicinal
products. EMA/622074/2013, pp.1-4. European Medicines Agency (EMA). 2013b. Orphan medicinal product designation. London:
European Medicines Agency. European Medicines Agency (EMA). 2004. EU Standard of Medicinal Product Registration:
Clinical Evaluation of Risk/Benefit--The Role of Comparator Studies, Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Position_statement/2009/12/WC500017660.pdf [Accessed 19 August 2014].
European Medicines Agency (EMA). N.d. Application for Orphan Medicinal Product
Designation. European Parliament. 2000. Charter of Fundamental Rights of the European Union. Eurodis –
The Voice of Rare Disease Patients in Europe. Available from: http://www.europarl.eu/charter/pdf/text_en.pdf [Accessed 18 July 2014].
European Parliament & The Council of the European Union. 1999. Regulation (EC) No
141/2000 of the European Parliament and of the Council. Official journal of the European Communities, 18, pp.1—5.
Heemstra, H. E., De Vrueh, R. L., Van Weely, S., Büller, H. A. and Leufkens, H. G. 2008.
Predictors of orphan drug approval in the European Union. European journal of clinical pharmacology, 64 (5), pp. 545—552.
Heemstra, H. E., Leufkens, H. G., Rodgers, R., Xu, K., Voordouw, B. C. and Braun, M. M.
2011. Characteristics of orphan drug applications that fail to achieve marketing approval in the USA. Drug discovery today, 16 (1), pp. 73—80.
Hughes, D., Tunnage, B. and Yeo, S. 2005. Drugs for exceptionally rare diseases: do they
deserve special status for funding?. Qjm, 98 (11), pp. 829—836. Human Medicines Development and Evaluation (HMDE). 2011. Onsenal (celecoxib). European

35
Medicines Committee. Issa, A. M. 2002. Ethical perspectives on pharmacogenomic profiling in the drug development
process. Nature Reviews Drug Discovery, 1 (4), pp. 300—308. Institute of Medicine (IOM). 2010. Rare Diseases and Orphan Products: Accelerating Research
and Development. Joppi, R., Bertele, V. and Garattini, S. 2006. Orphan drug development is progressing too
slowly. British journal of clinical pharmacology, 61 (3), pp. 355—360. Kesselheim, A.S., Myers, J.A., & Avorn, J. 2011. Characteristics of Clinical Trials to Support
Approval of Orphan vs Nonorphan Drugs for Cancer. JAMA, 305 (22), pp.2320—2326. Lichtenberg, F. R. and Waldfogel, J. 2003. Does misery love company? Evidence from
pharmaceutical markets before and after the Orphan Drug Act. Linertov'A, R., Serrano-Aguilar, P., Posada-De-La-Paz, M., Hens-P'Erez, M., Kanavos, P.,
Taruscio, D., Schieppati, A., Stefanov, R., P'Entek, M., Delgado, C. and Others. 2012. Delphi approach to select rare diseases for a European representative survey. The BURQOL-RD study. Health policy, 108 (1), pp. 19—26.
McNeilly, E. K. 2014. Orphan Products Grants Program Overview. Joint EMA/FDA?MHLW-
PMDA orphan medicinal product workshop, 10 March, London. http://www.ema.europa.eu/docs/en_GB/document_library/Presentation/2014/03/WC500164161.pdf [Accessed 29 July 2014].
Melnikova, I. 2012. Rare diseases and orphan drugs. Nature Reviews Drug Discovery, 11 (4),
pp. 267—268. Michel, M. & Toumi, M. 2012. Access to orphan drugs in Europe: current and future issues.
Review of Pharmacoeconomics & Outcomes Research, 12 (1), pp. 23—29. Milto, J. 2014. FDA Incentives. EMA/FDA/MHLW-PMDA Orphan Medicinal Product
Workshop, 10 March, London. http://www.ema.europa.eu/docs/en_GB/document_library/Presentation/2014/03/WC500164165.pdf [Accessed 5 August 2014].
