Embed Size (px)
Citation preview

Electrophilic Fluorophosphonium Cations – The Transition from Boron to Phosphorus Lewis Acids in Frustrated Lewis
Pair Chemistry
by
Christopher Blain Caputo
A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy
Department of Chemistry University of Toronto
© Copyright by Christopher Blain Caputo 2015

ii
Electrophilic Fluorophosphonium Cations – The Transition from Boron to Phosphorus Lewis Acids in Frustrated Lewis Pair Chemistry
Christopher Blain Caputo
Doctor of Philosophy
Department of Chemistry
University of Toronto
2015
Abstract
Catalytic transformations are of utmost importance for the synthesis of goods and materials.
Transition metal systems have traditionally been employed for catalytic processes, however this
notion has begun to change with the renaissance in metal-free catalysts. One subset of metal-free
catalysis is frustrated Lewis pairs (FLPs). These are combinations of Lewis acids and Lewis
bases that are precluded from generating classical adducts due to steric encumbrance. This thesis
expands the scope of FLP reactivity by investigating the chemistry with novel Lewis bases, acids
and substrates.
Sterically unencumbered amines form Lewis acid base adducts with B(C6F5)3. These amines
were encapsulated into a [2]rotaxane in order to impart steric protection without covalent
modifications. These combinations were shown to activate H2 in an FLP manner and led to the
discovery of an oxygen/boron hydrogenation catalyst.
Phosphorus/boron FLPs were reacted with a variety of fluoroalkanes resulting in the activation of
the C–F bond. From this it was shown that B(C6F5)3 can act as a competent hydrodefluorination
catalyst for fluoroalkanes. The reactions of P/B FLP systems with XeF2 were also explored,
resulting in the formation of fluorophosphonium fluoroborate salts. This led to the discovery of
highly electrophilic fluorophosphonium cations.

iii
Organophosphorus compounds are best known for their roles as Lewis bases. Considerably less
attention has been paid to Lewis acidic phosphonium cations. The reactivity of these EPCs is
described herein. They have found applications in CO2 sequestration, catalytic C–F bond
activation, hydrosilylation, dehydrocoupling and transfer hydrogenation reactions. Finally, they
have shown promise in FLP chemistry, with preliminary results indicating they are capable of
acting as a Lewis acid for H2 activation.

iv
Acknowledgments
I would like to acknowledge my supervisor, Prof. Doug Stephan. Above all, thank you for
always being open to discussing new ideas and providing advice when required. The freedom
which you give us to explore our own curiosities makes the lab such an exciting place and this
has truly made me become a better scientist. I would like to thank my committee members, Prof.
Bob Morris and Prof. Datong Song for their guidance. I would also like to acknowledge my
undergraduate supervisor and collaborator Prof. Steve Loeb for first getting me interested in
chemistry.
I have to thank all the Stephan group members, both past and present for your constant advice
and friendship throughout my time here. Particularly, I would like to thank Dr. Stephen Geier
for mentoring me when I first joined the lab and giving me a great foundation to build upon.
Thank you to Dr. Lindsay Hounjet, Dr. Roman Dobrovetsky and Dr. Manuel Pérez for working
with me on a project which we once thought was crazy, thank you for your advice and guidance,
I will always be grateful your help. Thanks to Dr. Chris Brown for teaching me about X-ray
crystallography and to my fellow X-ray group, Conor Pranckevicius, Lauren Longobardi, Dr.
Adam McKinty, Dr. Mike Boone, Dr. Fatme Dahcheh, and Dr. Mike Sgro for your help
whenever it was required.
I would like to thank Prof. Gerhard Erker for welcoming me into his group at the University of
Münster and for his supervision and insight during my exchange in his group. Thank you to
everyone in the Erker lab as well, in particular Dr. Juri Möbus and Dr. René Liedtke for your
friendship and help around Germany, Danke Schön!
Thank you to the fantastic support staff in the department. Thanks to Dr. Darcy Burns, Dmitry
Pichugian and Dr. Timothy Burrows for your help with all my NMR concerns. Thanks also to
Shanna Pritchard, Dr. Alan Lough, Anna-Liza Villavelez, John Ford, Ken Greaves and Rose
Balazs.
Finally, I would like to thank my family; Mom, Dad, and Nicole, thanks for your love and
support throughout this process, without it I would not be where I am today. Finally, to my
former desk-mate and best friend (sorry Conor, not you) Cheryl for your constant understanding,
support and encouragement throughout the last four years!

v
Table of Contents
Acknowledgments .......................................................................................................................... iv
Table of Contents ............................................................................................................................ v
List of Tables ................................................................................................................................. ix
List of Schemes ............................................................................................................................... x
List of Figures .............................................................................................................................. xiv
List of Symbols and Abbreviations ............................................................................................. xvii
Chapter 1 Introduction .................................................................................................................... 1
1.1 Chemistry is Everywhere – Impact on Society ................................................................... 1
1.2 History of Lewis Acid/Base Chemistry .............................................................................. 2
1.3 Discovery of Frustrated Lewis Pair Chemistry ................................................................... 4
1.3.1 Frustrated Lewis Pair Reactivity with H2 ............................................................... 4
1.3.1.1 Catalytic Hydrogenation with FLPs ......................................................... 8
1.3.2 Reactivity with other small molecules .................................................................... 9
1.4 Alternative Lewis acids ..................................................................................................... 12
1.4.1 Aluminium Based Lewis Acids ............................................................................ 12
1.4.2 Carbon Based Lewis Acids ................................................................................... 14
1.4.3 Silicon Based Lewis Acids ................................................................................... 15
1.4.4 Group 4 Metallocene Based Lewis Acids ............................................................. 16
1.5 Scope of Thesis ................................................................................................................. 17
1.6 References ......................................................................................................................... 19
Chapter 2 Using Mechanically Interlocked Molecules to Impart FLP Reactivity onto
Sterically Unencumbered Bases ............................................................................................... 27
2.1.1 The Border of Classical and Frustrated Lewis Pair Chemistry ............................. 27
2.1.2 A Brief Introduction to [2]Rotaxanes ................................................................... 28
2.2 Results and Discussion ..................................................................................................... 30

vi
2.2.1 A Supramolecular Approach to Steric Frustration ................................................ 30
2.2.2 Mechanism of H2 Activation ................................................................................ 38
2.2.3 Crown Ether Reactivity with B(C6F5)3 ................................................................. 39
2.3 Conclusions ....................................................................................................................... 42
2.4 Experimental Section ........................................................................................................ 43
2.4.1 General Considerations ......................................................................................... 43
2.4.2 Synthesis of Compounds ....................................................................................... 43
2.4.3 X-Ray Crystallography ......................................................................................... 57
2.4.3.1 X-Ray Collection and Reduction ............................................................ 57
2.4.3.2 X-Ray Solution and Refinement............................................................. 58
2.5 References ......................................................................................................................... 60
Chapter 3 Frustrated Lewis Pair Activation of Carbon–Fluorine Bonds ...................................... 64
3.1 Introduction ....................................................................................................................... 64
3.1.1 Interest in Carbon–Fluorine Bonds ....................................................................... 64
3.1.2 Metal Mediated C–F Bond Activation .................................................................. 65
3.1.3 Main Group C–F Bond Activation ....................................................................... 66
3.2 Results and Discussion ..................................................................................................... 67
3.2.1 Stoichiometric Reactions of Phosphines and Boranes with Alkyl Fluorides ....... 67
3.2.2 Stoichiometric Transformations of C–F bonds ..................................................... 73
3.2.3 Catalytic Hydrodefluorination of Alkyl Fluorides using B(C6F5)3 ....................... 75
3.3 Conclusions ....................................................................................................................... 78
3.4 Experimental Section ........................................................................................................ 79
3.4.1 General Considerations ......................................................................................... 79
3.4.2 Synthesis of Compounds ....................................................................................... 79
3.4.3 Hydrodefluorination Reaction Procedures ............................................................ 85
3.4.4 X-Ray Crystallography ......................................................................................... 85

vii
3.4.4.1 X-Ray Collection and Reduction ............................................................ 85
3.4.4.2 X-Ray Solution and Refinement............................................................. 85
3.5 References ......................................................................................................................... 88
Chapter 4 Frustrated Lewis Pair Activation of Xenon Difluoride – Uncovering Highly Lewis
Acidic Fluorophosphonium Cations ........................................................................................ 92
4.1 Introduction ....................................................................................................................... 92
4.1.1 A Brief History of Xenon Chemistry .................................................................... 92
4.2 Results and Discussion ..................................................................................................... 94
4.2.1 Reactions of Frustrated Lewis Pairs with Xenon Difluoride ................................ 94
4.2.2 Competitive Fluorophosphonium Lewis Acidity with B(C6F5)3 ........................ 101
4.2.3 Utilizing Different Lewis Acids to Generate Fluorophosphonium Cations ....... 104
4.2.4 Determining the Lewis Acidity of Fluorophosphonium Cations ........................ 110
4.3 Conclusions ..................................................................................................................... 114
4.4 Experimental Section ...................................................................................................... 115
4.4.1 General Considerations ....................................................................................... 115
4.4.2 Synthesis of Compounds ..................................................................................... 115
4.4.3 X-Ray Crystallography ....................................................................................... 124
4.4.3.1 X-Ray Data Collection and Reduction ................................................. 124
4.4.3.2 X-Ray Data Solution and Refinement .................................................. 124
4.5 References ....................................................................................................................... 127
Chapter 5 Uncovering Electrophilic Fluorophosphonium Reactivity ........................................ 131
5.1 Introduction ..................................................................................................................... 131
5.1.1 Phosphorus Compounds as Electron Acceptors ................................................. 131
5.1.2 Phosphonium Cations as Lewis Acids ................................................................ 133
5.2 Results and Discussion ................................................................................................... 136
5.2.1 Nucleophilic Aromatic Substitution Reactions on Phosphonium Cations ......... 136

viii
5.2.2 Exploiting Phosphonium Lewis Acids in FLP-like Activation of CO2 .............. 140
5.2.3 Realizing the Potential of Highly Lewis Acidic Phosphonium Cations ............. 147
5.2.3.1 Stoichiometric Reactions of [(C6F5)3PF] [B(C6F5)4] with C–F Bonds . 148
5.2.3.2 Catalytic Hydrodefluorination with [(C6F5)3PF] [B(C6F5)4] ................ 149
5.2.3.3 Mechanistic Insights into Fluorophosphonium-Catalyzed HDF .......... 156
5.2.3.4 Fluorophosphonium Catalyzed Hydrosilylation Reactions .................. 159
5.2.3.5 Phosphonium Catalyzed Dehydrocoupling Reactions ......................... 166
5.2.3.6 Phosphonium Catalyzed Transfer Hydrogenations of Olefins ............. 171
5.2.3.7 Dihydrogen Activation with Fluorophosphonium Lewis Acids .......... 176
5.3 Conclusions ..................................................................................................................... 181
5.4 Experimental Section ...................................................................................................... 181
5.4.1 General Considerations ....................................................................................... 181
5.4.2 Synthesis of Compounds ..................................................................................... 182
5.4.3 Catalytic Reaction Procedures ............................................................................ 190
5.4.4 X-Ray Crystallography ....................................................................................... 201
5.4.4.1 X-Ray Collection and Reduction .......................................................... 201
5.4.4.2 X-Ray Solution and Refinement........................................................... 202
5.5 References ....................................................................................................................... 205
Chapter 6 Conclusion .................................................................................................................. 213
6.1 Thesis Summary .............................................................................................................. 213
6.2 Future Work .................................................................................................................... 214
6.3 References ....................................................................................................................... 216

ix
List of Tables
Table 2.2.1 - Hydrogenation of 1,1-diphenylethylene with crown ethers and B(C6F5)3. .............. 41
Table 2.4.1 - Select Crystallographic Data for [2-224C6]. ........................................................ 59
Table 3.2.1 - Hydrodefluorination catalysis with B(C6F5)3. ......................................................... 76
Table 3.4.1 - Select Crystallographic Data for 3-3 and 3-5. ......................................................... 87
Table 4.2.1 - NMR spectroscopic data, calculated 31P chemical shift and FIA for compounds
4-1 – 4-6, 4-16, 4-17 in CD2Cl2 at ambient temperature. ........................................................... 113
Table 4.4.1 - Select Crystallographic Data for 4-1, 4-3, 4-12, 4-13 and 4-15. ........................... 125
Table 5.2.1 - Hydrodefluorination results using 4-17 as a catalyst. ........................................... 153
Table 5.2.2 - Hydrosilylation results using 4-17 as a catalyst. ................................................... 164
Table 5.2.3 - Catalytic dehydrocoupling reactions using 4-17. .................................................. 169
Table 5.2.4 - Transfer hydrogenation of olefins with concurrent dehydrocoupling catalysis using
4-17. ............................................................................................................................................ 173
Table 5.4.1 - Select Crystallographic Data for 5-1, 5-2, 5-5, 5-6 and 5-9. ................................. 203

x
List of Schemes
Scheme 1.2.1 - Depiction of classic Lewis pair reactivity. ............................................................. 2
Scheme 1.2.2 - Exceptions to classical Lewis acid and base reactivity. ......................................... 4
Scheme 1.3.1 - Hydrogen activation with an intramolecular phosphino-borane FLP. ................... 5
Scheme 1.3.2 - Depiction of a frustrated Lewis pair. ..................................................................... 5
Scheme 1.3.3 - Examples of FLP systems developed by the Erker group. .................................... 6
Scheme 1.3.4 - Reactions of carbenes with B(C6F5)3 and H2. ........................................................ 7
Scheme 1.3.5 - Examples of pyridine bases in FLP chemistry. ...................................................... 7
Scheme 1.3.6 - FLP catalyzed hydrogenation reactions. ................................................................ 9
Scheme 1.3.7 - CO2 and N2O activation with FLPs. .................................................................... 10
Scheme 1.3.8 - CO activation with FLPs. ..................................................................................... 11
Scheme 1.3.9 - Olefin and alkyne reactivity with FLPs ............................................................... 12
Scheme 1.4.1 - Examples of Al Lewis acids in FLP chemistry. ................................................... 13
Scheme 1.4.2 - Examples of carbon Lewis acids in FLP chemistry. ............................................ 15
Scheme 1.4.3 - H2 activation using a silylium Lewis acid. ........................................................... 16
Scheme 1.4.4 - Group 4 transition metal Lewis acids for FLP chemistry. All cations shown have
B(C6F5)4 anions. ............................................................................................................................ 17
Scheme 1.6.1 - Examples of Lewis acid base adducts which exhibit FLP reactivity. .................. 28
Scheme 1.6.2 - Depiction of a [2]pseudorotaxane and [2]rotaxane. ............................................. 28
Scheme 1.6.3 - Two general [2]rotaxane synthetic routes using a [2]pseudorotaxane (top) and a
ring closing reaction (bottom). ...................................................................................................... 29

xi
Scheme 2.2.1 - Reactions of 2-1 and 2-2 with B(C6F5)3. .............................................................. 31
Scheme 2.2.2 - Reactions probing the irreversible nature of adducts 2-3 and 2-4. ...................... 33
Scheme 2.2.3 - The reaction of [2-124C6] with B(C6F5)3. ........................................................ 34
Scheme 2.2.4 - The synthesis of [2-224C6] (left) and [2-222C6] (right). .............................. 35
Scheme 2.2.5 - FLP type H2 activation between [2-224C6] (top) and [2-222C6] (bottom) with
B(C6F5)3. ....................................................................................................................................... 37
Scheme 2.2.6 - Cyclic ether ring opening reactions with FLPs. ................................................... 39
Scheme 3.1.1 - Transition metal complexes for C–F bond activation. ......................................... 66
Scheme 3.1.2 - Main group C–F bond activation. ........................................................................ 67
Scheme 3.2.1 - C–F bond activation with the FLP, tBu3P and B(C6F5)3. ..................................... 68
Scheme 3.2.2 - C–F bond activation with tBu2PH and reactions with carbon Lewis bases. ........ 73
Scheme 3.2.3 - Reactions of fluoroalkanes with [tBu3PH] [HB(C6F5)3] and B(C6F5)3. ............... 74
Scheme 3.2.4 - Stoichiometric and catalytic heteroatom transfer via C–F activation. ................. 75
Scheme 3.2.5 - Two possible mechanisms for the B(C6F5)3 catalyzed hydrodefluorination
reaction. ......................................................................................................................................... 78
Scheme 4.1.1 - Reactions of platinum hexafluoride with oxygen and xenon. .............................. 93
Scheme 4.1.2 - Examples of fluorination reactions with XeF2. .................................................... 93
Scheme 4.1.3 - FLP reactivity with halogenating reagents. ......................................................... 94
Scheme 4.2.1 - Synthesis of 4-1, 4-2, 4-3, 4-4, 4-5 and 4-6. ........................................................ 96
Scheme 4.2.2 - Reaction of electron deficient phosphines with XeF2 and B(C6F5)3. ................. 100
Scheme 4.2.3 - Reaction mechanism for the reactivity of FLPs with XeF2. .............................. 100

xii
Scheme 4.2.4 - Reactions of fluorophosphonium fluoroborate salts with Et3PO. ...................... 104
Scheme 4.2.5 - Reactions of phosphines with XeF2 and TMSOTf. ........................................... 105
Scheme 4.2.6 - Formation of electrophilic fluorophosphonium salts 4-13 – 4-17. .................... 109
Scheme 4.2.7 - Childs (top) and Gutmann-Becket (bottom) methods for measuring the Lewis
acidity of 4-17. ............................................................................................................................ 111
Scheme 5.1.1 - PBr3 acting as an acceptor in the presence of alcohols, forming bromoalkanes.132
Scheme 5.1.2 - General diagram of phosphenium cations and NHPs. ....................................... 132
Scheme 5.1.3 - Phosphonium hypervalence acceptor properties. ............................................... 133
Scheme 5.1.4 - The Wittig reaction, an example of phosphonium acceptor capabilities. .......... 134
Scheme 5.1.5 - Phosphonium catalyzed addition reactions. ....................................................... 134
Scheme 5.1.6 - Phosphonium catalyzed Diels-Alder reaction. ................................................... 135
Scheme 5.1.7 - Cooperative fluoride ion binding between a borane and phosphonium cation. . 136
Scheme 5.2.1 - Formation of 5-1. ............................................................................................... 137
Scheme 5.2.2 - Formation of 5-2. ............................................................................................... 139
Scheme 5.2.3 - The formation of 5-3 and 5-4. ............................................................................ 141
Scheme 5.2.4 - Formation of 5-5 and 5-6. .................................................................................. 143
Scheme 5.2.5 - Formation of 5-8 and 5-9. .................................................................................. 145
Scheme 5.2.6 - Equilibrium and resonance structures of 5-5. .................................................... 147
Scheme 5.2.7 - Stoichiometric reactions of 4-17 with fluorocarbons. ........................................ 149
Scheme 5.2.8 - Hydrodefluorination reactions using 4-17. ........................................................ 150
Scheme 5.2.9 - Metal-free aromatic C–F bond activation. ......................................................... 155

xiii
Scheme 5.2.10 - Trityl cation initiated hydrodefluorination. ...................................................... 156
Scheme 5.2.11 - Competition experiment between 4-8 and octafluorotoluene with
[Et3Si][B(C6F5)4. ......................................................................................................................... 157
Scheme 5.2.12 - Proposed catalytic cycle for the fluorophosphonium catalyzed HDF. ΔH for
each reaction step reported in kcal/mol and the Gibbs free energies are reported in parenthesis
(kcal/mol). ................................................................................................................................... 158
Scheme 5.2.13 - Calculated interaction between 4-16 with silane and fluoroalkanes (top).
Isodesmic reaction to determine hydride or fluoride affinities (bottom). ................................... 159
Scheme 5.2.14 - Various catalysts used in hydrosilylation reactions. ........................................ 160
Scheme 5.2.15 - Proposed mechanism for the isomerization of 1-hexene with 4-17. ................ 162
Scheme 5.2.16 - Proposed hydrosilylation reaction mechanism with 4-17. ............................... 166
Scheme 5.2.17 - Proposed mechanistic pathways for dehydrocoupling of silane and amine with
4-17. Gibbs free energies and enthalpies in parentheses for every step are provided in kcal/mol.
..................................................................................................................................................... 171
Scheme 5.2.18 - Proposed mechanistic pathways for dehydrocoupling of silane and amine and
transfer hydrogenation of olefins with 4-17. Gibbs free energies and enthalpies in parentheses for
every step are provided in kcal/mol. ........................................................................................... 176
Scheme 5.2.19 - Calculated interaction between 4-17 and Ph2NH (A). HOMO and LUMO
depictions of this combination (B & C). ..................................................................................... 177
Scheme 5.2.20 - Exchange process between 4-17 and p-tol2NH. ............................................... 178
Scheme 6.2.1 - Proposed lignin degradation using 4-17. ........................................................... 215
Scheme 6.2.2 - Dicationic fluorophosphonium salt (top) and a potential P–O EPC with a Martin
type ligand (bottom). ................................................................................................................... 216

xiv
List of Figures
Figure 2.2.1 - Low temperature 19F NMR spectrum of 2-4. ......................................................... 32
Figure 2.2.2 - POV–Ray depiction of [2-224C6] (left), N: blue, O: red, C: black, H: white, C-H
atoms omitted for clarity. Space filling model of [2-224C6] (right), crown ether: red, axle:
blue, N: dark blue. ......................................................................................................................... 36
Figure 2.2.3 - Variable temperature 1H NMR experiment with 12C4 and B(C6F5)3. ................... 40
Figure 2.4.1 - 1H NMR spectrum of the reaction of [2-124C6] with B(C6F5)3. ........................ 51
Figure 2.4.2 - 11B NMR spectrum of the reaction of [2-124C6] with B(C6F5)3. ....................... 52
Figure 2.4.3 - 1H NMR spectrum of the reaction of [2-224C6] with B(C6F5)3. ......................... 53
Figure 2.4.4 - 11B NMR spectrum of the reaction of [2-224C6] with B(C6F5)3. ....................... 53
Figure 3.2.1 - POV–Ray depiction of 3-3, P: orange, B: green, F: pink, C: black. H-atoms
omitted for clarity. ........................................................................................................................ 69
Figure 3.2.2 – POV–Ray depiction of 3-5, P: orange, B: green, F: pink, C: black, H: white. H-
atoms omitted for clarity. .............................................................................................................. 72
Figure 3.2.3 - 1H and 19F NMR spectra of B(C6F5)3 catalyzed hydrodefluorination of
1-fluoroadamantane. ..................................................................................................................... 77
Figure 4.2.1 - Multinuclear NMR spectra of 4-1. ......................................................................... 96
Figure 4.2.2 - POV–ray depiction of 4-1, P: orange, B: green, F: pink, C: black. H-atoms omitted
for clarity. ...................................................................................................................................... 97
Figure 4.2.3 - POV–ray depiction of 4-3, P: orange, B: green, F: pink, C: black. H-atoms omitted
for clarity. ...................................................................................................................................... 99
Figure 4.2.4 - 31P{1H} NMR spectra of 4-6 in CD2Cl2 (–40 to 25 °C) and C6D5Br (25 to 60 °C).
..................................................................................................................................................... 102

xv
Figure 4.2.5 - 19F NMR Spectra of 4-6 in CD2Cl2 (–40 to 0 °C) and C6D5Br (25 to 60 °C). ..... 103
Figure 4.2.6 - POV–ray depiction of 4-12, P: orange, B: green, F: pink, C: black. H-atoms
omitted for clarity. ...................................................................................................................... 106
Figure 4.2.7 - POV–ray depiction of 4-13, P: orange, B: green, F: pink, C: black. H-atoms
omitted for clarity. ...................................................................................................................... 107
Figure 4.2.8 - POV–ray depiction of 4-15, P: orange, B: green, F: pink, C: black. H-atoms
omitted for clarity. ...................................................................................................................... 109
Figure 4.2.9 - 31P{1H} NMR stack plot of the 4-1 – 4-6, 4-16 and 4-17 synthesized. ............... 112
Figure 5.2.1 - POV–ray depiction of 5-1, P: orange, B: green, F: pink, C: black. H-atoms omitted
for clarity. .................................................................................................................................... 138
Figure 5.2.2 - POV–ray depiction of 5-2, P: orange, F: pink, C: black, S: yellow, O: red. H-atoms
omitted for clarity. ...................................................................................................................... 140
Figure 5.2.3 - POV–ray depiction of 5-5, P: orange, N: blue, F: pink, C: black. H-atoms omitted
for clarity. .................................................................................................................................... 142
Figure 5.2.4 - POV–ray depiction of 5-6, P: orange, N: blue, O: red, F: pink, C: black. H-atoms
omitted for clarity. ...................................................................................................................... 144
Figure 5.2.5 - POV–ray depiction of 5-9, P: orange, N: blue, O: red, C: black H-atoms omitted
for clarity. .................................................................................................................................... 146
Figure 5.2.6 - 11B NMR spectrum of the reaction of 4-17 with 1-fluoropentane. ...................... 149
Figure 5.2.7 - 19F NMR Spectra of the hydrodefluorination of 1-fluoroadamantane with 1 mol%
4-17 after 1 h. .............................................................................................................................. 151
Figure 5.2.8 - 19F NMR spectrum of the hydrodefluorination of octafluorotoluene with 1 mol%
4-17 after 24 h. ............................................................................................................................ 154
Figure 5.2.9 - 1-Hexene (#) isomerization to 2-hexene (*) with 4-17. ....................................... 161

xvi
Figure 5.2.10 - Low temperature 1H VT-NMR study between 4-16 and p-tol2NH. ................... 179
Figure 5.2.11 - Observed HD scrambling after 18 h with p-tol2NH and 4-17 at 100 °C............ 180
Figure 5.4.1 - 19F NMR spectrum of the reaction of 1-fluoropentane with Et3SiH in the presence
of 4-17 (1 mol%) after 2 h. ......................................................................................................... 191
Figure 5.4.2 - Transfer Hydrogenation of 1,1-diphenylethylene with tandem dehydrocoupling
between pentafluorophenyl carboxylic acid and Et3SiH using 4-17. ......................................... 201

xvii
List of Symbols and Abbreviations
Å angstrom
° degrees
°C degrees Celsius
η eta (bonding mode)
μ bridging
δ chemical shift
Δδp-m para-meta separation
∆ heat
G Gibbs free energy
ΔH enthalpy
pi
sigma
ν wavenumber
ν1/2 frequency difference at half maximum
μL microliters
μmol micromol
threaded through
12C4 12-crown-4
Ad adamantyl
Amu atomic mass unit
Anal analytical
Ar ortho-N-methylaniline
atm atmospheres
br broad
C6D5Br deuterated bromobenzene
Calcd. calculated
CD2Cl2 deuterated dichloromethane
CDCl3 deuterated chloroform
C7D8 deuterated toluene
C7H8 toluene

xviii
CF3 trifluoromethyl
C6F5 pentafluorophenyl
CFC chlorofluorocarbon
CN cyanide
CO carbon monoxide
COD 1,5-cyclooctadiene
Conv. conversion
d doublet
DB24C8 dibenzo-24-crown-8
dcalc calculated density
dd doublet of doublets
ddd doublet of doublet of doublets
dp doublet of pentets
DFT density functional theory
DMF dimethylformamide
DOSY diffusion ordered NMR spectroscopy
EPC electrophilic phosphonium cation
ESI electrospray ionization
eq. equivalent
Et ethyl
Et2O diethyl ether
Fc calculated structure factor
Fo observed structure factor
FIA fluoride ion affinity
FLP frustrated Lewis pair
g grams
GOF goodness of fit
h hour
HDF hydrodefluorination
HFC hydrofluorocarbon
HOESY heteronuclear overhauser effect NMR spectroscopy
Hz Hertz

xix
HOMO highest occupied molecular orbital
iPr isopropyl
ItBu 1,3-ditertbutylimidazole-2-ylidene
Idipp 1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene
IR infrared
nJxy n-bond scalar coupling constant between X and Y atoms
K Kelvin
kcal kilocalories
kJ kilojoule
LUMO lowest unoccupied molecular orbital
m meta
m multiplet
Me methyl
MeCN acetonitrile
Mes mesityl
MHz megahertz
MIM mechanically interlocked molecule
mg milligram
mL millilitre
mmol millimol
MS mass spectrometry
m/z mass-to-charge ratio
NBO natural bond order
NHC N-heterocyclic carbene
NHP N-heterocyclic phosphenium cation
NMR Nuclear Magnetic Resonance
o ortho
o-tol ortho-tolyl
OTf trifluoromethanesulfonate
p para
PFC perfluorocarbon
Ph phenyl

xx
POV-Ray Persistence of Vision Raytracer
ppb parts per billion
ppm parts per million
ppt parts per trillion
Rw weighted residual
r.t. room temperature
s singlet
sept septet
T temperature
t triplet
td triplet of doublets
THF tetrahydrofuran
tm triplet of multiplets
TMP 2,2,6,6-tetramethylpiperidine
TMS trimethylsilyl
tol toluene
tBu tert-butyl
q quartet
VT variable temperature

1
Chapter 1 Introduction
1
1.1 Chemistry is Everywhere – Impact on Society
Unprecedented growth in the Earth’s population over the last 100 years has been largely
facilitated by advances in science. For example, the “green revolution” of the 1940’s can be
viewed as a major event which has facilitated population growth. This green revolution
represented the introduction of nitrogen based fertilizers and chemical pesticides into farming;
leading to increased crop production. Such an increased access to food has sparked the
exponential population growth observed in recent decades. Prior to this advancement, nitrogen
based fertilizers were difficult to produce as there were no simple ways to synthesize ammonia
from nitrogen. However, the development of the Haber-Bosch process allowed for the mass
production of ammonia for the first time. Employing a heterogeneous iron catalyst and high
temperatures, nitrogen gas can be reduced to ammonia in the presence of H2, albeit at a high cost,
consuming nearly 2% of all the worlds energy supply.1 The Haber-Bosch process is arguably
one of the most important scientific discoveries for mankind due to its impact on the food
supply. The allowance of population growth notwithstanding, one could not go a single day
without being impacted by chemistry. An example of this is the development of advanced
pharmaceuticals. Such pharmaceuticals have drastically improved healthcare and in turn have
had a pronounced effect on life expectancy, with the average Canadian life expectancy
increasing from 58.8 years for men and 60.6 years for women in 1921 to 78.0 and 82.7 years for
men and women in 2005.2
The chemical science remains critical to humankind and chemistry will be vital in solving many
of the current issues facing the world. Due to exponential population growth and technological
advancements, humankind is consuming natural resources, such as fossil fuels, at an
unsustainable pace. Consequently, the result is an increased greenhouse gas emissions which in
turn leads to irreversible climate change.3-5 Chemistry has the opportunity to come up with
solutions to reduce emissions, develop cleaner energy sources and be a steward for the
responsible use of our limited natural resources. One approach to address the strains on the
world’s resources involves the investigation of less energy intensive processes. Catalysts

2
provide a solution for the decreasing of energy consumption and have been acknowledged as one
of the twelve principles of “Green Chemistry”.6 Innumerable catalysts have been developed
based on transition metals, including precious metals,7 but as the worldwide reserves of these
rare elements decrease and their prices increase dramatically, research has shifted focus to
incorporating more abundant metals into catalytic systems (Fe, Ni, Co, etc.).8-14 The work
outlined in this thesis takes catalyst development a step further by investigating the use of metal-
free catalysts in a number of organic transformations. In particular, examining the frustrated
Lewis pair (FLP) chemistry of boron based Lewis acids as well as the development of new
fluorophosphonium Lewis acids as catalysts for the remediation of environmentally persistent
fluorocarbons amongst other catalytic transformations will be detailed.
1.2 History of Lewis Acid/Base Chemistry
It was nearly 100 years ago that Gilbert Lewis published his seminal work entitled “The Atom
and the Molecule.” In it he classifies the reactivity of certain molecules as those that can donate
a pair of electrons and molecules which can accept a pair of electrons, later defined as Lewis
bases and acids, respectively (Scheme 1.2.1).15,16 The reactions of Lewis acids with Lewis bases
has been well explored. Generally, they interact and form a classical Lewis acid/base adduct,
with a famous example being ammonia-borane. Lewis acids have found numerous applications
in synthesis and catalysis, including the well-known Friedel-Crafts alkylation and acylation
reactions.17,18 In such reactions the Lewis acid abstracts a halide from an alkyl halide, generating
a carbocation in situ which is then attacked by a nucleophile. Lewis bases have found
applications as well; the donor capabilities of phosphines have been exploited as ligands in
transition metal catalysis,19,20 and more recently in organocatalysis.21
Scheme 1.2.1 - Depiction of classic Lewis pair reactivity.

3
The first exception to Lewis’ concept came in 1942 in a report from H.C. Brown.22 In this report
the lack of reactivity between 2,6-dimethylpyridine and BMe3 was described, while it was shown
that other Lewis acids such as BF3 will form a classical adduct. This result was attributed to the
steric hindrance that the methyl substituents impart on the boron and nitrogen centers, preventing
adduct formation. Furthermore, in 1950 Wittig and co-workers observed a ring opening reaction
of THF when bound to BPh3 in the presence of a bulky trityl anion, instead of the anticipated
solvent displacement and classical adduct formation.23 It was subsequently reported that PPh3
and BPh3 does not form a classical adduct in the presence of benzyne, but P/B addition occurs
across the C–C triple bond, resulting in the formation of an ortho-disposed phosphonium borate
zwitterion (Scheme 1.2.2).24 In related work, the reaction of trityl anion with Lewis acids (BPh3,
AlPh3, BePh2 and MgPh2) in the presence of butadiene was reported by Tochtermann in 1966.25
The goal of this research was to initiate anionic polymerization of the diene, however addition of
the Lewis acid and base across the olefin was observed. This result prompted Tochtermann to
name these combinations “antagonistisches Paar” or antagonistic pairs. Unconventional
reactivity in such systems continued to be observed; in the 1990’s Erker and co-workers reported
that a phosphorus ylide does not form an adduct with the Lewis acid B(C6F5)3, instead the Lewis
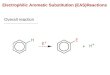
base attacks the pentafluorophenyl ring in the para-position. This nucleophilic aromatic
substitution reaction yields a linked phosphonium fluoroborate salt (Scheme 1.2.2).26 Similar
reactivity was subsequently observed with a number of phosphine Lewis bases with B(C6F5)327
and [Ph3C] [B(C6F5)4].28

4
Scheme 1.2.2 - Exceptions to classical Lewis acid and base reactivity.
1.3 Discovery of Frustrated Lewis Pair Chemistry
1.3.1 Frustrated Lewis Pair Reactivity with H2
The aforementioned reactivity indicates that steric constraints play a large role in the reactivity
observed between Lewis acids and bases. In 2006 our lab developed a sterically encumbered
phosphino-borane system, Mes2P(C6F4)B(C6F5)2, which reversibly activates hydrogen gas
forming the zwitterionic salt Mes2P(H)(C6F4)B(H)(C6F5)2 (Scheme 1.3.1).29 Heating this salt to
150 °C results in reformation of H2, regenerating the original linked system. This system was the
first metal-free compound to reversibly activate hydrogen. Such reactivity was coined as
“frustrated Lewis pair” (FLP) chemistry because the steric demand on the Lewis acidic and basic
centers prevent adduct formation, resulting in unquenched reactivity which in turn can be
exploited in the activation of small molecules (Scheme 1.3.2). The observation of the steric
requirement on the reactivity of such a system led our group to investigate intermolecular
systems and indeed it was found that complicated linked systems were not required to induce

5
FLP reactivity. It was subsequently found that the combination of tBu3P and B(C6F5)3 does not
form an adduct, and activation of H2 was observed under ambient conditions to form
[tBu3PH] [HB(C6F5)3].30
Scheme 1.3.1 - Hydrogen activation with an intramolecular phosphino-borane FLP.
Scheme 1.3.2 - Depiction of a frustrated Lewis pair.
Shortly after the initial publication on FLPs, the Erker group reported an intramolecular ethyl-
linked P/B FLP system, Mes2PCH2CH2B(C6F5)2.31 At room temperature this system forms a
classical P–B adduct, but it has the ability to activate H2 to yield the corresponding zwitterionic
phosphonium hydridoborate salt (Scheme 1.3.3). This chemistry has been extended to include
larger chains between the P and B centers,32 as well as the introduction of unsaturation into the
backbone, which is achieved by a carboboration reaction as opposed to hydroboration.33,34 In
addition to the intramolecular systems that have been developed, intermolecular systems which
reversibly activate H2 under mild conditions have been investigated. For example,
1,8-bis(diphenylphosphino)-naphthalene will activate hydrogen with B(C6F5)3 at room
temperature with loss of H2 occurring at 60 °C (Scheme 1.3.3).35

6
Scheme 1.3.3 - Examples of FLP systems developed by the Erker group.
Hydrogen activation with FLPs is not limited to the use of phosphorus Lewis bases. Bertrand et
al. have discovered that (alkyl)(amino)carbenes are capable of activating H2 and NH3 without the
use of a Lewis acid.36 They report that this reactivity results from the strong nucleophilicity and
accepting capabilities of the carbene, alternatively N-heterocyclic carbenes (NHCs) do not
undergo analogous reactivity. This result led our group37 and the group of Tamm38 to investigate
the reactivity of NHCs with B(C6F5)3 in FLP chemistry. The NHC Idipp forms a strong adduct
with B(C6F5)3 but ItBu does not form an adduct at low temperatures (–60 °C) and is able to
activate H2 to give the corresponding imidazolium hydridoborate salt (Scheme 1.3.4). In the
absence of H2, this combination forms an abnormal carbene adduct with B(C6F5)3 at room
temperature. Tamm et al have extended this chemistry to the Lewis acid B(3,5-(CF3)2C6H3)3 and
reported that it exhibits similar reactivity to B(C6F5)3.39 Furthermore, Arduengo et al have shown
that less electrophilic Lewis acids such as trialkylboranes can effect hydrogen activation with
NHCs. The electron rich borohydride can subsequently deliver the hydride to the imidazolium
cation, resulting in generation of the corresponding CH2 aminal and free borane.40

7
Scheme 1.3.4 - Reactions of carbenes with B(C6F5)3 and H2.
Various nitrogen Lewis bases have also been examined in FLP chemistry. Pyridine derivatives
have been shown to have a fine line between “classical” and “frustrated” behaviour. As opposed
to BMe3, B(C6F5)3 does indeed form an adduct with 2,6-dimethylpyridine. However, it is in an
equilibrium process between a “classical” adduct and a FLP species. Nevertheless, this
combination has been shown to activate H2 to form the pyridinium hydridoborate salt
[2,6-(CH3)2C5H3NH] [HB(C6F5)3] (Scheme 1.3.5).41 This chemistry was extended to a number of
other pyridine derivatives and were explored for their FLP reactivity.42,43 Certain N-heterocycles,
such as acridine and quinoline derivatives (Scheme 1.3.5), were shown form weak reversible
adducts with B(C6F5)3 and reduction of the heterocycle can be achieved upon addition of H2.44
Scheme 1.3.5 - Examples of pyridine bases in FLP chemistry.

8
1.3.1.1 Catalytic Hydrogenation with FLPs
The ability to activate hydrogen gas without the use of a transition metal was a revolutionary
discovery and a number of these systems have found use as hydrogenation catalysts for polarized
unsaturated substrates.45 For example, the original linked FLP system, Mes2P(C6F4)B(C6F5)2,
was found to be an effective catalyst for the hydrogenation of sterically encumbered imines.46
The mechanism for this hydrogenation reaction begins with H2 activation across the FLP
followed by hydride delivery to the unsaturated imine, then protonation to furnish the amine
product. The combination of 1,8-bis(diphenylphosphino)-naphthalene and B(C6F5)3 can
reversibly activate hydrogen under mild conditions (vide supra). This rapid reversibility led to
its use as an effective catalyst for the hydrogenation of silyl enol ethers.35 Subsequently, it was
then discovered that the substrate itself can act as the Lewis base, allowing for Lewis acid
catalyzed hydrogenations of sterically encumbered imines (Scheme 1.3.6).47 In these reactions
the H2 is split between the B and N atoms, with subsequent hydride delivery to the substrate.
A myriad of systems have since been developed, further expanding the scope of substrates which
can be hydrogenated.45,48 A notable example is the development of metal-free aromatic
hydrogenation systems (Scheme 1.3.6).49 This system takes advantage of the activation of
hydrogen between aniline Lewis bases and B(C6F5)3. Due to resonance, a negative charge is
localized on the para-carbon atom of the aniline which can be protonated (via direct H2
activation or through protonation from the generated ammonium cation) which breaks the
aromaticity and facilitates reduction of the ring. Another example of such reactivity is the use of
electron deficient Lewis bases with B(C6F5)3 to effect the hydrogenation of olefins.50 The
combination of (C6F5)Ph2P with B(C6F5)3 does not appear to activate H2 at room temperature,
however cooling the reaction to −80 °C resulted in an observable H2 activated product,
[(C6F5)Ph2PH] [HB(C6F5)3]. This implies that this reaction is rapidly reversible at room
temperature, and correspondingly it was exploited as a hydrogenation catalyst for a number of
olefinic substrates. The authors were able to show that the mechanism of olefin hydrogenation
differs from that of imine hydrogenation. The initial step in the hydrogenation involves the
protonation of the olefin from the phosphonium cation, generating an incipient carbocation to
which hydride is delivered from the hydridoborate anion.51 With slight modifications to the
system, the same group have been able to extend catalytic hydrogenation to include nitroolefins
and acrylates.52 Alternatively, borenium cations have attracted attention as boron based

9
hydrogenation catalysts. The reaction of an NHC with 9–BBN results in the formation of a
stable adduct and upon hydride abstraction, a borenium cation is generated.53 The borenium
cation, [(IiPr2)(BC8H24)] [B(C6F5)4], was found to activate hydrogen in an FLP manner and
further hydrogenate a number of imines and enamines under mild conditions (Scheme 1.3.6).
Scheme 1.3.6 - FLP catalyzed hydrogenation reactions.
1.3.2 Reactivity with other small molecules
Hydrogen is not the only small molecule that FLPs can activate. The reactivity of countless
small molecules has been investigated with FLP combinations. Due to the effect of CO2 on
climate change, the reactivity and sequestration of CO2 has been a focus of scientific research
and FLP’s have been shown to be applicable in this field of chemistry. Inter- and intra-
molecular FLPs were shown to activate CO2 (Scheme 1.3.7) with the latter being rapidly
reversible at low temperatures.54 Since this initial discovery a number of FLP combinations have
been investigated in the binding of CO2.55-60 Catalytic reduction of CO2 has been achieved; Piers
et al. have reported the use of a tandem FLP/hydrosilylation catalyst using TMP and B(C6F5)3 in
the catalytic reduction of CO2 to methane with the use of silanes.61 A number of carbonyl
functionalities have also been shown to be activated by FLPs in a similar fashion to CO2.33 For
example, the greenhouse gas N2O is also activated by FLPs, resulting in the addition product
tBu3P(N2O)B(C6F5)3 (Scheme 1.3.7).62

10
Scheme 1.3.7 - CO2 and N2O activation with FLPs.
Recently, both the Erker and Stephan labs have simultaneously investigated the reactivity of
FLPs with CO. These studies have demonstrated the ability of Lewis acidic boranes to interact
with the relatively inert CO molecule. Piers borane [HB(C6F5)2] has been shown to form an
adduct with CO, and when this combination is reacted with an in situ generated intramolecular
FLP system, the CO is reduced to a formyl functionality which is side-on bound to boron
(Scheme 1.3.8).63,64 Furthermore, the intermolecular combination of tBu3P and B(C6F5)3 were
shown to react with syngas (H2 and CO mixture) resulting in the formation of an epoxyborate
which can react with both additional CO or H2 (Scheme 1.3.8).65

11
Scheme 1.3.8 - CO activation with FLPs.
The reactivity of olefins with FLPs has also been examined. Initially, ethylene was reacted with
tBu3P and B(C6F5)3, resulting in the addition product, tBu3P(CH2CH2)B(C6F5)3 (Scheme 1.3.9).66
Bulky phosphines with pendant olefins were also found to react in the presence of B(C6F5)3 and
resulted in the formation of the corresponding cyclized product.66 The Erker group has utilized
their intramolecular FLP system to effect the addition of norbornene, resulting in the exclusively
exo addition product.67 Such olefin addition chemistry has been extended to N/B FLP
systems.68,69 Mechanistic insight into these addition reactions was realized by synthesizing a
borane containing a pendant olefin. These results indicated that there was an olefin “van der
Waals” complex formed with the Lewis acidic borane in solution.70 These olefin complexes
undergo FLP addition reactions in the presence of nucleophiles, forming boron heterocyclic
products.71 Parallels can be drawn between the reactivity of olefins with FLPs to the reactivity of
alkynes with FLPs. Depending on the basicity of the Lewis base, terminal alkynes either
undergo a 1,2-addition across the C–C triple bond or a deprotonation reaction resulting in a
phosphonium alkynylborate salt (Scheme 1.3.9).72,73 Alkynes tend to be the most reactive

12
substrates for FLP chemistry and have thus been synonymous with exploring the reactivity of
new FLP systems. Sulfur Lewis bases have been found to only react with alkynes, for example,
the adduct Me2SB(C6F5)3 will add across the C–C triple bond of phenylacetylene.72 The sulfur–
boron dimer, [PhSCH2B(C6F5)2]2 can be broken in the presence of terminal and internal alkynes
to yield the addition products and a new five-membered heterocycle.74 Certain P–B adducts
which were thought to be irreversible have been shown to react in an FLP fashion with
phenylacetylene, demonstrating the tunability of FLP systems.75
Scheme 1.3.9 - Olefin and alkyne reactivity with FLPs
1.4 Alternative Lewis acids
The Lewis base used in FLP chemistry has been varied significantly and despite the variation of
bases used the anticipated FLP reactivity is still observed. This has not been the case with the
Lewis acid and the majority of FLP chemistry thus far has utilized boron Lewis acids. B(C6F5)3
is a very strong Lewis acid, commercially available, stable in solution, and its reactivity can be
monitored by both 11B and 19F NMR handles. However, there are drawbacks to B(C6F5)3 which
include its limited functional group tolerance, in addition to its instability towards air and
moisture. Many research groups are currently developing alternative Lewis acids for FLP
chemistry in an attempt to uncover more favourable and robust reactivity.
1.4.1 Aluminium Based Lewis Acids
Investigating the chemistry of aluminium Lewis acids is an obvious extension from boron.
Aluminium is one row below boron in the periodic table and possesses many of the same

13
desirable properties of boron. Generally, the chemistry reported with Al based FLPs is similar to
that of B(C6F5)3 with minor variations. The first reported use of an aluminium Lewis acid in FLP
chemistry came from our lab in 2009, and utilized Al(C6F5)3 in combination with bulky
phosphines to activate terminal alkynes.73 Shortly thereafter the chemistry of aluminium halides
in FLP chemistry was explored. The combination of PMes3 with AlX3 (X = Cl, Br, I) was shown
to activate CO2 to yield the product, Mes3P(CO2)(AlX3)2 where each oxygen in CO2 is bound to
an aluminium center (Scheme 1.4.1).76 Treatment of this complex with ammonia borane led to
rapid reduction of CO2 to methanol. Interestingly enough, this FLP combination was found to
further react with CO2, facilitating the reduction of CO2 to CO.77,78 Olefins have been shown
undergo activation in the presence of PMes3 and AlX3. When ethylene is used the expected
addition product is observed, however isobutylene results in an unanticipated C–H bond
activation to yield a bis-Al allyl species.79,80 Al(C6F5)3 was shown to be able to activate H2 with
PR3 (R = tBu, Mes) to form the salt [R3PH] [(μ-H)-(Al(C6F5)3)2].81 This salt was inactive in
catalysis, however simple alkylaluminium compounds were proven to be competent
hydrogenation catalysts for imines.82 Lastly, the activation of N2O with Al(C6F5)3 did not yield
the expected FLP addition product, instead a radical reaction occurs resulting in the formation of
a phosphorus radical which can initiate C–H activation.83
Scheme 1.4.1 - Examples of Al Lewis acids in FLP chemistry.

14
Other groups have also investigated the chemistry of Al based FLPs. Uhl and co-workers have
developed geminal P/Al FLPs systems and showed that these can activate alkynes and CO2
(Scheme 1.4.1).84 This same system was shown to form classical adducts with BH3 and NH3, but
will dehydrocouple the amine-boranes H3NBH3 and Me2HNBH3.85 Subsequent reports by Uhl et
al. have described the reactivity of N/Al and additional P/Al systems and have demonstrated
their reactivity with nitriles, carbodiimides, acetylenes, and isocyanates.86-89 Furthermore,
aluminium FLPs have found applications in materials science. Chen and co-workers have
reported the polymerization of methyl methacrylate and methylene butyrolactones into high
molecular weight polymers with a number of Lewis bases in combination with alane Lewis
acids.90,91 More recently, they have extended the scope of such polymerizations to pyridine and
oxazoline functionalized vinyl polymers by using similar methods.92
1.4.2 Carbon Based Lewis Acids
Parallels can be drawn between the trityl cation [Ph3C] and B(C6F5)3. Trityl is isoelectronic with
B(C6F5)3 and is an inherently strong Lewis acid due to the empty p-orbital at the central carbon,
as well as the positive charge that the molecule possesses. However, as mentioned earlier in this
chapter, this cation is very susceptible to nucleophilic aromatic substitution by Lewis bases,
which inhibits FLP reactivity.26,28 Tricks had to be played in order to get trityl to participate in
FLP chemistry, and indeed it was shown to be able to bind N2O with tBu3P. This was only
accomplished via a Lewis acid exchange reaction. The N2O activated complex,
[tBu3PN2OB(p-C6H4F)3], was generated and addition of [Ph3C] [B(C6F5)4] displaced the weaker
Lewis acid and furnished the trityl bound N2O complex, [tBu3PN2OCPh3] [B(C6F5)4].93
Alcarazo and co-workers took a unique approach developing carbon based Lewis acids. They
note that the trityl cation is able to delocalize its positive charge into the aromatic rings. Such an
approach was used to design allene Lewis acids which can delocalize negative charge into the
bound fluorene rings, generating an electrophilic central carbon atom. The combination of this
allene with an NHC formed an all carbogenic FLP which was shown to cleave the S–S bond in
disulfides (Scheme 1.4.2).94,95 Our group pursued a different approach to the generation of a
carbon based Lewis acid. A Ru-η6-arene complex was shown to undergo nucleophilic attack at
the ortho and para positions of the aromatic ring bound to the Ru center with PCy3.96

15
Interestingly, PMes3 and PtBu3 did not attack the aromatic ring, forming a frustrated Lewis pair.
This unique combination was able to heterolytically cleave H2 between the P Lewis base and the
aromatic C Lewis acid (Scheme 1.4.2) and the arene complex was applied in the catalytic
hydrogenation of imines. Alternatively, Ingleson and co-workers have investigated the use of
acridine derivatives as carbon Lewis acids. It was initially shown that the borenium bound
acridine, [(acridine)BCl2]+, was able to act as both a boron and carbon based Lewis acid.97
Subsequently, they were able to show that simple methylated acridine can act as a Lewis acid in
the C9 position to effect H2 activation in combination with 2,6-lutidine as a base (Scheme
1.4.2).98
Scheme 1.4.2 - Examples of carbon Lewis acids in FLP chemistry.
1.4.3 Silicon Based Lewis Acids
Silicon compounds have often been exploited for their Lewis acidity in organic
transformations.99,100 The development and isolation of free silylium cations101-104 have
generated prime candidates for applications in FLP chemistry, however this research field is still
in its infancy. This is partially due to the extreme sensitivity and reactivity of silylium cations,

16
as they possess extreme Lewis acidity105 and they are also typically less sterically encumbered.
Nonetheless, researchers have overcome these barriers and have begun to investigate the
reactivity of silylium Lewis acids in FLP chemistry. This work has been pioneered by the group
of Müller and it has been shown that the reaction between a permethylated-triphenylsilylium
cation and Mes3P under an atmosphere of hydrogen results in H2 activation, forming the silane
and phosphonium borate salt (Scheme 1.4.3).106 Shortly thereafter they extended the scope of
reactivity of this system to demonstrate CO2 activation.107 Furthermore, they recently published a
report of an all silicon FLP, where they have shown that a bulky silylene can act as a Lewis base
with a silylium cation to effect H2 activation. Furthermore, Ashley and co-workers have recently
reported that silylium-phosphine adducts can in-fact activate H2 under elevated temperatures,
indicating that even though they are strong Lewis acids, reversible adducts are still possible.108
Scheme 1.4.3 - H2 activation using a silylium Lewis acid.
1.4.4 Group 4 Metallocene Based Lewis Acids
Another extension to FLP chemistry is exploring transition metal complexes as Lewis acids.
This field of research comes with the added benefit of decades of research into the reactivity and
behaviour of transition metal complexes. Early transition metal group 4 complexes have been
previously exploited as Lewis acids for a number of transformations, in particular olefin
polymerization.109 The group of Wass has initiated the effort to apply group 4 complexes in FLP
chemistry and they reported the formation of a cationic zirconocene-phosphinoaryloxide
complex which bears a Zr Lewis acid and pendant P Lewis base.110 A weak Zr–P adduct is
observed in the complex, however the steric bulk around the phosphorus center promotes
dissociation and this system is sufficiently reactive to react H2 in an FLP fashion (Scheme 1.4.4).
This seminal work was successively expanded upon and these systems were shown to react with
a variety of substrates, including CO2, alkenes, alkynes, and cyclic ethers resulting in the FLP

17
activated products.111,112 The Wass group has also extended this chemistry to titanocene
derivatives.113 The Erker group has developed a geminal P/Zr system (Scheme 1.4.4), which is
generated by an unusual carbozirconation reaction between a methyl zirconocene cation and a
phosphorus alkyne.114 This system was also found to react as an FLP, cleanly activating CO2,
isocyanates, aryl azides, enones and ynones. Activation of H2 does occur in this system,
however it leads to a disproportionation reaction and formation of a zirconium hydride
species.115 Furthermore, while investigating the reactivity of chelating phosphinoamine ligands
with Hf, our group discovered that the bound phosphorus center is labile enough to activate CO2
in an FLP fashion with the Hf center acting as the Lewis acid (Scheme 1.4.4).116
Scheme 1.4.4 - Group 4 transition metal Lewis acids for FLP chemistry. All cations shown have
B(C6F5)4 anions.
1.5 Scope of Thesis
As will be described throughout this dissertation, the objective of my graduate work was to
expand the scope of FLP chemistry and work to develop novel Lewis acids. This was achieved

18
by exploring the reactivity of previously unexplored fluorophosphonium Lewis acids in FLP
chemistry. A wide range of chemistry is investigated, including taking a supramolecular
approach to imparting FLP behaviour. This chemistry led to the development of a novel
hydrogenation catalyst based on a never before explored oxygen/boron system described in
Chapter 2. Inert carbon−fluorine activation is investigated with classical FLP systems in Chapter
3 and Chapter 4 uncovers the reactivity of FLP systems with the highly reactive compound,
XeF2. Observations in this chapter led to the discovery of highly electrophilic
fluorophosphonium cations. The reactivity of these new Lewis acids is described in Chapter 5.
Some of the reactivity draws parallels with B(C6F5)3, in particular nucleophilic aromatic
substitution and CO2 activation. In other cases the reactivity deviates from that of B(C6F5)3 in
part to the greater Lewis acidity of these fluorophosphonium Lewis acids. This reactivity
observed includes catalytic hydrodefluorination, hydrosilylation of olefins, dehydrocoupling of
molecules containing an E–H functionality (E = R2N, RS, RO) with silanes and subsequent
transfer hydrogenation to olefins, as well as the reactivity of H2 with fluorophosphonium cations.
Utilizing mechanically interlocked molecules as FLPs in Chapter 2 was undertaken in
collaboration with Professor Stephen Loeb at the University of Windsor. The synthesis and
characterization of the [2]rotaxanes utilized were performed at the University of Windsor by
Dr. Kelong Zhu and Dr. V. Nicholas Vukotic, while the FLP investigations were undertaken by
myself. All work done in Chapters 3 and 4, with exception of elemental analysis and the
computational study (Dr. Roman Dobrovetsky), was performed by myself. The work in Chapter
5 was done in collaboration with three postdoctoral fellows in the laboratory, Dr. Lindsay
Hounjet, Dr. Manuel Pérez and Dr. Roman Dobrovetsky. All chemistry reported in that chapter
were devised and executed collaboratively with the exception of the hydrodefluorination
chemistry, which was undertaken by myself.
Portions of each chapter have been published or have been drafted at the time of writing:
Chapter 2: 1) Caputo, C. B.; Zhu, K.; Vukotic, V. N.; Loeb S. J.; Stephan, D. W. “Heterolytic
Activation of H2 Using a Mechanically Interlocked Molecule as a Frustrated Lewis Base.”
Angewandte Chemie International Edition, 2013, 52, 960-963. 2) Hounjet, L. J.; Bannwarth, C.;
Garon, C. N.; Caputo, C. B.; Grimme, S.; Stephan, D. W. “Ether/B(C6F5)3 Combinations

19
Function as Hydrogenation Catalysts.” Angewandte Chemie International Edition, 2013, 52,
7492-7495.
Chapter 3: 1) Caputo, C. B.; Stephan D. W. “Activation of Alkyl C–F Bonds by B(C6F5)3:
Stoichiometric and Catalytic Transformations.” Organometallics, 2012, 31, 27-30.
Chapter 4: 1) Caputo, C. B.; Hounjet, L. J.; Dobrovetsky, R.; Stephan, D. W. “Lewis Acidity of
Organofluorophosphonium Salts: Hydrodefluorination by a Saturated Acceptor.” Science, 2013,
341, 1374-1377. 2) Caputo, C. B.; Dobrovetsky, R.; Hounjet L. J.; Stephan, D. W. “Reactions of
Frustrated Lewis Pairs with XeF2: Uncovering the Lewis acidity of Fluorophosphonium Cations”
Drafted.
Chapter 5: 1) Hounjet, L. J.; Caputo, C. B.; Stephan D. W. “The Lewis Acidity of
Fluorophosphonium Salts: Access to Mixed Valent Phosphorus(III)/(V) Species.” Dalton
Transactions, 2013, 42, 2629-2635. 2) Hounjet L. J.; Caputo, C. B.; Stephan D. W.
“Phosphorous as a Lewis Acid: CO2 Sequestration with Amidophosphoranes.” Angewandte
Chemie International Edition, 2012, 51, 4714-4717. 3) Caputo, C. B.; Hounjet, L. J.;
Dobrovetsky, R.; Stephan, D. W. “Lewis Acidity of Organofluorophosphonium Salts:
Hydrodefluorination by a Saturated Acceptor.” Science, 2013, 341, 1374-1377. 4) Pérez, M.;
Hounjet, L. J.; Caputo, C. B.; Dobrovetsky, R.; Stephan D. W. “Olefin Isomerization and
Hydrosilylation Catalysis by Lewis Acidic Organofluorophosphonium Salts.” Journal of the
American Chemical Society, 2013, 135, 18308-18310. 5) Pérez, M.; Caputo, C. B.; Dobrovetsky,
R.; Stephan, D. W. “Olefin Transfer Hydrogenation via Electrophilic Phosphonium Catalysis of
Heteroatom-Silane Dehydrocoupling.” Proceedings of the National Academy of Science, 2014,
111, 10917-10921.
1.6 References
(1) Smil, V. Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World
Food Production; MIT Press, 2004.
(2) Decady, Y.; Greenberg, L. Statistics Canada 2014.
(3) Edenhofer, O.; Pichs-Madruga, R.; Sokona, Y.; Farahani, E.; Kadner, S.; Seyboth, K.;
Adler, A.; Baum, I.; Brunner, S.; Eickemeier, P.; Kriemann, B.; Savolainen, J.; Schlömer, S.; von
Stechow, C.; Zwickel, T.; Minx, J. C. IPCC, 2014: Climate Change 2014: Mitigation of Climate
Change. Contribution of working Group III to the Firth Assessment Report of the

20
Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, United
Kingdom and New York, NY, USA, 2014.
(4) Loulergue, L.; Schilt, A.; Spahni, R.; Masson-Delmotte, V.; Blunier, T.; Lemieux, B.;
Barnola, J.-M.; Raynaud, D.; Stocker, T. F.; Chappellaz, J. Nature 2008, 453, 383-386.
(5) Luthi, D.; Le Floch, M.; Bereiter, B.; Blunier, T.; Barnola, J.-M.; Siegenthaler, U.;
Raynaud, D.; Jouzel, J.; Fischer, H.; Kawamura, K.; Stocker, T. F. Nature 2008, 453, 379-382.
(6) Anastas, P. T.; Warner, J. C. Green Chemistry: Theory and Practice; Oxford University
Press, 1998.
(7) Auer, E.; Freund, A.; Pietsch, J.; Tacke, T. Applied Catalysis A: General 1998, 173, 259-
271.
(8) Tondreau, A. M.; Atienza, C. C. H.; Weller, K. J.; Nye, S. A.; Lewis, K. M.; Delis, J. G.
P.; Chirik, P. J. Science 2012, 335, 567-570.
(9) Zuo, W.; Lough, A. J.; Li, Y. F.; Morris, R. H. Science 2013, 342, 1080-1083.
(10) Baker, R. T.; Gordon, J. C.; Hamilton, C. W.; Henson, N. J.; Lin, P.-H.; Maguire, S.;
Murugesu, M.; Scott, B. L.; Smythe, N. C. Journal of the American Chemical Society 2012, 134,
5598-5609.
(11) Yin, Q.; Tan, J. M.; Besson, C.; Geletii, Y. V.; Musaev, D. G.; Kuznetsov, A. E.; Luo, Z.;
Hardcastle, K. I.; Hill, C. L. Science 2010, 328, 342-345.
(12) Reece, S. Y.; Hamel, J. A.; Sung, K.; Jarvi, T. D.; Esswein, A. J.; Pijpers, J. J. H.;
Nocera, D. G. Science 2011, 334, 645-648.
(13) Helm, M. L.; Stewart, M. P.; Bullock, R. M.; DuBois, M. R.; DuBois, D. L. Science
2011, 333, 863-866.
(14) Enthaler, S.; Junge, K.; Beller, M. Angewandte Chemie International Edition 2008, 47,
3317-3321.
(15) Lewis, G. N. Journal of the American Chemical Society 1916, 38, 762-785.
(16) Lewis, G. N. Valence and the Structure of Atoms and Molecules; Chemical Catalogue
Company: New York, 1923.
(17) Groves, J. K. Chemical Society Reviews 1972, 1, 73-97.
(18) Rueping, M.; Nachtsheim, B. J. Beilstein Journal of Organic Chemistry 2010, 6, 6.
(19) Tolman, C. A. Chemical Reviews 1977, 77, 313-348.
(20) Tang, W.; Zhang, X. Chemical Reviews 2003, 103, 3029-3070.

21
(21) Denmark, S. E.; Beutner, G. L. Angewandte Chemie International Edition 2008, 47,
1560-1638.
(22) Brown, H. C.; Schlesinger, H. I.; Cardon, S. Z. Journal of the American Chemical Society
1942, 64, 325-329.
(23) Wittig, G.; Rückert, A. Justus Liebigs Annalen der Chemie 1950, 566, 101-113.
(24) Wittig, G.; Benz, E. Chemische Berichte 1959, 92, 1999-2013.
(25) Tochtermann, W. Angewandte Chemie International Edition in English 1966, 5, 351-371.
(26) Döring, S.; Erker, G.; Fröhlich, R.; Meyer, O.; Bergander, K. Organometallics 1998, 17,
2183-2187.
(27) Welch, G. C.; Cabrera, L.; Chase, P. A.; Hollink, E.; Masuda, J. D.; Wei, P.; Stephan, D.
W. Dalton Transactions 2007, 3407-3414.
(28) Cabrera, L.; Welch, G. C.; Masuda, J. D.; Wei, P.; Stephan, D. W. Inorganica Chimica
Acta 2006, 359, 3066-3071.
(29) Welch, G. C.; Juan, R. R. S.; Masuda, J. D.; Stephan, D. W. Science 2006, 314, 1124-
1126.
(30) Welch, G. C.; Stephan, D. W. Journal of the American Chemical Society 2007, 129,
1880-1881.
(31) Spies, P.; Erker, G.; Kehr, G.; Bergander, K.; Frohlich, R.; Grimme, S.; Stephan, D. W.
Chemical Communications 2007, 5072-5074.
(32) Wang, X.; Kehr, G.; Daniliuc, C. G.; Erker, G. Journal of the American Chemical Society
2014, 136, 3293-3303.
(33) Stute, A.; Kehr, G.; Daniliuc, C. G.; Frohlich, R.; Erker, G. Dalton Transactions 2013,
42, 4487-4499.
(34) Ekkert, O.; Kehr, G.; Fröhlich, R.; Erker, G. Journal of the American Chemical Society
2011, 133, 4610-4616.
(35) Wang, H.; Frohlich, R.; Kehr, G.; Erker, G. Chemical Communications 2008, 5966-5968.
(36) Frey, G. D.; Lavallo, V.; Donnadieu, B.; Schoeller, W. W.; Bertrand, G. Science 2007,
316, 439-441.
(37) Chase, P. A.; Stephan, D. W. Angewandte Chemie International Edition 2008, 47, 7433-
7437.
(38) Holschumacher, D.; Bannenberg, T.; Hrib, C. G.; Jones, P. G.; Tamm, M. Angewandte
Chemie International Edition 2008, 47, 7428-7432.

22
(39) Kolychev, E. L.; Bannenberg, T.; Freytag, M.; Daniliuc, C. G.; Jones, P. G.; Tamm, M.
Chemistry – A European Journal 2012, 18, 16938-16946.
(40) Runyon, J. W.; Steinhof, O.; Dias, H. V. R.; Calabrese, J. C.; Marshall, W. J.; Arduengo,
A. J. Australian Journal of Chemistry 2011, 64, 1165-1172.
(41) Geier, S. J.; Stephan, D. W. Journal of the American Chemical Society 2009, 131, 3476-
3477.
(42) Caputo, C. B.; Geier, S. J.; Winkelhaus, D.; Mitzel, N. W.; Vukotic, V. N.; Loeb, S. J.;
Stephan, D. W. Dalton Transactions 2012, 41, 2131-2139.
(43) Geier, S. J.; Gille, A. L.; Gilbert, T. M.; Stephan, D. W. Inorganic Chemistry 2009, 48,
10466-10474.
(44) Geier, S. J.; Chase, P. A.; Stephan, D. W. Chemical Communications 2010, 46, 4884-
4886.
(45) Stephan, D. W.; Greenberg, S.; Graham, T. W.; Chase, P.; Hastie, J. J.; Geier, S. J.;
Farrell, J. M.; Brown, C. C.; Heiden, Z. M.; Welch, G. C.; Ullrich, M. Inorganic Chemistry
2011, 50, 12338-12348.
(46) Chase, P. A.; Welch, G. C.; Jurca, T.; Stephan, D. W. Angewandte Chemie International
Edition 2007, 46, 8050-8053.
(47) Chase, P. A.; Jurca, T.; Stephan, D. W. Chemical Communications 2008, 1701-1703.
(48) Chen, D.; Klankermayer, J. In Frustrated Lewis Pairs II; Erker, G., Stephan, D. W., Eds.;
Springer Berlin Heidelberg: 2013; Vol. 334, p 1-26.
(49) Mahdi, T.; Heiden, Z. M.; Grimme, S.; Stephan, D. W. Journal of the American
Chemical Society 2012, 134, 4088-4091.
(50) Greb, L.; Oña-Burgos, P.; Schirmer, B.; Grimme, S.; Stephan, D. W.; Paradies, J.
Angewandte Chemie International Edition 2012, 51, 10164-10168.
(51) Greb, L.; Tussing, S.; Schirmer, B.; Ona-Burgos, P.; Kaupmees, K.; Lokov, M.; Leito, I.;
Grimme, S.; Paradies, J. Chemical Science 2013, 4, 2788-2796.
(52) Greb, L.; Daniliuc, C.-G.; Bergander, K.; Paradies, J. Angewandte Chemie International
Edition 2013, 52, 5876-5879.
(53) Farrell, J. M.; Hatnean, J. A.; Stephan, D. W. Journal of the American Chemical Society
2012, 134, 15728-15731.
(54) Mömming, C. M.; Otten, E.; Kehr, G.; Fröhlich, R.; Grimme, S.; Stephan, D. W.; Erker,
G. Angewandte Chemie International Edition 2009, 48, 6643-6646.

23
(55) Courtemanche, M.-A.; Légaré, M.-A.; Maron, L.; Fontaine, F.-G. Journal of the
American Chemical Society 2014, 136, 10708-10717.
(56) Lu, Z.; Wang, Y.; Liu, J.; Lin, Y.-j.; Li, Z. H.; Wang, H. Organometallics 2013, 32,
6753-6758.
(57) Sgro, M. J.; Domer, J.; Stephan, D. W. Chemical Communications 2012, 48, 7253-7255.
(58) Stephan, D. W.; Erker, G. Chemical Science 2014, 5, 2625-2641.
(59) Wang, T.; Stephan, D. W. Chemistry – A European Journal 2014, 20, 3036-3039.
(60) Boone, M. P.; Stephan, D. W. Organometallics 2013, 33, 387-393.
(61) Berkefeld, A.; Piers, W. E.; Parvez, M. Journal of the American Chemical Society 2010,
132, 10660-10661.
(62) Otten, E.; Neu, R. C.; Stephan, D. W. Journal of the American Chemical Society 2009,
131, 9918-9919.
(63) Sajid, M.; Elmer, L. M.; Rosorius, C.; Daniliuc, C. G.; Grimme, S.; Kehr, G.; Erker, G.
Angewandte Chemie International Edition 2013, 52, 2243-2246.
(64) Sajid, M.; Kehr, G.; Daniliuc, C. G.; Erker, G. Angewandte Chemie International Edition
2014, 53, 1118-1121.
(65) Dobrovetsky, R.; Stephan, D. W. Journal of the American Chemical Society 2013, 135,
4974-4977.
(66) McCahill, J. S. J.; Welch, G. C.; Stephan, D. W. Angewandte Chemie International
Edition 2007, 46, 4968-4971.
(67) Mömming, C. M.; Frömel, S.; Kehr, G.; Fröhlich, R.; Grimme, S.; Erker, G. Journal of
the American Chemical Society 2009, 131, 12280-12289.
(68) Sortais, J.-B.; Voss, T.; Kehr, G.; Frohlich, R.; Erker, G. Chemical Communications
2009, 7417-7418.
(69) Voss, T.; Chen, C.; Kehr, G.; Nauha, E.; Erker, G.; Stephan, D. W. Chemistry – A
European Journal 2010, 16, 3005-3008.
(70) Zhao, X.; Stephan, D. W. Journal of the American Chemical Society 2011, 133, 12448-
12450.
(71) Zhao, X.; Stephan, D. W. Chemical Science 2012, 3, 2123-2132.
(72) Dureen, M. A.; Brown, C. C.; Stephan, D. W. Organometallics 2010, 29, 6594-6607.
(73) Dureen, M. A.; Stephan, D. W. Journal of the American Chemical Society 2009, 131,
8396-8397.

24
(74) Tanur, C. A.; Stephan, D. W. Organometallics 2011, 30, 3652-3657.
(75) Caputo, C. B.; Geier, S. J.; Ouyang, E. Y.; Kreitner, C.; Stephan, D. W. Dalton
Transactions 2012, 41, 237-242.
(76) Ménard, G.; Stephan, D. W. Journal of the American Chemical Society 2010, 132, 1796-
1797.
(77) Ménard, G.; Stephan, D. W. Angewandte Chemie International Edition 2011, 50, 8396-
8399.
(78) Ménard, G.; Gilbert, T. M.; Hatnean, J. A.; Kraft, A.; Krossing, I.; Stephan, D. W.
Organometallics 2013, 32, 4416-4422.
(79) Ménard, G.; Stephan, D. W. Angewandte Chemie International Edition 2012, 51, 4409-
4412.
(80) Ménard, G.; Tran, L.; McCahill, J. S. J.; Lough, A. J.; Stephan, D. W. Organometallics
2013, 32, 6759-6763.
(81) Ménard, G.; Stephan, D. W. Angewandte Chemie International Edition 2012, 51, 8272-
8275.
(82) Hatnean, J. A.; Thomson, J. W.; Chase, P. A.; Stephan, D. W. Chemical Communications
2014, 50, 301-303.
(83) Ménard, G.; Hatnean, J. A.; Cowley, H. J.; Lough, A. J.; Rawson, J. M.; Stephan, D. W.
Journal of the American Chemical Society 2013, 135, 6446-6449.
(84) Appelt, C.; Westenberg, H.; Bertini, F.; Ehlers, A. W.; Slootweg, J. C.; Lammertsma, K.;
Uhl, W. Angewandte Chemie International Edition 2011, 50, 3925-3928.
(85) Appelt, C.; Slootweg, J. C.; Lammertsma, K.; Uhl, W. Angewandte Chemie International
Edition 2013, 52, 4256-4259.
(86) Roters, S.; Appelt, C.; Westenberg, H.; Hepp, A.; Slootweg, J. C.; Lammertsma, K.; Uhl,
W. Dalton Transactions 2012, 41, 9033-9045.
(87) Roters, S.; Hepp, A.; Slootweg, J. C.; Lammertsma, K.; Uhl, W. Chemical
Communications 2012, 48, 9616-9618.
(88) Holtrichter-Rößmann, T.; Isermann, J.; Rösener, C.; Cramer, B.; Daniliuc, C.-G.;
Kösters, J.; Letzel, M.; Würthwein, E.-U.; Uhl, W. Angewandte Chemie International Edition
2013, 52, 7135-7138.
(89) Holtrichter-Rößmann, T.; Rösener, C.; Hellmann, J.; Uhl, W.; Würthwein, E.-U.;
Fröhlich, R.; Wibbeling, B. Organometallics 2012, 31, 3272-3283.

25
(90) Chen, E. X. In Frustrated Lewis Pairs II; Erker, G., Stephan, D. W., Eds.; Springer
Berlin Heidelberg: 2013; Vol. 334, p 239-260.
(91) Zhang, Y.; Miyake, G. M.; Chen, E. Y. X. Angewandte Chemie International Edition
2010, 49, 10158-10162.
(92) He, J.; Zhang, Y.; Chen, E. Y. X. Synlett 2014, 25, 1534-1538.
(93) Neu, R. C.; Otten, E.; Lough, A.; Stephan, D. W. Chemical Science 2011, 2, 170-176.
(94) Inés, B.; Holle, S.; Goddard, R.; Alcarazo, M. Angewandte Chemie International Edition
2010, 49, 8389-8391.
(95) Palomas, D.; Holle, S.; Ines, B.; Bruns, H.; Goddard, R.; Alcarazo, M. Dalton
Transactions 2012, 41, 9073-9082.
(96) Boone, M. P.; Stephan, D. W. Journal of the American Chemical Society 2013, 135,
8508-8511.
(97) Clark, E. R.; Ingleson, M. J. Organometallics 2013, 32, 6712-6717.
(98) Clark, E. R.; Ingleson, M. J. Angewandte Chemie International Edition 2014, 53, 11306-
11309.
(99) Rendler, S.; Oestreich, M. Angewandte Chemie International Edition 2008, 47, 5997-
6000.
(100) Dilman, A. D.; Ioffe, S. L. Chemical Reviews 2003, 103, 733-772.
(101) Lambert, J. B.; Zhang, S. Journal of the Chemical Society, Chemical Communications
1993, 383-384.
(102) Lambert, J. B.; Zhang, S.; Ciro, S. M. Organometallics 1994, 13, 2430-2443.
(103) Nava, M.; Reed, C. A. Organometallics 2011, 30, 4798-4800.
(104) Reed, C. A.; Xie, Z.; Bau, R.; Benesi, A. Science 1993, 262, 402-404.
(105) Gusev, D. G.; Ozerov, O. V. Chemistry – A European Journal 2011, 17, 634-640.
(106) Schäfer, A.; Reißmann, M.; Schäfer, A.; Saak, W.; Haase, D.; Müller, T. Angewandte
Chemie International Edition 2011, 50, 12636-12638.
(107) Reißmann, M.; Schäfer, A.; Jung, S.; Müller, T. Organometallics 2013, 32, 6736-6744.
(108) Herrington, T. J.; Ward, B. J.; Doyle, L. R.; McDermott, J.; White, A. J. P.; Hunt, P. A.;
Ashley, A. E. Chemical Communications 2014, 50, 12753-12756.
(109) Yamamoto, H. Lewis Acids in Organic Synthesis; Wiley-VCH, 2000.
(110) Chapman, A. M.; Haddow, M. F.; Wass, D. F. Journal of the American Chemical Society
2011, 133, 8826-8829.

26
(111) Chapman, A. M.; Haddow, M. F.; Wass, D. F. European Journal of Inorganic Chemistry
2012, 2012, 1546-1554.
(112) Chapman, A. M.; Haddow, M. F.; Wass, D. F. Journal of the American Chemical Society
2011, 133, 18463-18478.
(113) Chapman, A. M.; Wass, D. F. Dalton Transactions 2012, 41, 9067-9072.
(114) Xu, X.; Frohlich, R.; Daniliuc, C. G.; Kehr, G.; Erker, G. Chemical Communications
2012, 48, 6109-6111.
(115) Xu, X.; Kehr, G.; Daniliuc, C. G.; Erker, G. Journal of the American Chemical Society
2013, 135, 6465-6476.
(116) Sgro, M. J.; Stephan, D. W. Chemical Communications 2013, 49, 2610-2612.

27
Chapter 2 Using Mechanically Interlocked Molecules to Impart FLP
Reactivity onto Sterically Unencumbered Bases
2
2.1.1 The Border of Classical and Frustrated Lewis Pair Chemistry
The systems developed at the onset of frustrated Lewis pair chemistry required the Lewis acid
and base to not interact in solution,1,2 which limited the compounds that could be used in this
chemistry. Classical Lewis acid-base adducts were thought to be a dead end to the reactivity and
had to be avoided at all cost. This notion began to change with the discovery by the Erker lab of
an intramolecular ethyl linked P/B FLP system.3 This molecule formed a four-membered P–B
heterocycle but still activated H2 gas under ambient conditions (Scheme 1.6.1). Shortly
thereafter our group began to investigate the reactivity of pyridine Lewis bases and their
propensity to react in an FLP fashion. It was discovered that the reaction of B(C6F5)3 with
2,6-dimethylpyridine results in an equilibrium between the classical adduct and a frustrated
system.4 This result is reminiscent of that from H.C. Brown in 1942, where they report that
2,6-dimethylpyridine does not form an adduct with BMe3.5 This reactivity was attributed to the
steric constraints around the Lewis acid and base, in fact, this system could be thought of as the
first FLP. Unlike the combination of 2,6-dimethylpyridine and BMe3, the use of
2,6-dimethylpyridine with B(C6F5)3 activates hydrogen to form the corresponding pyridinium
hydridoborate salt (Scheme 1.6.1). Four coordinate boron amidinates have also been shown to
react as masked FLPs allowing insertion of small molecules, such as CO2 or CO, into the B–N
bond (Scheme 1.6.1).6 This type of reactivity has been extended to B–S dimers, the compound
(PhSCH2B(C6F5)2)2 has been shown to open in the presence of alkynes to yield the FLP addition
product across the alkyne.7 The group of Repo have utilized this concept to develop an arene
bridged aminoborane system, where the B–N adduct can be broken by heating under an
atmosphere of H2, generating an effective catalyst for the reduction of internal alkynes to
cis-alkenes.8 These results show that the line between classical and frustrated Lewis pairs is not
so absolute and compounds that form adducts can still exhibit FLP reactivity.

28
Scheme 1.6.1 - Examples of Lewis acid base adducts which exhibit FLP reactivity.
2.1.2 A Brief Introduction to [2]Rotaxanes
A rotaxane is a mechanically interlocked molecule (MIM) consisting of a linear axle portion and
a macrocyclic wheel. The macrocycle is unable to slide off of the axle due to sterically
encumbering stopper groups at the ends of the axle (Scheme 1.6.2). There are no covalent bonds
between the two components and this mechanical interlocking is best described as how keys are
hung in a keychain. The keys are not physically attached to the ring of the keychain, but they
cannot be removed without breaking the ring.9 In the absence of bulky stopping groups, the
complexation of the axle and macrocycle is known as a pseudorotaxane (Scheme 1.6.2). The [2]
in [2]rotaxanes represents the number of components in the interlocked molecules; for example,
a rotaxane with one axle and two macrocycles would be referred to as a [3]rotaxane.
Scheme 1.6.2 - Depiction of a [2]pseudorotaxane and [2]rotaxane.

29
Interlocked molecules were first reported in 1964 by Schill,10 and the first reports of the
formation of a [2]rotaxane followed shortly thereafter in 1967.11 First synthesized by repeatedly
passing a solution of the axle and capping group across a resin with bound macrocycle.11 This
procedure only resulted in 6% yield of the interlocked molecule. The synthetic methods to
generate MIMs have improved significantly since this initial finding and a number of template
syntheses have been developed,12 predominantly taking advantage of π–π stacking interactions,13
hydrogen bonding,14 and ion pairing interactions,15 amongst others.16 There are a number of
ways to synthesize [2]rotaxanes, two examples of which are shown in Scheme 1.6.3. The first
involves the formation of a [2]pseudorotaxane followed by the addition of a stopper group in
order to lock the macrocycle in place. The second method utilizes a pre-capped axle which
interacts with a macrocycle precursor and upon a ring closing reaction the [2]rotaxane is formed.
Scheme 1.6.3 - Two general [2]rotaxane synthetic routes using a [2]pseudorotaxane (top) and a
ring closing reaction (bottom).
The macrocycle can undergo both rotational and translational movement because it is not
covalently bound to the axle. These unique motions make [2]rotaxanes and other related MIMs
ideal candidates for molecular machines.17 The translational motion has already been exploited
in numerous systems, including molecular shuttles,18,19 elevators,20 and ratchets.21 More
recently, [2]rotaxanes were integrated into a metal-organic framework and the rotational motion

30
of the macrocycle could be observed in the solid state.22 With all of these applications, MIMs
had yet to be investigated in FLP chemistry and those results will be discussed in this chapter.
2.2 Results and Discussion
2.2.1 A Supramolecular Approach to Steric Frustration
The steric bulk which inhibits Lewis acid-base adduct formation and allows for “frustration” to
occur has always been directly bound to the Lewis acidic or basic atoms.23 The phosphines,1,2
amines24-28 and boranes23 that have been used to elicit FLP reactivity all contain either bulky
alkyl or aryl substituents directly bonded to the heteroatom. Without this steric bulk restricting
access to the lone pair on the Lewis base or the vacant orbital on the Lewis acid, adduct
formation could not be avoided, reducing the likelihood of FLP reactivity. Since this
requirement can be synthetically challenging, and thus somewhat restrictive, an alternate strategy
to sterically encumber Lewis bases was sought. One potential avenue to restrict access to the
reactive center is to bury it inside some sort of cavity to preclude access from other reagents.
This strategy parallels that of Nature utilizing complex proteins to control the reactivity and
selectivity of enzymes.29,30 In an attempt to achieve this synthetically, a collaboration with the
Loeb lab from the University of Windsor was undertaken with the goal of encapsulating
sterically unencumbered secondary amines within a MIM13,31 such as a [2]rotaxane.32-34 This
would convert simple Lewis bases into bulky Lewis bases for FLP chemistry without any
covalent modification.
Amines have long been known to form strong adducts with boranes, one of the most famous
examples being ammonia borane, H3NBH3.35 A number of recent results have shown that
B(C6F5)3 adducts with amines can exhibit FLP reactivity (vide supra).4 We sought to determine
whether or not benzyl substituents bound to a nitrogen centre would be sterically bulky enough
to preclude adduct formation with B(C6F5)3. Two amine bases were chosen to test this
hypothesis, a dibenzylamine derivative (2-1) and a benzyl aniline derivative (2-2), the prior
being previously reported in literature.36 Compound 2-1 was reacted with a stoichiometric
amount of B(C6F5)3 in CH2Cl2 (Scheme 2.2.1). The reaction was allowed to stir for 2 h before
removing the solvent in vacuo and washing the remaining residue with hexanes to remove any
starting material. The product of this reaction (2-3) was isolated in 95% yield. The 1H NMR

31
spectrum of 2-3 shows two distinct benzyl CH2 resonances: a doublet resonance is seen at
4.55 ppm, with a 3JHH = 14 Hz and a multiplet resonance at 3.52 ppm. This splitting indicates
dissymmetry around N atom due to B−N bond formation. The 11B NMR spectrum shows a
broad singlet resonance at –3.51 ppm, indicating a 4-coordinate borate species and the 19F NMR
spectrum shows 8 sharp peaks corresponding to the pentafluorophenyl rings. This indicates the
locked conformation of the pentafluorophenyl rings, indicating restricted rotation around the
B−N bond. This data supports the formulation of 2-3 to be the adduct between B(C6F5)3 and 2-1.
A small side reaction occurred in this reaction (< 5%) where the α-hydride adjacent to the
nitrogen atom is abstracted by B(C6F5)3, forming the iminium hydridoborate salt. This is
reminiscent of other reactivity shown between amines and B(C6F5)3.37,38
Scheme 2.2.1 - Reactions of 2-1 and 2-2 with B(C6F5)3.
The benzyl aniline derivative 2-2 was prepared from the reaction of 3,5-dimethylaniline with
3,5-dimethylbenzylbromide. 2-2 was subsequently reacted with a stoichiometric amount of
B(C6F5)3 in CH2Cl2 (Scheme 2.2.1) under identical conditions to 2-1. The product, 2-4, was
isolated in 82% yield. The 1H NMR spectra of 2-4 showed a drastic change from the free
aniline, the NH resonance is shifted downfield to 7.67 ppm from 3.97 ppm in 2-2 and a distinct
splitting of the benzyl CH2 group occurs from a solitary resonance at 4.20 ppm into two sets of
resonances of equal integration: a doublet at 4.76 ppm with a 2JHH = 14 Hz and a triplet at

32
4.15 ppm with a 2JHH = 12 Hz. These results show that the benzylic protons are diastereotopic
and are indicative of B–N bond formation. The 11B NMR spectrum of 2-4 shows a broad singlet
at −2.50 ppm, consistent with B–N bond formation.39,40 Three broad resonances are observed in
the 19F NMR spectrum at –128.66 ppm, −158.32 ppm and –164.70 ppm, corresponding to the
ortho, para and meta fluorine atoms of the pentafluorophenyl rings. The broad resonances can
be attributed to restricted rotation around the B–N bond, resulting from the steric congestion
created by the large substituents bound to both the B and N atoms. The meta–para gap in 2-4 is
6.38 ppm, indicating strong adduct formation has occurred. Upon cooling to 233 K, the 19F
NMR spectrum became resolved to the extent that 13 of the 15 individual fluorine peaks could
be observed, there appears to be two peaks which overlap (Figure 2.2.1). This data confirms the
formulation of 2-4 to be the adduct between 2-2 and B(C6F5)3.
Figure 2.2.1 - Low temperature 19F NMR spectrum of 2-4.