Needleman, Katherine. 2011. Overview of the Office of Orphan Products Development:
Incentives for Rare Diseases. CDER Small Business Assistance Webinar, 18 October, Silver Spring. http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/SmallBusinessAssistance/UCM276029.pdf [Accessed 29 July 2014].
Orphanet. 2012. About Rare Disease. Orphanet, Available from:
http://www.orpha.net/consor/cgi-bin/Education_AboutRareDiseases.php?lng=EN)

36
[Accessed 5 August 2014]. Picavet, E., Cassiman, D. and Simoens, S. 2012. Evaluating and improving orphan drug
regulations in Europe: A Delphi policy study. Health policy, 108 (1), pp. 1—9. Press Office. 2012. Rilonacept Regeneron (Rilonacept). European Medicines Agency,
EMA/695977/2012. Reardon, S. 2014. Regulators adopt more orphan drugs. Nature, 508, pp. 16—17. Reese, J. H. 2014. FDA orphan drug designation 101. Worldwide Orphan Medicinal Designation
Workshop, 10 March, London. http://www.ema.europa.eu/docs/en_GB/document_library/Presentation/2014/03/WC500164160.pdf [Accessed 30 July 2014].
Regnstrom, J., Koenig, F., Aronsson, B., Reimer, T., Svendsen, K., Tsigkos, S., Flamion, B.,
Eichler, H. and Vamvakas, S. 2010. Factors associated with success of market authorisation applications for pharmaceutical drugs submitted to the European Medicines Agency. European journal of clinical pharmacology, 66 (1), pp. 39—48.
Stolk, P, Willenman, MJC & Leufkens, HGM. 2006. “Rare essentials”: drugs for rare diseases as
essential medicines. Bulletin of the World Health Organization, 84(9), pp. 745-751. Tambuyzer, E. 2010. Rare diseases, orphan drugs and their regulation: questions and
misconceptions. Nature Reviews Drug Discovery, 9 (12), pp. 921—929. Thorat, C., Xu, K., Freeman, S. N., Bonnel, R. A., Joseph, F., Phillips, M. I. and Imoisili, M. A.
2012. What the Orphan Drug Act has done lately for children with rare diseases: a 10-year analysis. Pediatrics, 129 (3), pp. 516—521.
U.S. Department of Health and Human Services (HHS). 2014a. Common EMEA/FDA Application for Orphan Medicinal Product Designation. Available from: http://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Forms/UCM048361.pdf [Accessed 01 August 2014]. U.S. Department of Health and Human Services (HHS), FDA, CDER & CBER. 2014b.
Guidance for industry: Expedited Programs for Serious Conditions – Drugs and Biologics. U.S. Department of Health and Human Services (HHS), FDA, CDER & CBER. 2001. Guidance
for industry: E 10 Choice of Control Group and Related Issues in Clinical Trials. The U.S. Food and Drug Administration (USFDA). 2014a. Search orphan designation drugs and
approvals. Available from: http://www.accessdata.fda.gov/scripts/opdlisting/oopd/ [Accessed 5 August 2014].
The U.S. Food and Drug Administration (USFDA). 2014b. Drugs@FDA: FDA Approved Drug

37
Products. Available from: http://www.accessdata.fda.gov/scripts/cder/drugsatfda/>. [Accessed 5 August 2014].
The U.S. Food and Drug Administration (USFDA). 2013a. New Drug Application (NDA)
Introduction. Available from: http://www.fda.gov/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved/approvalapplications/newdrugapplicationnda/default.htm [Accessed 5 August 2014].
The U.S. Food and Drug Administration (USFDA). 2013b. A valuable resource for drug
developers: the rare disease repurposing database. For Industry. Available from: http://www.fda.gov/ForIndustry/DevelopingProductsforRareDiseasesConditions/HowtoapplyforOrphanProductDesignation/ucm216147.htm [Accessed 5 August 2014].
Wastfelt, M., Fadeel, B. and Henter, J. 2006. A journey of hope: lessons learned from studies on
rare diseases and orphan drugs. Journal of internal medicine, 260 (1), pp. 1—10.
Published EPAR References:
Committee for Medicinal Products for Human Use (CHMP). 2014. Assessment report: Masican. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002670/WC500170722.pdf [Accessed 21 July 2014].
Committee for Medicinal Products for Human Use (CHMP). 2013. Assessment report: Istodax.
Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002122/WC500140233.pdf [Accessed 21 July 2014].
Committee for Medicinal Products for Human Use (CHMP). 2013. Assessment report: Opsumit.
Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002697/WC500160900.pdf [Accessed 21 July 2014].
Committee for Medicinal Products for Human Use (CHMP). 2013. Assessment report: Procysbi.
Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002465/WC500151314.pdf [Accessed 21 July 2014].
Committee for Medicinal Products for Human Use (CHMP). 2013. Assessment report: Sirturo.
Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002614/WC500163215.pdf [Accessed 21 July 2014].
Committee for Medicinal Products for Human Use (CHMP). 2012. Assessment report: Adcetris.
Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002455/WC500135054.pdf [Accessed 21 July 2014].
Committee for Medicinal Products for Human Use (CHMP). 2012. Assessment report: Cometriq.
Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002640/WC500163705.pdf [Accessed 21 July 2014].
Committee for Medicinal Products for Human Use (CHMP). 2012. Assessment report: Elelyso.

38
Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002250/WC500135112.pdf [Accessed 21 July 2014].
Committee for Medicinal Products for Human Use (CHMP). 2012. CHMP Assessment report:
Jakavi. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002464/WC500133226.pdf [Accessed 21 July 2014].
Committee for Medicinal Products for Human Use (CHMP). 2012. CHMP Assessment report:
Kalydeco. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002494/WC500130766.pdf [Accessed 21 July 2014].
Committee for Medicinal Products for Human Use (CHMP). 2012. Assessment report: Revestive.
Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002345/WC500132928.pdf [Accessed 21 July 2014].
Committee for Medicinal Products for Human Use (CHMP). 2012. Assessment report: Signifor.
Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002052/WC500128058.pdf [Accessed 21 July 2014].
Committee for Medicinal Products for Human Use (CHMP). 2012. Final CHMP Assessment
report: Folotyn. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002096/WC500129886.pdf [Accessed 21 July 2014].
Committee for Medicinal Products for Human Use (CHMP). 2010. Assessment report: Vpriv.
Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/001249/WC500096489.pdf [Accessed 21 July 2014].
Committee for Medicinal Products for Human Use (CHMP). 2009. Assessment report: Cayston.
Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/000996/WC500019995.pdf [Accessed 21 July 2014].
Committee for Medicinal Products for Human Use (CHMP). 2008. CHMP Assessment report:
Firazyr. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/000899/WC500022970.pdf [Accessed 21 July 2014].
Committee for Medicinal Products for Human Use (CHMP). 2008. CHMP Assessment report:
Sovrima. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/000908/WC500070576.pdf [Accessed 21 July 2014].
European Medicines Agency (EMEA). 2008. Refusal CHMP Assessment report for Mylotarg.
Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/000705/WC500070677.pdf [Accessed 21 July 2014].

39
European Medicines Agency (EMEA). 2008. Refusal CHMP Assessment report for Rhucin.
Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/000769/WC500070611.pdf [Accessed 21 July 2014].
European Medicines Agency (EMEA). 2007. Scientific Discussion: Revlimid. Available from:
http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000717/WC500056022.pdf [Accessed 21 July 2014].
European Medicines Agency (EMEA). 2007. Assessment report: Soliris. Available from:
http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000791/WC500054212.pdf [Accessed 21 July 2014].