33
As mentioned in Section 2.1.1. it has been shown a number of times that adducts of Lewis acids
and bases can still exhibit FLP reactivity and in order to ensure that these adducts are
irreversible, FLP activation reactions were attempted with both 2-3 and 2-4 (Scheme 2.2.2). The
activation of phenylacetylene was tested with 2-3 to remove the possibility of misleading results
due to the noted α-hydride abstraction and borohydride formation with 2-3. Phenylacetylene has
already been shown to be activated under facile conditions using both classical and frustrated
Lewis acid-base pairs.7,41 An excess of phenylacetylene was added to a CD2Cl2 solution of 2-3.
No indication of alkyne activation was observed, indicating the irreversible nature of this adduct.
The adduct 2-4 was dissolved in CD2Cl2 or d8-toluene and exposed to 4 atmospheres of H2 in a
J-Young NMR tube. At room temperature and upon heating to 100 °C for 24 h resulted in no
evidence for hydrogen activation, confirming the irreversible nature of this adduct.
Scheme 2.2.2 - Reactions probing the irreversible nature of adducts 2-3 and 2-4.
The sterically unencumbered amines (2-1 and 2-2) were then incorporated into a [2]rotaxane in
an attempt to induce FLP reactivity. The amines act as the axles of the rotaxane molecules and
are surrounded by a crown ether macrocyclic ring. The corresponding rotaxane of 2-1 has been
previously reported,42,43 and so was an ideal target for the initial investigation of this strategy to
protect the nitrogen center from forming an adduct with B(C6F5)3 (Scheme 2.2.3). The methoxy-
substituents bound to the aromatic rings provide sufficient steric bulk to prevent the macrocycle

34
from falling off of the axle. To test the ability of the crown ether in preventing adduct formation,
the previously reported rotaxane [2-124C6] was combined with one equivalent of B(C6F5)3 in
CD2Cl2. The 1H NMR spectrum of this reaction indicated that multiple reactions occurred and
unexpectedly, the major reaction product is the α-hydride abstraction by B(C6F5)3 from the
benzylic position. A typical imine CH resonance is observed as a doublet at 9.29 ppm, with a
3JHH = 17 Hz. This formulation is corroborated in the 11B NMR spectrum as a doublet
borohydride resonance is observed at –25.5 ppm with a 1JBH = 96 Hz. Heating this mixture
under an atmosphere of H2 or D2 did not result in the FLP activated product. The increase in the
α-hydride abstraction product indicates that the crown ether is indeed providing the required
steric encumbrance to prevent B–N bond formation, but not large enough to prevent the observed
α-hydride abstraction from the benzylic position.
Scheme 2.2.3 - The reaction of [2-124C6] with B(C6F5)3.
After ruling out the FLP reactivity of [2-124C6], we turned our attention to the aniline 2-2.
Scheme 2-2-4 outlines the synthesis of two different [2]rotaxanes using 2-2, [2-224C6] and
[2-222C6]. The close proximity of the xylyl groups to the nitrogen center should restrict the
translational motion of the macrocyclic wheel, sterically shielding the N atom. The difference
between these two rotaxanes is the size of the macrocyclic wheels, with hopes of showing the
smaller crown ether can better protect the nitrogen atom. The key feature of the synthesis
utilizes a ring-closing metathesis reaction enabled by Grubbs’ first-generation catalyst.42 This
ring closing occurs while hydrogen bonding and ion-dipole templating interactions between the
protonated axle and the crown ether oxygen atoms hold the rotaxane components in close
proximity, favouring [2]rotaxane formation. Subsequent reduction of the olefin and
neutralization yielded the [2]rotaxanes, [2-224C6] and [2-222C6], in moderate yields
(Scheme 2.2.4).

35
Scheme 2.2.4 - The synthesis of [2-224C6] (left) and [2-222C6] (right).
The 1H NMR spectra of [2-224C6] and [2-222C6] show very significant downfield shifts for
the NH protons as compared to 2-2 due to NH···O hydrogen bonding between the NH group and
crown ether oxygen atoms. The NH resonance for 2-1 is observed at 3.96 ppm but shifts to 5.12
and 5.61 ppm for [2-224C6] and [2-222C6], respectively. The benzyl and the ortho-
resonances are also affected by rotaxane formation as these protons are involved in weaker
CH···O interactions. It is noted that resonances shift to a greater extent in [2-222C6]
compared to [2-224C6] and can be attributed to more significant hydrogen-bonding between
the amine and the smaller macrocycle. Rotaxane [2-224C6] was characterized
crystallographically (Figure 2.2.2). The crown ether completely encircles the central NHCH2
fragment, and is held in place by a single NH···O hydrogen-bond (N···O 3.27 Å, N-H···O 161o).
No covalent interactions are observed between the two components. The space filling model of

36
the structure shows the aniline N-atom is only partially visible, emphasizing the steric protection
that the macrocycle provides to the close approach of a Lewis acid (Figure 2.2.2).
Figure 2.2.2 - POV–Ray depiction of [2-224C6] (left), N: blue, O: red, C: black, H: white, C-
H atoms omitted for clarity. Space filling model of [2-224C6] (right), crown ether: red, axle:
blue, N: dark blue.
Rotaxane [2-224C6] was then reacted with one equivalent of B(C6F5)3 in the hope of providing
FLP-type reactivity. Initial 1H NMR spectral analysis indicated that the two species were slowly
reacting to yield a complex mixture of unidentifiable products. This is presumed to be an
equilibrium mixture of a weak Lewis acid-base adduct with the crown ether oxygen atoms or the
aniline base; this observation is corroborated in the 11B NMR spectrum, with a resonance at
−2.60 ppm. This notion was further supported by the observation that this mixture does indeed
react when exposed to 4 atm of H2 at 100 °C, resulting in the clean formation of the protonated
[2]rotaxane and the hydridoborate anion [HB(C6F5)3]–. This reactivity is consistent with FLP
activation of H2 and when the reaction is performed in hexanes at 100 °C the
[2-2-H24C6] [HB(C6F5)3] salt cleanly precipitates from solution in 60% yield (Scheme 2.2.5).
The 1H NMR spectrum shows a broad NH2 resonance at 9.15 ppm and a benzyl CH2 signal at
4.81 ppm, matching an authentic sample of the closely related [2-2-H24C6][BF4]. The 11B
NMR spectrum showed a distinctive doublet at –25.46 ppm with a 1JBH = 92 Hz, corresponding

37
to the formation of the hydridoborate anion, which can also be detected in the 19F NMR spectrum
by the observance in a small para-meta gap of 2.84 ppm. To ensure that the proton and hydride
were coming from hydrogen gas, the same experiment was carried out under analogous
conditions under 4 atm of D2. The product was isolated from hexanes as an oil and was
determined to be the corresponding deuterium activated product, [2-2-D24C6] [DB(C6F5)3].
The 1H NMR spectrum showed an NHD resonance at 9.18 ppm but the peak integrated to less
than 1H, indicating deuterium incorporation while the remaining chemical shifts indicated clean
deuteration of the [2]rotaxane. Deuterium incorporation into the ortho position of the aniline
ring is also observed and is in accordance with the known H/D exchange reactivity with
anilines.44 The corresponding 2H NMR spectrum showed resonances at 9.06 and 3.47 ppm,
indicating ND and BD formation. The 11B NMR spectrum also showed a broad resonance at
−25.61 ppm, the same region where the borohydride anion resonances are normally found.
These data unambiguously demonstrates that [2-224C6] can in fact act as a bulky Lewis base
and effect heterolytic cleavage of H2 with B(C6F5)3.
Scheme 2.2.5 - FLP type H2 activation between [2-224C6] (top) and [2-222C6] (bottom)
with B(C6F5)3.
The success of using a [2]rotaxane as a frustrated Lewis base was followed up by studying how
the macrocycle size affected the reactivity. It was envisioned that using a smaller crown ether

38
might reduce the flexibility of the ring and concomitantly increase the steric constraints around
the nitrogen center. Indeed, the reaction of [2-222C6] containing a smaller 22-membered
crown ether ring with B(C6F5)3 without an atmosphere of H2 led to similar results as observed
with the larger crown, likely a B–O or B–N interaction. Upon pressuring the reaction at room
temperature with 4 atm of H2 an oily product, [2-2-H22C6] [HB(C6F5)3], immediately
precipitated from solution (Scheme 2.2.5). The 1H NMR spectrum shows the NH2 resonance as
a broad singlet at 9.33 ppm and the benzyl CH2 resonances are observed at 4.92 ppm. The 11B
NMR spectrum is indicative of H2 activation, as a borohydride signal was observed as a doublet
at –25.48 ppm with a 1JBH = 90 Hz. The reaction with [2-222C6] with B(C6F5)3 is in stark
contrast with [2-224C6], in the latter case H2 activation is only observed at elevated
temperature, whereas the smaller crown ether provides more steric bulk around the nitrogen
center to allow for rapid activation of H2 at room temperature.
2.2.2 Mechanism of H2 Activation
The mechanism of H2 splitting in this system was presumed to take place between the nitrogen
Lewis base and B(C6F5)3. As of this point there were no reports of H2 activation in an FLP
fashion with oxygen donors as Lewis bases. Cyclic ethers have long been known to undergo
ring opening reactions, first being noted in 1950 by Wittig and Rückert,45 where they described
the reaction of [Ph3C]– with THF(BPh3) which resulted in the ring opening product
[Ph3C(CH2)4OBPh3]–. Cyclic ethers, such as THF, dioxane, thioxane, lactones and lactides have
been shown to undergo ring opening reactions with FLP’s (Scheme 2.2.6).46,47 There have been
subtle indications that combinations of boranes and oxygen Lewis bases could potentially
activate H2, Nikonov et al.48 and Nolan et al.49 have shown that simple ether-borane adducts,
such as THF-BD3, can undergo H/D exchange under an atmosphere of H2. Stoichiometric CO
reduction has also recently been achieved using FLPs and syngas, with a proposed H2 activation
step between a boron Lewis acid and an oxygen Lewis base.50

39
Scheme 2.2.6 - Cyclic ether ring opening reactions with FLPs.
Following our report, the Paul group published a thorough computational study on this system.51
They calculated the reaction of [2-222C6] with B(C6F5)3 and observed that two reaction
minima exist. The first being B–N adduct formation; they show the crown ether is flexible
enough to move out of the way and allow B(C6F5)3 to coordinate, with a calculated bond length
of 1.76 Å. The other minimum with an energy lower by ~ 1 kcal/mol is the frustrated
combination of the two compounds. These results support the experimental observations of a
mixture of species forming when the [2]rotaxanes are mixed with B(C6F5)3. Strong π-stacking
interactions between the xylyl substituents and the C6F5 rings lead to favouring the “frustrated”
form as well as weak interactions between the boron center and the crown ether oxygen atoms.
The distance between the boron and the nitrogen atoms increase to 4.83 Å, while the distance
between the boron and the closest oxygen atom is 3.85 Å. Most interestingly, they showed that it
is actually more favourable for H2 activation to occur between B and O with a barrier of 6.5
kcal/mol versus between B and N with a barrier of 10.8 kcal/mol. The observed product [2-
2-H22C6] [HB(C6F5)3] is then generated via a proton transfer. Their studies concurred with
the experimental findings that the larger crown ether in [2-224C6] resulted in a more stable
B−N adduct and required higher energy to effect H2 activation.
2.2.3 Crown Ether Reactivity with B(C6F5)3
This computational study opened the question whether it is possible for etheral Lewis bases to
activate hydrogen with B(C6F5)3. Previous studies in our lab had investigated the reactivity of

40
B(C6F5)3 with crown ethers in attempts to effect ring opening reactions. The combinations were
unreactive and room temperature NMR studies had shown no interaction between crown ethers
and B(C6F5)3. However, the results presented in the computational study led us to revisit this
reaction. This work was done simultaneously with others in the lab investigating the use of
diethyl ether as a Lewis base.52 A stoichiometric combination of 12C4 and B(C6F5)3 was
dissolved in CD2Cl2 at room temperature, and again it appeared no interaction was occurring
whatsoever. A variable temperature NMR study was undertaken in the presence of H2 in an
attempt to observe a B–O adduct or an H2 activated product (Figure 2.2.3). The crown ether
protons begin to broaden out as the solution is cooled and four broad peaks begin to appear at
−80 °C. At this temperature the oxygen exchange at the boron center slows enough to observe
different proton environments on the crown ether. This helps explain the room temperature
result, where the oxygen exchange is too rapid to be observed on the NMR time scale.
Figure 2.2.3 - Variable temperature 1H NMR experiment with 12C4 and B(C6F5)3.
This result is reminiscent of the reactivity of electron deficient phosphines with B(C6F5)3 where
the H2 activated product was only observed at very low temperatures, and allowed for the

41
hydrogenation of olefins.53 We applied this approach to the crown ether/B(C6F5)3 combination
in hopes of generating a competent hydrogenation catalyst. Two different macrocyclic crown
ethers were utilized in this study, 12C4 and a larger macrocycle, DB24C8. A catalytic amount of
these macrocycles (20 mol%) were combined with B(C6F5)3 (20 mol%) in the presence of
1,1-diphenylethylene in CD2Cl2 and exposed to 4 atm of H2 in a J-Young NMR tube. The results
of these reactions are summarized in Table 2.2.1. Both crown ethers were shown to activate H2
with B(C6F5)3 and deliver the hydrogen to the unsaturated substrate. The size of the macrocycle
did not appear to have an effect on the conversion of 1,1-diphenylethylene to 1,1-diphenylethane
as both 12C4 and DB24C8 resulted in 56% and 57% conversion after 48 h at room temperature,
respectively. These reactions can be pushed to completion by gently heating the reaction
mixtures to 50 °C for 72 h. Attempts do hydrogenate other olefins resulted in minimal reactivity.
Table 2.2.1 - Hydrogenation of 1,1-diphenylethylene with crown ethers and B(C6F5)3.
Substrate Product B(C6F5)3
(mol%)
Crown Ether
(mol%)
T (°C) t (h) Conv. %a
Ph2C=CH2 Ph2CHCH3 20 12C4 (20) 25 48 56
Ph2C=CH2 Ph2CHCH3 20 12C4 (20) 50 72 >99
Ph2C=CH2 Ph2CHCH3 20 DB24C8 (20) 25 48 57
Ph2C=CH2 Ph2CHCH3 20 DB24C8 (20) 50 72 >99
a Percent conversion determined by 1H NMR spectroscopy.
It should be noted that the combination of diethyl ether and B(C6F5)3 was found to be a more
effective catalyst. No Friedel-Crafts dimerization product of 1,1-diphenylethylene54 was
observed in this reaction, demonstrating that there is no significant interaction between the Lewis

42
acid and the olefin. These observations indicate that the boron center is interacting more
favourably with the crown ether. The mechanism of this hydrogenation is thought to proceed via
rapid and reversible H2 activation between the crown ether O and B(C6F5)3. The protonated
crown ether is quite acidic, and parallels to Jutzi’s acid55 can be drawn, which then protonates the
olefin, generating an incipient carbocation. The borohydride then delivers hydride to the
carbocation, forming the alkane and turning the catalytic cycle over.
2.3 Conclusions
This chapter has presented the reactivity of [2]rotaxanes as sterically encumbered Lewis bases in
FLP chemistry. The amines 2-1 and 2-2 form irreversible adducts with B(C6F5)3 (2-3 and 2-4,
respectively), which effectively precludes any potential FLP reactivity. Non-covalent
modifications were made to the amines 2-1 and 2-2 to impart steric bulk and three [2]rotaxanes
were synthesized: two with a larger 24 membered macrocycle, [2-124C6] and [2-224C6],
and one with a smaller 22 membered macrocycle, [2-222C6]. The [2]rotaxane [2-124C6]
had been previously reported in literature and was the initial focus of the present study.
However, the reaction of [2-124C6] with B(C6F5)3 did not yield an FLP, but the major product
resulted from α-hydride abstraction from the amine by the borane. The [2]rotaxanes [2-224C6]
and [2-222C6] were shown to form FLPs with B(C6F5)3 to activate H2, with the smaller crown
ether allowing for room temperature activation. These results showed that the crown ether was
sufficiently encumbering enough to “frustrate” the small amine bases. Mechanistic studies
performed by the Paul group gave insight into the H2 activation, and showed that the H2
activation likely occurred between the boron and oxygen atoms, not the typical boron/nitrogen
activations as previously reported. This led to the study of the reactivity of crown ethers with
B(C6F5)3 and the combination of 12C4 and DB24C8 with B(C6F5)3 proved to be active catalysts
for the hydrogenation of 1,1-diphenylethylene.

43
2.4 Experimental Section
2.4.1 General Considerations
All preparations and manipulations were carried out under an anhydrous N2 atmosphere using
standard Schlenk and glovebox techniques. All glassware was oven-dried and cooled under
vacuum before use. Solvents were purified with a Grubbs-type column system manufactured by
Innovative Technologies and dispensed into thick-walled Schlenk glass flasks equipped with
Teflon valve stopcocks. All solvents were degassed prior to use via repeated freeze-pump-thaw
cycles. CD2Cl2 (Aldrich) and d8-toluene (Aldrich) were deoxygenated, distilled over CaH2, then
stored over 4 Å molecular sieves before use. NMR spectra were obtained on a Bruker
AvanceIII-400 MHz and Bruker Avance 500 MHz spectrometers. In selective cases
ipso-carbons were not observed in the 13C{1H} NMR spectra. Combustion analyses were
performed in house at Analest, employing a Perkin Elmer 2400 Series II CHNS Analyzer.
B(C6F5)3 was purchased from Boulder Scientific and used without further purification. Grubbs
Catalyst1st, 3,5-dimethylaniline, 3,5-dimethylbenzyl bromide, 5-bromopentane, pentaethylene
glycol, 1,4-diiodobutane, and tetrafluoroboric acid diethyl ether complex were purchased from
Sigma-Aldrich and used without further purification. Pentaethyleneglycol-dipent-4-enyl ethers42
and pentaethyleneglycol-dibut-3-enyl ether56 were prepared according to literature. The
syntheses of the rotaxanes were performed at the University of Windsor by Dr. Kelong Zhu and
Dr. V. Nicholas Vukotic, and their procedures reported below.
2.4.2 Synthesis of Compounds
Compound: 2-1
2-1 was synthesized with slight modifications to literature procedures.36
3,5-dimethoxybenzylamine (1.00 g, 5.98 mmol), 3,5-dimethoxybenzaldehyde (994 mg,
5.98 mmol) and MgSO4 (720 mg, 5.98 mmol) were combined under nitrogen in 50 mL of
CH2Cl2 in a 100 mL Schlenk flask. The condensation reaction was complete after 16 h, and the
solution was filtered and dried in vacuo producing the imine as an oil. The imine was dissolved
in 50 mL of MeOH and NaBH4 (452 mg, 11.96 mmol) were added. The reaction was complete
after 24 h and the MeOH was removed in vacuo. CH2Cl2 (50 mL) and H2O (50 mL) was added

44
to the crude product. The layers were separated and the aqueous phase extracted with CH2Cl2
(3 50 mL). The organic extracts dried over MgSO4. The product was obtained as a white solid
(1.8 g, 5.67 mmol, 95%).
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 6.51 (s, 4H, o-CH), 6.34 (s, 2H, p-CH), 3.77 (s, 12 H,
OCH3), 3.75 (s, 1H, NH), 3.72 (s, 4H, CH2).
Compound: 2-2
2-2 was synthesized according to a similar reported method.57 A mixture of 3,5-dimethylaniline
(940 mg, 4.72 mmol) and 3,5-dimethylbenzyl bromide (670 mg, 2.43 mmol) in acetonitrile
(50 mL) was stirred at room temperature overnight. The resulted solid anilinium bromide was
filtered off. The filtrate was evaporated and the residue was purified by column chromatography
on SiO2 using dichloromethane/hexanes (1: 8). A colorless liquid was obtained. (460 mg,
1.92 mmol, 80%).
MS (ESI): Calculated Mass (positive ion): 240.17522 amu, Obtained: 240.17474 amu.
1H NMR (CD2Cl2, 500 MHz, Me4Si): δ 6.98 (s, o-CHbenzyl, 2H), 6.91 (s, p-CHbenzyl, 1H), 6.34 (s,
p-CHaniline, 1H), 6.26 (s, o-CHaniline, 2H), 4.20 (s, CH2, 2H), 3.97 (br. s, NH, 1H), 2.30 (s, CH3,
6H), 2.20 (s, CH3, 6H).
13C{1H} NMR (CD2Cl2, 125 MHz, Me4Si): δ 148.97 (CH aromatic), 140.25 (CH aromatic),
139.17 (CH aromatic), 138.54 (CH aromatic), 129.12 (CH aromatic), 125.77 (CH aromatic),
119.76 (CH aromatic), 111.16 (CH aromatic), 48.64 (CH2N), 21.68 (CH3), 21.48 (CH3).
Compound: 2-3
To a solution of B(C6F5)3 (100 mg, 195 μmol) under N2 in 5 mL of CH2Cl2 was added 2-1
(0.062 g, 195 μmol) in 5 mL of CH2Cl2. The reaction mixture was allowed to stir for 2 hours at
room temperature and the volatiles were removed in vacuo and the residue washed with pentane

45
(2 2 mL). The remaining solid was dried in vacuo yielding the product. (154 mg, 185 μmol,
95%).
Anal. Calcd. for C36H23BF15NO4: C, 52.13; H, 2.80; N: 1.69 %. Found: C, 52.24; H: 3.13; N:
1.77 %.
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 6.38 (br. s, NH, 1H), 6.23 (t, 4JHH = 2 Hz, p-CH, 2H),
6.14 (d, 4JHH = 2 Hz, o-CH, 4H), 4.55 (d, 2JHH = 15 Hz, CH2, 2H), 3.68 (s, OCH3, 12H), 3.52 (m,
CH2, 2H)
11B NMR (CD2Cl2, 128 MHz, BF3·OEt2): δ –3.51 (s)
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –126.61 (d, 3JFF = 20 Hz, o-F, 2F), –129.68 (s, o-F,
2F), –137.31 (s, o-F, 2F), –155.97 (t, 3JFF = 21 Hz, p-F, 1F), –158.53 (t, 3JFF = 21 Hz, p-F, 2F),
−163.23 (s, m-F, 2F), –163.92 (t, 3JFF = 21 Hz, m-F, 2F), –164.56 (s, m-F, 2F)
13C{1H} NMR (CD2Cl2, 100 MHz, Me4Si): δ 160.93 (CH aromatic), 149.23 (br. d, 1JCF = 246
Hz), 147.64 (br. d, 1JCF = 248 Hz), 139.85 (br. d, 1JCF = 246 Hz), 134.57 (CH aromatic), 107.22
(CH aromatic), 106.84 (CH aromatic), 100.40 (CH aromatic), 56.84 (CH2N), 55.15 (CH3)
Compound: 2-4
To a solution of B(C6F5)3 (100 mg, 195 μmol) under N2 in CH2Cl2 (5 mL) was added 2-2 (47
mg, 195 μmol) in CH2Cl2 (5 mL). The reaction mixture was allowed to stir for 2 hours at room
temperature and the volatiles were removed in vacuo and the residue washed with pentane
(2 2 mL). The remaining solid was again dried in vacuo yielding the product. (120 mg,
160 μmol, 82%).
Anal. Calcd. for C35H21BF15N: C, 55.95; H, 2.82; N: 1.86 %. Found: C, 55.92; H: 3.20; N:
2.24 %.
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 7.67 (br. s, NH, 1H), 6.90 (s, p-CHbenzy, 1H), 6.85 (s, p-
CHaniline, 1H), 6.73 (s, o-CHbenzyl, 2H), 6.41 (s, o-CHaniline, 2H), 4.76 (d, 2JHH = 14 Hz, CHbenzyl,
1H), 4.15 (t, 2JHH = 12 Hz, CHbenzyl, 1H), 2.21 (s, CH3, 6H), 2.13 (s, CH3, 6H)
11B NMR (CD2Cl2, 128 MHz, BF3·OEt2): δ –2.50 (s)
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –128.66 (br. s, o-F, 6F), –158.32 (br. s, p-F, 3F),
−164.70 (br. s, m-F, 6F)

46
19F NMR (CD2Cl2, 377 MHz, CFCl3, 233 K) δ: –124.34 (s, o-F, 1F), –127.96 (d, 3JFF = 23 Hz,
o-F, 1F), –128.31 (m, o-F, 2F), –129.93 (m, o-F, 1F), –137.07 (dd, 3JFF = 54 Hz, 4JFF = 22.6 Hz,
o-F, 1F), –154.78 (t, 3JFF = 20 Hz, p-F, 1F), –157.15 (t, 3JFF = 21 Hz, p-F, 1F), –157.39 (t,
3JFF = 21 Hz, p-F, 1F), –161.87 (p, 3JFF = 24 Hz, m-F, 2F), –162.29 (t, 3JFF = 23 Hz, m-F, 1F),
−163.17 (t, 3JFF = 21 Hz, m-F, 1F), –163.92 (t, 3JFF = 22 Hz, m-F, 1F), –164.87 (t, 3JFF = 21 Hz,
m-F, 1F)
13C{1H} NMR (CD2Cl2, 100 MHz, Me4Si): δ 149.30 (br. d, 1JCF = 220 Hz), 139.57 (CH
aromatic), 139.12 (CH aromatic), 137.71 (br. d, 1JCF = 248 Hz), 137.33 (CH aromatic), 132.00
(CH aromatic), 130.96 (CH aromatic), 130.44 (CH aromatic), 127.12 (CH aromatic), 121.32 (CH
aromatic), 55.81 (CH2N), 21.24 (CH3)
Compound: [2-124C6]
This compound was prepared as it was in the literature42 and the ammonium salt generated
deprotonated using the techniques outlined described by Takata et al.43 [2-1-H][BF4] (500 mg,
1.24 mmol) and pentaethyleneglycol-dipent-4-enyl ether (554 mg, 1.49 mmol) were added to a
250 mL Schlenk flask. Degassed and backfilled with N2. Dichloromethane (100 mL) was added
to the mixture under N2 atmosphere. After stirred for 10 min, Grubbs Catalyst1st (51 mg,
0.062 mmol) was added. The resulted mixture was heated at 42 °C under N2 atmosphere for 24 h.
After the mixture was cooled down to room temperature, vinyl ether (1 mL) was added. The
mixture was further stirred for 1 h and the dichloromethane solution was poured into diethyl
ether and the product was recovered by filtration. The product was subsequently
recrystallization from EtOAc followed by hydrogenation, anion exchange and neutralization. The
solid was dissolved in MeOH (100 mL) and added to a Schlenk flask which was equipped with
10% Pd/C (150 mg) under N2 atmosphere. The flask was slightly degassed and flushed with H2,
introduced via a balloon. The mixture was stirred for 3 h under ambient conditions. Filtration and
removal of solvent gave the desired hydrogenated product as a BF4 salt which was subsequently
anion exchanged to fluoride salt via TBAF and neutralized with NaHCO3 (312 mg, 0.47 mmol,
45%).

47
MS (ESI): Calculated Mass (positive ion): 560.39511 amu, Obtained: 560.39374 amu).
1H NMR (CD2Cl2, 500 MHz, Me4Si): δ 6.77 (d, 4JHH = 2 Hz, o-CH, 4H), 6.27 (t, 4JHH = 2 Hz
p-CH, 2H), 3.94 (s, CH2, 4H), 3.78 (s, CH3 12H), 3.42 (m, CH2O, 24H), 1.47 (m, CH2, 2H), 1.27
(m, CH2, 10H), NH resonance not observed.
13C{1H} NMR (CD2Cl2, 125 MHz, Me4Si): δ 159.93 (s, CH aromatic), 143.93 (s, CH aromatic),
106.43 (s, s, CH aromatic), 99.89 (s, CH aromatic), 70.69 (s, CH2O), 70.03 (s, CH2O), 69.73 (s,
CH2O), 69.71 (s, CH2O), 69.64 (s, CH2O), 69.38 (s, CH2O), 54.51 (s, OCH3), 53.75 (s, CH2N),
28.80 (s, CH2), 28.14 (s, CH2) 25.03 (s, CH2).
Compound: [2-2-H][BF4]
2-2 (240 mg, 1.01 mmol) was dissolved in hexane (50 mL) and to the solution was added 1.0
equivalent of tetrafluoroboric acid diethyl ether complex to yield [2-2-H][BF4] as a white solid.
The solid was filtered off and air dried. (310 mg, 0.948 mmol, 94%).
MS (ESI): Calculated Mass (positive ion): 240.17522 amu, Obtained: 240.17474 amu.
1H NMR (CD2Cl2, 500 MHz, Me4Si): δ 8.51 (s, NH, 2H), 7.07 (s, p-CHaniline, 1H), 6.98 (s,
p-CHbenzyl, 1H), 6.82 (s, o-CHaniline, 2H), 6.77 (s, o-CHbenzyl, 2H), 4.40 (s, CH2, 2H), 2.28 (s, CH3,
6H), 2.23 (s, CH3, 6H).
13C{1H} NMR (CD2Cl2, 125 MHz, Me4Si): δ 140.60 (CH aromatic), 139.06 (CH aromatic),
134.02 (CH aromatic), 131.71 (CH aromatic), 131.69 (CH aromatic), 129.25 (CH aromatic),
128.75 (CH aromatic), 121.11 (CH aromatic), 57.62 (CH2N), 21.19 (CH3), 21.07 (CH3).

48
Compound: [2-2-H24C6][BF4]
[2-2-H][BF4] (750 mg, 2.29 mmol) and pentaethyleneglycol-dipent-4-enyl ether (1.29 g,
3.44 mmol) were added to a 250 mL Schlenk flask. Degassed and backfilled with N2.
Dichloromethane (100 mL) and nitromethane (11 mL) were added to the mixture under N2
atmosphere. After stirred for 10 min, Grubbs Catalyst1st (189 mg, 0.229 mmol) was added. The
resulted mixture was heated at 45 °C under N2 atmosphere for 24 hr. Then another 5 mol%
catalyst was added and heated for another 24 hr. After the mixture was cooled down to room
temperature, vinyl ether (1 mL) was added. The mixture was stirred for 1 h and dried on a rotary
evaporator. The oily residue was washed once with hot hexane (50 mL) and dried under vacuum.
The E/Z mixture was obtained after flash column chromatography (SiO2, CH2Cl2/hexanes = 1:1
then ethyl acetate) (751 mg, 50 %). This mixture was dissolved in MeOH (100 mL) and added to
a Schlenk flask which was equipped with 10% Pd/C (150 mg) under N2 atmosphere. The flask
was slightly degassed and flushed with H2, introduced via a balloon. The mixture was stirred for
3 hr under ambient conditions. Filtration and removal of solvent gave the desired product
(713 mg, 1.06 mmol, 95%).
MS (ESI): Calculated Mass (positive ion): 588.42720 amu, Obtained: 588.42641 amu.
1H NMR (CD2Cl2, 500 MHz, Me4Si): δ 9.17 (s, NH, 2H), 7.29 (s, o-CHaniline, 2H), 7.18 (s,
o-CHbenzyl, 2H), 7.18, (s, p-CHaniline, 1H), 7.12 (s, p-CHbenzyl, 1H), 4.80 (m, CH2, 2H), 3.39 (m,
CH2O, 24H), 2.42 (s, CH3, 6H), 2.38 (s, CH3, 6H), 1.35 (m, CH2, 4H), 1.24 (m, CH2, 4H), 1.18
(m, CH2, 4H).
13C{1H} NMR (CD2Cl2, 125 MHz, Me4Si): δ 140.56 (CH aromatic), 139.10 (CH aromatic),
135.49 (CH aromatic), 131.92 (CH aromatic), 131.81 (CH aromatic), 131.34 (CH aromatic),
127.41 (CH aromatic), 120.72 (CH aromatic), 72.12 (CH2O), 71.85 (CH2O), 71.43 (CH2O),
71.00 (CH2O), 70.82 (CH2O), 70.67 (CH2O), 53.31 (CH2N), 29.82 (s), 28.22 (s), 25.75 (s), 21.54
(s), 21.51 (s).

49
Compound: [2-224C6]
Triethylamine (1 mL, 7.2 mmol) was added to a solution of [2-2-H24C6][BF4] (300 mg,
0.44 mmol) in MeOH (50 mL). The mixture was stirred at room temperature for 5 min. The
solvent was removed on a rotary evaporator. The crude product was purified by column
chromatography (SiO2, EtOAc/petroleum ether 1:4). X-ray suitable single crystals were obtained
from slow evaporation of a hexane solution. (230 mg, 0.39 mmol, 89%).
MS (ESI): Calculated Mass (positive ion): 588.42720 amu, Obtained: 588.42641 amu.
1H NMR (CD2Cl2, 500 MHz, Me4Si): δ 7.34 (s, o-CHaniline, 2H), 6.87 (s, p-CHaniline, 1H), 6.55 (s,
o-CHbenzyl, 2H), 6.23 (s, p-CHbenzyl, 1H), 5.11 (t, 3JHH = 5 Hz, NH, 1H), 4.38 (d, 3JHH = 5 Hz,
CH2, 2H), 3.39 (m, CH2O, 24H), 2.31 (s, CH3, 6H), 2.23 (s, CH3, 6H), 1.43 (m, CH2, 4H), 1.28
(m, CH2, 4H), 1.12 (m, CH2, 4H).
13C{1H} NMR (CD2Cl2, 125 MHz, Me4Si): δ 148.07 (s, CH aromatic), 139.85 (s, CH aromatic),
136.55 (s, s, CH aromatic), 136.00 (s, CH aromatic), 126.79 (s, CH aromatic), 126.71 (s, CH
aromatic), 115.98 (s, CH aromatic), 110.77 (s, CH aromatic), 70.72 (s, CH2O), 70.18 (s, CH2O),
69.79 (s, CH2O), 69.75 (s, CH2O), 69.43 (s, CH2O), 46.58 (s, CH2N), 29.03 (CH2), 28.22 (CH2),
24.98 (CH2), 20.61 (CH3), 20.30 (CH3).
Compound: [2-2-H22C6][BF4]
[2-2-H][BF4] (220 mg, 0.672 mmol) and pentaethyleneglycol-dibut-4-enyl ether (350 mg,
1.01 mmol) were added to a 100-mL Schlenk flask. Degassed and backfilled with N2.
Dichloromethane (45 mL) and nitromethane (15 mL) were added to the mixture under N2
atmosphere. After stirred for 10 min, Grubbs Catalyst1st (55.3 mg, 0.0672 mmol) was added. The
resulted mixture was heated at 45 °C under N2 atmosphere for 24 hr. Then another 5 mol%
catalyst was added and heated for another 2 days. After the mixture was cooled down to room

50
temperature, vinyl ether (0.5 mL) was added. The mixture was stirred for 1 h and dried on a
rotary evaporator. The oily residue was washed once with hot hexane (50 mL) and dried under
vacuum. The E/Z mixture was obtained after flash column chromatography (SiO2,
CH2Cl2/hexanes = 1:1 then ethyl acetate) (130 mg, 0.201 mmol, 30 %). This mixture was
dissolved in MeOH (50 mL) and added to a Schlenk flask which was equipped with 10% Pd/C
(40 mg) under N2 atmosphere. The flask was slightly degassed and flushed with H2, introduced
via a balloon. The mixture was stirred for 6 hr under ambient conditions. Filtration and removal
of solvent gave the desired product. (122 mg, 0.188 mmol, 94%).
MS (ESI): Calculated Mass (positive ion): 560.39511 amu, Obtained: 560.39374 amu.
1H NMR (CD2Cl2, 500 MHz, Me4Si): δ 9.34 (s, NH, 2H), 7.40 (s, o-CHaniline, 2H), 7.22 (s,
o-CHbenzyl, 2H), 7.17, (s, p-CHaniline, 1H), 7.12 (s, p-CHbenzyl, 1H), 4.92 (m, CH2, 2H), 3.53 (m,
CH2O, 24H), 2.41 (s, CH3, 6H), 2.39 (s, CH3, 6H), 1.30 (m, CH2, 8H).
13C{1H} NMR (CD2Cl2, 500 MHz, Me4Si): δ 140.01 (CH aromatic), 138.57 (CH aromatic),
134.93 (CH aromatic), 131.86 (CH aromatic), 131.22 (CH aromatic), 130.92 (CH aromatic),
127.54 (CH aromatic), 120.63 (CH aromatic), 71.66 (CH2O), 71.14 (CH2O), 70.87 (CH2O),
70.65 (CH2O), 51.28 (CH2N), 29.00 (CH2), 25.18 (CH2), 21.07 (CH3), 21.00 (CH3).
Compound: [2-222C6]
A mixture of [2-2-H22C6][BF4] (95 mg, 147 μmol), 0.2 M NaOH (60 mL), and hexane
(60 mL) was stirred vigorously at room temperature for 1 hr. The hexane phase was separated
the aqueous phase was extracted with hexanes (60 mL). The organic phases were combined and
dried over anhydrous MgSO4, and concentrated under vacuum to give the target compound as a
white solid (64 mg, 114 μmol, 79%).
MS (ESI): Calculated Mass (positive ion): 560.39511 amu, Obtained: 560.39374 amu.
1H NMR (CD2Cl2, 500 MHz, Me4Si): δ 7.39 (s, o-CHaniline, 2H), 6.84 (s, p-CHaniline, 1H), 6.66 (s,
o-CHbenzyl, 2H), 6.15 (s, p-CHbenzyl, 1H), 5.61 (t, 3JHH = 4 Hz, NH, 1H), 4.57 (d, 3JHH = 4 Hz,
CH2, 2H), 3.44 (m, CH2O, 24H), 2.31 (s, CH3, 6H), 2.22 (s, CH3, 6H), 1.28 (m, CH2, 8H).
13C{1H} NMR (CD2Cl2, 125 MHz, Me4Si): δ 149.98 (CH aromatic), 140.88 (CH aromatic),

51
135.96 (CH aromatic), 135.64 (CH aromatic), 126.87 (CH aromatic), 126.27 (CH aromatic),
115.01 (CH aromatic), 111.34 (CH aromatic), 70.75 (CH2O), 70.36 (CH2O), 70.24 (CH2O),
70.06 (CH2O), 70.02 (CH2O), 69.83 (CH2O), 45.94 (CH2N), 28.02 (CH2), 24.87 (CH2), 20.72
(CH3), 20.36 (CH3).
Reactions of [2]rotaxanes with B(C6F5)3
Reaction of [2-124C6] with B(C6F5)3
The reaction of B(C6F5)3 (8 mg, 15 μmol) with [2-124C6] (10 mg, 15 μmol) in CD2Cl2 yields a
mixture of products. However, the predominant product results from the α-hydride abstraction
from the benzylic positions forming an iminium hydridoborate salt (Scheme 2.2.3). The other
products are difficult to elucidate and separate. The 1H and 11B NMR spectra of this reaction are
shown in Figure 2.4.1 and Figure 2.4.2.
Figure 2.4.1 - 1H NMR spectrum of the reaction of [2-124C6] with B(C6F5)3.

52
Figure 2.4.2 - 11B NMR spectrum of the reaction of [2-124C6] with B(C6F5)3.
Reaction of [2-224C6] with B(C6F5)3
The reaction of B(C6F5)3 (9 mg, 17 μmol) with [2-224C6] (10 mg, 17 μmol) in either CD2Cl2
or C7D8 yields a mixture of products. These products are difficult to elucidate and separate,
however, they are likely three things, the free species, [2-224C6], as well as weak B–N or B–O
interactions and the reaction of the compounds with trace water from the starting materials. The
1H and 11B NMR spectra of this reaction are shown below.