European Medicines Agency (EMEA). 2007. Refusal CHMP Assessment report for Mycograb.
Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/000658/WC500070523.pdf [Accessed 21 July 2014].
Published FDA Report References:
Beitz, J. 2010. Application Number: 22-575. Center for Drug Evaluation and Research. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/022575s000sumr.pdf [Accessed 21 July 2014].
Chowdhury, B.A. 2012. Application Number: 203188Orig1s000. Center for Drug Evaluation
and Research. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203188Orig1s000SumR.pdf [Accessed 21 July 2014].
Chowdhury, B.A. 2011. Application Number: 022150 Orig1s000. Center for Drug Evaluation
and Research. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/022150Orig1s000SumR.pdf [Accessed 21 July 2014].
Cox, E.M. 2012. NDA 204384. Department of Health and Human Services. Available from:
http://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/204384Orig1s000ltr.pdf [Accessed 21 July 2014].
Farrell, A.T. 2011. Application Number: 125399Orig1s000. Center for Drug Evaluation and
Research. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/125399Orig1s000SumR.pdf [Accessed 21 July 2014].
He, R. 2012. Application Number: 203441Orig1s000. Center for Drug Evaluation and Research.
Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203441Orig1s000SumR.pdf

40
[Accessed 21 July 2014]. Justice, R. 2006. NDA 21-880/S-001. Department of Health & Human Services. Available from:
http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2006/021880s001LTR.pdf [Accessed 21 July 2014].
Kaminskas, E. 2011. Application Number: 202192Orig1s000. Center for Drug Evaluation and
Research. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202192Orig1s000SumR.pdf [Accessed 21 July 2014].
Keegan, P. 2012. Application Number: 203756Orig1s000. Center for Drug Evaluation and
Research. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203756Orig1s000SumR.pdf [Accessed 21 July 2014].
Laessig, K.A. 2010. Application Number: 050814Orig1s000. Center for Drug Evaluation and
Research. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/125399Orig1s000SumR.pdf [Accessed 21 July 2014].
Mulberg, A.E. 2013. Application Number: 203389Orig1s000. Center for Drug Evaluation and
Research. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/203389Orig1s000SumR.pdf [Accessed 21 July 2014].
Parks, M.H. 2012. Application Number: 200677Orig1s000. Center for Drug Evaluation and
Research. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/200677Orig1s000SumR.pdf [Accessed 21 July 2014].
Pazdur, R. 2007. Application Number: 125166. Center for Drug Evaluation and Research.
Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2007/125166s0000_APPROV.pdf [Accessed 21 July 2014].
Southworth, M.R. 2013. Application Number: 204410Orig1s000. Center for Drug Evaluation
and Research. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/204410Orig1s000SumR.pdf [Accessed 21 July 2014].
Published HTA References:
Haute Autorité de Santé (HAS). 2013a. Transparency Committee Opinion Adcetris, Available from: http://www.has-sante.fr/portail/jcms/c_1517924/en/adcetris?xtmc=&xtcr=1 [Accessed 6 August 2014].

41
Haute Autorité de Santé (HAS). 2013b. Transparency Committee Opinion Jakavi 5 mg tablet, Available from: http://www.has-sante.fr/portail/upload/docs/application/pdf/2013-07/jakavi_ct_12530.pdf [Accessed 6 August 2014].
Haute Autorité de Santé (HAS). 2012a. Transparency Committee Opinion Kalydeco 150 mg
film-coated tablet, Available from: http://www.has-sante.fr/portail/upload/docs/application/pdf/2013-05/kalydeco_ct_12474.pdf [Accessed 6 August 2014].
Haute Autorité de Santé (HAS). 2012b. Transparency Committee Opinion Lenalidomide,
Available from: http://www.has-sante.fr/portail/upload/docs/application/pdf/2013-01/revlimid_ct_11991.pdf [Accessed 6 August 2014].
Haute Autorité de Santé (HAS). 2012c. Transparency Committee Opinion Pasireotide, Available
from: http://www.has-sante.fr/portail/upload/docs/application/pdf/2013-02/signifor_ct_12258.pdf [Accessed 6 August 2014].
Haute Autorité de Santé (HAS). 2012d. Transparency Committee Opinion Soliris 300 mg,
concentrate for solution for infusion, Available from: http://www.has-sante.fr/portail/upload/docs/application/pdf/2013-04/soliris_ct_12290.pdf [Accessed 6 August 2014].
Haute Autorité de Santé (HAS). 2011. Transparency Committee Opinion Vpriv, Available from:
http://www.has-sante.fr/portail/jcms/c_1014915/en/vpriv?xtmc=&xtcr=1 [Accessed 6 August 2014].
Haute Autorité de Santé (HAS). 2009. Transparency Committee Opinion Firazyr, Available
from: http://www.has-sante.fr/portail/jcms/c_724460/en/firazyr?xtmc=&xtcr=1 [Accessed 6 August 2014].
National Institute for Clinical Excellence (NICE). 2014a. Atypical haemolytic uraemic syndrome (aHUS) – eculizumab, Available from: http://www.nice.org.uk/guidance/indevelopment/GID-ATYPICALHAEMOLYTICURAEMICSYNDROMEAHUSECULIZUMAB [Accessed 6 August 2014].
National Institute for Clinical Excellence (NICE). 2014b. Lymphoma (Hodgkin's, CD30-
positive) - brentuximab vedotin (after autologous stem cell transplant), Available from: http://www.nice.org.uk/guidance/indevelopment/GID-TAG467 [Accessed 6 August 2014].
National Institute for Clinical Excellence (NICE). 2013a. Colistimethate sodium and tobramycin
dry powders for inhalation for treating pseudomonas lung infection in cystic fibrosis, Available from: http://www.nice.org.uk/guidance/TA276 [Accessed 6 August 2014].
National Institute for Clinical Excellence (NICE). 2013b. Ruxolitinib for disease-related
splenomegaly or symptoms in adults with myelofibrosis, Available from: http://www.nice.org.uk/guidance/TA289 [Accessed 6 August 2014].

42
National Institute for Clinical Excellence (NICE). 2009. Lenalidomide for the treatment of
multiple myeloma in people who have received at least one prior therapy. Available from: http://www.nice.org.uk/guidance/TA171/chapter/1-guidance [Accessed 6 August 2014].
Scottish Medical Consortium (SMC). 2014a. Lenalidomide (Revlimid), Available from: http://www.scottishmedicines.org.uk/SMC_Advice/Advice/441_08_lenalidomide_Revlimid/lenalidomide_Revlimid_Resubmission [Accessed 6 August 2014].
Scottish Medical Consortium (SMC). 2014b. Macitentan (Opsumit), Available from:
http://www.scottishmedicines.org.uk/SMC_Advice/Advice/952_14_macitentan_Opsumit/macitentan_Opsumit [Accessed 6 August 2014].
Scottish Medical Consortium (SMC). 2013a. Ivacaftor (Kalydeco), Available from:
http://www.scottishmedicines.org.uk/SMC_Advice/Advice/827_12_ivacaftor_Kalydeco/ivacaftor_Kalydeco_Resubmission [Accessed 6 August 2014].
Scottish Medical Consortium (SMC). 2013b. Ruxolitinib (Jakavi), Available from:
http://www.scottishmedicines.org.uk/SMC_Advice/Advice/867_13_ruxolitinib_Jakavi/ruxolitinib_Jakavi [Accessed 6 August 2014].
Scottish Medical Consortium (SMC). 2012a. Aztreonam (Cayston), Available from:
http://www.scottishmedicines.org.uk/SMC_Advice/Advice/753_12_aztreonam_Cayston/aztreonam_Cayston [Accessed 6 August 2014].