53
Figure 2.4.3 - 1H NMR spectrum of the reaction of [2-224C6] with B(C6F5)3.
Figure 2.4.4 - 11B NMR spectrum of the reaction of [2-224C6] with B(C6F5)3.
Reaction of [2-222C6] with B(C6F5)3
The reaction of B(C6F5)3 (9 mg, 17 μmol) with [2-222C6] (10 mg, 17 μmol) in either CD2Cl2
or C7D8 yields a mixture of products similarly to the analogous reaction with [2-224C6].

54
Reactions of [2]rotaxanes with B(C6F5)3 and Hydrogen
Compound: [2-2-H24C6] [HB(C6F5)3]
To a solution of B(C6F5)3 (26 mg, 51 μmol) under N2 in hexanes (10 mL) was added [2-224C6]
(30 mg, 51 μmol) and sealed in a bomb. The reaction mixture was subjected to three freeze
pump thaw cycles and refilled with 4 atm of H2. The reaction mixture was stirred for overnight at
100 °C which resulted in an oil precipitating from solution. The hexanes were decanted off and
the oil was dried in vacuo. The resulting oil was washed with pentane (2 2 mL) and again
dried in vacuo. (34 mg, 31 μmol, 61%).
MS (ESI): Calculated Mass (positive ion): 588.4258 amu, Obtained: 588.4276 amu,
Calculated Mass (negative ion): 512.9937 amu, Obtained: 512.9930 amu.
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 9.15 (br. s, NH2, 2H), 7.30 (s, o-CHaniline, 2H), 7.18 (s,
CH, 3H), 7.11 (s, p-CHbenzyl, 1H), 4.81 (m, CH2, 2H), 3.44 (m, OCH2, 20H), 2.45 (s, CH3, 6H),
2.42 (s, CH3, 6H), 1.38 (m, CH2, 4H), 1.29 (m, CH2 12H)
11B NMR (CD2Cl2, 128 MHz, BF3·OEt2): δ –25.46 (d, 1JBH = 92 Hz, BH)
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –134.99 (d, 3JFF = 21 Hz, o-F, 6F), –165.88 (t, 3JFF =
21 Hz, p-F, 3F), –168.72 (td, 3JFF = 22 Hz, 4JFF = 7 Hz, m-F, 6F)
13C{1H} NMR (partial, CD2Cl2, 100 MHz, Me4Si): δ 148.34 (br. d, 1JCF = 235 Hz), 140.61 (CH
aromatic), 139.14 (CH aromatic), 137.63 (br. d, 1JCF = 251 Hz), 136.11 (br. d, 1JCF = 232 Hz),
135.42 (CH aromatic), 131.89 (CH aromatic), 131.84 (CH aromatic), 131.36 (CH aromatic),
127.39 (CH aromatic), 120.39 (CH aromatic), 72.15 (CH2O), 71.86 (CH2O), 71.42 (CH2O),
70.95 (CH2O), 70.74 (CH2O), 51.44 (CH2N), 29.80 (CH2), 28.19 (CH2), 25.74 (CH2), 21.47
(CH3), 21.43 (CH3).

55
Compound: [2-2-H22C6] [HB(C6F5)3]
To a solution of B(C6F5)3 (12 mg, 23 μmol) under N2 in hexanes (10 mL) was added [2-222C6]
(13 mg, 23 μmol) and sealed in a bomb. The reaction mixture was subjected to three freeze
pump thaw cycles and refilled with 4 atm. of H2. The reaction mixture was stirred for overnight
at room temperature, which resulted in an oil precipitating from solution. The hexane was
decanted off and the oil was dried in vacuo. The resulting oil was washed with pentane
(2 2 mL) and again dried in vacuo. (15 mg, 14 μmol, 60%).
MS (ESI): Calculated Mass (positive ion): 560.3945 amu, Obtained: 560.3940 amu,
Calculated Mass (negative ion): 512.9937 amu, Obtained: 512.9948 amu.
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 9.33 (br. s, NH2, 2H), 7.39 (s, o-CHaniline, 2H), 7.21 (s,
o-CHbenzyl, 2H), 7.16 (s, p-CHaniline, 1H), 7.11 (s, p-CHbenzyl, 1H), 4.92 (m, CH2, 2H), 3.41 (m,
OCH2, 24H), 2.40 (s, CH3, 6H), 2.38 (s, CH3, 6H), 1.30 (m, CH2, 8H).
11B NMR (CD2Cl2, 128 MHz, BF3·OEt2): δ –25.48 (d, 1JBH = 90 Hz. BH)
19F NMR (CD2Cl2): δ –134.02 (d, 3JFF = 23 Hz, o-F, 6F), –164.88 (t, 3JFF = 21 Hz, p-F, 3F),
−167.72 (tm, 3JFF = 23 Hz, m-F, 6F)
13C{1H} NMR (partial, CD2Cl2, 100 MHz, Me4Si): δ 148.22 (br. d, 1JCF = 233 Hz), 140.01 (CH
aromatic), 138.57 (CH aromatic), 137.75 (br. d, 1JCF = 253 Hz), 136.53 (br. d, 1JCF = 235 Hz),
134.93 (CH aromatic), 131.86 (CH aromatic), 131.22 (CH aromatic), 130.92 (CH aromatic),
127.54 (CH aromatic), 120.63 (CH aromatic), 71.66 (CH2O), 71.14 (CH2O), 70.87 (CH2O),
70.65 (CH2O), 51.28 (CH2N), 29.00 (CH2), 25.18 (CH2), 21.07 (CH3), 21.00 (CH3).

56
Compound: [2-2-D24C6] [HB(C6F5)3]
To a solution of B(C6F5)3 (26 mg, 51 μmol) under N2 in hexanes (10 mL) was added [2-224C6]
(30 mg, 51 μmol) and sealed in a bomb. The reaction mixture was subjected to three freeze
pump thaw cycles and refilled with 4 atm. of D2. The reaction mixture was stirred for overnight
at 100 °C, which resulted in an oil precipitating from solution. The hexane was decanted and the
oil was dried in vacuo. The resulting oil was washed with pentane (2 2 mL) and again dried in
vacuo. (35 mg, 32 μmol, 63%).
MS (ESI): Calculated Mass (positive ion): 589.4321 amu, Obtained: 589.4328 amu,
Calculated Mass (negative ion): 513.9937 amu, Obtained: 512.9962 amu.
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 9.18 (br. s, NHD, 0.2H), 7.31 (s, o-CHaniline, 1H), 7.18
(s, CH aromatic, 2H), 7.11 (s, p-CHbenzyl, 1H), 4.81 (s, CH2, 2H), 3.30 (m, OCH2, 24H), 2.41 (s,
CH3, 6H), 2.38 (s, CH3, 6H), 1.35 (m, CH2, 4H), 1.30 (m, CH2, 8H)
2D NMR (CH2Cl2, 400 MHz): δ 9.06 (br. s), 7.32 (br. s, o-CDaniline), 3.47 (br. s, BD)
11B NMR (CD2Cl2, 128 MHz, BF3·OEt2): δ –25.61 (br. s)
19F NMR (CD2Cl2): δ –134.99 (d, 3JFF = 21 Hz, o-F, 6F), –165.87 (t, 3JFF = 20 Hz, p-F, 3F),
−168.72 (tm, 3JFF = 22 Hz, m-F, 6F).
General Procedure for Catalytic Hydrogenation with Crown Ethers and B(C6F5)3
In a glovebox, in separate 5 mL vials, B(C6F5)3 (11 mg, 22 μmol), DB24C8 (10 mg, 22 μmol)
and Ph2CCH2 (20 mg, 111 μmol) was weighed. The crown ether and B(C6F5)3 were dissolved in
CD2Cl2 (~1 mL) and the solution was transferred to the vial containing Ph2CCH2. The solution
was quickly transferred to the J-Young NMR tube using a pipette. The tube was sealed and
removed from the glovebox. The reaction mixture was subjected to three freeze pump thaw
cycles and refilled with 4 atm. of H2, then allowed to react at RT or heated in an oil bath at
50 °C. The mixture was analyzed by 1H NMR spectroscopy after 24 h, 48 h, and 72 h showing
slow conversion of Ph2CCH2 to Ph2C(H)CH3 (major product). An example of the 1H NMR
analysis is seen in Figure 2.3.5.

57
Figure 2.3.5 – 1H NMR spectrum of the reaction of 20 % DB24C8 and B(C6F5)3 with
1,1-diphenyletheylene in CD2Cl2 after 48 h at r.t., showing 57% conversion of Ph2CCH2 to
Ph2C(H)CH3.
2.4.3 X-Ray Crystallography
2.4.3.1 X-Ray Collection and Reduction
Crystals were coated in Paratone-N oil in the glovebox, mounted on a MiTegen Micromount and
placed under an N2 stream, thus maintaining a dry, O2-free environment for each crystal. The
data were collected on a Bruker Kappa Apex II diffractometer. Data collection strategies were
determined using Bruker Apex 2 software and optimized to provide >99.5% complete data to a
2θ value of at least 55°. The data were collected at 150(±2) K for all. The data integration and
absorption correction were performed with the Bruker Apex 2 software package.58

58
2.4.3.2 X-Ray Solution and Refinement
Non-hydrogen atomic scattering factors were taken from the literature tabulations.59,60 The heavy
atom positions were determined using direct methods employing the SHELX-2013 direct
methods routine. The remaining non-hydrogen atoms were located from successive difference
Fourier map calculations. The refinements were carried out by using full-matrix least squares
techniques on F, minimizing the function ω (Fo-Fc)2 where the weight ω is defined as 4Fo2/2σ
(Fo2) and Fo and Fc are the observed and calculated structure factor amplitudes, respectively. In
the final cycles of each refinement, all non-hydrogen atoms were assigned anisotropic
temperature factors in the absence of disorder or insufficient data. In the latter cases atoms were
treated isotropically. C-H atom positions were calculated and allowed to ride on the carbon to
which they are bonded assuming a C-H bond length of 0.95 Å. H-atom temperature factors were
fixed at 1.20 times the isotropic temperature factor of the C-atom to which they are bonded. The
H-atom contributions were calculated, but not refined. The locations of the largest peaks in the
final difference Fourier map calculation as well as the magnitude of the residual electron
densities in each case were of no chemical significance.

59
Table 2.4.1 - Select Crystallographic Data for [2-224C6].
[2-224C6]
Formula C35H57NO6
Formula weight 587.82
Crystal System Monoclinic
Space group P2(1)/c
a(Å) 20.613(5)
b(Å) 19.349(5)
c(Å) 18.562(4)
α(deg) 90
β(deg) 105.105(3)
γ(deg) 90
V(Å3) 7148(3)
Z 8
Temp. (K) 150
d(calc)gcm-1 1.092
Abs coeff,μ,mm-1 0.073
Data collected 12574
DataFO2
>3(FO2) 5891
Variables 757
R 0.1066
Rw 0.3549
GOF 1.014

60
2.5 References
(1) Welch, G. C.; Juan, R. R. S.; Masuda, J. D.; Stephan, D. W. Science 2006, 314, 1124-
1126.
(2) Welch, G. C.; Stephan, D. W. Journal of the American Chemical Society 2007, 129,
1880-1881.
(3) Spies, P.; Erker, G.; Kehr, G.; Bergander, K.; Frohlich, R.; Grimme, S.; Stephan, D. W.
Chemical Communications 2007, 5072-5074.
(4) Geier, S. J.; Stephan, D. W. Journal of the American Chemical Society 2009, 131, 3476-
3477.
(5) Brown, H. C.; Schlesinger, H. I.; Cardon, S. Z. Journal of the American Chemical Society
1942, 64, 325-329.
(6) Dureen, M. A.; Stephan, D. W. Journal of the American Chemical Society 2010, 132,
13559-13568.
(7) Tanur, C. A.; Stephan, D. W. Organometallics 2011, 30, 3652-3657.
(8) Chernichenko, K.; Madarász, Á.; Pápai, I.; Nieger, M.; Leskelä, M.; Repo, T. Nat Chem
2013, 5, 718-723.
(9) Zhu, K.; Loeb, S. In Molecular Machines and Motors; Credi, A., Silvi, S., Venturi, M.,
Eds.; Springer International Publishing: 2014; Vol. 354, p 213-251.
(10) Schill, G.; Lüttringhaus, A. Angewandte Chemie International Edition in English 1964, 3,
546-547.
(11) Harrison, I. T.; Harrison, S. Journal of the American Chemical Society 1967, 89, 5723-
5724.
(12) Amabilino, D. B.; Stoddart, J. F. Chemical Reviews 1995, 95, 2725-2828.
(13) Barin, G.; Coskun, A.; Fouda, M. M. G.; Stoddart, J. F. ChemPlusChem 2012, 77, 159-
185.
(14) Kay, E.; Leigh, D. In Molecular Machines; Kelly, T. R., Ed.; Springer Berlin Heidelberg:
2005; Vol. 262, p 133-177.
(15) Hoffart, D. J.; Tiburcio, J.; de la Torre, A.; Knight, L. K.; Loeb, S. J. Angewandte Chemie
International Edition 2008, 47, 97-101.
(16) Aricó, F.; Badjic, J.; Cantrill, S.; Flood, A.; Leung, K. F.; Liu, Y.; Stoddart, J. F. In
Templates in Chemistry II; Schalley, C., Vögtle, F., Dötz, K., Eds.; Springer Berlin Heidelberg:
2005; Vol. 249, p 203-259.

61
(17) Stoddart, J. F. Angewandte Chemie International Edition 2014, 53, 11102-11104.
(18) Anelli, P. L.; Spencer, N.; Stoddart, J. F. Journal of the American Chemical Society 1991,
113, 5131-5133.
(19) Bissell, R. A.; Cordova, E.; Kaifer, A. E.; Stoddart, J. F. Nature 1994, 369, 133-137.
(20) Badjić, J. D.; Balzani, V.; Credi, A.; Silvi, S.; Stoddart, J. F. Science 2004, 303, 1845-
1849.
(21) Serreli, V.; Lee, C.-F.; Kay, E. R.; Leigh, D. A. Nature 2007, 445, 523-527.
(22) Vukotic, V. N.; Harris, K. J.; Zhu, K.; Schurko, R. W.; Loeb, S. J. Nature Chemistry
2012, 4, 456-460.
(23) Stephan, D. W.; Erker, G. Angew. Chem. 2010, 122, 50-80. 2010, 49, 46-76.
(24) Sumerin, V.; Chernichenko, K.; Nieger, M.; Leskela, M.; Rieger, B.; Repo, T. Advanced
Synthesis & Catalysis 2011, 353, 2093-2110.
(25) Sumerin, V.; Schulz, F.; Atsumi, M.; Wang, C.; Nieger, M.; Leskela, M.; Repo, T.;
Pyykko, P.; Rieger, B. Journal of the American Chemical Society 2008, 130, 14117-14118.
(26) Sumerin, V.; Schulz, F.; Nieger, M.; Atsumi, M.; Wang, C.; Leskela, M.; Pyykko, P.;
Repo, T.; Rieger, B. Journal of Organometallic Chemistry 2009, 694, 2654-2660.
(27) Sumerin, V.; Schulz, F.; Nieger, M.; Leskela, M.; Repo, T.; Rieger, B. Angewandte
Chemie International Edition 2008, 47, 6001-6003.
(28) Schwendemann, S.; Frohlich, R.; Kehr, G.; Erker, G. Chemical Science 2011, 2, 1842-
1849.
(29) Garcia-Viloca, M.; Gao, J.; Karplus, M.; Truhlar, D. G. Science 2004, 303, 186-195.
(30) Knappe, T. A.; Linne, U.; Zirah, S.; Rebuffat, S.; Xie, X.; Marahiel, M. A. Journal of the
American Chemical Society 2008, 130, 11446-11454.
(31) Beves, J. E.; Blight, B. A.; Campbell, C. J.; Leigh, D. A.; McBurney, R. T. Angewandte
Chemie International Edition 2011, 50, 9260-9327.
(32) Loeb, S. J. Chemical Society Reviews 2007, 36, 226-235.
(33) Faiz, J. A.; Heitz, V.; Sauvage, J.-P. Chemical Society Reviews 2009, 38, 422-442.
(34) Dichtel, W. R.; Miljanić, O. Š.; Zhang, W.; Spruell, J. M.; Patel, K.; Aprahamian, I.;
Heath, J. R.; Stoddart, J. F. Accounts of Chemical Research 2008, 41, 1750-1761.
(35) Shore, S. G.; Boddeker, K. W. Inorganic Chemistry 1964, 3, 914-915.
(36) Cantrill, S. J.; Fyfe, M. C. T.; Heiss, A. M.; Stoddart, J. F.; White, A. J. P.; Williams, D.
J. Organic Letters 1999, 2, 61-64.

62
(37) Millot, N.; Santini, Catherine C.; Fenet, B.; Basset, Jean M. European Journal of
Inorganic Chemistry 2002, 2002, 3328-3335.
(38) Farrell, J. M.; Heiden, Z. M.; Stephan, D. W. Organometallics 2011, 30, 4497-4500.
(39) Geier, S. J.; Gille, A. L.; Gilbert, T. M.; Stephan, D. W. Inorganic Chemistry 2009, 48,
10466-10474.
(40) Caputo, C. B.; Geier, S. J.; Winkelhaus, D.; Mitzel, N. W.; Vukotic, V. N.; Loeb, S. J.;
Stephan, D. W. Dalton Transactions 2012, 41, 2131-2139.
(41) Caputo, C. B.; Geier, S. J.; Ouyang, E. Y.; Kreitner, C.; Stephan, D. W. Dalton
Transactions 2012, 41, 237-242.
(42) Kilbinger, A. F. M.; Cantrill, S. J.; Waltman, A. W.; Day, M. W.; Grubbs, R. H.
Angewandte Chemie International Edition 2003, 42, 3281-3285.
(43) Nakazono, K.; Takata, T. Chemistry – A European Journal 2010, 16, 13783-13794.
(44) Best, A. P.; Wilson, C. L. Journal of the Chemical Society (Resumed) 1938, 28-29.
(45) Wittig, G.; Rückert, A. Justus Liebigs Annalen der Chemie 1950, 566, 101-113.
(46) Kreitner, C.; Geier, S. J.; Stanlake, L. J. E.; Caputo, C. B.; Stephan, D. W. Dalton
Transactions 2011, 40, 6771-6777.
(47) Birkmann, B.; Voss, T.; Geier, S. J.; Ullrich, M.; Kehr, G.; Erker, G.; Stephan, D. W.
Organometallics 2010, 29, 5310-5319.
(48) Nikonov, G. I.; Vyboishchikov, S. F.; Shirobokov, O. G. Journal of the American
Chemical Society 2012, 134, 5488-5491.
(49) Nelson, D. J.; Egbert, J. D.; Nolan, S. P. Dalton Transactions 2013, 42, 4105-4109.
(50) Dobrovetsky, R.; Stephan, D. W. Journal of the American Chemical Society 2013, 135,
4974-4977.
(51) Bhunya, S.; Paul, A. Chemistry – A European Journal 2013, 19, 11541-11546.
(52) Hounjet, L. J.; Bannwarth, C.; Garon, C. N.; Caputo, C. B.; Grimme, S.; Stephan, D. W.
Angewandte Chemie International Edition 2013, 52, 7492-7495.
(53) Greb, L.; Oña-Burgos, P.; Schirmer, B.; Grimme, S.; Stephan, D. W.; Paradies, J.
Angewandte Chemie International Edition 2012, 51, 10164-10168.
(54) Basavaiah, D.; Reddy, K. R. Organic Letters 2006, 9, 57-60.
(55) Jutzi, P.; Müller, C.; Stammler, A.; Stammler, H.-G. Organometallics 2000, 19, 1442-
1444.
(56) Dasgupta, S.; Wu, J. Chemical Science 2012, 3, 425-432.

63
(57) Loeb, S. J.; Tiburcio, J.; Vella, S. J. Organic Letters 2005, 7, 4923-4926.
(58) Kraft, B. M.; Lachicotte, R. J.; Jones, W. D. Journal of the American Chemical Society
2001, 123, 10973-10979.
(59) Choi, J.; Wang, D. Y.; Kundu, S.; Choliy, Y.; Emge, T. J.; Krogh-Jespersen, K.;
Goldman, A. S. Science 2011, 332, 1545
(60) Kiplinger, J. L.; Richmond, T. G.; Osterberg, C. E. Chem. Rev. 1994, 94, 373-431.

64
Chapter 3 Frustrated Lewis Pair Activation of Carbon–Fluorine Bonds
3
3.1 Introduction
3.1.1 Interest in Carbon–Fluorine Bonds
Fluorocarbons are unique molecules, the changes in the physical properties of molecules when a
hydrogen atom is replaced by a fluorine atom are quite drastic. A bond to fluorine is the
strongest which carbon can form with a bond dissociation energy of up to 130 kcal/mol.1
However, fluorocarbons are naturally the rarest of the halocarbons, due to the fact that most
terrestrial fluorine sources are found in an insoluble form and cannot be taken up biologically.2
A number of synthetic protocols have been developed for the formation of carbon–fluorine
bonds involving nucleophilic3-5 and electrophilic fluorinating reagents,6 transition-metal
catalysis,7 as well as trifluoromethylating reagents.8 Fluorine forms a strongly polarized bond to
carbon due to its inherent electronegativity (3.98 for F, vs 2.55 for C);9 in addition the small size
of the fluorine atom makes it an excellent substitute for a hydrogen atom. Fluorine substituents
affect the physical properties of a compound, in particular enhancing the bioavailability of the
molecule.10 This has drawn the attention of the pharmaceutical and agrochemical industry and
now nearly 20% of all pharmaceuticals and 30% of all agrochemicals contain a C–F bond,
including the important drugs Lipitor and Prozac.10
While there are clearly a number of benefits for the introduction of carbon–fluorine bonds into
molecules, a number of these molecules pose significant threats to the environment. The most
eminent examples of the detrimental side effects of fluorocarbons originate from
chlorofluorocarbons (CFCs) as coolants and propellants. As these gasses are released into the
stratosphere, the photo-induced scission of the carbon chlorine bond occurs, generating a
chlorine radical which reacts with ozone (O3), to produce oxygen gas.11 Stratospheric ozone is
an important molecule as it protects the surface of the Earth from harmful ultra-violet radiation.
The degradation of the ozone layer became so severe that these chemicals were banned in 1989
under the Montreal Protocol on Substances that Deplete the Ozone Layer. CFCs have
subsequently been replaced with hydrofluorocarbons (HFCs) or perfluorocarbons (PFCs), but

65
there are other major drawbacks to the use of these chemicals. HFCs and PFCs can also degrade
in the atmosphere to cause deleterious effects, but the more pressing threat is that these
compounds are extremely potent greenhouse gasses.12 The long lifetime of fluorocarbons, in
some cases up to 10,000 years,12 essentially imparts permanent damage to the atmosphere’s
ability to absorb infrared radiation, increasing the greenhouse effect on Earth. Luckily, the
concentration of fluorocarbons in the atmosphere is low; for example, there is only 70 ppt of CF4
in the atmosphere. However it has taken only 50 years to double its previous concentration.12-14
Due to the robustness of these HFCs and PFCs, it is crucial for scientists to develop synthetic
methodologies which can degrade these fluorocarbons in order to prevent a runaway greenhouse
effect.
3.1.2 Metal Mediated C–F Bond Activation
A number of metal complexes (alkali, alkaline earth and transition metals) have been
investigated for their propensity to interact with carbon–fluorine bonds. The alkali and alkaline
earth metals tend to interact with fluorocarbons via weak secondary bonding interactions.15,16
The hard acid/hard base nature of the complexes favour this interaction which is exemplified in
the barium−copper alkoxide complex, Ba[Cu(OCCH3(CF3)2)3]2, where the twelve coordinate
barium cation is bound by four oxygen atoms and eight fluorine atoms.17 Early transition metals,
which can act as strong Lewis acids, have also been shown to be effective at cleaving carbon–
fluorine bonds. For example, zirconocene complexes are best known for their use in olefin
polymerization,18 however, these hard Lewis acids have also been shown to activate C–F bonds.
The complex [(η5-C5Me5)2Zr(CH3)] [B(p-C6H4F)4] exhibits coordination of the anion to the
zirconium center via an aromatic C–F bond, as evidenced through a drastic upfield chemical shift
of the para-fluorine resonance in the 19F NMR spectrum.19 The group of William Jones has
investigated a number of Group 4 metal hydride species, in particular zirconocene hydride
complexes and their ability to stoichiometrically reduce aromatic, aliphatic, and olefinic C–F
bonds (Scheme 3.1.1).20-23 The mechanism of these processes vary depending on substrate, from
radical chain reactions in fluoroalkanes, to SNAr attack in fluoroaromatics or olefin insertion,
followed by β-fluoride elimination to generate new olefins.24 A number of nickel and iron
complexes have been shown to effect aromatic C–F bond cleavage.25-28 For example, Ni(COD)2
(COD = cyclooctadiene) has been shown to insert into a carbon–fluorine bond in

66
pentafluoropyridine (Scheme 3.1.1). A number of these species have even been exploited
catalytically.29-32 Late transition metals are generally more suitable for these catalytic processes
because their bonds to fluorine are not as strong and they favour metal–carbon bond formation.30
Milstein and Aizenberg were able to exploit the rhodium complex (Me3P)3Rh(C6F5) as an
effective hydrodefluorination catalyst using both silane33 and hydrogen34 as the hydride source
(Scheme 3.1.1). One example of an oxidative addition of fluoroalkanes to a late transition metal
exists; the iridium pincer compound (PCP)Ir(NBE) (PCP = κ3-C6H3-2,6-[CH2P(tBu)2]2; NBE =
norbornene) can add alkyl C–F bonds via an initial C–H bond activation.35
Scheme 3.1.1 - Transition metal complexes for C–F bond activation.
3.1.3 Main Group C–F Bond Activation
A drawback of transition metal C–F bond activation is the relative inability to activate
fluoroalkanes catalytically. However, main group Lewis acids have been shown to activate
fluoroalkanes, first being noted by the group of Olah in 1964.36 In this report, BF3 was used as a
catalyst to activate alkylfluorides to facilitate Friedel-Crafts reactions with benzene and benzene
derivatives (Scheme 3.1.2, top). While one example of stoichiometric C–F bond activation with
B(C6F5)3 is known, it requires a powerful carbodiphosphorane Lewis base.37 Furthermore,
aluminium Lewis acids have been exploited as initiators for similar Friedel-Crafts type

67
reactions38 as well as in hydrodefluorination catalysis.39,40 The strong fluoride affinity which
aluminum possesses has also been exploited stoichiometrically using reagents of the form R2AlX
(X = Cl, C, H, O, S, Se, Te, N) to form C–X bonds from C–F bonds (Scheme 3.1.2, middle).41
However, the most effective and well-studied main group systems have been based on silicon
species, in particular silylium cations.42-45 These systems exploit the thermodynamic
favourability to form Si–F bonds. The active silylium species are generated in situ using a
carbenium cation initiator, generally trityl borate or trityl carborane (Scheme 3.1.2, bottom).
These are highly effective processes, in some cases achieving nearly 3000 TONs and a number
of these cations can even effect the hydrodefluorination of PFCs. Aromatic C–F bond activation
has generally been the realm of transition metal chemistry, and in fact there is only one report of
a main group promoted aromatic C–F bond activation. In this case, the triaryl systems are
predisposed to cyclization via a Friedel-Crafts reaction, and the silylium species is able to
activate the aromatic C–F bond to trigger C–C bond formation (for a more detailed mechanism,
please see Chapter 5, Scheme 5.2.9).46
Scheme 3.1.2 - Main group C–F bond activation.
3.2 Results and Discussion
3.2.1 Stoichiometric Reactions of Phosphines and Boranes with Alkyl Fluorides
The library of substrates which frustrated Lewis pair systems are able to activate continues to
grow.47 Fluoroalkanes had yet to be investigated as substrates for activation by P/B FLPs and it
was the goal of this project to investigate their reactivity. Initially, the FLP combination of

68
tBu3P and B(C6F5)3 was dissolved in CD2Cl2 and added to the substrate 1-fluoropentane (Scheme
3.2.1). The reaction was quite rapid, being complete in a matter of hours. Upon workup a white
solid was isolated in 88% yield (3-1). The 1H NMR spectrum of this sample no longer showed
the doublet of triplets resonance at 4.19 ppm for the CH2F moiety. In addition, a new doublet
was seen for the tert-butyl resonance at 1.58 ppm with a 3JPH = 14 Hz. The 19F NMR spectrum
showed the consumption of the fluoroalkanes with the absence of the fluoroalkanes C–F
resonance at –217.06 ppm. Furthermore, the pentafluorophenyl resonances indicated the
formation of a 4-coordinate borate product, with a meta-para gap of 4.35 ppm. The formation of
a B–F bond is evident as a broad resonance at –191.65 ppm is seen in the 19F NMR spectrum and
a doublet at –0.60 ppm with a 1JBF = 70 Hz is observed in the 11B NMR spectrum, this data
suggests the presence of a fluoroborate anion, [FB(C6F5)3]. Lastly, the 31P NMR spectrum had a
new resonance at 48.5 ppm indicating the formation of an alkylated phosphonium center. These
data supports the formulation of the C–F bond activated product, [tBu3P(C5H11)] [FB(C6F5)3]
(3-1).
Scheme 3.2.1 - C–F bond activation with the FLP, tBu3P and B(C6F5)3.
Having shown the ability of FLPs to promote fluoroalkane activation, the scope was expanded to
investigate the limits of this reactivity. The substrate 1-fluoroadamantane was examined with the
FLP combination of tBu3P and B(C6F5)3. Under identical conditions to the previous reaction, the
C–F bond activated product [tBu3PAd] [FB(C6F5)3] (3-2) was isolated as a white solid in 89%
yield. Multi-nuclear NMR analysis was valuable in identifying the product; the 11B NMR
spectrum showed a doublet at –0.6 ppm with a 1JBF = 67 Hz. The 19F NMR spectrum once again
indicated the formation of a 4-coordinate fluoroborate anion, with a similar meta-para gap of

69
4.32 ppm and a broad singlet at –191.87 ppm representing the newly formed B–F bond. The 31P
NMR spectrum showed an alkylated phosphonium resonance at 48.7 ppm, similarly to that of
3-1. The analogous reaction was attempted with 1,3-difluoropropane. The 1H NMR spectrum
showed the presence of a remaining CH2F functionality at 4.54 ppm with observable coupling to
the fluorine and the neighbouring protons resulting in a 2JHF = 46 Hz and 3JHH = 5 Hz. The 11B
and 19F NMR spectra support the formation of a 4-coordinate fluoroborate anion, with a B−F
resonance at –0.45 ppm and –190.54 ppm respectively. The 19F NMR spectrum also showed a
fluoroalkanes resonance at –222.61 ppm, supporting the notion that one CH2F functionality was
activated while the second remained intact. The 31P NMR spectrum showed the alkylated
phosphonium product as a multiplet at 48.9 ppm. This data supported the formulation of
[tBu3PCH2CH2CH2F] [FB(C6F5)3] (3-3) and this was unambiguously confirmed via X-ray
crystallography (Figure 3.2.1).
Figure 3.2.1 - POV–Ray depiction of 3-3, P: orange, B: green, F: pink, C: black. H-atoms
omitted for clarity.
Solid state structural analysis of 3-3 shows a B–F bond length of 1.432(2) Å and the expected
tetrahedral geometries around both P and B centers with the sum of the bond angles totalling

70
335.8° and 334.3°, respectively. Using two equivalents of the Lewis acid and base did not result
in the double activation of the fluoroalkanes, even at elevated reaction temperatures. The
reaction of 1-fluorocyclohexane with tBu3P and B(C6F5)3 did not proceed analogously to the
aforementioned reactions. The 1H NMR spectrum showed the formation of a new resonance at
5.66 ppm, which indicated the formation of cyclohexene. The 11B, 19F and 31P NMR spectra also
corroborate this elimination result, with clear evidence for the formation of the salt [tBu3PH]
[FB(C6F5)3].48
1,1,1,3,3,3-Hexafluoro-2-(fluoromethoxy)propane was subsequently investigated due to the
presence of both CH2F and CF3 functionalities in an attempt to get a better understanding of the
reactivity and potential chemoselectivity. The reaction with this molecule was undertaken with
tBu3P, B(C6F5)3 and the substrate combined in a 1:1:1 ratio in CH2Cl2. Monitoring the
hexafluoroisopropyl CH resonance in the 1H NMR spectrum gave strong indication that the
trifluoromethyl functionalities remained intact throughout this reaction, as the peak remained as
a heptet at 4.99 ppm with observed 3-bond coupling to the fluorine atoms of 6 Hz. The 19F NMR
spectrum of this reaction displayed the disappearance of the CH2F resonance at –152.42 ppm
without changes to the trifluoromethyl doublet resonance at –73.16 ppm with a 3JHF = 6 Hz. The
expected resonance for the newly formed B–F bond was observed as a broad singlet at
−185.27 ppm and the meta–para gap has once again been reduced to 4.74 ppm, indicating
4-coordinate borate formation. The 11B and 31P NMR spectra showed resonances at –0.36 ppm
(1JBF = 62 Hz) and 45.5 ppm, respectively. This data supports the selective formation of the
product [tBu3P(CH2OCH(CF3)2)] [FB(C6F5)3] (3-4). This selective reactivity likely occurs
because of the polarized nature of the C–F bond in the CH2F functionality, allowing the Lewis
acid to interact with the directed carbon–fluorine dipole.49 Whereas the dipole moment is more
diffuse in the trifluoromethyl functionality, preventing B(C6F5)3 from polarizing the C–F bond
enough for phosphine attack. This result also indicates that the reactivity of B(C6F5)3 differs
from other Lewis acids used in C–F bond activation, the complete C–F bond scission and
carbocation formation seen with other main group catalysts is not observed here with B(C6F5)3.
It is not unreasonable to propose that activation proceeds through a weak interaction between the
substrate fluorine atoms and B(C6F5)3, as evidence for these weak van der Waals interactions has
been previously reported with B(C6F5)3.50 In attempts to force trifluoromethyl activation, a 1:1
ratio of tBu3P and B(C6F5)3 were dissolved in CD2Cl2 and added to an equivalent of

71
α,α,α-trifluorotoluene and heated to 60 °C overnight. However, this reaction did not result in any
observable C–F bond activation.
A number of other Lewis bases were investigated for their ability to activate carbon–fluorine
bonds. The utilization of secondary phosphines could potentially lead to a novel synthetic route
to form substituted phosphines via a deprotonation of the activated product. Reactions with
secondary phosphines were performed but showed a limited scope for C–F activation. The
combination of tBu2PH and B(C6F5)3 was reacted with 1,3-difluoropropane in CH2Cl2 and
resulted in the product [tBu2PH(CH2CH2CH2F)] [FB(C6F5)3] (3-5) (Scheme 3.2.2). The 1H
NMR spectrum again gave evidence for the single activation product, with a resonance for the
remaining CH2F moiety seen at 4.54 ppm as a doublet of triplets with coupling to the
neighbouring fluorine and hydrogen atoms of 2JHF = 46 Hz, and 3JHH = 5 Hz, respectively. The
P–H moiety was evident in the 1H NMR spectrum with a doublet resonance at 5.64 ppm with a
1JPH = 458 Hz. The 31P NMR spectrum verified the formation of the alkylated product, as a
doublet resonance was observed at 48.3 ppm with a 1JPH = 458 Hz. The 11B and 19F NMR
spectra substantiated the formation of a fluoroborate anion. This combined data supported the
proposed formulation of product [tBu2PH(CH2CH2CH2F)] [FB(C6F5)3] (3-5) and this was
unambiguously confirmed via X-ray crystallography (Figure 3.2.2). The molecular structure
shows a B–F bond length of 1.435(4) Å. In addition, the tetrahedral nature of the phosphorus
and boron centers with the sum of the bond angles totalling 339.1° and 334.1°, respectively.

72
Figure 3.2.2 – POV–Ray depiction of 3-5, P: orange, B: green, F: pink, C: black, H: white. H-
atoms omitted for clarity.
The use of alternate secondary phosphines was challenging due to of competitive nucleophilic
aromatic substitution reactions between the phosphine and B(C6F5)3,51 and the expansion of this
chemistry proved to be difficult. A potential extension of this methodology may be the
formation of carbon–carbon bonds. A number of FLP systems have utilized nucleophilic carbon
centers as the Lewis bases, namely enamines, and pyrroles,52 amongst others.37,53 Attempts to
use 1,2,5−trimethylpyrrole, N-tertbutylpyrrole and the enamine, 1-piperidino-1-cyclohexene
under a number of conditions as the Lewis base did not afford any C–F bond activation and or
C–C bond formation (Scheme 3.2.2).

73
Scheme 3.2.2 - C–F bond activation with tBu2PH and reactions with carbon Lewis bases.
3.2.2 Stoichiometric Transformations of C–F bonds
Expanding upon the reactivity of B(C6F5)3 mediated C–F bond activation led to the use of
alternative nucleophiles to phosphines. These nucleophiles were various borate anions generated
from classical FLP reactions. To that end, the reaction with a hydridoborate anion as an
alternative nucleophile was investigated. To a 1:1 mixture of B(C6F5)3 and 1-fluoropentane was
added an equivalent of the salt [tBu3PH] [HB(C6F5)3], which was generated from the reaction of
H2 with tBu3P and B(C6F5)3.54 This reaction resulted in the formation of pentane, [tBu3PH]
[FB(C6F5)3] and an equivalent of B(C6F5)3 (Scheme 3.2.3). It should be noted that the
phosphonium hydridoborate salt does not react with 1-fluoropentane in the absence of B(C6F5)3,
inferring that borane activation of the C–F bond is required for hydride attack. In the same vein,
treatment of 1-fluoroadamantane with the salt, [tBu3PH] [HB(C6F5)3], in the presence of
B(C6F5)3 also cleanly yields the stoichiometric production of adamantane, [tBu3PH] [FB(C6F5)3],
and B(C6F5)3 (Scheme 3.2.3). In both of the aforementioned reactions, the 11B NMR spectra
showed a broad resonance at ~25 ppm (ν1/2 = 1038 Hz). The 19F NMR spectra also showed
broad resonances for the ortho, para and meta fluorine atoms of the pentafluorophenyl rings with
no observable B–F resonance. Two possible explanations for these observations are imaginable.
This resonance could either represent the bridging fluoride anion [(C6F5)3B–F···B(C6F5)3]– or a

74
dynamic exchange process is occurring between the fluoride ion between and the two borane
centers. Independent experiments between a fluoroborate anion and free B(C6F5)3 supported
these observations. This exchange process implied that this reaction might be able to proceed
with a catalytic amount of B(C6F5)3. Therefore, revisiting the reaction of 1-fluoropentane with
[tBu3PH] [HB(C6F5)3] using 10 mol% B(C6F5)3 resulted in complete conversion to pentane
(Scheme 3.2.3).
Scheme 3.2.3 - Reactions of fluoroalkanes with [tBu3PH] [HB(C6F5)3] and B(C6F5)3.
In a similar fashion, [tBu3PSPh] [PhSB(C6F5)3] was utilized as a nucleophile. This salt is readily
generated via the reaction of diphenyl disulfide with the FLP combination tBu3P and B(C6F5)3.55
Addition of this salt to a mixture of 1-fluoropentane and B(C6F5)3 results in the sulfur atom
acting as a nucleophile towards the activated fluoroalkanes. The thioether, C5H11SPh, was
generated as well as the salt [tBu3SPh] [FB(C6F5)3] (Scheme 3.2.4). The salt gives rise to the
expected resonances in the various multinuclear 31P, 19F and 11B NMR spectra,55 while the 1H
NMR spectrum shows a diagnostic resonance for the SCH2 protons at 2.91 ppm with a 3JHH =
7 Hz, which is consistent with the reported data for this thioether.56
Boron–oxygen bonds are notoriously hard to break, and in many cases this prevents catalytic
turnover in FLP systems.57,58 We sought to use this methodology to break B–O bonds in a
similar fashion to the B–S bonds. The reactivity of alcohols with FLPs had been previously
investigated,59 and the salt [tBu3PH] [MeOB(C6F5)3] can be easily generated from the reaction of
anhydrous methanol with the FLP combination tBu3P/B(C6F5)3. Using this salt as the
nucleophile, the combinations of 1-fluoropentane with a stoichiometric amount of B(C6F5)3
resulted in the transfer of a methoxy moiety to form the new ether product (Scheme 3.2.4). The
salt by-product, [tBu3PH] [FB(C6F5)3] was once again observed and the newly formed

75
1-methoxypentane was confirmed by 1H NMR analysis.60 This process can also be effected by a
catalytic amount of B(C6F5)3 (10 mol%).
Scheme 3.2.4 - Stoichiometric and catalytic heteroatom transfer via C–F activation.
3.2.3 Catalytic Hydrodefluorination of Alkyl Fluorides using B(C6F5)3
The above reactions demonstrate the utility of B(C6F5)3 promoted C–F bond activation in a
variety of transformations. Extensions this type of reactivity to further catalytic processes was
sought. As previously mentioned, a small number of main group Lewis acids have been utilized
as hydrodefluorination catalysts for fluoroalkanes, but B(C6F5)3 had not been previously
investigated. Initially, 5 mol% of B(C6F5)3 was used as a catalyst and combined with Et3SiH and
1-fluoroadamantane in CD2Cl2 (Table 3.2.1). Unlike in other Lewis acid catalyzed
hydrodefluorination reactions, minimal effervescence of hydrogen gas could be observed. In
prior cases, the hydrogen gas is produced from rapid Friedel-Crafts alkylation between the
carbocation and solvent. This reaction was extremely rapid, and within 5 minutes complete
conversion to adamantane and Et3SiF was observed in the 1H and 19F NMR spectra (Figure
3.2.3). This catalytic process was extended to additional fluoroalkanes such as 1-fluoropentane,
1,3-difluoropropane, and 1-fluorocyclohexane (Table 3.2.1). In all cases, rapid
hydrodefluorination occurred and the corresponding alkane product was observed in the 1H
NMR spectrum. The 19F NMR spectrum of the reactions showed free B(C6F5)3 as well as the
formation of Et3SiF at –176.07 ppm. In the example of 1,1,1,3,3,3-hexafluoro-
2-(fluoromethoxy)-propane, the reaction was extremely sluggish at 25 °C and required warming
to 60 °C for 18 h, resulting in selective reduction of the fluoromethoxy- fragment, affording
CH3OCH(CF3)2 in 72% conversion (Table 3.2.1). The resonance corresponding to the (CF3)2CH
proton is still observed in the 1H NMR spectrum as a heptet at 4.01 ppm, with coupling seen to

76
the two trifluoromethyl groups of 3JHF = 6 Hz and a new methoxy- resonance is observed at
3.73 ppm. The 19F NMR spectrum shows the trifluoromethyl resonance at –74.73 ppm and the
formation of Et3SiF at –176.07 ppm. The slower reaction time is attributed to the presence of the
etheral oxygen, which offers the potential of a weak donor–acceptor complex with B(C6F5)3,
while rendering the C–F bond less polar. Subsequently, a large excess of silane was used in this
reaction in an effort to reduce the trifluoromethyl groups, but to no avail. Thus it can be
concluded that B(C6F5)3 is not an effective catalyst for the degradation of CF3 functionalities.
Table 3.2.1 - Hydrodefluorination catalysis with B(C6F5)3.
Substrates Products Time Temp. °C Conversion (%)a
5 min 25 >95
5 min 25 >95
18 h 60 72
5 min 25 >95
5 min 25 >95
a Conversions determined by 1H NMR analysis.