Scottish Medical Consortium (SMC). 2012b. Eculizumab (Soliris), Available from:
http://www.scottishmedicines.org.uk/SMC_Advice/Advice/767_12_eculizumab_Soliris/eculizumab_Soliris [Accessed 6 August 2014].
Scottish Medical Consortium (SMC). 2012c. Icatibant (Firazyr), Available from:
http://www.scottishmedicines.org.uk/SMC_Advice/Advice/476_08_icatibant/icatibant_Firazyr_Resubmission [Accessed 6 August 2014].
Scottish Medical Consortium (SMC). 2012d. Pasireotide (Signifor), Available from:
http://www.scottishmedicines.org.uk/SMC_Advice/Advice/815_12_pasireotide_Signifor/pasireotide_Signifor [Accessed 6 August 2014].
Scottish Medical Consortium (SMC). 2012e. Velaglucerase (Vpriv), Available from:
http://www.scottishmedicines.org.uk/SMC_Advice/Advice/681_11_velaglucerase_Vpriv/velaglucerase_Vpriv_Full [Accessed 6 August 2014].
Tandvårds- och läkemedelsförmånsverket (TLV). 2014a. Jakavi ingår i högkostnadsskyddet med
begränsning, Available from: http://www.tlv.se/beslut/beslut-lakemedel/begransad-subvention/Jakavi-ingar-i-hogkostnadsskyddet-med-begransning/ [Accessed 6 August 2014].
Tandvårds- och läkemedelsförmånsverket (TLV). 2014b. Opsumit ingår i högkostnadsskyddet

43
med begränsning, Available from: http://www.tlv.se/beslut/beslut-lakemedel/begransad-subvention/Opsumit-ingar-i-hogkostnadsskyddet-med-begransning/ [Accessed 6 August 2014].
Tandvårds- och läkemedelsförmånsverket (TLV). 2013. Adcetris ingår i högkostnadsskyddet
med begränsning, Available from: http://www.tlv.se/beslut/beslut-lakemedel/begransad-subvention/adcetris-ingar-i-hogkostnadsskyddet-med-begransning/ [Accessed 6 August 2014].
Tandvårds- och läkemedelsförmånsverket (TLV). 2012. Cayston ingår i högkostnadsskyddet,
Available from: http://www.tlv.se/beslut/beslut-lakemedel/generell-subvention/cayston-ingar-i-hogkostnadsskyddet/ [Accessed 6 August 2014].
Tandvårds- och läkemedelsförmånsverket (TLV). 2011. Vi beslutar att Vpriv inte ska ingå i
högkostnadsskyddet, Available from: http://www.tlv.se/beslut/beslut-lakemedel/avslag-uteslutningar/vi-beslutar-att-vpriv-inte-ska-inga-i-hogkostnadsskyddet/ [Accessed 6 August 2014].
Tandvårds- och läkemedelsförmånsverket (TLV). 2010a, Firazyr ingår i högkostnadsskyddet
med begränsning, Available from: http://www.tlv.se/beslut/beslut-lakemedel/begransad-subvention/firazyr-ingar-i-hogkostnadsskyddet-med-begransning/ [Accessed 6 August 2014].
Tandvårds- och läkemedelsförmånsverket (TLV). 2010b. Revlimid i högkostnadsskyddet utan
begränsning, Available from: http://www.tlv.se/beslut/beslut-lakemedel/generell-subvention/revlimid-i-hogkostnadsskyddet-utan-begransning/ [Accessed 6 August 2014].

44
Appendix Appendix I. Orphan approvals and designations per year in the EU and US
Year MA / Approvals Designations
US EU US EU prior 198 0 987 0 2000 15 0 70 5 2001 6 0 78 26
2002 14 2 64 28 2003 13 1 95 33 3004 13 5 132 49
2005 20 3 123 63 2006 24 7 142 57
2007 16 13 119 79 2008 17 6 165 64 2009 20 6 164 88 2010 15 3 195 112 2011 26 5 203 93
2012 26 10 189 140 2013 33 7 260 116 2014 17 8 136 72
’00-‘14 275 76 2135 1025 Total 473 76 3122 1025
Source: USFDA 2014a; EMA 2014d; EMA 2014b Appendix II. Refused EMA orphan designation decisions, EMA and FDA Dates
Indication Active Substance EMA
decision FDA
designated FDA MA
A Graft rejection after liver transplantation
4-[[[4-(4-Chlorophenoxy)phenyl]sulfonyl]-methyl]tetrahydro-N-hydroxy-2H-pyran-4-carboxamide
10.09.2008 No No
B painful HIV-associated neuropathy Capsaicin
21.09.2006 02.05.2003 No
C Pancreatic cancer Chelidonii radix special liquid extract 03.12.2007 No No
D
Acute uncomplicated plasmodium falciparum malaria
Chlorproguanil hydrochloride and dapsone
26.07.2002 No No
E Pancreatic cancer Gastrin 17C diphtheria toxoid conjugate 16.07.2010 07.07.2009 No
F Malignant melanoma Histamine dihydrochloride 24.08.2004 No No
G B-cell non-Hodgkin`s lymphoma
Ibritumomab tiuxetan for use with 90Yttrium
31.01.2002 06.09.1994 19.02.2002
H
Patent ductus arteriosus in premature neonates of less than 34 week of gestational age (prevention of) Ibuprofen L-Lysinate
07.07.2005 29.10.1996 No

45
I
Patent ductus arteriosus in premature neonates of less than 34 week gestational age (treatment of) Ibuprofen L-Lysinate
24.06.2005 29.10.1996 13.04.2006
J
‘Off’-periods in adult patients with advanced Parkinson’s disease
Lentiviral vector expressing the truncated form of human tyrosine hydroxylase gene, human aromatic L amino-acid decarboxylase gene, human GTP-cyclohydrolase 1 gene
09.09.2010 No No
K Status epilepticus Midazolam Hydrochloride 29.02.2004 No No
L Cystic fibrosis Molgramostim 16.07.2010 No No
M Carcinoma in situ of the urinary bladder Mycobacterial cell wall complex
17.12.2002 No No
N Amyotrophic lateral sclerosis Nabilone
16.01.2011 No No
O Active phase of Peyronie's disease Sudismase
16.06.2005 12.03.1996 06.12.2013
P P-gp positive breast cancer Tariquidar
21.03.2012 No No
Q Painful HIV-associated neuropathy Tramadol hydrochloride
21.09.2006 28.01.2005 No
R Complex regional pain syndrome Zoledronic acid
17.04.2013 06.05.2013 No
Source: USFDA 2014a; EMA 2014d; EMA 2014b
Appendix III. EMA MA Orphan Drugs
Product EMA MA date FDA orphan FDA MA FDA MA date CA EC
brentuximab vedotin 25/10/2012
riociguat 27/03/2014 08/10/2013
ofatumumab 19/04/2010 17/04/2014
nelarabine 22/08/2007 28/10/2005
bosutinib 27/03/2013 04/09/2012
mannitol 13/04/2012 N/A
carglumic acid 24/01/2003 18/03/2010
aztreonam lysine 21/09/2009 22/02/2010
histamine dihydrochloride 07/10/2008 N/A
cholic acid 04/04/2014 N/A
cabozantinib 21/03/2014 29/11/2012
betaine anhydrous 15/02/2007 25/10/1996