77
Figure 3.2.3 - 1H and 19F NMR spectra of B(C6F5)3 catalyzed hydrodefluorination of
1-fluoroadamantane.
This selective hydrodefluorination of fluoroalkanes is unique compared to the metal-free
methods previously reported. In the previous metal-free cases, the hydrodefluorination catalysts
are strong enough Lewis acids to completely cleave all alkyl C–F bonds, leading to carbocationic
rearrangement products and Friedel-Crafts type side products.42 Using B(C6F5)3 as a catalyst
addresses these issues, as it selectively produces the corresponding hydrocarbon without any side
reactions. This implies that a different mechanism from the previously described processes
occurs. This finding is conceptually related to the work from the groups of Piers,61 Gevorgyan,62

78
Rubinstajn63 and Brook64-67 where B(C6F5)3 is used as a catalyst for the hydrosilylation of
ketones and the formation of silicone oligomers and polymers. The hydrosilylation reaction
mechanism proceeds via Lewis acid Si–H bond activation followed by subsequent attack of the
unsaturated substrate to the silicon center. A similar reaction mechanism could proceed for the
HDF reaction, where the fluoroalkanes attacks the activated silicon center (Mechanism A,
Scheme 3.2.5). Another possible mechanism for this process is one where the borane activates
the polarized C–F bond, as it did in the stoichiometric FLP activations, followed by hydride
attack from the silane. The transient hypervalent silicon center abstracts fluoride from the
fluoroborate anion in a concerted fashion, releasing B(C6F5)3 back into the catalytic cycle
(Mechanism B, Scheme 3.2.5). Both of these mechanisms are plausible and can both explain the
hydrodefluorination reactivity seen.
Scheme 3.2.5 - Two possible mechanisms for the B(C6F5)3 catalyzed hydrodefluorination
reaction.
3.3 Conclusions
This chapter has presented the reactivity of frustrated Lewis pairs with fluoroalkanes. A number
of these substrates were readily activated with the combination of tBu3P and B(C6F5)3, resulting
in the formation of the alkylated phosphonium fluoroborate salts. These reactions proceeded
under ambient conditions, which was encouraging due to the strength of the carbon–fluorine
bond. These activations proved to be chemoselective, leaving trifluoromethyl substituents

79
untouched in the presence of monofluoroalkanes. Functionalization of fluoroalkanes has been
achieved stoichiometrically and catalytically using borate nucleophiles derived from FLP
activated substrates, resulting in alkane, thioether and ether products. Catalytic
hydrodefluorination of fluoroalkanes was achieved using 5 mol% B(C6F5)3, with the majority of
these reactions being complete within 5 minutes. The hydrodefluorination reactions cleanly
produced the corresponding alkane and were also selective, leaving trifluoromethyl groups
untouched. Two possible mechanisms are proposed for the catalytic process, one where
B(C6F5)3 activates the silane, and the other involving activation of the substrate C–F bond by
B(C6F5)3.
3.4 Experimental Section
3.4.1 General Considerations
All preparations and manipulations were carried out under an anhydrous N2 atmosphere using
standard Schlenk and glovebox techniques. All glassware was oven-dried and cooled under
vacuum before use. Solvents were purified with a Grubbs-type column system manufactured by
Innovative Technologies and dispensed into thick-walled Schlenk glass flasks equipped with
Teflon valve stopcocks. All solvents were degassed prior to use via repeated freeze-pump-thaw
cycles. CD2Cl2 (Aldrich) was deoxygenated, distilled over CaH2, then stored over 4 Å molecular
sieves before use. NMR spectra were obtained on a Bruker AvanceIII-400 MHz spectrometer.
In selective cases the resonance for the ipso-carbon in the 13C{1H} NMR is not located.
Combustion analyses were performed in house at Analest, employing a Perkin Elmer 2400 Series
II CHNS Analyzer. tBu3P, and tBu2PH were purchased from Strem and used without further
purification. The fluoroalkanes used were purchased from Alfa Aesar or Sigma Aldrich and
used without further purification. Finally, B(C6F5)3 was purchased from Boulder Chemicals and
used without further purification.
3.4.2 Synthesis of Compounds
[tBu3P(C5H11)] [FB(C6F5)3] (3-1)
tBu3P (40 mg, 200 μmol) and B(C6F5)3 (100 mg, 195 μmol) were added to a solution of
1-fluoropentane (18 mg, 195 μmol) in 5 mL of dichloromethane. The solution was allowed to
stir for four hours; the solvent was removed in vacuo and the residue was washed with hexanes
(2 2 mL) yielding the product as a white powder. (138 mg, 172 μmol, 88%).

80
Anal. Calcd. for C35H38BF16P (%) C: 52.26, H: 4.78; found C: 52.04, H: 5.15.
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 2.08 (m, CH2, 2H), 1.90 (m, CH2, 2H), 1.58 (d, 3JPH =
14 Hz, tBu, 27H), 1.42 (m, CH2 4H), 0.92 (t, 3JHH = 7 Hz, CH3, 3H).
11B NMR (CD2Cl2, 128 MHz, BF3·OEt2): δ –0.6 (d, 1JBF = 70 Hz).
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –136.55 (p, 3JFF = 5 Hz, 6F, o-C6F5), –163.60 (t, 3JFF =
20 Hz, 3F, p-C6F5 ), –167.95 (m, 6F, m-C6F5); –191.65 (br. s, BF).
31P NMR (CD2Cl2, 162 MHz, H3PO4): δ 48.5 (m).
13C{1H} NMR (CD2Cl2, 100 MHz, Me4Si): δ 148.30 (br. d, 1JCF = 241 Hz), 139.04 (br. d, 1JCF =
256 Hz), 137.01 (br. d, 1JCF = 264 Hz), 39.6 (d, 1JPC = 29 Hz), 34.4 (d, 2JPC = 12 Hz), 30.0, 24.9
(d, 3JPC = 7 Hz), 22.4 (d, 4JPC = 2 Hz), 19.2 (d, 1JPC = 35 Hz), 13.9.
[tBu3P(adamantyl)] [FB(C6F5)3] (3-2)
tBu3P (20 mg, 98 μmol) and B(C6F5)3 (50 mg, 98 μmol) were added to a solution of
1-fluoroadamantane (14 mg, 98 μmol) in 5 mL of dichloromethane. The solution was allowed to
stir for four hours; the solvent was removed in vacuo and the residue was washed with hexanes
(2 2 mL) resulting in a white solid. (75 mg, 86 μmol, 88%).
Anal. Calcd. for C40H42BF16P (%) C: 55.32, H: 4.87; found C: 54.84, H: 4.96.
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 2.61 (t, 3JHH = 4Hz, CH2, 6H), 2.18 (br.s., CH, 3H), 1.81
(m, 24H, 18H tBu, 6H Ad), 1.73 (d, 3JPH = 9 Hz, tBu 9H).
11B NMR (CD2Cl2, 128 MHz, BF3·OEt2): δ –0.60 (d, 1JBF = 67 Hz).
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –136.57 (p, 3JFF = 13 Hz, 6F, o-C6F5), –163.64 (t, 3JFF =
20 Hz, 3F, p-C6F5), –167.96 (m, 6F, m-C6F5); –191.87 (br. s, BF).
31P NMR (CD2Cl2, 162 MHz, H3PO4): δ 48.7 (m).
13C{1H} NMR (CD2Cl2, 100 MHz, Me4Si): δ 148.42 (br. d, 1JCF = 243 Hz), 139.13 (br. d, 1JCF =
241 Hz), 137.07 (br. d, 1JCF = 251 Hz), 45.17, (d, 1JPC = 18 Hz), 39.91 (d, 2JPC = 4 Hz), 35.88 (d,
3JPC = 2 Hz), 32.59, 32.21, 30.25, 29.48 (d, 1JPC = 8 Hz).
[tBu3P(CH2CH2CH2F)] [FB(C6F5)3] (3-3)
tBu3P (40 mg, 200 μmol) and B(C6F5)3 (100 mg, 195 μmol) were added to a solution of
1,3-difluoropropane (16 mg, 200 μmol) in 5 mL of dichloromethane. The solution was allowed
to stir for four hours; the solvent was removed in vacuo and the residue was washed with

81
hexanes (2 2 mL). X-ray quality crystals were grown from a CH2Cl2/hexane mixture. (147 mg,
185 μmol, 94 %).
Anal. Calcd. for C33H33BF17P (%) C: 49.90, H: 4.19; found C: 49.86, H: 4.11.
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 4.54 (dt, 2JHF = 46 Hz, 3JHH = 5 Hz, CH2F, 2H), 2.30 (m,
CH2CH2, 4H), 1.60 (d, 3JHP = 14 Hz, tBu, 27H).
11B NMR (CD2Cl2, 128 MHz, BF3·OEt2): δ –0.45 (br.s).
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –136.58 (p, 3JFF = 13 Hz, 6F, o-C6F5), –162.45 (t,
3JFF = 20 Hz, 3F, p-C6F5), –166.88 (m, 6F, m-C6F5); –190.57 (br. s, BF), –222.61 (m, 1F, CH2F).
31P NMR (CD2Cl2, 162 MHz, H3PO4): δ 48.9 (m).
13C{1H} NMR (CD2Cl2, 100 MHz, Me4Si): δ 148.38 (br. d, 1JCF = 238 Hz), 139.18 (br. d,
1JCF = 251 Hz), 137.06 (br. d, 1JCF = 252 Hz), 83.28 (dd, 1JCF = 168 Hz, 3JPC = 13 Hz), 39.84 (d,
1JPC = 29 Hz), 29.96 (Calkyl), 26.30 (dd, 2JPC= 20 Hz, 2JCF = 6 Hz), 15.21 (dd, 1JCP = 40 Hz,
3JCF = 5 Hz).
[tBu3P(CH2OCH(CF3)2)] [FB(C6F5)3] (3-4)
tBu3P (40 mg, 200 μmol) and B(C6F5)3 (100 mg, 195 μmol) were added to a solution of
1,1,1,3,3,3-hexafluoro-2-(fluoromethoxy)propane (38 mg, 200 μmol) in 5 mL of
dichloromethane. The solution was allowed to stir overnight; the solvent was removed in vacuo
and the residue was washed with hexanes (2 2 mL). (154 mg, 168 μmol, 86 %).
Anal. Calcd. for C35H32BF22OP (%) C: 45.28, H: 3.47; found C: 45.90, H: 3.65.
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 4.99 (h, 3JHF = 6 Hz, CH, 1H), 4.95 (d, 2JHP = 16 Hz,
CH2, 2H), 1.59 (d, 3JHP = 15 Hz, tBu, 27H).
11B NMR (CD2Cl2, 128 MHz, BF3·OEt2): δ –0.36 (d, 1JBF = 62 Hz).
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –73.16 (d, 3JFH = 6 Hz, 6F, CF3), –135.80 (p, 3JFF =
13 Hz, 6F, o-C6F5), –161.93 (t, 3JFF = 20 Hz, 3F, p-C6F5), –166.67 (m, 6F, m-C6F5); –185.27 (br.
s, BF).
31P NMR (CD2Cl2, 162 MHz, H3PO4): δ 45.5 (m).
13C{1H} NMR (CD2Cl2, 100 MHz, Me4Si): δ 148.27 (br. d, 1JCF = 244 Hz), 139.26 (br. d, 1JCF =
249 Hz), 137.12 (br. d, 1JCF = 242 Hz), 122.95 (q, 1JCF = 285 Hz), 76.60 (CH(CF3)2), 63.03 (d,
1JCP = 52 Hz), 39.66 (d, 1JCP = 26 Hz), 30.29, 29.51.

82
[tBu2PH(CH2CH2CH2F)] [FB(C6F5)3] (3-5)
tBu2PH (29 mg, 200 μmol) and B(C6F5)3 (100 mg, 200 μmol) were added to a solution of
1,3-difluoropropane (16 mg, 200 μmol) in 5 mL of dichloromethane. The solution was allowed
to stir overnight; the solvent was removed in vacuo and the residue was washed with hexanes
(2 2 mL), resulting in the product as a white solid (130 mg, 176 μmol, 88 %).
Anal. Calcd. for C29H25BF17P (%) C: 47.18, H: 3.41; found C: 47.03, H: 3.38.
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 5.64 (d, 1JHP = 458 Hz, PH, 1H), 4.54 (dt, 2JHF = 46 Hz,
3JHH = 5 Hz, CH2F, 2H), 2.22 (d, 3JHP= 16 Hz, tBu, 18H).
11B NMR (CD2Cl2, 128 MHz, BF3·OEt2): δ –0.46 (d, 1JBF = 68 Hz);
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –136.55 (p, 3JFF = 13 Hz, 6F, o-C6F5), –162.03 (t, 3JFF =
20 Hz, 3F, p-C6F5), –166.68 (m, 6F, m-C6F5), –186.0 (br. s, BF), –221.13 (m, CH2F).
31P NMR (CD2Cl2, 162 MHz, H3PO4): δ 48.3 (d, 2JPH = 458 Hz).
13C{1H} NMR (CD2Cl2, 100 MHz, Me4Si): δ 148.26 (br. d, 1JCF = 242 Hz), 139.25 (br. d, 1JCF =
237 Hz), 137.04 (br. d, 1JCF = 253 Hz), 83.16 (dd, 1JCF= 168 Hz, 3JCP = 11 Hz), 33.57 (d, 1JCP =
34 Hz), 27.28, 26.02, 11.71 (dd, 1JCP = 43 Hz, 3JCF = 5Hz).
Stoichiometric Reactions
tBu3P (40 mg, 200 μmol) and B(C6F5)3 (100 mg, 200 μmol) were added to a solution of
1-fluorocyclohexane (20 mg, 200 μmol) in 1 mL of CD2Cl2. The 1H NMR spectrum showed
resonances attributable to cyclohexene. These signals were identical to those from an authentic
sample. The 11B{1H}, 19F, and 31P NMR spectra were consistent with the complete consumption
of the 1-fluorocyclohexane and the formation of the known salt [tBu3PH] [FB(C6F5)3].48
Selective diagnostic NMR chemical shifts are reported below.
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 5.66 (s, cyclohexene), 5.43 (d, 1JPH = 442 Hz, PH), 1.98
(m, CH2 cyclohexane), 1.61 (d, 3JPH= 16 Hz, tBu), 1.61 (m, CH2 cyclohexane, overlapped with
tBu resonance).
11B NMR (CD2Cl2, 128 MHz, BF3·OEt2): δ –0.52 (d, 1JBF = 70 Hz).
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –187.32 (br. s).
31P NMR (CD2Cl2, 162 MHz, H3PO4): δ 57.13 (d, 1JPH = 442 Hz).

83
[tBu3PH] [HB(C6F5)3] (14 mg, 20 μmol) and B(C6F5)3 (10 mg, 20 μmol) were added to a solution
of 1-fluoropentane (2 mg, 20 μmol) in 1 mL of CD2Cl2. The 1H NMR spectrum shows the
emergence of resonances attributable to pentane. The 19F NMR spectrum was consistent with
the complete consumption of the 1-fluoropentane. The 31P NMR spectrum is unchanged showing
the resonance arising from the phosphonium cation [tBu3PH]. The 19F NMR spectrum was
consistent with the complete consumption of the 1-fluorocyclohexane. The 11B and 19F NMR
spectra showed broad resonances, indicating fluoride exchange between the two boron centers.
Identical resonances were observed in an independent reaction of [tBu3PH] [FB(C6F5)3] and
B(C6F5)3, consistent with fluoride exchange. Selective diagnostic NMR chemical shifts are
reported below. This reaction also proceeded using 10 mol% of B(C6F5)3 as a catalyst.
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 5.41 (d, 1JPH = 440 Hz, PH), 1.30 (m, CH2, 6H), 0.89 (t,
3JHH = 7Hz, CH3, 6H).
11B NMR (CD2Cl2, 128 MHz, BF3·OEt2): δ 25 (br.).
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –132.55 (br., o-F), –154.32 (br., p-F) and –164.35 (br.,
m-F).
31P NMR (CD2Cl2, 162 MHz, H3PO4): δ 57.35 (d, 1JPH = 440 Hz).
[tBu3PH] [HB(C6F5)3] (14 mg, 20 μmol) and B(C6F5)3 (10 mg, 20 μmol) were added to a solution
of 1-fluoroadamantane (3 mg, 20 μmol) in 1 mL of CD2Cl2. The 1H NMR spectrum shows the
emergence of resonances attributable to adamantane. This was confirmed by comparison to an
authentic sample. The 31P{1H} NMR spectrum is unchanged showing the resonance arising from
the phosphonium cation [tBu3PH]. The 19F NMR spectrum was consistent with the complete
consumption of 1-fluoroadamantane. The borate resonances in the 11B and 19F spectra showed
identical broad resonances to those previously mentioned. These same resonances were observed
in an independent solution of [tBu3PH] [FB(C6F5)3] and B(C6F5)3, consistent with exchange of
fluoride between the anion and the borane as described above. Selective diagnostic NMR
chemical shifts are reported below.

84
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 5.43 (d, 1JPH = 442 Hz, 1H, PH), 1.86 (br. s, 4 H,
adamantane CH), 1.76 (br. s, 12 H, adamantane, CH2).
PhSSPh (5 mg, 24 μmol) was added to a solution of tBu3P (5 mg, 24 μmol) and B(C6F5)3 (25 mg,
49 μmol) in 1 mL of CD2Cl2. To this solution, 1-fluoropentane (2 mg, 24 μmol) was
immediately added. The reaction was allowed to react for 30 minutes before the multinuclear
NMR spectra were obtained. The 1H NMR spectrum is consistent with the complete
consumption of the 1-fluoropentane and the formation of PhSCH2(CH2)3CH368 and
[tBu3PSPh]+.55 The 31P{1H} NMR spectrum showed the resonance arising from the phosphonium
cation [tBu3PSPh]+. The 19F NMR spectrum was consistent with the complete consumption of
1-fluoropentane. The 11B and 19F NMR spectra showed resonances attributable to the
aforementioned fluoride ion exchange between the B(C6F5)3 molecules. Selective diagnostic
NMR chemical shifts are reported below.
1H NMR (CD2Cl2, 400 MHz, Me4Si) δ: 7.30 (m, 5H, Ph), 2.91 (t, 3JHH = 7 Hz, 2H, SCH2), 1.64
(m, overlapped with tBu peaks, 2H, CH2), 1.35 (m, 4H, CH2CH2), 0.87 (t, 3JHH = 7 Hz, 3H, CH3)
[tBu3PH] [MeOB(C6F5)3] (22 mg, 29 μmol) was added to a solution of B(C6F5)3 (15 mg,
29 μmol) in 1 mL of CD2Cl2. To this solution, 1-fluoropentane (2 mg, 29 μmol) was added and
the mixture was allowed to react for 30 minutes before multinuclear NMR analysis was
undertaken. The 1H NMR spectrum showed the complete consumption of 1-fluoropentane as
well as the [MeOB(C6F5)3]- functionality with concomitant formation of 1-methoxypentane.60
The 11B and 19F NMR spectra again indicate a process where the fluoride ion is rapidly
exchanging between the two boron centers. The 31P NMR spectrum shows that the phosphonium
cation remained intact. Selective diagnostic NMR chemical shifts are reported below. This
reaction also proceeded using 10 mol% B(C6F5)3 as a catalyst.

85
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 5.41 (d, 1JPH = 441 Hz, 1H, PH), 3.33 (t, 2H, 3JHH =
7 Hz, CH2O), 3.28 (s, 3H, OCH3), 1.61 (d, 27H, 3JPH = 16 Hz C(CH3)3) 1.54 (m, 2H CH2), 1.31
(m, 4H, CH2) 0.90 (m, 3H, CH3).
3.4.3 Hydrodefluorination Reaction Procedures
The hydrodefluorination reactions of 1-fluoropentane, 1,3-difluoropropane, 1-fluoroadamantane
and 1-fluorocyclohexane were performed under identical conditions and thus only one sample
methodology is described. 5 mol% B(C6F5)3 (5 mg, 10 μmol) and 1-fluoropentane (18 mg,
195 μmol) were added to a solution of Et3SiH (23 mg, 195 μmol) in 1 mL of CD2Cl2. The
reaction was complete in approximately 5 minutes, the time it took to go down to the NMR
spectrometer. Products were identified by comparison of spectral data to those of authentic
samples. Yields were determined by integration of 1H and or 19F NMR resonances from residual
fluoroalkane to hydrocarbon product. Note: The reaction of 1,1,1,3,3,3-hexafluoro-2-
(fluoromethoxy)propane was performed in an analogous fashion although the mixture was
heated 60 oC for 18h.
3.4.4 X-Ray Crystallography
3.4.4.1 X-Ray Collection and Reduction
Crystals were coated in Paratone-N oil in the glovebox, mounted on a MiTegen Micromount and
placed under an N2 stream, thus maintaining a dry, O2-free environment for each crystal. The
data were collected on a Bruker Kappa Apex II diffractometer. Data collection strategies were
determined using Bruker Apex 2 software and optimized to provide >99.5% complete data to a
2θ value of at least 55°. The data were collected at 150(±2) K for all. The data integration and
absorption correction were performed with the Bruker Apex 2 software package.21
3.4.4.2 X-Ray Solution and Refinement
Non-hydrogen atomic scattering factors were taken from the literature tabulations.16,35 The heavy
atom positions were determined using direct methods employing the SHELX-2013 direct
methods routine. The remaining non-hydrogen atoms were located from successive difference
Fourier map calculations. The refinements were carried out by using full-matrix least squares
techniques on F, minimizing the function ω (Fo-Fc)2 where the weight ω is defined as 4Fo2/2σ
(Fo2) and Fo and Fc are the observed and calculated structure factor amplitudes, respectively. In

86
the final cycles of each refinement, all non-hydrogen atoms were assigned anisotropic
temperature factors in the absence of disorder or insufficient data. In the latter cases atoms were
treated isotropically. C-H atom positions were calculated and allowed to ride on the carbon to
which they are bonded assuming a C-H bond length of 0.95 Å. H-atom temperature factors were
fixed at 1.20 times the isotropic temperature factor of the C-atom to which they are bonded. The
H-atom contributions were calculated, but not refined. The locations of the largest peaks in the
final difference Fourier map calculation as well as the magnitude of the residual electron
densities in each case were of no chemical significance.

87
Table 3.4.1 - Select Crystallographic Data for 3-3 and 3-5.
3-3 3-5
Formula C33H33BF17P C29H24BF17P
Formula weight 794.37 737.26
Crystal System Monoclinic Monoclinic
Space group P2(1)/c P2(1)/n
a(Å) 11.7609(3) 11.3914(5)
b(Å) 24.5495(6) 25.2126(10
c(Å) 12.6226(3) 11.5847(5)
α(deg) 90 90
β(deg) 114.154(1) 114.310(2)
γ(deg) 90 90
V(Å3) 3325.37(14) 3032.3(2)
Z 4 4
Temp. (K) 150 150
d(calc)gcm-1 1.587 1.615
Abs coeff,μ,mm-1 0.204 0.216
Data collected 7664 6946
DataFO2
>3(FO2) 5821 4622
Variables 477 437
R 0.0380 0.0700
Rw 0.0952 0.2224
GOF 1.017 1.392

88
3.5 References
(1) Lemal, D. M. The Journal of Organic Chemistry 2004, 69, 1-11.
(2) Harper, D.; O’Hagan, D.; Murphy, C. In Natural Production of Organohalogen
Compounds; Gribble, G., Ed.; Springer Berlin Heidelberg: 2003; Vol. 3P, p 141-169.
(3) Middleton, W. J. The Journal of Organic Chemistry 1975, 40, 574-578.
(4) Lal, G. S.; Pez, G. P.; Pesaresi, R. J.; Prozonic, F. M.; Cheng, H. The Journal of Organic
Chemistry 1999, 64, 7048-7054.
(5) Hayashi, H.; Sonoda, H.; Fukumura, K.; Nagata, T. Chemical Communications 2002,
1618-1619.
(6) Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications; Wiley,
2006.
(7) Li, Y.; Wu, Y.; Li, G.-S.; Wang, X.-S. Advanced Synthesis & Catalysis 2014, 356, 1412-
1418.
(8) Joubert, J.; Roussel, S.; Christophe, C.; Billard, T.; Langlois, B. R.; Vidal, T. Angewandte
Chemie International Edition 2003, 42, 3133-3136.
(9) Pauling, L. The Nature of the Chemical Bond and the Structure of Molecules and
Crystals: An Introduction to Modern Structural Chemistry; Cornell University Press, 1960.
(10) Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881-1886.
(11) Tang, X.; Madronich, S.; Wallington, T.; Calamari, D. Journal of Photochemistry and
Photobiology B: Biology 1998, 46, 83-95.
(12) Shine, K. P.; Sturges, W. T. Science 2007, 315, 1804-1805.
(13) Harnisch, J.; Borchers, R.; Fabian, P.; Gaggeler, H. W.; Schotterer, U. Nature 1996, 384,
32-32.
(14) Worton, D. R.; Sturges, W. T.; Gohar, L. K.; Shine, K. P.; Martinerie, P.; Oram, D. E.;
Humphrey, S. P.; Begley, P.; Gunn, L.; Barnola, J.-M.; Schwander, J.; Mulvaney, R.
Environmental Science & Technology 2007, 41, 2184-2189.
(15) Kulawiec, R. J.; Crabtree, R. H. Coordination Chemistry Reviews 1990, 99, 89-115.
(16) Kiplinger, J. L.; Richmond, T. G.; Osterberg, C. E. Chemical Reviews 1994, 94, 373-431.
(17) Purdy, A. P.; George, C. F. Inorganic Chemistry 1991, 30, 1969-1970.
(18) Yang, X.; Stern, C. L.; Marks, T. J. Journal of the American Chemical Society 1994, 116,
10015-10031.
(19) Horton, A. D.; Orpen, A. G. Organometallics 1991, 10, 3910-3918.

89
(20) Clot, E.; Mégret, C.; Kraft, B. M.; Eisenstein, O.; Jones, W. D. Journal of the American
Chemical Society 2004, 126, 5647-5653.
(21) Kraft, B. M.; Lachicotte, R. J.; Jones, W. D. Journal of the American Chemical Society
2001, 123, 10973-10979.
(22) Kraft, B. M.; Lachicotte, R. J.; Jones, W. D. Journal of the American Chemical Society
2000, 122, 8559-8560.
(23) Rieth, R. D.; Brennessel, W. W.; Jones, W. D. European Journal of Inorganic Chemistry
2007, 2007, 2839-2847.
(24) Jones, W. D. Dalton Transactions 2003, 3991-3995.
(25) Braun, T.; Perutz, R. N. Chemical Communications 2002, 2749-2757.
(26) Doster, M. E.; Johnson, S. A. Angewandte Chemie International Edition 2009, 48, 2185-
2187.
(27) Johnson, S. A.; Huff, C. W.; Mustafa, F.; Saliba, M. Journal of the American Chemical
Society 2008, 130, 17278-17280.
(28) Johnson, S. A.; Mroz, N. M.; Valdizon, R.; Murray, S. Organometallics 2011, 30, 441-
457.
(29) Braun, T.; Perutz, R. N.; Sladek, M. I. Chemical Communications 2001, 2254-2255.
(30) Kuehnel, M. F.; Lentz, D.; Braun, T. Angewandte Chemie International Edition 2013, 52,
3328-3348.
(31) Saeki, T.; Takashima, Y.; Tamao, K. Synlett 2005, 2005, 1771-1774.
(32) Vela, J.; Smith, J. M.; Yu, Y.; Ketterer, N. A.; Flaschenriem, C. J.; Lachicotte, R. J.;
Holland, P. L. Journal of the American Chemical Society 2005, 127, 7857-7870.
(33) Aizenberg, M.; Milstein, D. Science 1994, 265, 359-361.
(34) Aizenberg, M.; Milstein, D. Journal of the American Chemical Society 1995, 117, 8674-
8675.
(35) Choi, J.; Wang, D. Y.; Kundu, S.; Choliy, Y.; Emge, T. J.; Krogh-Jespersen, K.;
Goldman, A. S. Science 2011, 332, 1545-1548
(36) Olah, G. A.; Kuhn, S. Journal of Organic Chemistry 1964, 29, 2317-2320.
(37) Alcarazo, M.; Gomez, C.; Holle, S.; Goddard, R. Angewandte Chemie International
Edition 2010, 49, 5788-5791.
(38) Kronenthal, D. R.; Muller, R. H.; Kuester, P. L.; Kissick, T. P.; Johnson, E. J.
Tetrahedron Letters 1990, 31, 1241-1244.

90
(39) Klahn, M.; Fischer, C.; Spannenberg, A.; Rosenthal, U.; Krossing, I. Tetrahedron Letters
2007, 48, 8900-8903.
(40) Gu, W.; Haneline, M. R.; Douvris, C.; Ozerov, O. V. Journal of the American Chemical
Society 2009, 131, 11203-11212.
(41) Terao, J.; Begum, S. A.; Shinohara, Y.; Tomita, M.; Naitoh, Y.; Kambe, N. Chemical
Communications 2007, 855-857.
(42) Douvris, C.; Ozerov, O. V. Science 2008, 321, 1188-1190.
(43) Douvris, C.; Nagaraja, C. M.; Chen, C.-H.; Foxman, B. M.; Ozerov, O. V. Journal of the
American Chemical Society 2010, 132, 4946-4953.
(44) Scott, V. J.; Çelenligil-Çetin, R.; Ozerov, O. V. Journal of the American Chemical
Society 2005, 127, 2852-2853.
(45) Panisch, R.; Bolte, M.; Müller, T. Journal of the American Chemical Society 2006, 128,
9676-9682.
(46) Allemann, O.; Duttwyler, S.; Romanato, P.; Baldridge, K. K.; Siegel, J. S. Science 2011,
332, 574-577.
(47) Stephan, D. W.; Erker, G. Angewandte Chemie International Edition 2010, 49, 46-76.
(48) Marwitz, A. J. V.; Dutton, J. L.; Mercier, L. G.; Piers, W. E. Journal of the American
Chemical Society 2011, 133, 10026-10029.
(49) O'Hagan, D. Chemical Society Reviews 2008, 37, 308-319.
(50) Zhao, X.; Stephan, D. W. Journal of the American Chemical Society 2011, 133, 12448-
12450.
(51) Welch, G. C.; Holtrichter-Roessmann, T.; Stephan, D. W. Inorganic Chemistry 2008, 47,
1904-1906.
(52) Dureen, M. A.; Brown, C. C.; Stephan, D. W. Organometallics 2010, 29, 6422-6432.
(53) Inés, B.; Holle, S.; Goddard, R.; Alcarazo, M. Angewandte Chemie International Edition
2010, 49, 8389-8391.
(54) Welch, G. C.; Stephan, D. W. Journal of the American Chemical Society 2007, 129,
1880-1881.
(55) Dureen, M. A.; Welch, G. C.; Gilbert, T. M.; Stephan, D. W. Inorganic Chemistry 2009,
48, 9910-9917.
(56) Gul, K.; Narayanaperumal, S.; Dornelles, L.; Rodrigues, O. E. D.; Braga, A. L.
Tetrahedron Letters 2011, 52, 3592-3596.

91
(57) Longobardi, L. E.; Tang, C.; Stephan, D. W. Dalton Transactions 2014, 43, 15723-
15726.
(58) Lindqvist, M.; Sarnela, N.; Sumerin, V.; Chernichenko, K.; Leskela, M.; Repo, T. Dalton
Transactions 2012, 41, 4310-4312.
(59) Neu, R. C., University of Toronto, 2012.
(60) Van Dyke Tiers, G. Acta Chemica Scandinavica 1998, 52, 1223-1233.
(61) Parks, D. J.; Blackwell, J. M.; Piers, W. E. The Journal of Organic Chemistry 2000, 65,
3090-3098.
(62) Gevorgyan, V.; Rubin, M.; Benson, S.; Liu, J.-X.; Yamamoto, Y. The Journal of Organic
Chemistry 2000, 65, 6179-6186.
(63) Chojnowski, J.; Rubinsztajn, S.; Cella, J. A.; Fortuniak, W.; Cypryk, M.; Kurjata, J.;
Kazmierski, K. Organometallics 2005, 24, 6077.
(64) Kamino, B. A.; Grande, J. B.; Brook, M. A.; Bender, T. P. Organic Letters 2011, 13,
154-157.
(65) Grande, J. B.; Thompson, D. B.; Gonzaga, F.; Brook, M. A. Chemical Communications
2010, 46, 4988-4990.
(66) Grande, J. B.; Gonzaga, F.; Brook, M. A. Dalton Transactions 2010, 39, 9369-9378.
(67) Thompson, D. B.; Brook, M. A. Journal of the American Chemical Society 2008, 130,
32-33.
(68) Reddy, V. P.; Kumar, A. V.; Swapna, K.; Rao, K. R. Organic Letters 2009, 11, 1697-
1700.

92
Chapter 4 Frustrated Lewis Pair Activation of Xenon Difluoride – Uncovering
Highly Lewis Acidic Fluorophosphonium Cations
4
4.1 Introduction
4.1.1 A Brief History of Xenon Chemistry
There have long been indicators of unreactive gases besides nitrogen existing in the atmosphere.
Cavendish reported in 1784 that upon removal of nitrogen and oxygen from an atmospheric
sample, there still remained a small residue of unreactive gas.1 Originally attributed to
experimental error, it was nearly 80 years until concrete evidence for a noble gas was made, with
the observation of unknown spectral lines in the solar chromosphere during a solar eclipse. This
new compound was coined helium, after the Greek word for the sun.1 In the 1890’s Rayleigh
and Ramsay discovered argon, neon, krypton and xenon from the fractional distillation of air.1
These gases were originally termed Edelgas2, which is German for noble gas. Their extreme
unreactivity is owing to the fact that these elements have a complete shell of electrons. Despite
the general inertness of these gases, they have found many practical applications in everyday life.
Helium, the lightest of the noble gas elements, is useful for many purposes since it is lighter than
air and unreactive (unlike hydrogen). It is best known for its use in balloons or blimps but it has
found a very important use as a liquid coolant for superconducting magnets in both NMR and
MRI instruments. The next lightest noble gas, neon, has found uses in certain lamps. Argon is an
extremely useful gas for protecting air sensitive reactions in the laboratory setting since it is
heavier than air. Krypton and xenon gas have found fewer uses in everyday life.
The unreactivity of these elements was also seen as a challenge; it is human nature is to push the
boundaries and challenge what is known in order to discover something new. Predictions from
Von Antropoff3,4 and Pauling5 indicated that the heavier noble gas elements would be able to
participate in chemical reactions under oxidative conditions. However, it wasn’t until the 1960s
that the boundaries were pushed far enough and reactivity of a noble gas was discovered. Neil
Bartlett, from the University of British Columbia, discovered the reaction of oxygen with
platinum hexafluoride to give dioxygenyl hexafluoroplatinate, [O2][PtF6] (Scheme 4.1.1).6 He

93
realized that the first ionization energy for molecular oxygen and xenon were quite similar,7 and
xenon does indeed oxidize platinum hexafluoride, resulting in the product [Xe][PtF6].8
Scheme 4.1.1 - Reactions of platinum hexafluoride with oxygen and xenon.
Shortly thereafter, xenon difluoride was first synthesized by the reaction of xenon gas and
fluorine gas in the presence of an electric arc.9 Xenon difluoride is arguably one of the most
important noble gas compounds ever created as it has found uses in materials science10,11 as well
as numerous applications in organic chemistry as a highly efficient fluorinating reagent.12 Initial
reports of fluorination of unsaturated compounds followed shortly after the discovery of XeF2; in
1964 Chernick et al. discovered the reactions of olefins with XeF2 yielded the fluorinated
hydrocarbons.13 In the case of ethylene they note the formation of both 1,1- and 1,2-
difluorinated alkanes, but with propylene the major product is 1,1-difluoropropane. The scope of
the fluorination was subsequently expanded to other unsaturated substrates including
acetylenes,14 aromatic systems15 and peptides16 (Scheme 4.1.2). However, fluorination reactions
using XeF2 are quite indiscriminate and in recent years its usefulness as a fluorinating reagent
has decreased with the introduction of softer and more selective fluorinating reagents, including
Selectfluor.17
Scheme 4.1.2 - Examples of fluorination reactions with XeF2.
Interestingly, xenon does not only make bonds to fluorine, it was shown to form bonds with
other electronegative elements such as oxygen18 and nitrogen19 and recently the scope has been
expanded to xenon–carbon bonds. Evidence for these bonds were first reported in 1979 with the
formation of Xe(CF3)2. While this product was observed spectroscopically, it was never

94
isolable.20 Unexpectedly, the reaction of B(C6F5)3 with an excess of XeF2 in CH2Cl2 yielded the
salt [(C6F5)Xe] [F3B(C6F5)], however this product proved to be relatively unstable and
decomposed rapidly.21 Fine tuning the reaction conditions in acetonitrile and using a
stoichiometric amount of XeF2 and B(C6F5)3 at –35 °C results in the formation of an acetonitrile
stabilized xenonium cation, [(C6F5)Xe(MeCN)] [F2B(C6F5)2]. This was the first example of a X-
ray crystallographically characterized Xe–C bond.22 XeF2 is also effective at oxidizing heavier
pnictogen molecules, readily forming the corresponding pentavalent Pn(V) compound.23,24
While the reaction of FLP systems with XeF2 has not been studied, the Erker lab has recently
shown reactions of an intramolecular FLP system with reactive substrates such as Cl2, Br2 and
thionyl chloride.25 These reactions result in the halogenation of the FLP and formation of the
zwitterionic salt R2P(X)CH2CH2B(X)(C6F5)2 where X = Cl or Br (Scheme 4.1.3).
Scheme 4.1.3 - FLP reactivity with halogenating reagents.
4.2 Results and Discussion
4.2.1 Reactions of Frustrated Lewis Pairs with Xenon Difluoride
While reports of difluorophosphoranes reacting with Lewis acids such as BF3 or AsF526,27 exist,
the combination of a frustrated Lewis pair with xenon difluoride has never been investigated.
Initiating these efforts, we decided to investigate the reaction of tBu3P and B(C6F5)3 with XeF2.
A 1:1 mixture of phosphine and borane were dissolved in dichloromethane and the solution was
then added to an equivalent of premeasured XeF2. Immediate effervescence could be observed,
indicating that xenon gas was being liberated from the reaction mixture. After approximately
five minutes the reaction was complete and the solvent was removed in vacuo. The residue was
washed with pentane to yield a white powder, identified as the product [tBu3PF] [FB(C6F5)3]
(4-1) in 95% yield (Scheme 4.2.1). The 1H NMR spectrum exhibited a doublet of doublets for
the tBu resonance at 1.63 ppm with a 3JPH = 15.9 Hz and a 4JFH = 1.4 Hz. The 11B NMR
spectrum showed a doublet resonance consistent with boron fluorine bond formation at –

95
0.60 ppm with a coupling constant of 1JBF = 70 Hz. The corresponding B–F resonance was also
seen in the 19F NMR spectrum as a broad quartet at –190.3 ppm with a coupling constant of
1JBF = 70 Hz. The 19F NMR spectrum also gave indication of four coordinate borate formation as
the ortho-fluorine resonances of the C6F5 rings appeared as a multiplet at –136.6 ppm, the para-
fluorine resonances appeared as a triplet at –162.7 ppm with a 3JFF = 20 Hz and the meta-fluorine
resonances appeared as a multiplet at −167.0 ppm. The meta–para gap was 4.3 ppm which
indicates a four coordinate borate species. The final peak seen in the 19F NMR spectrum
corresponded to the PF moiety as a doublet at −171.6 ppm with a large coupling constant of
1JPF = 1019 Hz. This doublet was also seen in the 31P{1H} NMR spectrum at 148.5 ppm with the
same coupling constant of 1JPF = 1019 Hz. This large coupling constant is consistent with other
fluorophosphonium cations which have been generated in the literature.26 These data supported
the proposed formulation of [tBu3PF] [FB(C6F5)3] and the multinuclear NMR spectra can be seen
in Figure 4.2.1. This was unambiguously confirmed via X-ray crystallography (Figure 4.2.2).
X-ray structural analysis of 4-1 shows the expected tetrahedral geometry around both P and B
centers. The P–F and B–F bond lengths are 1.628(2) Å and 1.427(3) Å, respectively, and there
appear to be no strong interactions between the cation and the anion.