decitabine 20/09/2012 N/A
defibrotide 18/10/2013 N/A
delamanid 28/04/2014 N/A
stiripentol 04/01/2007 N/A
idursulfase 08/01/2007 24/07/2006
pirfenidone 28/02/2011
clofarabine 29/05/2006 28/12/2004
deferasirox 28/08/2006 02/11/2005
icatibant 11/07/2008 25/11/2003

46
amifampridine 23/12/2009 N/A 5-aminolevulinic acid
hydrochloride 07/09/2007 N/A
alipogene tiparvovec 25/10/2012 N/A
para-aminosalicylic acid 07/04/2014 N/A
ponatinib 01/07/2013 20/11/2009
pomalidomide 05/08/2013 08/02/2013
mecasermin 03/08/2007 30/08/2005
rufinamide 16/01/2007 14/11/2008
ruxolitinib 23/08/2012
ivacaftor 23/07/2012
sapropterin dihydrochloride 02/12/2008 13/12/2007
cladribine 14/04/2004 26/02/1993
mitotane 28/04/2004
mifamurtide 06/03/2009
plerixafor 31/07/2009 15/12/2008
alglucosidase alfa 29/03/2006 24/05/2010
galsulfase 24/01/2006 31/05/2005
sorafenib 19/07/2006 20/12/2005 concentrate of proteolytic
enzymes enriched in bromelain
18/12/2012 N/A
romiplostim 04/02/2009 22/08/2008
macitentan 20/12/2013
nitisinone 21/02/2005 18/01/2002
cholic acid 12/09/2013 N/A
ibuprofen 29/07/2004 13/04/2006
caffeine citrate 02/07/2009 09/21/1999
hydrocortisone 03/11/2011 N/A
ziconotide 21/02/2005
mercaptamine bitartrate 06/09/2013
sildenafil 28/10/2005 N/A
teduglutide 30/08/2012
lenalidomide 14/06/2007
dexrazoxane hydrochloride 28/07/2006 06/09/2007
pasireotide diaspartate 24/04/2012 14/12/2012
hydroxycarbamide 29/06/2007 N/A
bedaquiline fumarate 05/03/2014
eculizumab 20/06/2007
dasatinib 20/11/2006 28/06/2006
siltuximab 22/05/2014 23/04/2014
nilotinib 19/11/2007 29/10/2007
thiotepa 15/03/2010 N/A
thalidomide 16/04/2008 25/05/2006
tobramycin 20/07/2011 N/A
temsirolimus 19/11/2007 30/05/2007
bosentan monohydrate 15/05/2002 20/11/2001
azacitidine 17/12/2008 19/05/2004
rhgalns 28/04/2014 14/02/2014
47
ambrisentan 21/04/2008 15/06/2007
everolimus 02/09/2011 26/04/2012
velaglucerase alfa 26/08/2010
tafamidis 16/11/2011 N/A
zinc 13/10/2004 28/01/1997
anagrelide 16/11/2004 14/03/1997 6-mercaptopurine
monohydrate 09/03/2012 28/04/2014
trabectedin 17/09/2007 N/A
miglustat 20/11/2002 31/07/2003
Source: EMA 2014b Appendix IV. FDA Status of EMA Refused MAA
Active Substance Indication FDA orphan designated
FDA MA
A taliglucerase alfa Gaucher disease 03.09.2009 01.05.2012 B pralatrexate T-cell lymphoma No 24.09.2009 C romidepsin Peripheral T-cell lymphoma No 05.11.2009
D Recombinant human monoclonal
antibody to hsp Candidiasis No No
E gemtuzumab ozogamicin Acute myeloid leukaemia 24.11.1999 Withdrawn F Recombinant human C1 inhibitor Angioedema No No G idebenone Friedreich Ataxia 25.03.2004 No
Source: EMA 2014b, USFDA 2014a

For further information on this or any of theHealth publications contact:
Anthony KellyManaging EditorLSE HealthThe London School of Economics and Political Science Houghton StreetLondon WC2A 2AE
Tel: + 44 (0)20 7955 7392 Fax: + 44 (0)20 7955 6090
Email: [email protected]: www.lse.ac.uk/collections/LSEHealth/