96
Figure 4.2.1 - Multinuclear NMR spectra of 4-1.
Scheme 4.2.1 - Synthesis of 4-1, 4-2, 4-3, 4-4, 4-5 and 4-6.

97
Figure 4.2.2 - POV–ray depiction of 4-1, P: orange, B: green, F: pink, C: black. H-atoms
omitted for clarity.
The aforementioned reactivity was extended to a series of phosphine precursors. The reaction of
Mes3P and B(C6F5)3 with XeF2 proceeds cleanly and efficiently, yielding the product
[Mes3PF] [FB(C6F5)3] (4-2) in 84% yield. The diagnostic doublet appeared in the 31P{1H} NMR
spectrum at 92.9 ppm with a coupling constant of 1JPF = 940 Hz as well as in the 19F NMR
spectrum at –116.7 ppm with a coupling constant of 1JFP = 940 Hz indicating the formation of a
P–F bond. The fluoroborate peak is also observed in the 19F NMR spectrum as a broad quartet at
–190.9 ppm with a coupling constant of 1JBF = 69 Hz. The 11B NMR spectrum supports
fluoroborate formation as a doublet resonance at –0.6 ppm with a 1JBF = 69 Hz is observed. The
scope of triaryl phosphines was further expanded to include (o-tol)3P, Ph3P and (p-C6H4F)3P.
The reactions with these compounds proceeded cleanly to the desired products
[(o-tol)3PF] [FB(C6F5)3] (4-3), [Ph3PF] [FB(C6F5)3] (4-4) and [(p-C6H4F)3PF] [FB(C6F5)3] (4-5)
in high yields. The diagnostic doublets indicating P–F formation could be seen in the 31P{1H}
NMR spectra at 104.3 ppm with a 1JPF = 994 Hz, 94.7 ppm with a 1JPF = 996 Hz and 93.3 ppm
with a 1JPF = 998 Hz, respectively. The 19F NMR spectra were also quite informative with

98
characteristic doublets appearing at –125.5 ppm with a 1JPF = 994 Hz, –128.3 ppm with a 1JPF =
996 Hz and –123.8 ppm with a 1JPF = 994 Hz, respectively. The fluoroborate chemical shifts in
the 19F NMR spectra were consistent with what was previously observed, all showing a broad
resonance around –191 ppm. Compound 4-3 was unambiguously characterized via X-ray
crystallography (Figure 4.2.3). To compare, a crystallographic analysis of 4-3 also shows a B–F
bond length of 1.418(6) Å, although it has a P−F bond length of 1.554(3) Å which is
substantially shorter than that of 4-1 at 1.628(2) Å. This discrepancy can perhaps be best
explained by considering the steric attributes of the phosphonium ions. The larger tBu groups in
4-1 cannot as easily accommodate a tetrahedral geometry at P, thereby resulting in a lengthening
its P–F bond in response to steric crowding, whereas 4-3, containing the less sterically
cumbersome o-tolyl groups, can more readily stabilize the additional fluoro substituent. The
differing geometries at P can be appropriately illustrated by comparing the sum of the C–P–C
angles: The more sterically encumbered phosphonium ion, 4-1 (344.4°), is perhaps more
reminiscent of a distorted trigonal pyramid, while that of the less bulky 4-3 (337.2°) better
resembles a true tetrahedron (328.4°). Compound 4-3 also seems to exhibit a weak cation-anion
interaction, although the (B)F···P(F) separation of ca. 3.55 Å is greater than the sum of the van
der Waals radii of these atoms (3.24 Å),28 suggesting that favorable π-stacking and Coulombic
interactions between these ions instead stabilize their mutual orientation in the solid state.
It should be noted that the formation of 4-4 was hindered by the known adduct formation
between Ph3P and B(C6F5)3.29 This adduct initially precipitated out of solution but it slowly
reacted with the XeF2 which led to the formation of the product. An alternate approach to form
the desired product is to initially react Ph3P with XeF2 to yield the difluorophosphorane, Ph3PF2.
This can then be reacted with B(C6F5)3 to cleanly yield 4-4. It is interesting to note that
introduction of a para-fluorine substituent on the aromatic rings in 4-5 allows the reaction to
proceed without the detrimental adduct formation.

99
Figure 4.2.3 - POV–ray depiction of 4-3, P: orange, B: green, F: pink, C: black. H-atoms
omitted for clarity.
We began to introduce increasingly electron withdrawing groups by substituting phenyl rings for
pentafluorophenyl rings. The reaction of (C6F5)Ph2P with B(C6F5)3 and XeF2 resulted in the
formation of the anticipated salt [(C6F5)Ph2PF] [FB(C6F5)3] (4-6) in 84% yield. In contrast to the
aforementioned products, the resonances in the 11B, 19F and 31P{1H} NMR spectra appeared
broadened. The P–F resonance appeared as a broad doublet at 87.2 ppm with a coupling
constant of 1JPF = 1020 Hz in the 31P{1H} NMR spectrum and at –123.4 ppm in the 19F NMR
spectrum with a coupling to the phosphorus center of 1JPF = 1020 Hz and a small coupling to the
ortho-fluorine atoms on the pentafluorophenyl ring of 4JFF = 17 Hz. The 11B NMR spectrum also
showed a broad singlet resonance for the B–F moiety at 1.9 ppm as opposed to the usual doublet
at ~ 0 ppm. These data suggests that there is an equilibrium process involving fluoride ion
exchange occurring. We will revisit this unique result later in this chapter.
Decreasing the phosphine basicity further by using (C6F5)2PhP and (C6F5)3P yielded an
unforeseen result. Reactions of those phosphines with XeF2 and B(C6F5)3 only produced the

100
respective difluorophosphoranes (C6F5)2PhPF2 (4-7) and (C6F5)3PF2 (4-8) (Scheme 4.2.2). These
compounds are easily identified by the high field triplet seen in the 31P{1H} NMR spectrum with
chemical shifts of –57.3 ppm with a 1JPF = 687 Hz and –48.0 ppm with a 1JPF = 695 Hz,
respectively. This product can be observed in the 19F NMR spectrum as a low field doublet at
−20.2 ppm with a 1JPF = 694 Hz, and 0.5 ppm with a 1JPF = 695 Hz, respectively. The Lewis acid
in this reaction is just a bystander and it does not abstract the fluoride.
Scheme 4.2.2 - Reaction of electron deficient phosphines with XeF2 and B(C6F5)3.
These results shine light into the mechanism of the reaction of FLPs with XeF2, as well as the
properties of fluorophosphonium cations. The fact that B(C6F5)3 does not participate in the
reaction with electron deficient phosphines and XeF2 indicate that the mechanism of this reaction
likely proceeds through the known oxidation of the phosphine Lewis base to the
difluorophosphorane followed by fluoride abstraction with B(C6F5)3 (Scheme 4.2.3). These
results also insinuate that if fluorophosphonium cations could be generated from 4-7 and 4-8 that
they should be more Lewis acidic than B(C6F5)3.
Scheme 4.2.3 - Reaction mechanism for the reactivity of FLPs with XeF2.
Phosphines are not the only Lewis bases that can be utilized in FLP chemistry, and to that end
the reactivity of bulky nitrogen bases such as 2,6-dimethylpyridine and bulky phosphites such as
tris(2,6-dimethylphenyl)phosphite and tris(2,4-ditertbutylphenyl)phosphite was explored with
B(C6F5)3 and XeF2. However, these reactions do not yield the desired products. Pyridine
derivatives resulted in intractable mixtures of products. It is likely that the XeF2 reacted with the
pyridine ring on its own, as has been noted in literature.30 Phosphites have also been shown to
react with XeF2 to afford a variety of disproportionation products.31

101
4.2.2 Competitive Fluorophosphonium Lewis Acidity with B(C6F5)3
The broadening of the resonances in both the 31P{1H} and 19F NMR spectra which was observed
in [(C6F5)Ph2PF] [FB(C6F5)3] (4-6) was further probed. Both low and high temperature NMR
experiments were undertaken to test whether or not this observation was the result of an
equilibrium in solution. To facilitate this experiment two samples were prepared, the low
temperature experiment was performed in CD2Cl2 and the high temperature experiment in
C6D5Br. The signals corresponding to the P–F moiety sharpen with decreasing temperature such
that the P–F couplings are resolved at −40 °C. This signal broadens dramatically upon
increasing the temperature to 60 °C (Figure 4.2.4 and Figure 4.2.5). The temperature dependent
behaviour suggests that an equilibrium exists between the fluorophosphonium fluoroborate salt
and the difluorophosphorane and free borane. This result is corroborated by the results from the
Gabbaï group. They have shown that pendent phosphonium cations are capable of interacting
with fluoroborate anions to enhance fluoride ion binding of simple boranes.32

102
Figure 4.2.4 - 31P{1H} NMR spectra of 4-6 in CD2Cl2 (–40 to 25 °C) and C6D5Br (25 to 60 °C).

103
Figure 4.2.5 - 19F NMR Spectra of 4-6 in CD2Cl2 (–40 to 0 °C) and C6D5Br (25 to 60 °C).
The observed lack of reactivity between B(C6F5)3 and electron deficient difluorophosphoranes
further supports these results. The concept of exploiting phosphonium Lewis acidity has largely
been unexplored in literature and will be discussed in greater detail in the following chapter. A
method to gauge the relative Lewis acidities of these phosphonium cations is known as the
Gutmann-Beckett method.33-35 This technique utilizes Et3PO as a donor to interact with the
Lewis acidic center and the difference in the 31P NMR chemical shift is used to correlate the
relative Lewis acidities. Since it appeared the synthesized fluorophosphonium cations had the
propensity to be Lewis acids, we decided to test them with this method.
Compounds 4-1 to 4-3 showed very little change by 31P NMR spectroscopy when reacted with
Et3PO, just a slight broadening of the Et3PO peak. Interestingly, when compounds 4-4 and 4-5
were reacted with Et3PO, very drastic chemical shift changes can be observed. Free Et3PO has a
31P{1H} NMR chemical shift of 50.7 ppm but in these reactions we can see a large downfield

104
shift to 77.0 ppm. This chemical shift is nearly identical to that of the Et3POB(C6F5)3 adduct.36
The phosphonium chemical shifts also change accordingly; the usual doublets at 94.7 ppm and
93.3 ppm are shifted upfield and become triplets at –57.3 ppm and –58.8 ppm, respectively
(Scheme 4.2.4). These chemical shifts correspond to the difluorophosphorane products. The
reactivity of 4-4 and 4-5 with Et3PO indicates that a similar equilibrium to that observed in 4-6
must be occurring.
Scheme 4.2.4 - Reactions of fluorophosphonium fluoroborate salts with Et3PO.
This equilibrium process could be thought of as a way to “protect” B(C6F5)3 with a fluoride ion
and allow it to be exploited under conditions which may have not been previously possible. The
Piers group has developed a protected analogue of B(C6F5)3 using a carbazole based derivative
which can release free B(C6F5)3 upon irradiation of ultraviolet light at 254 nm.37 Attempts were
made to exploit this equilibrium as a novel way to protect B(C6F5)3 in hydrogenation catalysis.
However, using compound 4-6 as a surrogate for free borane in the hydrogenation of
N-benzylidene-tert-butylamine did not result in appreciable hydrogenation of the imine, even
under elevated temperatures and pressures.
4.2.3 Utilizing Different Lewis Acids to Generate Fluorophosphonium Cations
A variety of other Lewis acids were explored in order to access the potentially more Lewis acidic
fluorophosphonium cations from the difluorophosphoranes 4-7 and 4-8. The reaction protocol
had to be slightly manipulated by initially oxidizing the phosphines to the corresponding
difluorophosphoranes in order to prevent side reactions between the phosphines and the Lewis
acids. Initially, a commercially available Lewis acid, trimethylsilyl trifluoromethanesulfonate
(TMSOTf) was used. This Lewis acid has well-known halide abstracting capabilities, including

105
the recent utilization by the Burford group to access a variety of antimony based acceptors via
fluoride ion abstraction.38 To study this reactivity, PPh3 and P(C6F5)Ph2 were chosen because the
corresponding fluorophosphonium fluoroborate salts existed in equilibrium with the neutral
phosphorane and free borane. The difluorophosphorane Ph3PF2 (4-9) is a known compound,
originally prepared using SF4.39 However, in this case it was prepared using XeF2 and Ph3P.
Similarly, (C6F5)Ph2PF2 (4-10) was prepared and was confirmed by 19F and 31P{1H} NMR
analysis with a chemical shift as a doublet of triplets at –34.5 ppm (1JPF = 688 Hz, 4JFF = 14 Hz)
and a triplet of doublets –57.3 ppm (1JPF = 688 Hz, 3JPF = 14 Hz), respectively, with coupling
being observed with the ortho-fluorine atoms of the pentafluorophenyl ring. Reactions of 4-9
and 4-10 with TMSOTf resulted in clean fluoride abstraction and formation of the corresponding
fluorophosphonium triflate salt and trimethylsilylfluoride. Isolation and purification required
removal of the dichloromethane solvent and washing with pentane or hexanes to give white
solids [Ph3PF] [OTf] (4-11) and [(C6F5)Ph2PF] [OTf] (4-12) in 96% and 75% yields respectively
(Scheme 4.2.5). Compound 4-12 was unambiguously characterized by X-ray crystallography
(Figure 4.2.6) and it shows the tetrahedral nature of the cation with a sum of the bond angles
around phosphorus totaling 338.8° and a P–F bond length of 1.547(2) Å, which are in agreement
with the fluorophosphonium cations reported earlier. There is no interaction between the triflate
anion and the cation in the solid state. Thanks to the rapid oxidation of the phosphine with XeF2
this reaction can be performed without the isolation of 4-9 or 4-10 and can be performed in a
two-step, one pot procedure. It should be noted that Et3SiH was unsuccessful in exchanging a
fluoride with a hydride in order to generate an unsymmetrical hydridofluorophosphorane.
Scheme 4.2.5 - Reactions of phosphines with XeF2 and TMSOTf.

106
Figure 4.2.6 - POV–ray depiction of 4-12, P: orange, B: green, F: pink, C: black. H-atoms
omitted for clarity.
This protocol was then applied to electron deficient phosphoranes 4-7 and 4-8. However,
reactions of these phosphoranes with TMSOTf did not yield the desired fluorophosphonium
triflate salts but resulted in no reaction. It was clear that stronger Lewis acids would be required
if we were to access these highly electrophilic fluorophosphonium cations.
Furthermore, Al(C6F5)3·tol40 has been extensively used in our laboratory41 and others42 as a
Lewis acid in FLP type chemistry. Hydrogen activation,41 carbon dioxide reduction,43 olefin
activation44 and C–H activation45 have all been observed using Al(C6F5)3 as a Lewis acid.
Interestingly, the initial report of the alane toluene adduct40 indicated that it was in fact a weaker
Lewis acid than B(C6F5)3; the study used the corresponding benzonitrile adducts and measured
the CN bond stretching frequencies to correlate the Lewis acidity. While this seems
counterintuitive to the FLP reactivity which has been observed, the Krossing group has recently
calculated the fluoride ion affinities (FIA) of a variety of Lewis acids and using this scale
Al(C6F5)3 is a significantly stronger Lewis acid than B(C6F5)3 with a FIA of 530 kJ/mol vs.
444 kJ/mol.46

107
The reaction of 4-7 with either one or two equivalents of Al(C6F5)3·tol yielded the same product
(Scheme 4.2.6). These reagents were mixed at –35 °C in toluene and the reaction mixture
immediately turned a light pink colour. After an hour the solvent was removed in vacuo and a
pink oil was obtained, which was dissolved in a small amount of CH2Cl2 and layered with
pentane to yield light pink X-ray quality crystals. The 31P{1H} NMR chemical shift appears as a
doublet of multiplets at 77.7 ppm with an observed one bond P–F coupling of 1042 Hz as well as
small coupling to the o-C6F5 fluorine atoms. The 19F NMR spectrum showed a corresponding
doublet at –121.9 ppm with a 1JPF = 1042 Hz. The spectrum also indicated fluoride abstraction
has occurred with a broad resonance at –170.6 ppm which is indicative of a bridging
fluoroaluminate species.47 This data indicates that Al(C6F5)3 is indeed a strong enough Lewis
acid to abstract the fluoride from 4-7 to generate the fluorophosphonium salt and the formulation
was unambiguously confirmed via X-ray crystallography to be [(C6F5)2PhPF] [F(Al(C6F5)3)2] (4-
13) (Figure 4.2.7).
Figure 4.2.7 - POV–ray depiction of 4-13, P: orange, B: green, F: pink, C: black. H-atoms
omitted for clarity.

108
The X-ray structure of 4-13 clearly reveals that the geometry around the fluorophosphonium
cation is tetrahedral with a sum of the angles being 339.2° and a P–F bond length of 1.533(2) Å.
The anion contains an approximately linear Al–F–Al angle of 171.84(11)°, with Al–F and Al–F
bond lengths of 1.788(2) Å and 1.780(2) Å, respectively. This data is consistent with known
bridging fluoroaluminate anions reported by Marks et al. with a reported Al–F–Al angle of
167.10(12)° and an Al–F bond length of 1.7945(9) Å.48 The molecular structure of 4-13 is
reminiscent of 4-3 where the cation and anion in 4-13 pack in the solid state such that the P–F
bond is oriented along the Al–F–Al vector with an P–F∙∙∙Al interatomic separation of 3.677(2) Å,
well within the sum of the van der Waals radii of these nuclei (3.98 Å).28 This interesting feature
may be suggestive of π-stacking interactions between arenes of the cation and anion, and/or a
weak dative F–Al attraction, consistent with the well-known hypervalency which aluminum can
participate in.49
We then sought to see if Al(C6F5)3·tol would be a strong enough Lewis acid to abstract a fluoride
from the most electron deficient phosphorane, 4-8. Following similar procedures for the
synthesis of 4-13, two equivalents of Al(C6F5)3·tol were reacted with 4-8 at –35 °C and upon
completion the solvent was removed and the solid dried in vacuo. Isolation of the product
proved to be difficult and the sample was only stable long enough for a multinuclear NMR
analysis to be undertaken. The 31P{1H} and 19F NMR spectra showed a distinctive doublet at
68.0 ppm with a 1JPF = 1062 Hz and –120.7 ppm with a 1JPF = 1062 Hz, respectively. No
evidence of the difluorophosphorane was observed in the either NMR spectra. This data
supports that the product formed in this reaction was indeed the fluorophosphonium salt,
[(C6F5)3PF] [F(Al(C6F5)3)2] (4-14) (Scheme 4.2.6).
The instability of 4-13 and 4-14 can be attributed to the bridging fluoroaluminate anion and in
order to enhance the stability of these electrophilic fluorophosphonium cations a new fluoride
abstracting reagent with a stable anion was utilized. As noted before, silylium cations are a
stable class of Lewis acids which are well known to exhibit a strong affinity for fluoride
binding.50 [Et3Si·tol] [B(C6F5)4] is readily synthesized from Et3SiH and [Ph3C] [B(C6F5)4].51
The reactions of the electron deficient phosphoranes 4-7, 4-8 and 4-10 with [Et3Si·tol] [B(C6F5)4]
proceeded cleanly and effectively to yield the corresponding fluorophosphonium borate salts
[(C6F5)Ph2PF] [B(C6F5)4] (4-15), [(C6F5)2PhPF] [B(C6F5)4] (4-16) and [(C6F5)3PF] [B(C6F5)4]

109
(4-17) in 83%, 89% and 89% yields, respectively (Scheme 4.2.6). Compound 4-15 was
unambiguously characterized by X-ray crystallography and is seen in Figure 4.2.8. The P–F
bond length is 1.540(3) Å, which is consistent with other aryl substituted fluorophosphonium
bond lengths observed thus far. The tetrahedral nature of the phosphonium center can be seen
with a sum of the bond angles around phosphorus totalling 336.2°. Alternatively, 4-15 can be
prepared using 4-17 as a fluoride abstracting reagent in lieu of silylium.
Scheme 4.2.6 - Formation of electrophilic fluorophosphonium salts 4-13 – 4-17.
Figure 4.2.8 - POV–ray depiction of 4-15, P: orange, B: green, F: pink, C: black. H-atoms
omitted for clarity.

110
The 31P{1H} NMR spectra of these compounds again show the diagnostic doublet at 77.7 ppm
with a one bond P–F coupling of 1042 Hz, and 67.8 ppm with a one bond P–F coupling of
1062 Hz, respectively. It can be seen that the anion exchange had minimal effect on the
phosphorus chemical shift. This similarity can also be seen in the 19F NMR where the only
spectral changes originate from the introduction of the pentafluorophenyl rings in the anion. An
interesting trend can be observed in the magnitude of the gap between meta and para fluorine
resonances in the 19F NMR spectra for the cations of 4-15, 4-16 and 4-17. It has been shown
with other compounds, and in particular boranes,52 that the Δδm-p is an indication of the
π-conjugation from the pentafluorophenyl ring to a heteroatom which it is bound to.53 This is
indicative of the availability of the empty orbital in a Lewis acid and thus the relative strength of
the Lewis acid. The Δδm-p in B(C6F5)3 has been reported to be 16.3 ppm,52 the Δδm-p is 22.3 ppm
for 4-15, 25.5 ppm for 4-16 and 28.1 ppm for 4-17. The pattern can be observed that as we add
more electron withdrawing substituents to a fluorophosphonium cation that the Δδm-p increases,
indicating more conjugation to the δ* orbital on the phosphorus.
4.2.4 Determining the Lewis Acidity of Fluorophosphonium Cations
With a variety of Lewis acidic fluorophosphonium cations in hand, we needed an accurate way
to compare the relative strengths with other known Lewis acids. There are a number of well-
known experimental techniques including the Child’s method,54 which utilizes crotonaldehyde
binding to the Lewis acidic center and monitoring the change in 1H NMR chemical shifts.
Another technique is the aforementioned Gutmann-Beckett method33-35 for determining the
Lewis acidity of a species. We proceeded to experimentally investigate the Lewis acidity of 4-17
using these techniques. Initially using the method prescribed by Childs, the reaction of
crotonaldehyde with 4-17 at –35 °C in CD2Cl2 resulted in a variety of unidentifiable products
and the reaction solution changing from colourless to black (Scheme 4.2.7). This was
unexpected but not surprising; results in the next chapter will describe the reactivity of 4-17 with
olefins and oxygen donors. Utilizing the Gutmann-Beckett method yields an improved results
and the reaction of 4-17 with Et3PO in CD2Cl2 indicated strong adduct formation (Scheme 4.2.7).

111
Scheme 4.2.7 - Childs (top) and Gutmann-Becket (bottom) methods for measuring the Lewis
acidity of 4-17.
The 31P{1H} NMR spectrum of this reaction showed a doublet of doublet resonance for the
Et3PO center at 91.1 ppm with observable coupling to both the phosphorus and fluorine on 4-17
with a 2JPP = 66 Hz and a 3JPF = 7 Hz. The phosphonium resonance of 4-17 shifted drastically
upfield to –51.3 ppm, a region where the corresponding difluorophosphoranes are usually
observed. This resonance was a doublet of doublets arising from coupling to the Et3PO fragment
with a 1JPF = 674 Hz and 2JPP = 66 Hz. Free Et3PO has a 31P{1H} chemical shift of 50.7 ppm, the
Et3PO adduct of B(C6F5)3 has a chemical shift of 77.3 ppm,36 resulting in a difference of Δ =
26.6 ppm. As mentioned, the adduct of Et3PO with 4-17 resonates at 91.1 ppm which results in a
chemical shift difference of Δ = 40.4 ppm. This drastic shift implies that 4-17 is nearly 1.52
times as Lewis acidic as B(C6F5)3. This impressive result explains the inability of B(C6F5)3 to
abstract the fluoride from the difluorophosphorane 4-8 and gives a good estimate for the Lewis
acidity of 4-17. However, these results should be taken with a note of caution. The
Gutmann-Beckett method has some drawbacks which need to be taken into consideration. The
method does not take steric factors into account on the chemical shift change since larger steric
hindrance would impede adduct formation. An interesting observation of a possible way to scale
the relative Lewis acidities of the proceeding fluorophosphonium cations is using the 31P NMR
chemical shift (Figure 4.2.9). It can be seen that generally as the number electron withdrawing
groups on the phosphorus centre is increased, the resultant 31P NMR chemical shift moves

112
upfield. The exception to this pattern is 4-2, likely due to the sterically encumbering mesityl
groups bound to the phosphorus centre.
Figure 4.2.9 - 31P{1H} NMR stack plot of the 4-1 – 4-6, 4-16 and 4-17 synthesized.
A more accurate method of determining the relative Lewis acidities is using quantum chemical
calculations. Following the approached laid out by Neil Bartlett55 where he states that the
fluoride ion affinity (FIA) is a reliable measure of the Lewis acidity. The FIA can be evaluated
by combining the strength of a Lewis acid, in this case a fluorophosphonium cation, with the
energy that is released upon binding a fluoride ion F–.55-57 The FIA is defined as the negative of
the enthalpy, ΔH, therefore the strength of the Lewis acid corresponds to the absolute value of
the FIA. The simplest and most general approach to access reliable FIA values now involves the
use of quantum chemical calculations of the F– addition reaction.57 The FIA of the cations 4-1 –
4-6, 4-16 and 4-17 were calculated using the reaction in which F– is added to cation and forming
the corresponding difluorophosphoranes [Equation 1]46 at the WB97XD/def2TZV level of

113
theory58,59 in conjunction with the conductor-like polarizable continuum solvation model
(CPCM)60-63 in dichloromethane. The FIA values are presented below in Table 4.2.1.
Table 4.2.1 - NMR spectroscopic data, calculated 31P chemical shift and FIA for compounds
4-1 – 4-6, 4-16, 4-17 in CD2Cl2 at ambient temperature.
(Equation 1)
31P (δ / ppm) 19F, PF (δ / ppm) Calc. 31P
(δ / ppm)
FIA
[kJ mol-1]
4-1 148.5 (d, 1JPF = 1019 Hz) –171.6 (d) 181.6 163
4-2 92.9 (d, 1JPF = 940 Hz) –116.7 (d) - -
4-3 104.3 (d, 1JPF = 994 Hz) –125.5 (d) - -
4-4 94.7 (d, 1JPF = 996 Hz) –128.3 (d) 108.1 200
4-5 93.3 (d, 1JPF = 998 Hz) –123.8 (d) 103.7 220
4-6 87.2 (d/br, 1JPF = 1020 Hz) –123.4 (dt/br, 4JFF = 17 Hz) 90.9 238
4-16 77.7 (d, 1JPF = 1042 Hz) –121.9 (dp, 4JFF = 17 Hz) 71.6 275
4-17 68.0 (d, 1JPF = 1062 Hz) –120.7 (dsept, 4JFF = 15 Hz) 53.2 311
The calculated FIA values support the experimental observations and follow similar trends.
Substitution of the phosphorus center by stronger electron withdrawing group leads to a higher
FIA value, and thus a more Lewis acidic fluorophosphonium cation. Furthermore, in order to get
an accurate comparison to B(C6F5)3, the FIA was also calculated using the same level of theory
and was found to be 260 kJ/mol. These results are in good agreement with the experimental
observation that a fluoride anion can be abstracted by B(C6F5)3 from the difluorophosphoranes
with up to one pentafluorophenyl substituent, for which the FIA values are lower than that of the
B(C6F5)3. 4-7 and 4-8 have higher FIA’s than that of B(C6F5)3, explaining the resultant
unreactivity. The calculated Lewis acidities compare well to the experimentally determined
results. FIA indicates that 4-17 is ~1.2 times as Lewis acidic as B(C6F5)3, and as mentioned
above the Gutmann-Beckett method implies a factor of 1.52. The discrepancy in values can be
attributed to the fact that the Gutmann-Beckett method is more of a measure of oxophilicity and
it does not take steric factors into account.

114
To obtain a relation between the FIA and experimental 31P NMR chemical shifts, NMR spectra
of 4-1, 4-4 – 4-6, 4-16 and 4-17 were calculated using gauge-including atomic orbital method
(GIAO)64,65 at WB97XD/def2TZV level of theory (Table 4.2.1). Calculated 31P NMR chemical
shifts were referenced to chemical shift of [Me3PF]+,24 and are in a good agreement with
experimental observations. Hence the calculations support the experimental evidence that more
Lewis acidic fluorophosphonium cations absorb in a higher field in the 31P NMR, and electron
donating substituents on phosphorus center give rise to a lower field 31P NMR chemical shift.
4.3 Conclusions
This chapter has presented the reactivity of frustrated Lewis pairs with xenon difluoride. In the
majority of cases the resulting fluorophosphonium fluoroborate salt was formed. However,
when electron withdrawing substituents, such as pentafluorophenyl rings, were appended to the
phosphorus center the resulting reactivity was altered. An equilibrium was observed in 4-6
where the species was fluxional between the salt and the difluorophosphorane plus free borane.
This result is indicative of the Lewis acidity of the fluorophosphonium cation. As the number of
pentafluorophenyl rings were increased on phosphorus, the same reaction yielded only the
oxidized difluorophosphorane and free B(C6F5)3, signifying that if accessible, the
fluorophosphonium cations would have greater Lewis acidity than B(C6F5)3. Fluoride
abstraction was achieved using stronger Lewis acids such as Al(C6F5)3 or [Et3Si] [B(C6F5)4]
yielding the fluorophosphonium salts 4-13 – 4-17. Using the Gutmann–Beckett method, a
standard experimental technique to determine Lewis acidity, it was uncovered that 4-17 was
nearly 1.5 times more Lewis acidic than B(C6F5)3. The relative Lewis acidity of the
fluorophosphonium cations could also be estimated experimentally via 31P NMR spectroscopy,
as there is a correlation between how many electron withdrawing substituents on phosphorus and
the relative upfield shift of the peak. A more accurate technique to measure the Lewis acidity is
by measuring the relative FIAs. These were calculated and showed that 4-17 was a stronger
Lewis acid than B(C6F5)3. The Lewis acidic nature of 4-17 has been exploited and will be
discussed in the following chapter.

115
4.4 Experimental Section
4.4.1 General Considerations
All preparations and manipulations were carried out under an anhydrous N2 atmosphere using
standard Schlenk and glovebox techniques. All glassware was oven-dried and cooled under
vacuum before use. Solvents were purified with a Grubbs-type column system manufactured by
Innovative Technologies and dispensed into thick-walled Schlenk glass flasks equipped with
Teflon valve stopcocks. All solvents were degassed prior to use via repeated freeze-pump-thaw
cycles. CD2Cl2 (Aldrich) was deoxygenated, distilled over CaH2, then stored over 4 Å molecular
sieves before use. C6D5Br (Aldrich) was deoxygenated and stored over 4 Å molecular sieves
before use. NMR spectra were obtained on a Bruker AvanceIII-400 MHz spectrometer and an
Agilent DD2 600 MHz spectrometer. In selective cases the resonance for the ipso-carbon in the
13C{1H} NMR is not located. DFT calculations were performed using Gaussian 09.66
Combustion analyses were performed in house at Analest, employing a Perkin Elmer 2400 Series
II CHNS Analyzer. Al(C6F5)3·C7H840 and [Et3Si·C7H8)2][B(C6F5)4]
51 were prepared following
literature reports. tBu3P, Mes3P, (o-tol)3P, (p-C6H4F)3P, (C6F5)Ph2P, (C6F5)3P and Et3PO were
purchased from Strem and used without further purification. (C6F5)2PhP was purchased from
Alfa Aesar, TMSOTf, and Ph3P were purchased from Sigma Aldrich and used without further
purification. XeF2 was purchased from Apollo Scientific and used without further purification.
Finally, B(C6F5)3 was purchased from Boulder Chemicals and used without further purification.
4.4.2 Synthesis of Compounds
[tBu3PF] [FB(C6F5)3] (4-1)
A solution of tBu3P (40 mg, 195 μmol) in 5 mL of dichloromethane was added to B(C6F5)3
(100 mg, 195 μmol). This solution was added to XeF2 (33 mg, 195 μmol) in 5 mL of
dichloromethane, resulting in immediate effervescence. The effervescence ceased within one
minute but the reaction was allowed to stir for an additional 5 minutes and the solvent was then
removed in vacuo producing a colorless solid that was washed with pentane (3 2 mL) and was
dried in vacuo. Diffraction quality crystals were grown from a saturated solution of
dichloromethane and n-pentane. (139 mg, 185 μmol, 95%).
Anal. Calcd. for C30H27BF17P: C, 47.90; H: 3.62%. Found C, 47.78; H: 3.73%.

116
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 1.63 (dd, 3JPH = 15.9 Hz, 4JFH = 1.4 Hz, 27H, CH3).
11B NMR (CD2Cl2, 128 MHz, BF3·OEt2): δ –0.60 (d, 1JBF = 70 Hz, BF).
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –136.6 (m, 6F, o-C6F5), –162.7 (t, 3JFF = 20 Hz, 3F
p-C6F5), –167.0 (m, 6F, m-C6F5), –171.6 (d, 1JPF = 1019 Hz, 1F, PF), –190.3 (q/br, 1JBF = 70 Hz,
1F, BF).
31P{1H} NMR (CD2Cl2, 162 MHz, H3PO4): δ 148.5 (d, 1JPF = 1019 Hz, PF).
13C{1H} NMR (CD2Cl2, 100 MHz, Me4Si): δ 148.2 (dm, 1JFC = 240 Hz, C6F5), 139.1 (dm, 1JFC =
206 Hz, p-C6F5), 136.8 (dm, 1JFC = 231 Hz, C6F5), 41.4 (dd, 1JPC = 26 Hz, 2JFC = 7 Hz,
C(CH3)3)3), 27.9 (dd, 2JPC = 2 Hz, 3JFC = 1 Hz, C(CH3)3)3).
[Mes3PF] [FB(C6F5)3] (4-2)
The compound was prepared in a manner similar to that of 4-1 using Mes3P (76 mg, 195μmol),
XeF2 (33 mg, 195 μmol), B(C6F5)3 (100 mg, 195 μmol), and was isolated as a white solid
(153 mg, 163 μmol, 84%).
Anal. Calcd. for C45H33BF17P: C, 57.59; H, 3.54%. Found: C, 57.54; H: 3.76 %.
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 7.22 (d, 4JPH = 3.8 Hz, 3H, m-Mes), 7.08 (d, 4JPH =
6.6 Hz, 3H, m-Mes), 2.41 (s, 9H, o-CH3), 2.34 (d, 4JPH = 6.1 Hz, 9H, o-CH3), 1.96 (s, 9H, p-
CH3).
11B NMR (CD2Cl2, 128 MHz, BF3·OEt2): δ –0.60 (d, 1JBF = 69 Hz, BF).
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –116.7 (d, 1JFP = 940 Hz, 1F, PF), –136.6 (m, 6F,
o-C6F5), –163.7 (t, 3JFF = 20 Hz, 3F p-C6F5), –168.0 (m, 6F, m-C6F5), –190.9 (q/br, 1JFB = 69 Hz,
1F, BF).
31P{1H} NMR (CD2Cl2, 162 MHz, H3PO4): δ 92.9 (d, 1JPF = 940 Hz, PF).
13C{1H} NMR (CD2Cl2, 100 MHz, Me4Si): δ 149.4 (dd, JPC = 3 Hz, JFC = 1 Hz, Mes), 148.3
(dm, 1JFC = 240 Hz, C6F5), 145.5 (dd, JPC = 8 Hz, JFC = 1 Hz, Mes), 144.0 (dd, JPC = 18 Hz, JFC =
3 Hz, Mes), 139.1 (dm, 1JFC = 206 Hz, p-C6F5), 136.8 (dm, 1JFC = 231 Hz, C6F5), 133.5 (d, JPC =
14 Hz, Mes), 133.5 (d, 11 Hz, Mes), 117.1 (dd, 1JPC = 99 Hz, 2JFC = 13 Hz, i-Mes), 23.3 (d,
3JPC = 6 Hz, o-Me), 22.5 (dd, 3JPC = 7 Hz, 4JFC = 5 Hz, o-Me), 21.7 (p-Me).
[(o-Tol)3PF] [FB(C6F5)3] (4-3)
The compound was prepared in a manner similar to that of 4-1 using (o-tol)3P (59 mg,
195 μmol), XeF2 (33 mg, 195 μmol), B(C6F5)3 (100 mg, 195 μmol), and was isolated as a white
solid (151 mg, 130 μmol, 91%).

117
Anal. Calcd. for C39H21BF17P: C, 54.83; H, 2.48%. Found: C, 54.26; H, 2.46%.
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 7.92 (m, 3H, Tol), 7.68 (m, 3H, Tol), 7.51 (m, 3H, Tol),
7.22 (m, 3H, Tol), 2.47 (d, 3JHH = 2.6 Hz, 9H, CH3).
11B NMR (CD2Cl2, 128 MHz, BF3·OEt2): δ –0.61 (d/br, 1JFB = 70 Hz, BF)
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –125.5 (d, 1JFP = 994 Hz, 1F, PF), –135.6 (m, 6F,
o-C6F5), –162.7 (t, 3JFF = 21 Hz, 3F p-C6F5), –167.0 (m, 6F, m-C6F5), –190.9 (s/br, 1F, BF).
31P{1H} NMR (CD2Cl2, 162 MHz, H3PO4): δ 104.3 (d, 1JPF = 994 Hz, PF).
13C{1H} NMR (CD2Cl2, 100 MHz, Me4Si): δ 148.2 (dm, 1JFC = 240 Hz, C6F5), 145.4 (dd, JPC =
9 Hz, JFC = 1 Hz, Tol), 139.2 (dm, 1JFC = 206 Hz, p-C6F5), 138.8 (dd, JPC = 3 Hz, JFC = 1 Hz,
Tol), 136.9 (dm, 1JFC = 231 Hz, C6F5), 135.6 (dd, JPC = 18 Hz, JFC = 2 Hz, Tol), 134.4 (d, JPC =
12 Hz, Tol), 128.3 (d, JPC = 16 Hz, Tol), 115.7 (dd, 1JPC = 105 Hz, 2JFC = 13 Hz, i-Tol), 22.0 (dd,
3JPC = Hz, 4JFC = Hz, o-Me).
[Ph3PF] [FB(C6F5)3] (4-4)
The compound was prepared in a manner similar to that of 4-1 using Ph3P (51 mg, 195 μmol),
XeF2 (33 mg, 195 μmol), B(C6F5)3 (100 mg, 195 μmol), and was isolated as a white solid
(157 mg, 193 μmol, 99%).
Anal. Calcd. for C36H15BF17P: C, 53.23; H, 1.86%. Found: C, 52.80; H, 1.71%.
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 8.04 (m, 3H, Ph), 7.85 - 7.74 (12H, Ph).
11B NMR (CD2Cl2, 128 MHz, BF3·OEt2): δ –0.60 (d/br, 1JFB = 57 Hz, BF).
19F NMR (CD2Cl2, CFCl3, 377 MHz): δ –128.3 (d, 1JPF = 996 Hz, 1F, PF), –135.6 (d, 3JFF =
20 Hz, 6F, o-C6F5), –162.4 (t, 3JFF = 19 Hz, 3F, p-C6F5), –166.9 (m, 6F, m-C6F5), –190.9 (s/br,
1F, BF).
31P{1H} NMR (CD2Cl2, 162 MHz, H3PO4): δ 94.7 (d, 1JPF = 996 Hz, PF).
13C{1H} NMR (CD2Cl2, 100 MHz, Me4Si): δ 148.4 (dm, 1JFC = 240 Hz, C6F5), 139.2 (dm, 1JFC =
206 Hz, p-C6F5), 138.9 (dd, 4JPC = 3 Hz, 5JFC = 2 Hz, p-Ph), 136.9 (dm, 1JFC = 231 Hz, C6F5),
134.3 (dd, JPC = 13 Hz, JFC = 1 Hz, Ph), 131.2 (d, JPC = 14 Hz, Ph), 116.5 (dd, 1JPC = 109 Hz,
2JFC = 15 Hz, i-Ph).
[(p-C6H4F)3PF] [FB(C6F5)3] (4-5)
The compound was prepared in a manner similar to that of 4-1 using (p-C6H4F)3P (62 mg,
195 μmol), XeF2 (33 mg, 195 μmol), B(C6F5)3 (100 mg, 195 μmol), and was isolated as a white
solid (149 mg, 172 μmol, 88%).

118
Anal. Calcd. for C36H12BF20P: C, 49.92; H, 1.40%. Found: C, 49.36; H, 1.60%.
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 7.84 (m, 6H, C6H4F), 7.48 (m, 6H, C6H4F).
11B NMR (CD2Cl2, 128 MHz, BF3·OEt2): δ –0.60 (d/br, 1JFB = 62 Hz, BF).
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –93.9 (s, 3F, C6H4F), –123.8 (d, 1JPF = 994 Hz, 1F, PF),
–135.6 (d, 3JFF = 20 Hz, 6F, o-C6F5), –162.4 (t, 3JFF = 19 Hz, 3F, p-C6F5), –166.9 (m, 6F,
m-C6F5), –190.9 (s/br, 1F, BF).
31P{1H} NMR (CD2Cl2, 162 MHz, H3PO4): δ 93.3 (d, 1JPF = 998 Hz, PF).
13C{1H} NMR not obtained due to insolubility.
[(C6F5)Ph2PF] [FB(C6F5)3] (4-6)
The compound was prepared in a manner similar to that of 4-1 using (C6F5)Ph2P (137 mg,
351 μmol), XeF2 (59 mg, 350 μmol), B(C6F5)3 (179 mg, 350 μmol) and was isolated as a white
solid (307 mg, 292 μmol, 84%).
Anal. Calcd. for C36H10BF22P: C, 47.92; H, 1.12%. Found: C, 47.19; H, 1.38%).
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 8.10 (m, 2H, p-C6H5), 7.97 – 7.79 (8H, o,m-C6H5).
11B NMR (CD2Cl2, 128 MHz, BF3•OEt2): δ 1.91 (s/br, BF).
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –123.4 (dt/br, 1JPF = 1020 Hz, 4JFF = 17 Hz, 1F, PF),
−124.1 (s/br, 2F, P(o-C6F5)), –131.3 (s/br, 1F, P(p-C6F5)), –135.6 (d/br, 3JFF = 16 Hz, 6F,
B(o-C6F5)), –153.9 (m/br, 2F, P(m-C6F5)), –161.6 (s/br, 3H, B(p-C6F5)), –166.7 (m/br, 6F,
B(m-C6F5)), –190.4 (s/br, 1F, BF).
31P{1H} NMR (CD2Cl2, 162 MHz, H3PO4): δ 87.2 (d/br, 1JPF = 1020 Hz, PF).
13C{1H} NMR (CD2Cl2, 100 MHz, Me4Si): δ 148.4 (d, 1JFC = 240 Hz, B(o-C6F5)), 140.0
(p-C6H5), 139.2 (d, 1JFC = 206 Hz, B(p-C6F5)), 136.9 (d, 1JFC = 231 Hz, B(m-C6F5)), 134.2 (d,
2JPC = 15 Hz, o-C6H5), 131.5 (d, 3JPC = 16 Hz, m-C6H5).
(C6F5)2PhPF2 (4-7)
This reaction could take place with and without the presence of B(C6F5)3.
(C6F5)2PhP (140 mg, 317 μmol) was measured into a vial and dissolved in 5 mL of CH2Cl2. This
was added to solid XeF2 (54 mg, 317 μmol) in a separate vial. The reaction was allowed to stir
overnight and the solvent removed in vacuo. Washing the crude material with pentane yielded
the product was isolated as a white powder (150 mg, 312 μmol, 98%).
Anal. Calcd. for C18H5F12P: C, 45.02; H, 1.05%. Found: C, 44.69; H, 1.52%.

119
1H NMR (CD2Cl2, Me4Si, 400 MHz): δ 8.21 (m, 2H, o-C6H5), 7.75 (m, 1H, p-C6H5), 7.63 (m,
2H, m-C6H5).
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –20.2 (dp, 1JPF = 694 Hz, 4JFF = 15 Hz, 2F, PF2), –133.9
(m, 4F, o-C6F5), –150.4 (t, 3JFF = 21 Hz, 2F, p-C6F5), –161.7 (m, 4F, m-C6F5).
31P{1H} NMR (CD2Cl2, 162 MHz, H3PO4): δ –54.8 (tp, 1JPF = 694 Hz, 3JPF = 10 Hz, PF2).
13C{1H} NMR (CD2Cl2, 100 MHz, Me4Si): δ 146.2 (dm, 1JFC = 254 Hz, o-C6F5), 143.7 (dm, 1JFC
= 264 Hz, p-C6F5), 138.0 (d, 1JFC = 254 Hz, m-C6F5), 137.5 (dt, 2JPC = 15 Hz, 3JFC = 11 Hz,
o-C6H5), 134.7 (dt, 4JPC = 4 Hz, 5JPF = 1 Hz, p-C6H5), 129.6 (dt, 3JPC = 18 Hz, 4JFC = 2 Hz,
m-C6H5), 128.9 (dt, 1JPC = 184 Hz, 2JFC = 21 Hz, C6H5).
(C6F5)3PF2 (4-8)
The compound was prepared in a manner similar to 4-7 using (C6F5)3P (418 mg, 785 μmol) and
XeF2 (133 mg, 785 μmol), except that the reaction mixture was allowed to stir for 8 h before
work up. The product was isolated as a white solid (432 mg, 757 μmol, 96%).
Anal. Calcd. for C18F17P: C, 37.92%. Found: C, 38.26%.
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ 0.5 (dsept, 1JPF = 695 Hz, 4JFF = 16 Hz, 2F, PF2), –133.6
(m, 6F, o-C6F5), –147.3 (t, 3JFF = 21 Hz, 3F, p-C6F5), –160.3 (m, 6F, m-C6F5).
31P{1H} NMR (CD2Cl2, 162 MHz, H3PO4): δ –48.0 (tsept, 1JPF = 695 Hz, 3JPF = 10 Hz, PF2).
13C{1H} NMR (CD2Cl2, 100 MHz, Me4Si): δ 146.4 (d, 1JFC = 257 Hz, o-C6F5), 144.5 (d, 1JFC =
263 Hz, p-C6F5), 138.2 (d, 1JFC = 253 Hz, m-C6F5).
Ph3PF2 (4-9)39,67
A slightly modified literature procedure was used for this compound, using XeF2 as an oxidant.
A solution of Ph3P (100 mg, 381 μmol) in 5 mL of CH2Cl2 was added to a solution XeF2 (65 mg,
381 μmol) in 5 mL of CH2Cl2, resulting in immediate effervescence. After approx. 1 min,
effervescence had ceased and the solvent volume was removed in vacuo. The product was then
washed with pentane (2x2 mL) then dried again in vacuo (110 mg, 366 μmol, 99%).
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 8.00 (m, 6H, o-C6H5), 7.49 (m, 9H, p-C6H5, m-C6H5).
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –39.5 (d, 1JPF = 659 Hz, 2F, PF2).
31P{1H} NMR (CD2Cl2, 162 MHz, H3PO4): δ –54.9 (t, 1JPF = 659 Hz, PF2).

120
(C6F5)Ph2PF2 (4-10)
A solution of (C6F5)Ph2P (48 mg, 136 μmol) in 5 mL of CH2Cl2 was added to a solution XeF2
(23 mg, 136 μmol) in 5 mL of CH2Cl2, resulting in immediate effervescence. After approx.
1 min, effervescence had ceased and the solvent volume was reduced to approx. 1 mL in vacuo,
then 2 mL of n-pentane were added. Slow evaporation of solvent from the colorless solution
yielded diffraction-quality crystals (53 mg, 135 μmol, 99%).
Anal. Calcd. for C18H10F7P: C, 55.40; H, 2.58%. Found: C, 55.50; H, 2.82%.
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 8.12 (m, 4H, o-C6H5), 7.61 (m, 2H, p-C6H5), 7.52 (m,
4H, m-C6H5).
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –34.5 (dt, 1JPF = 688 Hz, 4JFF = 14 Hz, 2F, PF2), –134.2
(m, 2F, o-C6F5), –153.3 (t, 3JFF = 20 Hz, 1F, p-C6F5), –162.6 (m, 2F, m-C6F5).
31P{1H} NMR (CD2Cl2, 162 MHz, H3PO4): δ –57.3 (td, 1JPF = 687 Hz, 3JPF = 14 Hz, PF2).
13C{1H} NMR (CD2Cl2, 100 MHz, Me4Si): δ 145.8 (dm, 1JCF = 245 Hz, o-C6F5), 142.5 (dm, 1JCF
= 272 Hz, p-C6F5), 137.9 (dm, 1JCF = 250 Hz, m-C6F5), 135.8 (dt, 2JCP = 14 Hz, 3JCF = 11 Hz,
o-C6H5), 133.5 (dt, 1JCP = 182 Hz, 2JCF = 25 Hz, i-C6H5), 133.2 (dt, 4JCP = 4 Hz, 5JCF = 1 Hz,
p-C6H5), 129.1 (dt, 3JCP = 17 Hz, 4JCF = 2 Hz, m-C6H5).
[Ph3PF] [OTf] (4-11)
A 20 mL vial was charged with Ph3P (100 mg, 381 μmol), a stir bar and 10 mL of toluene. XeF2
(64 mg, 381 μmol) was weighed in a separate vial. The Ph3P solution was carefully transferred
via pipette onto the XeF2 and the colorless effervescing mixture was stirred for 30 min.
TMSOTf (86 mg, 381 μmol) was added and a precipitate immediately began to form in the
colorless solution. The solvent was then decanted and the solid dried in vacuo and the product
isolated as a white powder (158 mg, 367 μmol, 96 %).
Anal. Calcd. for C19H15F4O3PS: C, 53.03; H, 3.51%. Found: C, 52.96; H, 3.72%.
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 8.08 (t, 3JHH = 8 Hz, 1H, p-C6H5), 7.85 (m, 4H,
o,m-C6H5).
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –78.9 (s, 3F, CF3), –128.6 (d, 1JPF = 995 Hz, 1F, PF).
31P{1H} NMR (CD2Cl2, 81 MHz, H3PO4): δ 94.8 (d, 1JPF = 995 Hz PF).
13C{1H} NMR (CD2Cl2, 100 MHz, Me4Si): δ 138.9 (p-C6H5), 134.5 (d, 2JCP = 14 Hz, o-C6H5),
131.3 (d, 3JCP = 15 Hz, m-C6H5).

121
[(C6F5)Ph2PF] [OTf] (4-12)
In a glovebox, a 20 mL flask was charged with (C6F5)PPh2 (221 mg, 626 μmol), a stir bar and
10 mL of CH2Cl2. XeF2 (106 mg, 626 μmol) was weighed in a separate flask. The XeF2 solution
was carefully transferred to the (C6F5)PPh2 solution and the colorless effervescing mixture was
stirred for 30 min. The solution was then transferred to a vial containing TMSOTf (140 mg,
626 μmol) and the colorless effervescing solution was stirred for another 30 min. The solvent
volume was then reduced to approx 2 mL in vacuo and 10 mL of n-pentane was added to the
solution resulting in the formation of a white, oily precipitate. The mixture was stirred for
5 minutes before the precipitate was allowed to settle. The supernatant was decanted and
discarded, and the solid was dried in vacuo and isolated as a white powder (243 mg, 467 μmol,
75%).
Anal. Calcd. for C19H10F9O3PS: C, 43.86; H, 1.94%. Found: C, 43.59; H, 2.23%.
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 8.08 (m, 6H, o,p-C6H5), 7.87 (m, 4H, m-C6H5).
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –79.1 (s, 3F, CF3), –123.0 (dt, 1JPF = 1016 Hz, 4JFF =
17 Hz, 1F, PF), –125.2 (m, 2F, o-C6F5), –133.5 (m, 1F, p-C6F5), –155.8 (m, 2F, m-C6F5).
31P{1H} NMR (CD2Cl2, 81 MHz, H3PO4): δ 87.1 (dt, 1JPF = 1016 Hz, 3JPF = 6 Hz, PF).
13C{1H} NMR (CD2Cl2, 100 MHz, Me4Si): δ 149.4 (dm, 1JCF = 273 Hz, p-C6F5), 149.3 (dm, 1JCF
= 261 Hz, o-C6F5), 139.6 (dm, 1JCF = 257 Hz, m-C6F5), 139.6 (dd, 4JCP = 3 Hz, 5JCF = 2 Hz,
p-C6H5), 134.6 (d, 2JCP = 15 Hz, o-C6H5), 131.4 (d, 3JCP = 16 Hz, m-C6H5), 121.0 (q, 1JCF =
321 Hz, CF3), 115.8 (dd, 1JCP = 114 Hz, 2JCF = 14 Hz, i-C6H5).
[(C6F5)2PhPF] [F(Al(C6F5)3)2] (4-13)
A solution of 4-7 (10 mg, 21 μmol) in 1 mL of toluene was added to a solution of
Al(C6F5)3·C7H8 (26 mg, 42 μmol) in 1 mL of toluene at –35 °C, resulting in a slightly pink-
colored solution. The solvent was removed in vacuo yielding a pink oil that was then dissolved
in 1 mL of CH2Cl2. 1 mL of n-pentane was then added and slow evaporation of solvents from the
solution yielded light-pink, diffraction-quality crystals (32 mg, 21 μmol, 99%).
Anal. Calcd. for C54H5Al2F42P: C, 42.21; H, 0.33%. Found: C, 41.79; H, 0.81%.
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 8.26 (m, 1H, p-C6H5), 8.01 – 7.92 (4H, o,m-C6H5).
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –121.9 (dp, 1JPF = 1042 Hz, 4JFF = 17 Hz, 1F, PF),
−123.9 (m, 12F, Al(o-C6F5)), –124.7 (dd, 3JFF = 30 Hz, 4JFF = 16 Hz, 4F, P(o-C6F5)), –126.6 (m,

122
2F, P(p-C6F5)), –152.1 (m, 4F, P(m-C6F5)), –156.0 (t, 3JFF = 19 Hz, 6F, Al(p-C6F5)), –164.7 (m,
12F, Al(m-C6F5)), –170.6 (s/br, 1F, Al2F).
31P{1H} NMR (CD2Cl2, 162 MHz, H3PO4): δ 77.7 (dm, 1JPF = 1042 Hz, PF).
13C{1H} NMR: Could not be obtained due to low solubility of the compound in all common
NMR solvents.
[(C6F5)3PF] [F(Al(C6F5)3)2] (4-14)
The compound was prepared in a manner similar to that of 4-13 using 4-8 (31 mg, 54 μmol), and
Al(C6F5)3·C7H8 (67 mg, 109 μmol). The resulting off-white solid was then dissolved in CD2Cl2,
and analyzed by NMR spectroscopy after approx 30 min.
Anal. Calcd. for C54Al2F47P – Could not be obtained due to compound instability.
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –120.7 (dsept, 1JPF = 1062 Hz, 4JFF = 15 Hz, 1F PF),
−122.5 (m, 3F, p-PC6F5), –124.0 (m, 12F, o-AlC6F5), –125.0 (m, 6F, o-PC6F5), –150.5 (m, 6F,
m-PC6F5), –156.0 (t, 3JFF = 19 Hz, 6F, p-AlC6F5), –164.7 (m, 12F, m-AlC6F5), –170.6 (s/br, 1F,
AlFAl).
31P{1H} NMR (CD2Cl2, 162 MHz, H3PO4): δ 68.0 (d, 1JPF = 1062 Hz, PF).
13C{1H} NMR: Could not be obtained due to low solubility of the compound in all common
NMR solvents.
[(C6F5)Ph2PF] [B(C6F5)4] (4-15)
This compound was prepared in two ways, the first was in a similar fashion to 4-16, and the
second was as follows. A solution of 4-10 (32 mg, 820 μmol) in 2 mL of CH2Cl2 was added to a
vial containing 4-17 (100 mg, 813 μmol) at room temperature. The mixture was allowed to stir
for 1 h, allowing 4-17 to completely dissolve and react. The colourless solution was then dried
in vacuo resulting in a white crude powder. The crude product was then washed with toluene
and pentane to remove difluorophosphorane side products. The supernatant was decanted, the
solid was dried in vacuo and isolated as a white powder (71 mg, 676 μmol, 83%).
Anal. Calcd. for C42H10BF26P: C, 48.03; H, 0.96%. Found: C, 47.45; H, 1.17%).
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 8.15 (m, 2H, p-C6H5), 7.89 (m, 8H, o,m-C6H5).
11B NMR (CD2Cl2, 128 MHz, BF3·OEt2): δ –16.7 (s, B(C6F5)4)
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –123.54 (dt, 1JPF = 1023 Hz, 4JFF = 19 Hz, 1F, PF),
–123.83 (m, 2F, P(o-C6F5)), –130.13 (m, 1F, P(p-C6F5)), –133.18 (m, 8F, B(o-C6F5)), –152.80

123
(m, 2F, P(m-C6F5)), –163.75 (t, 3JFF = 20 Hz, 4F, B(p-C6F5)), –167.65 (t/br, 3JFF = 19 Hz, 8F,
B(m-C6F5)).
31P{1H} NMR (CD2Cl2, 162 MHz, H3PO4): δ 87.6 (dt, 1JPF = 1023 Hz, 3JPF = 7 Hz PF).
13C{1H} NMR: (CD2Cl2, 100 MHz, Me4Si): δ 148.5 (d, 1JCF = 240 Hz, C6F5), 140.59 (CH
aromatic), 138.6 (d, 1JCF = 246 Hz, C6F5), 136.7 (d, 1JCF = 245 Hz, C6F5), 134.0 (d, 2JPC =
14 Hz), 131.8 (d, 3JPC = 15 Hz, CH aromatic).
[(C6F5)2PhPF] [B(C6F5)4] (4-16)
A solution of 4-7 (55 mg, 115 μmol) in 2 mL of toluene was added to a vial containing
[Et3Si·(C7H8)2] [B(C6F5)4] (101 mg, 103 μmol) under 2 mL of toluene at room temperature
resulting in precipitation of a colorless, glassy solid from a colorless supernatant. The mixture
was agitated for 5 min, 1 mL of CH2Cl2 was added, and agitation was resumed for an additional
2 min, resulting in a fine, white precipitate, which was allowed to settle from the colorless
solution. The supernatant was decanted, the solid was dried in vacuo and isolated as a white
powder (104 mg, 91 μmol, 89%).
Anal. Calcd. for C42H5BF31P: C, 44.24; H, 0.44%. Found: C, 44.04; H, 0.73%.
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 8.26 (m, 1H, p-C6H5), 8.01 – 7.92 (4H, o,m-C6H5).
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –121.9 (dp, 1JPF = 1042 Hz, 4JFF = 17 Hz, 1F, PF),
−124.7 (dd, 3JFF = 30 Hz, 4JFF = 16 Hz, 4F, P(o-C6F5)), –126.6 (m, 2F, P(p-C6F5)), –134.5 (m/br,
8F, B(o-C6F5)), –152.1 (m, 4F, P(m-C6F5)), –164.9 (t, 3JFF = 20 Hz, 4F, B(p-C6F5)), –168.9
(m/br, 8F, B(m-C6F5)).
11B NMR (CD2Cl2, 128 MHz, BF3·OEt2): δ –16.7 (s, B(C6F5)4).
31P{1H} NMR (CD2Cl2, 162 MHz, H3PO4): δ 77.7 (d, 1JPF = 1042 Hz, PF).
13C{1H} NMR: Could not be obtained due to low solubility of the compound in all common
NMR solvents.
[(C6F5)3PF] [B(C6F5)4] (4-17)
A solution of 4-8 (579 mg, 1.02 mmol) in 5 mL of toluene was added to a slurry of
[Et3Si·(C7H8)2][B(C6F5)4] (900 mg, 920 μmol) in toluene 5 mL, and the resulting suspension was
agitated vigorously for 5 min before allowing the precipitate to settle, after which point the
supernatant was decanted. The white solid was washed with CH2Cl2 (5 mL) then dried in vacuo
and isolated as a fine white powder (1.01 g, 821 μmol, 89%).

124
Anal. Calcd. for C42BF36P: C, 41.01%. Found: C, 40.55%.
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –120.5 (dsept, 1JPF = 1062 Hz, 4JFF = 14 Hz, 1F, PF),
−122.5 (m, 3F, P(p-C6F5)), –125.3 (ddd, 3JFF = 29 Hz, 4JFF = 14 Hz, 4JFF = 6 Hz, 6F, P(o-C6F5)),
–134.5 (m/br, 8F, B(o-C6F5)), –150.6 (m, 6F, P(m-C6F5)), –164.9 (t, 3JFF = 20 Hz, 4F,
B(p-C6F5)), –168.9 (m/br, 8F, B(m-C6F5)).
11B NMR (CD2Cl2, 128 MHz, BF3·OEt2): δ –16.7 (s, B(C6F5)4).
31P{1H} NMR (CD2Cl2, 162 MHz, H3PO4): δ 67.8 (dm, 1JPF = 1062 Hz, PF).
13C{1H} NMR: Could not be obtained due to low solubility of the compound in all common
NMR solvents.
4.4.3 X-Ray Crystallography
4.4.3.1 X-Ray Data Collection and Reduction
Crystals were coated in Paratone-N oil in the glovebox, mounted on a MiTegen Micromount and
placed under an N2 stream, thus maintaining a dry, O2-free environment for each crystal. The
data were collected on a Bruker Kappa Apex II diffractometer. Data collection strategies were
determined using Bruker Apex 2 software and optimized to provide >99.5% complete data to a
2θ value of at least 55°. The data were collected at 150(±2) K for all. The data integration and
absorption correction were performed with the Bruker Apex 2 software package.68
4.4.3.2 X-Ray Data Solution and Refinement
Non-hydrogen atomic scattering factors were taken from the literature tabulations.69 The heavy
atom positions were determined using direct methods employing the SHELX-2013 direct
methods routine. The remaining non-hydrogen atoms were located from successive difference
Fourier map calculations. The refinements were carried out by using full-matrix least squares
techniques on F, minimizing the function ω (Fo-Fc)2 where the weight ω is defined as 4Fo2/2σ
(Fo2) and Fo and Fc are the observed and calculated structure factor amplitudes, respectively. In
the final cycles of each refinement, all non-hydrogen atoms were assigned anisotropic
temperature factors in the absence of disorder or insufficient data. In the latter cases atoms were
treated isotropically. C-H atom positions were calculated and allowed to ride on the carbon to
which they are bonded assuming a C-H bond length of 0.95 Å. H-atom temperature factors were
fixed at 1.20 times the isotropic temperature factor of the C-atom to which they are bonded. The
H-atom contributions were calculated, but not refined. The locations of the largest peaks in the

125
final difference Fourier map calculation as well as the magnitude of the residual electron
densities in each case were of no chemical significance.
Table 4.4.1 - Select Crystallographic Data for 4-1, 4-3, 4-12, 4-13 and 4-15.
4-1 4-3 4-12
Formula C30H27F17PB C39H21F17PB C19H10F9O3PS
Formula weight 752.30 854.34 520.30
Crystal System Triclinic Triclinic Orthorhombic
Space group P-1 P-1 Pbca
a(Å) 11.1422(6) 11.0189(8) 17.5800(11)
b(Å) 12.0791(7) 11.9251(9) 12.4210(7)
c(Å) 12.5740(7) 15.5762(11) 18.6519(12)
α(deg) 80.041(3) 68.914(4) 90
β(deg) 84.639(3) 69.688(4) 90
γ(deg) 68.173(3) 73.804(4) 90
V(Å3) 1546.53(15) 1763.0(2) 4072.9(4)
Z 2 2 8
Temp. (K) 150 150 150
d(calc)gcm-1 1.616 1.609 1.697
Abs coeff,μ,mm-1 0.214 0.199 0.339
Data collected 7077 7930 5704
DataFO2
>3(FO2) 4322 5243 3355
Variables 442 523 298
R 0.0475 0.0838 0.0446
Rw 0.1253 0.2708 0.1051
GOF 0.996 1.072 1.000

126
4-13 4-15
Formula C54H5F42PAl2 C42H10F26PB
Formula weight 1536.51 3768.7(3)
Crystal System Monoclinic Monoclinic
Space group P2(1)/n Cc
a(Å) 17.7521(10) 24.1133(10)
b(Å) 15.5005(8) 10.8799(4)
c(Å) 20.4431(10) 16.1215(7)
α(deg) 90 90
β(deg) 107.630(2) 916.995(2)
γ(deg) 90 90
V(Å3) 5361.0(5) 3768.7(3)
Z 4 4
Temp. (K) 150 150
d(calc)gcm-1 1.904 1.851
Abs coeff,μ,mm-1 0.273 0.238
Data collected 9445 8536
DataFO2
>3(FO2) 6539 7467
Variables 892 631
R 0.0438 0.0362
Rw 0.1051 0.1057
GOF 1.013 0.508

127
4.5 References
(1) Ozima, M.; Podosek., F. A. Noble Gas Geochemistry; Cambridge University Press, 2001.
(2) Renouf, E. Science 1901, 13, 268-270.
(3) A, A. Angewandte Chemie 1924, 37, 217-218.
(4) von Antropoff, A. Angewandte Chemie 1924, 37, 695-696.
(5) Pauling, L. Journal of the American Chemical Society 1932, 54, 3570-3582.
(6) Bartlett, N.; Lohmann, D. H. Proceedings of the Chemical Society 1962, 97-132.
(7) Haynes, W. M. L. D. R. CRC handbook of chemistry and physics : a ready-reference
book of chemical and physical data; CRC Press: Boca Raton, Fla., 2011.
(8) Bartlett, N. Proceedings of the Chemical Society 1962, 197-236.
(9) Hoppe, R. Angewandte Chemie 1964, 76, 455-463.
(10) Unger, E.; Liebau, M.; Duesberg, G. S.; Graham, A. P.; Kreupl, F.; Seidel, R.; Hoenlein,
W. Chemical Physics Letters 2004, 399, 280-283.
(11) Wheale, S. H.; Badyal, J. P. S. Journal of Adhesion Science and Technology 2012, 26,
2229-2237.
(12) Filler, R. Israel Journal of Chemistry 1978, 17, 71-79.
(13) Shieh, T.-C.; Yang, N. C.; Chernick, C. L. Journal of the American Chemical Society
1964, 86, 5021-5022.
(14) Zupan, M.; Pollak, A. The Journal of Organic Chemistry 1974, 39, 2646-2647.
(15) Bardin, V. V.; Shchegoleva, L. N.; Frohn, H. J. Journal of Fluorine Chemistry 1996, 77,
153-159.
(16) Garrett, G. S.; Emge, T. J.; Lee, S. C.; Fischer, E. M.; Dyehouse, K.; McIver, J. M. The
Journal of Organic Chemistry 1991, 56, 4823-4826.
(17) Singh, R. P.; Shreeve, J. n. M. Accounts of Chemical Research 2003, 37, 31-44.
(18) Grochala, W. Chemical Society Reviews 2007, 36, 1632-1655.
(19) Smith, G. L.; Mercier, H. P. A.; Schrobilgen, G. J. Inorganic Chemistry 2007, 46, 1369-
1378.
(20) Turbini, L. J.; Aikman, R. E.; Lagow, R. J. Journal of the American Chemical Society
1979, 101, 5833-5834.
(21) Frohn, H. J.; Jakobs, S. Journal of the Chemical Society, Chemical Communications
1989, 625-627.
(22) Frohn, H. J.; Jakobs, S.; Henkel, G. Angewandte Chemie 1989, 101, 1534-1536.

128
(23) Tyrra, W.; Naumann, D. Canadian Journal of Chemistry 1989, 67, 1949-1951.
(24) Forster, A. M.; Downs, A. J. Polyhedron 1985, 4, 1625-1635.
(25) Frömel, S.; Fröhlich, R.; Daniliuc, C. G.; Kehr, G.; Erker, G. European Journal of
Inorganic Chemistry 2012, 2012, 3774-3779.
(26) Seel, F.; Bassler, H. J. Zeitschrift für anorganische und allgemeine Chemie 1975, 418,
263-272.
(27) Seel, F.; Bassler, H. J. Zeitschrift für anorganische und allgemeine Chemie 1976, 423,
67-74.
(28) Bondi, A. The Journal of Physical Chemistry 1964, 68, 441-451.
(29) Welch, G. C.; Holtrichter-Roessmann, T.; Stephan, D. W. Inorganic Chemistry 2008, 47,
1904-1906.
(30) Anand, S. P.; Filler, R. Journal of Fluorine Chemistry 1976, 7, 179-184.
(31) Lermontov, S. A.; Popov, A. V.; Zavorin, S. I.; Sukhojenko, I. I.; Kuryleva, N. V.;
Martynov, I. V.; Zefirov, N. S.; Stang, P. Journal of Fluorine Chemistry 1994, 66, 233-235.
(32) Hudnall, T. W.; Kim, Y.-M.; Bebbington, M. W. P.; Bourissou, D.; Gabbai, F. Journal of
the American Chemical Society 2008, 130, 10890-10891.
(33) Mayer, U.; Gutmann, V.; Gerger, W. Monatshefte für Chemie 1975, 106, 1235-1257.
(34) Gutmann, V. Coordination Chemistry Reviews 1976, 18, 225-255.
(35) Beckett, M. A.; Strickland, G. C.; Holland, J. R.; Sukumar Varma, K. Polymer 1996, 37,
4629-4631.
(36) Ullrich, M.; Lough, A. J.; Stephan, D. W. Organometallics 2010, 29, 3647-3654.
(37) Khalimon, A. Y.; Piers, W. E.; Blackwell, J. M.; Michalak, D. J.; Parvez, M. Journal of
the American Chemical Society 2012, 134, 9601-9604.
(38) Chitnis, S. S.; Burford, N.; Ferguson, M. J. Angewandte Chemie International Edition
2013, 52, 2042-2045.
(39) Smith, W. C. Journal of the American Chemical Society 1960, 82, 6176-6177.
(40) Lee, C. H.; Lee, S. J.; Park, J. W.; Kim, K. H.; Lee, B. Y.; Oh, J. S. Journal of Molecular
Catalysis A: Chemical 1998, 132, 231-239.
(41) Ménard, G.; Stephan, D. W. Angewandte Chemie International Edition 2012, 51, 8272-
8275.
(42) Zhang, Y.; Miyake, G. M.; Chen, E. Y. X. Angewandte Chemie International Edition
2010, 49, 10158-10162.

129
(43) Ménard, G.; Gilbert, T. M.; Hatnean, J. A.; Kraft, A.; Krossing, I.; Stephan, D. W.
Organometallics 2013, 32, 4416-4422.
(44) Ménard, G.; Tran, L.; McCahill, J. S. J.; Lough, A. J.; Stephan, D. W. Organometallics
2013, 32, 6759-6763.
(45) Ménard, G.; Hatnean, J. A.; Cowley, H. J.; Lough, A. J.; Rawson, J. M.; Stephan, D. W.
Journal of the American Chemical Society 2013, 135, 6446-6449.
(46) Müller, L. O.; Himmel, D.; Stauffer, J.; Steinfeld, G.; Slattery, J.; Santiso-Quiñones, G.;
Brecht, V.; Krossing, I. Angewandte Chemie International Edition 2008, 47, 7659-7663.
(47) Chen, M.-C.; Roberts, J. A. S.; Marks, T. J. Organometallics 2004, 23, 932-935.
(48) Chen, M.-C.; Roberts, J. A. S.; Seyam, A. M.; Li, L.; Zuccaccia, C.; Stahl, N. G.; Marks,
T. J. Organometallics 2006, 25, 2833-2850.
(49) Chen, I. C.; Ho, S.-M.; Chen, Y.-C.; Lin, C.-Y.; Hu, C.-H.; Tu, C.-Y.; Datta, A.; Huang,
J.-H.; Lin, C.-H. Dalton Transactions 2009, 8631-8643.
(50) Douvris, C.; Ozerov, O. V. Science 2008, 321, 1188-1190.
(51) Lambert, J. B.; Zhang, S.; Stern, C. L.; Huffman, J. C. Science 1993, 260, 1917-1918.
(52) Massey, A. G.; Park, A. J. Journal of Organometallic Chemistry 1966, 5, 218-225.
(53) Hogben, M. G.; Graham, W. A. G. Journal of the American Chemical Society 1969, 91,
283-291.
(54) Childs, R. F.; Mulholland, D. L.; Nixon, A. Canadian Journal of Chemistry 1982, 60,
801-808.
(55) Mallouk, T. E.; Rosenthal, G. L.; Mueller, G.; Brusasco, R.; Bartlett, N. Inorganic
Chemistry 1984, 23, 3167-3173.
(56) Krossing, I.; Raabe, I. Chemistry – A European Journal 2004, 10, 5017-5030.
(57) Christe, K. O.; Dixon, D. A.; McLemore, D.; Wilson, W. W.; Sheehy, J. A.; Boatz, J. A.
Journal of Fluorine Chemistry 2000, 101, 151-153.
(58) Chai, J.-D.; Head-Gordon, M. Physical Chemistry Chemical Physics 2008, 10, 6615-
6620.
(59) Grimme, S. Journal of Computational Chemistry 2006, 27, 1787-1799.
(60) Andzelm, J.; Kölmel, C.; Klamt, A. The Journal of Chemical Physics 1995, 103, 9312-
9320.
(61) Klamt, A.; Schuurmann, G. Journal of the Chemical Society, Perkin Transactions 2 1993,
799-805.

130
(62) Barone, V.; Cossi, M. The Journal of Physical Chemistry A 1998, 102, 1995-2001.
(63) Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Journal of Computational Chemistry
2003, 24, 669-681.
(64) Schreckenbach, G.; Ziegler, T. The Journal of Physical Chemistry 1995, 99, 606-611.
(65) Wolinski, K.; Hinton, J. F.; Pulay, P. Journal of the American Chemical Society 1990,
112, 8251-8260.
(66) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman,
J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li,
X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara,
M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.;
Nakai, H.; Vreven, T.; Montgomery, J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.;
Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.;
Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene,
M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.
E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.;
Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.;
Daniels, A. D.; Farkas; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Wallingford CT,
2009.
(67) Arisawa, M.; Suzuki, T.; Ishikawa, T.; Yamaguchi, M. Journal of the American Chemical
Society 2008, 130, 12214-12215.
(68) Bruker AXS Inc. : 2013.
(69) D. T. Cromer, J. T. W. Int. Tables X-Ray Crystallography, 1974; Vol. 4.

131
Chapter 5 Uncovering Electrophilic Fluorophosphonium Reactivity
5
5.1 Introduction
5.1.1 Phosphorus Compounds as Electron Acceptors
Phosphorus compounds have long been synonymous with their donor capability, in particular for
transition metal coordination and organometallic chemistry.1,2 An often overlooked property of
phosphorus compounds is the acceptor capabilities that they possess. With the renaissance of
research in the reactivity of main group compounds, the electrophilic nature of these compounds
have been re-examined, resulting in small molecule activation and catalysis. The p-block
elements exploited for their Lewis acidic properties thus far have mainly consisted of boron,3
aluminium,4 silicon,5 and other elements to a lesser extent.6 These group 13 and 14 electrophiles
have found utility in the domain of frustrated Lewis pair chemistry, while group 15 compounds
have only been exploited for their Lewis basic properties. A serious drawback to current FLP
systems is the sensitivity of the Lewis acids to air, moisture and donor functional groups, and
alternatives to the status quo are highly desirable. Phosphorus compounds represent a potentially
feasible replacement, as the ability of phosphorus to become hypervalent allows this non-metal
to support coordination numbers between 2 and 6. Furthermore, the high natural abundance of
the 31P nucleus facilitates the study of organophosphorus compounds by nuclear magnetic
resonance (NMR) spectroscopy, which is beneficial for characterization purposes.
A simple demonstration of phosphorus acceptor capabilities is the reaction between phosphorus
tribromide and alcohols.7 Nucleophilic attack of the alcohol on the phosphorus center occurs and
induces bromide ion dissociation, transiently forming an intermediate alcohol-phosphenium
adduct. Bromide subsequently attacks the α-carbon to liberate a bromoalkane and phosphonic
dibromide (Scheme 5.1.1). Subsequent reactions ultimately result in the formation of
phosphorous acid.

132
Scheme 5.1.1 - PBr3 acting as an acceptor in the presence of alcohols, forming bromoalkanes.
Researchers have taken advantage of the widely acknowledged “diagonal relationship” between
P and C in the periodic table8 to generate analogues to N-Heterocyclic Carbenes (NHCs). These
N-Heterocyclic Phosphenium ions (NHPs) are similarly aromatic with the phosphorus center
bearing a lone pair of electrons orthogonal to a vacant p-orbital (Scheme 5.1.2). These species
were first reported nearly simultaneously about 15 years ago from the Cowley group9 and the
Denk group.10 NHPs are usually generated via halide abstraction using a strong Lewis acid, such
as AlCl3, from the corresponding chlorophosphine.
Scheme 5.1.2 - General diagram of phosphenium cations and NHPs.
Experimental and computational studies have suggested that there is a greater degree of
π-delocalization in the cationic NHPs compared to NHCs11 and they are poorer σ-donors, but
better π-acceptors to their carbon analogues. These studies have also suggested that NHPs tend
to be more stable but less electrophilic than saturated or non-cyclic analogous phosphenium
cations. These systems show reactivity akin to both donors and acceptors, and they can effect
P−P bond activation and can act as a catalyst in the formation of P–C bonds.12,13 The Jones
group has developed an NHP derivative with a bis(imino)acenapthene backbone with a pendant
pyridine donor and have shown the ability of this compound to be an internal Lewis pair, acting
as both a donor and acceptor.14 Isolable phosphenium cations are not limited to NHPs, as
demonstrated by the Burford group. They have found success generating a number phosphenium
cations stabilized by donors such as carbenes, amines, or phosphines (Scheme 5.1.2).15 These
phosphenium cations still retain the lone pair at the phosphorus center but the empty orbital is

133
stabilized by the donor atom. Subsequent results have led to the formation of catena-phosphorus
compounds which contain phosphenium cations.16 The coordination chemistry of phosphenium
cations has been investigated as well, showing that the lone pair is still sufficiently σ-donating to
bind metal centers.17 The FIA of certain free phosphenium cations has recently been calculated
by Slattery and the results show that free phosphenium cations have the potential to be as Lewis
acidic as silylium cations.18
5.1.2 Phosphonium Cations as Lewis Acids
Phosphonium cations are another class of phosphorus acceptor compounds. They are most
commonly used in phase transfer catalysis19,20 in addition to ionic liquids.21,22 Often overlooked
is the ability of phosphonium cations to go hypervalent and act as an acceptor (Scheme 5.1.3).23
Most Lewis acids generate their Lewis acidity from a vacant p-orbital,24 however phosphonium
cations have an available σ*-orbital in the apical position which can accept an electron lone pair
from a donor.25 The energy level of the σ*-orbital on the phosphonium center is lowered with
the introduction of an electron withdrawing substituent in the apical position of the trigonal
bipyramidal arrangement, leading to a stabilization of the hypervalent molecule.25,26
Scheme 5.1.3 - Phosphonium hypervalence acceptor properties.
Phosphonium cations of this nature have been exploited as stoichiometric reagents as well as
organocatalysts. One of the most widely employed P(V) Lewis acids in organic synthesis are the
Wittig reagents, used for the olefination of aldehydes and ketones.27 These phosphorus ylides
have a resonance form where the positively charged phosphorus center is bound to a carbanion.
This carbanion then undergoes nucleophilic attack at the carbonyl carbon center, in turn causing
the oxygen to bind to the acceptor phosphonium. This four-membered ring intermediate
subsequently collapses with concomitant formation of alkene and phosphine oxide (Scheme
5.1.4). The Staudinger reaction proceeds in a similar manner and involves the initial binding of a
tertiary phosphine to the terminal N atom of an alkyl azide. Nucleophilic attack of the internal N

134
atom at the transient phosphonium center occurs to release N2 and yield an alkylated
phosphinimine.28
Scheme 5.1.4 - The Wittig reaction, an example of phosphonium acceptor capabilities.
The group of Mukaiyama have investigated the use of simple phosphonium cations of the form
[R3POPR3] [OTf] (R = tBu, Ph) in a number of carbon-carbon bond forming reactions. It was
determined that these salts are active catalysts in Mukaiyama-aldol reactions of aldehydes with
silyl enol ethers and ketene silyl acetals.29 In addition, they are effective catalysts for the
formation of β-aminoesters from imines and ketene silyl acetals (Scheme 5.1.5, top).30
Phosphonium ionic liquids have shown encouraging results as mild Lewis acids for the Henry
reaction. McNulty et al. described the reaction where 2 mol% of the phosphonium ionic liquid,
[(C6H13)3PC14H29] [C9H19CO2] catalyzed the Henry reaction between aromatic aldehydes and
nitromethane under mild conditions (Scheme 5.1.5, middle).31 The authors proposed that the
phosphonium center activated the carbonyl moiety via complexation with the σ*-acceptor
orbital.
Scheme 5.1.5 - Phosphonium catalyzed addition reactions.

135
Recently, Terada and Kouchi reported the synthesis of a variety of geometrically strained
phosphonium cations with mono- or bicyclic moieties.25 These compounds bear polarized P–O
bonds which act as an electron withdrawing group to enhance the Lewis acidity at the
phosphorus center. They utilized DMF as a probe to determine the acceptor abilities of the
phosphonium cations and these results showed that the most Lewis acidic phosphonium cation
bore a chelating fluorene and catechol group (Scheme 5.1.6). Further utilization of this Lewis
acid was described by catalyzing a Diels-Alder reaction between α,β-unsaturated amides with
cyclopentadiene, resulting in the bicyclic product in excellent yields (Scheme 5.1.6). Others
have shown that such complicated phosphonium cations are not necessary for catalytic purposes;
the groups of Plumet and Tian have shown that simple phosphonium salts such as [Ph3PMe] [I]
and [Ph3PCH2Ph] [Cl] can catalyze the addition of TMSCN to aldehydes32 and ketones (Scheme
5.1.5, bottom).33
Scheme 5.1.6 - Phosphonium catalyzed Diels-Alder reaction.
Fluoride ion sensing has become an area of active investigation due to the potential toxicity of
the anion in high concentrations.34 Lewis acidic receptors have been explored in the capture and
sensing of fluoride ions, most commonly organoboron compounds.35-38 The group of Gabbaï
have been leaders in this field, first noting that a borane species with a para-disposed
phosphonium cation drastically increases the fluoride ion binding affinity, compared to the free
borane.39 Phosphonium cation have also been shown to participate in fluoride ion binding.
Gabbaï and co-workers have shown that the ortho-disposed phosphonium-borane, [1-Mes2B-2-
MePh2P-(C6H4)] [I], has a significantly enhanced fluoride ion affinity due to the cooperative
effects arising from the proximity of the Lewis acidic phosphonium moiety to the borane center
(Scheme 5.1.7).40 The Gabbaï group have also extended the scope of the research by moving
towards heavier pnictogen cations, which has led to drastic enhancements in the Lewis acidity of

136
these species. The generation of these heavier Lewis acid analogues has led to the activation of
relatively inert bonds such as fluoroalkanes and fluoroborate anions.41,42
Scheme 5.1.7 - Cooperative fluoride ion binding between a borane and phosphonium cation.
To this end, the reactivity of the newly developed electrophilic fluorophosphonium cations
described in the previous chapter has never been explored. This chapter seeks to explore the
potential reactivity of these unique Lewis acids, as well as apply them in frustrated Lewis pair
chemistry. The research performed in this chapter was done in collaboration with Dr. Lindsay
Hounjet, Dr. Manuel Pérez, and Dr. Roman Dobrovetsky.
5.2 Results and Discussion
5.2.1 Nucleophilic Aromatic Substitution Reactions on Phosphonium Cations
The reactivity of B(C6F5)3 with Lewis bases has been widely studied,43,44 and generally results in
adduct formation, no reaction (FLP reactivity), or nucleophilic aromatic substitution, depending
on the steric bulk. B(C6F5)3 is not the only Lewis acid known to undergo nucleophilic aromatic
substitution. For example, triphenylmethyl cations are well known to undergo para–attack
reactions.45 As mentioned in Chapter 4, compound 4-6, [(C6F5)Ph2PF] [FB(C6F5)3], exhibited an
equilibrium process between the phosphonium fluoroborate salt and difluorophosphorane in
solution, indicating that the phosphonium cation is of at least comparable Lewis acidity to
B(C6F5)3. In seeking to uncover new reactivity with Lewis acidic fluorophosphonium cations,
we set out to investigate the behaviour of Lewis bases with 4-6 and [(C6F5)Ph2PF] [OTf] (4-12)
in hopes of discovering an all phosphorus FLP.
In an initial attempt to understand the behaviour, 4-6 was reacted with Ph3P. Room temperature
addition of Ph3P to 4-6 in CD2Cl2 immediately generated a white precipitate which was

137
identified as the Ph3P–B(C6F5)3 adduct. 31P{1H} NMR analysis of the supernatant showed a
triplet resonance at –57.3 ppm with a coupling constant of 1JPF = 687 Hz, confirming the
formation of the difluorophosphorane 4-10 (Scheme 5.2.1). This result, while slightly
disappointing, is not unexpected as in the previous chapter it was shown that small donors, such
as Et3PO, initiated similar reactivity.
To drive the equilibrium towards the free phosphonium, the reaction mixture was gently heated
at 40 °C for 10 – 15 minutes. The white precipitation began to dissolve and multinuclear NMR
analysis indicated the formation of a new species. The 31P{1H} spectrum of this product shows
two new triplet resonances at 16.1 ppm with a small coupling constant, 3JPF = 6 Hz and at
−58.4 ppm with a large coupling constant, 1JPF = 699 Hz, in which the latter peak is indicative of
a new difluorophosphorane species. The 19F NMR spectrum exhibited seven resonances; a
doublet for the difluorophosphorane species at –36.8 ppm with a 1JPF = 699 Hz, four resonances
at −136.6 ppm, –163.5 ppm, –167.9 ppm and –191.4 ppm suggestive of the presence of a
fluoroborate anion, and two multiplet resonances at –123.5 ppm and –128.5 ppm which integrate
to four fluorine atoms. These two resonances imply the presence of a p-C6F4 moiety and these
data suggest the formulation of the new product is the para–attack species [Ph3P(C6F4)PF2Ph2]
[FB(C6F5)3] (5-1) (Scheme 5.2.1). Compound 5-1 was unambiguously characterized using X-ray
crystallography and can be seen in Figure 5.2.1. The axially disposed fluorine atoms of the
phosphorane moiety give rise to an F–P–F angle of 175.20(14)°. The P–F bond lengths of
1.656(3) and 1.657(3) Å, respectively, are typical of difluorotriarylphosphorane species.46,47
Scheme 5.2.1 - Formation of 5-1.

138
Figure 5.2.1 - POV–ray depiction of 5-1, P: orange, B: green, F: pink, C: black. H-atoms
omitted for clarity.
The reaction producing 5-1 is believed to proceed by thermally breaking the P–B adduct,
liberating B(C6F5)3, which abstracts a fluoride ion from 4-10. Subsequent attack of Ph3P at the
para–carbon of the pentafluorophenyl ring in 4-6 occurs with simultaneous fluoride ion
migration to the Lewis acidic fluorophosphonium center, producing 5-1. This reactivity is
analogous to the thermally-induced nucleophilic aromatic substitution reaction of Ph3P with
B(C6F5)3 affording Ph3P(C6F4)B(F)(C6F5)2.44
The original FLP system was generated from a nucleophilic aromatic substitution reaction of
B(C6F5)3 with a bulky secondary phosphine, generating a linked phosphonium fluoroborate salt.
Subsequent fluoride-hydride exchange using a silane generated the first metal free species which
could liberate and uptake hydrogen gas.48 We had found that difluorophosphoranes do not react
with silanes in a similar fashion, therefore a new route was required to generate an
intramolecular all-phosphorus FLP and this approach was to use Ph2PSiMe3.49,50
The
silylphosphine can attack the pentafluorophenyl ring of an electrophile with subsequent loss of
Me3SiF, resulting in the formation of a new phosphine. To remove the risk of side reactions with

139
the borate anion, compound 4-12 was used for this transformation, which possesses a more inert
OTf anion. In C6H5Br, the silylphosphine was added to 4-12 which immediately resulted in a
brilliant yellow solution. This sample exhibited a doublet resonance in the 31P{1H} NMR
spectrum at 86.8 ppm with a 1JPF = 1011 Hz and a triplet resonance at –15.0 ppm with a 3JPF =
23 Hz. The 19F NMR spectrum again showed resonances indicative of the formation of a para-
disposed C6F4 ring, as two multiplets are observed at –123.1 ppm and –125.9 ppm, each
integrating to two fluorine atoms. These data suggest that the product formed is
[Ph2P(C6F4)P(F)Ph2] [OTf] (5-2) (Scheme 5.2.2), which was unambiguously confirmed using
X-ray crystallography as seen in Figure 5.2.2.
Scheme 5.2.2 - Formation of 5-2.
The structure of 5-2 is of particular interest due to its similarity to the original FLP system,
R2P(C6F4)B(C6F5)2, and as such 5-2 can be described as an all-phosphorus intramolecular FLP.
Bond angles around the free phosphine show that the atom is highly pyramidalized with the sum
of the bond angles totalling 309.9°. The Lewis acidic phosphonium center has a P–F bond
length of 1.546(1) Å and has a tetrahedral geometry with the sum of the bond angles being
340.4°. The bright yellow colour of 5-2 can presumably be attributed to charge transfer from the
phosphine through the arene to the para-disposed phosphonium electrophile, further supporting
the Lewis acidity of the fluorophosphonium cation. Attempts to activate small molecules such as
H2, CO2, and alkynes were attempted with 5-2 but to no avail. It appeared that 5-2 did not
possess sufficient Lewis acidity or basicity to effect FLP reactivity.

140
Figure 5.2.2 - POV–ray depiction of 5-2, P: orange, F: pink, C: black, S: yellow, O: red. H-
atoms omitted for clarity.
5.2.2 Exploiting Phosphonium Lewis Acids in FLP-like Activation of CO2
While the reactivity of a para disposed intramolecular all-phosphorus frustrated Lewis pair did
not produce the anticipated results, we were still inspired by the possibility of a versatile
phosphorus acceptor system and we sought to find a way to generate a more reactive species.
We decided to utilize o-phosphinoanilines as target molecules, since the nitrogen may be more
basic than the prior phosphorus center. In addition, the intramolecular ortho disposition between
the nitrogen and phosphorus center could lead to a potential interaction between the Lewis basic
and acidic sites. Generation of a Lewis acidic phosphorus center was achieved by initial
oxidation of the o-phosphinoaniline, Ph2P(o-C6H4NHMe),51 with XeF2 at –35 °C to prevent side
reactions from occurring on the aniline ring (Scheme 5.2.3). This reaction cleanly yielded the
difluorophosphorane Ph2(o-C6H4NHMe)PF2 (5-3) and this is indicated by the 31P{1H} NMR
spectrum of 5-3, which reveals a high field triplet at –45.6 ppm with a 1JPF = 625 Hz. The 19F
NMR spectrum revealed a corresponding resonance as a low field doublet at –35.9 ppm with a
1JPF = 625 Hz. The 1H NMR spectrum showed that fluorination did not affect the amine
functional group due to an NH resonance observed at 4.68 ppm as a broad quartet with a
coupling constant to the methyl protons of 5 Hz. The methyl resonance can also be observed at
2.67 ppm. TMSOTf was utilized as a fluoride abstracting agent to generate the corresponding

141
phosphonium cation (B(C6F5)3 was avoided out of concern for side reactions with the amine
functional group). One equivalent of TMSOTf was cooled to –35 °C and treated with a solution
of 5-3 in CH2Cl2. Upon concentration and subsequent washes with pentane, an off white solid
was isolated in 65% yield (Scheme 5.2.3).
Scheme 5.2.3 - The formation of 5-3 and 5-4.
The 31P{1H} NMR spectrum revealed a deshielded phosphorus nucleus as a diagnostic doublet at
94.4 ppm with a 1JPF = 980 Hz. A corresponding resonance was also observed in the 19F NMR
spectrum at –125.3 ppm with the same 1JPF = 980 Hz, in addition to the CF3 resonance from the
triflate anion at –79.9 ppm. The 1H NMR resonances were similar to those of 5-3, with only a
downfield shift of the NH resonance to 5.20 ppm observed. These data suggest that the
formulation of 5-4 is the phosphonium salt [Ph2(o-C6H4NHMe)PF] [OTf]. Compound 5-4 was a
candidate for potential FLP reactivity because the Lewis acidic phosphonium center showed no
interaction with the pendant Lewis basic aniline. Reactions of 5-4 with both H2 and CO2 were
performed in hopes of seeing dihydrogen activation or carbon dioxide capture but these reactions
failed to produce any FLP reactivity. The apparent lack of reactivity was attributed to
insufficient Lewis acidity and basicity and therefore the P/N intramolecular FLP needed to be
more reactive.
Deprotonation of the aniline with concurrent fluoride abstraction was attempted in order to
generate a more reactive species, as this should increase the basicity of the N center. Using
standard bases such as Et3N or n-BuLi to deprotonate 5-3 resulted in intractable mixtures, but the
slow addition of t-BuLi in pentane to 5-3 in THF at –78 °C cleanly generated a new, bright
yellow species 5-5 in 89% yield (Scheme 5.2.4). The 1H NMR spectrum of the product indicates
P–N bond formation, due to the disappearance of the NH resonance and the N–methyl signal
being split into a doublet of doublets at 2.28 ppm with observed coupling to both the P and F

142
atoms (3JPH = 4.4 Hz and 4JFH = 2.4 Hz). The 31P{1H} NMR signal of 3 was observed as a
doublet at –44.6 ppm with a 1JPF = 679 Hz and this peak is within the region of other
difluorophosphorane species reported in the previous chapter. The corresponding 19F NMR
resonance was observed at –44.4 ppm with a 1JPF = 679 Hz. These data suggests the formulation
of 5-5 to be an amidofluorophosphorane, Ph2PF(o-C6H4NMe), and this was subsequently
confirmed via X-ray crystallographic analysis and can be seen in Figure 5.2.3.
Figure 5.2.3 - POV–ray depiction of 5-5, P: orange, N: blue, F: pink, C: black. H-atoms omitted
for clarity.
The X-ray structure of 5-5 demonstrated a distorted trigonal bipyramidal geometry at P,
contained within a strained four-membered ring. Although rare, a few amidofluorophosphoranes
have previously been prepared,52-55 however compound 5-5 is believed to be the first
crystallographically characterized. The P–N bond length was found to be exceptionally long
(1.842(7) Å and 1.839(6) Å)52,53 within each of two crystallographically independent molecules.
The N atom occupies an axial position trans to F, although the N–P–F angles (164.4(3)° and
165.1(3)°) are somewhat distorted from linearity. The geometry of the amido unit is nearly
planar, as the sum of the angles about N equal 356° and the N–Cortho distances of 1.439(9) Å and
1.442(10) Å are typical of single bonds. The C–C bonds within the anilido ring are similar to
those within neighbouring phenyl rings, in contrast to the dearomatization observed within metal
complexes of the parent amidophosphine ligand.56-58

143
Scheme 5.2.4 - Formation of 5-5 and 5-6.
Exposure of a THF solution of 5-5 to an atmosphere of CO2 at room temperature resulted in an
immediate colour change from bright yellow to colourless. Multinuclear NMR analysis of the
solution indicated that CO2 had been incorporated into 5-5 (Scheme 5.2.4). The 1H NMR
spectrum of the new product, 5-6, showed a singlet N-methyl resonance at 3.15 ppm. The loss of
its coupling to both the P and F centers is consistent with the cleavage of the P–N bond within
5-5 to incorporate CO2. The 13C{1H} NMR spectrum illustrated the CO2 unit as a doublet at
153.1 ppm with a notable 2JPC = 8 Hz, while the 31P{1H} NMR spectrum showed a doublet at
−57.0 ppm, exhibiting 1JPF = 664 Hz. The corresponding 19F NMR signal was seen at –34.2 ppm
with an identical coupling constant as seen in the 31P{1H} spectrum. IR data for 5-6 show a
characteristic carbonyl absorption band at 1696 cm-1. Collectively, these data suggest that 5-6 is
the carbamatofluorophosphorane Ph2PF(o-C6H4N(Me)CO2) and this formulation was
unambiguously confirmed via X-ray crystallographic analysis as seen in Figure 5.2.4.

144
Figure 5.2.4 - POV–ray depiction of 5-6, P: orange, N: blue, O: red, F: pink, C: black. H-atoms
omitted for clarity.
The structure of 5-6 revealed the insertion of CO2 into the strained P–N bond of 5-5 and resulted
in a six-membered ring with a P–O bond (1.775(1) Å and 1.778(1) Å) in each of two molecules
in the asymmetric unit. Within 5-6, the geometry about P is again trigonal bipyramidal with the
carbamato O occupying an axial position trans to F. The ring strain of the 4-membered ring has
been relieved with F–P–O bond angles of 174.88(7) Å and 175.73(7) Å. The C–O and C=O
bonds in the CO2 fragment of 5-6 average 1.330(2) and 1.212(2) Å, respectively, which are
slightly longer than those seen in tBu3PCO2B(C6F5)3 (1.299(2) Ả, 1.208(2) Å) and
(Me3C6H2)2PCH2CH2B(C6F5)2(CO2) (1.284(4) Ả, 1.209(4) Å).59 As mentioned earlier, it has
previously been reported from Cavell and co-workers that kinetically hindered insertion of CO2
into the phosphorane P–N bond of Me(CF3)3P−NMe2 is possible, but this process suffers from
the relative inert nature of the P–N bond and proceeds over several days to give a
hexacoordinate, zwitterionic carbamatophosphate.52-55 The facile insertion of CO2 into the P–N
bond of 5-5 relieves strain within the four-membered ring, thereby dramatically accelerating the
reaction. It is noteworthy that 5-6 is thermally robust as it is unchanged upon heating to 120 °C
in toluene for 1 h.
Incorporating a second o-N-methylaniline substituent on the phosphorus center generated the
opportunity for forming a diamidophosphorane, which would have the potential to uptake two

145
equivalents of CO2. The phosphine, PhP(o-C6H4NHMe)2 was synthesized following literature
procedures51 and oxidized with XeF2 in a similar fashion to 5-3, generating the
difluorophosphorane 5-7 in excellent yields. The 31P{1H} NMR spectrum displays an indicative
resonance at –38.8 ppm as a triplet with a 1JPF = 588 Hz. A corresponding peak is also observed
in the 19F NMR spectrum at –32.1 ppm as a doublet with an identical coupling constant to the
resonances observed in the phosphorus NMR spectrum. Consistent with the reactivity described
above, the addition of two equivalents of t-BuLi to 5-7 resulted in the loss of both fluorine atoms
and the formation of a new product, 5-8, as a yellow solid in 75% yield. The 31P{1H} NMR
spectrum showed a singlet resonance, devoid of fluorine coupling, at –58.7 ppm and this is
supported by an absence of resonances in the 19F NMR spectrum. The 1H NMR spectrum
showed evidence of an N–P bond formation as the NMe resonance at 2.55 ppm is split into a
doublet with a 3JPH = 6.8 Hz. These data support the conclusion that 5-8 as the
diamidophosphorane, PhP(o-C6H4NMe)2 (Scheme 5.2.5).
Scheme 5.2.5 - Formation of 5-8 and 5-9.
Analogously, exposure of 5-8 to an atmosphere of CO2 immediately resulted in discoloration of
the solution, indicating CO2 incorporation into the P–N bond and an off white solid, 5-9, was
isolated in 80% yield from the reaction. The 31P{1H} NMR spectrum of the product showed a
single peak at –68.6 ppm, slightly upfield from the shift in 5-6. The lack of coupling observed in
the NMe protons to phosphorus in the 1H NMR spectrum also indicates the insertion of CO2 into
both of the P–N bonds. Consequently, the 13C{1H} NMR spectrum revealed a peak at 152.8 ppm
for the CO2 fragment, this peak is nearly identical to that in 5-6. The infrared spectroscopy of
5-9 also showed the expected carbonyl absorption at 1697 cm-1. Collectively, these data suggest
that 5-9 is the dicarbamatophosphorane, PhP(o-C6H4N(Me)CO2)2 (Scheme 5.2.5) and this

146
formulation was unambiguously confirmed via X-ray crystallographic analysis as seen in Figure
5.2.5.
Figure 5.2.5 - POV–ray depiction of 5-9, P: orange, N: blue, O: red, C: black H-atoms omitted
for clarity.
The structural analysis shows a pseudo-C2-symmetric geometry for 5-9, wherein the oxygen
atoms from the CO2 fragments occupy both axial positions, while the Cipso atoms of the aryl
groups lie in the equatorial plane. The lengths of the trans P–O bonds were found to be
1.7635(8) and 1.7773(8) Å, with an O–P–O angle of 174.27(4)°. The C–O and C=O bonds in
5-9 average 1.339(1) Å and 1.215(2) Å, respectively, and are slightly longer than those in 5-6.
Moreover, its generation from 5-8 represents the first example of double CO2 activation by a
phosphorane.
The formation of compounds 5-6 and 5-9 result from the insertion of CO2 into the P–N bonds of
5-5 and 5-8, respectively. While the mechanism for the insertion remains unconfirmed, one can
imagine an FLP type mechanism of CO2 insertion into the P–N bond through the resonance
forms of 5-5 as seen in Scheme 5.2.6. While a concerted process is a possibility, the activation
likely proceeds through a ring opened charge-separated FLP with an amido Lewis base and a

147
phosphonium Lewis acid. This view of 5-5 is reminiscent of an
intramolecular B/P FLP, (C6H2Me3)2PCH2CH2B(C6F5)2, developed by
Erker and coworkers.60 To support this notion, the removal of fluoride
from 5-5 resulted in the formation of the phosphonium cation,
[Ph2P(o-C6H4NMe)] [OTf] (5-10). This product does not react with
CO2, suggesting that a ring-opened form is required to allow for CO2 insertion. In this regard,
the CO2 insertion chemistry discussed herein is also analogous to that reported for the four-
membered rings of boron amidinates, which are also thought to react with small molecules via an
“open” FLP form.61 This system and its reaction with CO2 was investigated computationally by
the group of Dr. J. Zhu at Xiamen University in China.62 At the level of theory,
M062X/6-31+G(d), they showed that there is an activation barrier for the insertion of CO2 of
22.7 kcal/mol, with the reaction pathway proceeding through an open “FLP” type form. Their
computations also give insight into why the cationic four-membered ring 5-10 does not react
with CO2, as they show it has a much higher activation barrier of 51.8 kcal/mol. NBO analysis
of the P–N bond in both 5-5 and 5-10 shows the bond order changes from 0.51 to 0.78 with the
removal of fluoride. Further calculations from the group support the notion that the fluoride
labilizes the P–N bond to allow for facile ring opening followed by “FLP” type activation of
CO2.
Scheme 5.2.6 - Equilibrium and resonance structures of 5-5.
5.2.3 Realizing the Potential of Highly Lewis Acidic Phosphonium Cations
The aforementioned chemistry illustrates the capability of phosphonium cations to act as Lewis
acids in FLP chemistry. However, the remainder of this chapter will focus on the reactivity of
the most electrophilic phosphonium cation generated, [(C6F5)3PF] [B(C6F5)4], 4-17.

148
5.2.3.1 Stoichiometric Reactions of [(C6F5)3PF] [B(C6F5)4] with C–F Bonds
It was discussed in Chapter 3 that carbon–fluorine bonds constitute the strongest bonds that
carbon can form, with a bond dissociation energy of up to 130 kcal/mol.63 The ability to cleave
these bonds and degrade these compounds is of utmost importance as chlorofluorocarbons
(CFCs), hydrofluorocarbons (HFCs) and perfluorocarbons (PFCs) are persistent greenhouse
gases.64 Initially, Ph3CF65 was looked at as a substrate to gauge the fluoride abstraction abilities
of 4-17 since the corresponding carbocation would be stable in solution and would not
participate in unwanted side reactions. Combining a stoichiometric amount of 4-17 with Ph3CF,
both of which are colourless solids, in CD2Cl2 results in an instantaneous colouration of the
solution to yellow-orange, the colour of [Ph3C] [B(C6F5)4] (Scheme 5.2.7). The 1H NMR
spectrum of the reaction confirms the formation of trityl borate, with resonances at 8.27 ppm,
7.88 ppm and 7.66 ppm as a triplet of triplets (3JHH = 7.5 Hz and 4JHH = 1.3 Hz), doublet of
doublets (3JHH = 8.4 Hz and 4JHH = 7.5 Hz) and a doublet of doublets (3JHH = 8.4 Hz and 4JHH =
1.4 Hz), respectively. The 19F NMR spectrum showed the absence of the Ph3CF resonance at
−126.6 ppm and the formation of the difluorophosphorane, 4-8. The 31P{1H} NMR spectrum
also supports the formation of 4-8. Interestingly enough, reactions of 4-17 with fluoroalkanes
are not limited to systems that generate stable carbocations as a product. Upon dissolving 4-17
in neat 1-fluoropentane, a gradual colour change occurred from the initial colourless solution to
an orange-red solution within half an hour. The 1-fluoropentane was allowed to slowly
evaporate in the glovebox, which produced crystals and upon analysis confirmed the formation
of 4-8 (Scheme 5.2.7).47 The orange-red residue was dissolved in C6D5Br and 31P NMR analysis
confirmed that the residue contained 4-8 with a triplet resonance at –48.0 ppm, with a 1JPF =
697 Hz. The 1H, 11B, and 19F NMR spectra are quite complicated and indicate a reaction
between the pentyl cation and the borate anion to produce a complex mixture of decomposition
products. The decomposition of the borate anion [B(C6F5)4]– via reactions with carbocations has
been previously reported by Ozerov66 and the 11B NMR spectrum generated from the reaction of
4-17 with 1-fluoropentane matches identically (Figure 5.2.6). Multiple boron species, including
B(C6F5)3, can be seen in addition to the borate anion in the 11B NMR spectrum, indicating the
generation of a pentyl cation in situ which reacts with the anion. Other alkylfluorides were also
observed to react with 4-17. For instance, reaction of 4-17 with α,α,α–trifluorotoluene yields
identical results, albeit at a slower pace. Attempts to affect stoichiometric FLP type C–F bond

149
activation was attempted with 4-17, similarly to that reported in Chapter 3, however these
reactions were unsuccessful because 4-17 decomposes in the presence of nucleophilic
phosphines via two possible pathways. One of these pathways is nucleophilic aromatic
substitution, and the other route is fluoride ion transfer from 4-17 to the more basic phosphine,
resulting in formation of P(C6F5)3.
Scheme 5.2.7 - Stoichiometric reactions of 4-17 with fluorocarbons.
Figure 5.2.6 - 11B NMR spectrum of the reaction of 4-17 with 1-fluoropentane.
5.2.3.2 Catalytic Hydrodefluorination with [(C6F5)3PF] [B(C6F5)4]
The fluoride ion affinity of 4-17 can be attributed to the electrophilicity of the cation, as well as a
phenomenon known as apicophilicity.67 This was initially described by Muetterties et al.
wherein, in the absence of steric restraints, the most electronegative substituents occupy the
apical positions of a trigonal bipyramidal phosphorane. We sought to exploit this reactivity as

150
was described in Chapter 3, utilizing 4-17 as a catalyst for the hydrodefluorination (HDF) of
fluoroalkanes with silane. Previous reports have described C–F bond cleavage and catalytic
HDF of fluoroalkanes by conventional, main group Lewis acids, including B(C6F5)3,68 silylium,66
carbenium,69 and alumenium cations.70 These Lewis acids garnered their Lewis acidity from
electronic unsaturation, each having an empty p-orbital to accept electrons. Phosphonium Lewis
acids are electronically saturated and access their Lewis acidity from a low-lying σ* anti-bonding
orbital, and this concept had yet to be explored in hydrodefluorination catalysis.
Initially, the fluoroalkanes chosen for this study were ones shown to undergo reactions with
B(C6F5)3 (Chapter 3). The general procedure for the catalytic reactions was as follows: the
chosen fluoroalkane was combined with Et3SiH and an internal standard, fluorobenzene. These
reagents were then dissolved in C6D5Br and added to 1 mol% of 4-17 (Scheme 5.2.8). These
reactions were all performed in J-Young NMR tubes because they vigorously react and release
hydrogen gas, a phenomenon noted in silylium-catalyzed hydrodefluorination and discussed in
Chapter 3.66 At the outset, 1-fluoroadamantane was utilized as a substrate and after 1 h an NMR
analysis was undertaken. The 19F NMR spectrum was the most diagnostic for these reactions as
the resonance for 1-fluoroadamantane at –128 ppm had completely disappeared and Et3SiF
generation was observed, with a chemical shift of –175 ppm (Figure 5.2.7). The 1H NMR
spectrum also indicated the formation of adamantane. The results of all the hydrodefluorination
reactions are summarized in Table 5.2.1.
Scheme 5.2.8 - Hydrodefluorination reactions using 4-17.

151
Figure 5.2.7 - 19F NMR Spectra of the hydrodefluorination of 1-fluoroadamantane with 1 mol%
4-17 after 1 h.
Subsequently, 1-fluoropentane was used as a substrate and after 2 h the 19F NMR spectrum
indicated the complete defluorination of 1-fluoropentane with the absence of a resonance at
−217 ppm and the formation of Et3SiF at –175 ppm (see Figure 5.4.1 in the experimental
section). Accurate determination of the product mixture proved to be difficult, as 1H NMR
analysis indicated a complex mixture of products and GC-MS and ESI-MS attempts did not
provide further insight. However, as previously reported with hydrodefluorination reactions,66
the transient carbocations which are formed can undergo rearrangements, as well as
Friedel-Crafts side reactions with the aromatic solvent. These side reactions are prevalent for all
of the hydrodefluorination reactions performed, with the exception of 1-fluoroadamantane, due
to the stability of the adamantyl cation.71 Hydrodefluorination of fluorocyclohexane and
α,α,α-trifluorotoluene also proceeded very rapidly using 1 mol% of 4-17, with completion in one
and three hours, respectively. The 19F NMR spectrum for the reaction with fluorocyclohexane
only showed a resonance at –175 ppm with the absence of a peak for the starting material at
−172 ppm, signifying the defluorination of fluorocyclohexane. The formation of cyclohexane
was observed in using ESI-MS (m/z = 84) along with several other unidentified products. The
19F NMR spectrum for the reaction with α,α,α-trifluorotoluene indicated virtually complete
consumption of all aliphatic fluorine atoms with the absence of a resonance at –62 ppm and the
formation of Et3SiF at –175 ppm. Again, MS results indicate the formation of toluene (m/z 92),
but as a minor product along with a number of unidentifiable side products.

152
Increasing the number of carbon–fluorine bonds resulted in an increase in required reaction time.
1,4-Bis(difluoromethyl)benzene was nearly completely consumed after 24 hours as evidenced by
the 19F NMR spectrum with the disappearance of the resonance at –111 ppm and the appearance
of Et3SiF at –175 ppm. Other substrates with increasing numbers of carbon–fluorine bonds were
tested to see if they would undergo complete hydrodefluorination.
1-Bromo-3,5-bis(trifluoromethyl)benzene and 1,3,5-tris(trifluoromethyl)benzene were reacted
with Et3SiH and 1 mol% of 4-17, however this led to rapid decomposition of the catalyst which
prevented hydrodefluorination from reaching completion. Increasing the catalyst loading to
5 mol% did have a significant effect on the reaction, resulting in the consumption of 91% and
69% of the C–F bonds, respectively. In order to achieve complete hydrodefluorination, 10 mol%
of 4-17 was required and this complete reaction was observed in the respective 19F NMR spectra
with the absence of trifluoromethyl resonances at –62 ppm and the appearance of Et3SiF at
−175 ppm. In these cases, substituent redistribution affords a small amount of Et2SiF2 and Et4Si,
which is consistent with previously observed results.72

153
Table 5.2.1 - Hydrodefluorination results using 4-17 as a catalyst.
a Relative to fluoroalkane substrate. b Calculated from the proportion of C–F bonds originally
present relative to Si–F bonds formed. c Calculated from the proportion of C–F bonds consumed
after t (h). Conversions were determined by 19F NMR spectroscopy using fluorobenzene as an
internal standard.
Lastly, the hydrodefluorination of octafluorotoluene proceeds with preferential activation of the
trifluoromethyl substituent. After 24 h, the CF3 resonance at –55 ppm is nearly completely
consumed and resonances at –142 ppm and –175 ppm indicate the formation of Et2SiF2 and
Et3SiF. New resonances in the 19F NMR spectrum at –143 ppm, –156 ppm and –162 ppm
correspond to the formation of pentafluorotoluene (Figure 5.2.8).73 ESI-MS analysis of the
product mixture does indicate pentafluorotoluene formation (m/z 182) as well as other
unidentified products. Attempts were also made to halt the reaction after one C–F bond was
activated, in hopes of leading to a new method of generating difluoromethyl groups from
trifluoromethyl substituents, however regardless of the conditions complete defluorination was
observed.
Substrate Cat. (mol %)a t (h) %Si–F conv.b %C–F conv.c
Adamantyl-F 1 1 84 100
C5H11F 1 2 88 100
C6H11F 1 1 >95 100
PhCF3 1 3 >95 >99
1,4-C6H4(CF2H)2 1 24 81 87
C6F5CF3 1 24 71 98
3,5-C6H3Br(CF3)2 5
10
24 67
77
91
>98
1,3,5-C6H3(CF3)3 5
10
24 49
73
69
>99

154
Figure 5.2.8 - 19F NMR spectrum of the hydrodefluorination of octafluorotoluene with 1 mol%
4-17 after 24 h.
Transition metal systems have been shown to activate aromatic carbon–fluorine bonds. Milstein
et al. initially showed that the rhodium complex, (Me3P)3RhSiPhMe2 was able to activate
hexafluorobenzene with subsequent loss of Me2PhSiF, forming the complex (Me3P)3RhC6F5.
Interestingly, this system is able to turn over catalytically, forming pentafluorobenzene in the
presence of additional silane.74 Johnson and coworkers have shown that less expensive and
more abundant metals such as nickel can effect aromatic C–F bond cleavage.75,76 Early
transition metal metallocenes have been utilized for both aromatic and olefinic C–F bond
activation77,78 and more recently the group of Wass has shown an extension of FLP type
chemistry with cationic zirconocenes which have the ability to activate 1-fluoropentane.79 There
are only two examples of a main group system which can induce aromatic carbon–fluorine bond
activation, recently reported by Siegel and co-workers.80,81 One example is able to turnover
catalytically and they utilize a dimethylmesityl silylium cation reagent to perform an
intramolecular Friedel-Crafts reaction on a number of arylfluoride molecules (Scheme 5.2.9).

155
Scheme 5.2.9 - Metal-free aromatic C–F bond activation.
Attempts to activate aromatic carbon–fluorine bonds were also undertaken. Unsurprisingly, a
variety of substituted fluorobenzene molecules were unreactive towards hydrodefluorination.
Pentafluoropyridine has been shown to be a more reactive substrate for aromatic carbon–fluorine
bond activation,82 but it does not undergo hydrodefluorination with 4-17. The lack of reactivity
can be attributed to the mechanism of aromatic C–F bond activation with transition metals.
These reactions require coordination of the aromatic ring to the metal center (in the case of Ni
and Pt), followed by a three-centered oxidative addition or a phosphine assisted C–F bond
activation.83 Aromatic C–F bonds are also stronger than alkyl C–F bonds,84 and fluoride
abstraction with 4-17 would result in an aromatic carbocation, which would be unfavourable.81
Environmentally detrimental CFCs were also targeted for degradation using 4-17.
Trichlorofluoromethane, also known as Freon, was an ideal candidate to examine. It has a single
carbon–fluorine bond and if hydrodefluorination could be controlled, a useful by-product in
chloroform would be generated. A variety of reaction conditions were tested, including varying
the amount of catalyst and the equivalents of silane, but hydrodefluorination was not attainable
with this substrate. A number of other environmentally persistent fluorocarbons were also tried

156
in hydrodefluorination reactions, including perfluorinated alkyl chains, and a recently discovered
potent greenhouse gas, perfluorinatedtributylamine,85 but to no avail. The difficulties of
activating these perfluorinated alkanes is attributed to the non-polar nature of these compounds.
4-17 is not a strong enough Lewis acid to polarize these inert substrates and fluoride abstraction
does not occur.
5.2.3.3 Mechanistic Insights into Fluorophosphonium-Catalyzed HDF
The mechanism of the fluorophosphonium-catalyzed hydrodefluorination was probed. Previous
mechanisms for hydrodefluorination involve the use of a trityl cation as an initiator, which
abstracts a hydride from silane, generating a transient silylium cation which can activate the
carbon–fluorine bonds (Scheme 5.2.10).66
Scheme 5.2.10 - Trityl cation initiated hydrodefluorination.
It was crucial to determine whether or not our process was initiated or catalyzed by 4-17. A
number of reactions were hypothesized to help probe the mechanism, and the first observation
noted is that there is no reaction between 4-17 and Et3SiH. The hydride peak, which resonates as
a septet at 3.70 ppm in the 1H NMR spectrum shows broadening in the presence of 4-17 but
hydride abstraction is never observed, even over the course of several weeks. Nevertheless, this
interaction between a strong Lewis acid and silanes is not unprecedented; the research of Piers,86
and Oestreich,87 amongst others,88 have shown that B(C6F5)3 and Et3SiH react in this manner.
This evidence implies that 4-17 does not act as an initiator for the hydrodefluorination reaction.
Further support for this is the reactivity of 4-17 with fluoroalkanes in which irreversible
activation occurs, generating a carbocationic species and the difluorophosphorane 4-8. Lastly, a
competition experiment was devised to probe how silylium cation would react in the catalytic
cycle. Adding 1 eq. of [Et3Si] [B(C6F5)4]·2C7H8 to a 1:1 mixture of 4-8 and C6F5CF3 in C6H5Br
led to the immediate precipitation of 4-17 with no spectroscopic evidence for C–F bond

157
activation after 10 minutes (Scheme 5.2.11). Most fluoroalkanes will instantaneously react with
4-17 and so perfluorotoluene was utilized in order to observe the formation of 4-17. This
observation demonstrates that the fluoride ion in 4-8 is more labile than the alkyl C–F bond,
supporting the proposition that catalyst 4-17 is regenerated by fluoride ion abstraction from 4-8.
Scheme 5.2.11 - Competition experiment between 4-8 and octafluorotoluene with
[Et3Si] [B(C6F5)]4.
These experiments led us to propose a mechanism where initial fluoride ion abstraction occurs
from a fluoroalkane by 4-17, forming a carbocationic species and the difluorophosphorane 4-8.
Hydride is then delivered from silane to the carbocation, generating an incipient silylium cation,
which then abstracts a fluoride from 4-8, regenerating the catalyst 4-17 and turning the cycle
over. This catalytic cycle can be seen below in Scheme 5.2.12.

158
Scheme 5.2.12 - Proposed catalytic cycle for the fluorophosphonium catalyzed HDF. ΔH for
each reaction step reported in kcal/mol and the Gibbs free energies are reported in parenthesis
(kcal/mol).
Calculations were also undertaken to support the proposed mechanism. The energies of each
step in the proposed mechanism were calculated in the gas phase at the wB97XD/def2-TZVPP
level of theory and the values for each step in kcal/mol are reported in Scheme 5.2.12, with the
Gibbs free energies given in parentheses. The calculations indicate that none of the steps are
energetically unreasonable. Additional calculations were used to probe whether it was more
favourable for silanes or fluoroalkanes to interact with a fluorophosphonium center.
Calculations were performed on the strength of the interactions between [(C6F5)2PhP]+ and tBuF
vs. Me3SiH at the wB97XD/def2-TZV level of theory (Scheme 5.2.13). We found that it is
energetically more favourable for the fluorophosphonium cation 4-16 to interact with a
fluoroalkane over a silane. This is also clear from experimental evidence that silanes do not
react irreversibly with 4-17, while fluoroalkanes do. Isodesmic reactions were also calculated at
the wB97XD/def2-TZVPP level of theory which also supported the fact that 4-17 has a higher
affinity for fluoride than hydride (Scheme 5.2.13).

159
Scheme 5.2.13 - Calculated interaction between 4-16 with silane and fluoroalkanes (top).
Isodesmic reaction to determine hydride or fluoride affinities (bottom).
5.2.3.4 Fluorophosphonium Catalyzed Hydrosilylation Reactions
Pushing forward with the notion of using 4-17 as a catalyst, we sought to explore its ability in
hydrosilylation reactions. Hydrosilylation is the addition of a Si–H bond across an unsaturated
bond and is an extremely important industrial reaction for the production of a number of
organosilicon compounds.89 Typically, precious metal catalysts are required for this process:
Karstedt has developed a catalyst based on a platinum vinyl siloxane complex,90 Speier91 and
Declercq92 subsequently developed other homogeneous platinum systems (Scheme 5.2.14). The
prevalence for the use of platinum catalysts in industry is clear as nearly 6 tonnes of platinum are
consumed annually for this process.93 A number of other precious metal catalysts have been
developed based on Pd94 and Ru95,96 to name a few. Recently Chirik et al. have developed a
highly effective catalyst based on Fe as a competent alternative to Pt (Scheme 5.2.14).97 As the
understanding of catalysis has evolved, metal-free variants have been developed. Initial work
was based on Al,98,99 but these Al-based systems suffered from limited substrate or silane scope.
Lambert et al. have developed a protocol based on silylium cations to isolate β-silyl carbocations
from olefins, with subsequent hydrosilylation.100 Despite these efforts, efficient metal free
variants were not known until the seminal work by Piers and co-workers. They discovered that
the strong Lewis acid B(C6F5)3 can effect hydrosilylation of carbonyl functions.86 The substrate
scope of B(C6F5)3-catalyzed hydrosilylation reactions has subsequently been expanded upon.88

160
Given the extreme Lewis acidity of 4-17, we sought to see how it compares to more conventional
Lewis acids in the hydrosilylation of olefins and alkynes.
Scheme 5.2.14 - Various catalysts used in hydrosilylation reactions.
It was evident from the hydrodefluorination catalysis that 4-17 reversibly interacts with Et3SiH.
However, its reactivity with alkenes remained unexplored. Reacting 1 mol% of 4-17 with
1-hexene in CD2Cl2 yielded nearly quantitative conversion to 2-hexene in less than 1 h under
ambient conditions. The 1H NMR spectrum showed the disappearance of 1-hexene and the
formation of 2-hexene (Figure 5.2.9). This process can be slowed down under dilute conditions.

161
Figure 5.2.9 - 1-Hexene (#) isomerization to 2-hexene (*) with 4-17.
These results indicate that even though the σ*-orbital of 4-17 is sterically protected, it is still
capable of interacting with olefinic substrates. This result is reminiscent of electrophilic boranes,
which are less Lewis acidic than 4-17 but have been shown to exhibit van der Waals interactions
with olefins.101,102 Thus, isomerization of the terminal olefin likely proceeds via this interaction
with 4-17, generating an incipient carbocation that can undergo a 1,3-proton migration (Scheme
5.2.15). Investigating the analogous reaction with 1,1-diphenylethylene resulted in a Friedel-
Crafts-type cyclodimerization product. Such reactivity is known to be catalyzed by Lewis acids,
including B(C6F5)3.103,104

162
Scheme 5.2.15 - Proposed mechanism for the isomerization of 1-hexene with 4-17.
Having observed interactions of 4-17 with both olefins and silanes, attempts were made to effect
catalytic olefin hydrosilylation. Initially, the hydrosilylation of 1-hexene was explored,
combining 1.5 mol% of 4-17 with 1-hexene and Et3SiH in CD2Cl2. No olefin isomerization was
observed and within 30 minutes, 1H NMR spectroscopic analysis indicated the disappearance of
the Si−H and olefinic resonances and indicated quantitative conversion. Isolation of the product,
triethylhexylsilane, was achieved by simple filtration through a silica plug (Table 5.2.2, Entry 1).
This process is not limited to one silane, which was an issue other metal free systems have
suffered from, and 1-hexene can be readily hydrosilylated using Me2ClSiH or Ph3SiH (Table
5.2.2, Entries 2 and 3). Attempts to isolate the product of the hydrosilylation of 1-hexene with
the chlorosilane were unsuccessful due to hydrolysis of the reactive Si–Cl bond. However,
confirmation of the product, chlorodimethylhexylsilane, was achieved by comparing the 1H
NMR spectrum with literature reports.105 This protocol was expanded to other olefins;
1,1-diphenylethylene does not undergo dimerization under these conditions but rapid
hydrosilylation was observed in nearly quantitative yield (Table 5.2.2, Entry 4). The product
from this reaction is easily identified by 1H NMR analysis, as the CH resonance appears at
3.99 ppm as a triplet with a 3JHH = 8 Hz and 1.33 ppm with a 3JHH = 8 Hz.
Styrenes are compatible under these hydrosilylation conditions (Table 5.2.2, Entries 5-8) and do
not undergo cationic polymerization reactions. Methyl- and chloro- substituted styrenes are
tolerated, the latter having potential functionalization implications. In the case of p-methoxy-α-
methylstyrene an interesting side reaction occurs with two equivalents of Et3SiH, where not only
does hydrosilylation occur, but ether cleavage with concurrent loss of methane, resulting in the

163
installation of a silyl ether functionality (Table 5.2.2, Entry 8). Internal olefins also underwent
hydrosilylation with 4-17; cyclohexene was hydrosilylated using Et3SiH in good yields at room
temperature, however, trans-2-hexene required heating to 60 °C for 10 h (Table 5.2.2, Entries 9
and 10). This protocol can also be extended to internal and terminal alkynes, as is shown with
the hydrosilylation of 1-decyne and diphenylacetylene with 4-17, which selectively produces the
cis-isomer in both cases (Table 5.2.2, Entries 11 and 12).

164
Table 5.2.2 - Hydrosilylation results using 4-17 as a catalyst.
Entry Unsaturated Substrate Silane Product Time (h) Yielda
1 Et3SiH
0.5 88 %
2 Me2ClSiH
1 99 %b
3 Ph3SiH
1 99 %
4
Et3SiH
0.5 99 %
5
Et3SiH
1 99 %
6
Et3SiH
1 99 %
7
Et3SiH
1 99 %
8
2 eq.
Et3SiH
1 89 %
9
Et3SiH
1 89 %
10 Et3SiH
10 89 %
11
Et3SiH
1 99 %

165
12
Et3SiH
1 99 %
13 iPr3SiH
24 0
14
Et3SiH
1 96 %
a Isolated yields reported. b Conversion determined by 1H NMR spectroscopy.
Efforts were undertaken to shed light on the mechanism of this reaction. As mentioned above,
there is a relatively strong Si···H···P interaction between 4-17 and Et3SiH (Scheme 5.2.13). This
feature is consistent with previously reported data and is reminiscent of observations made by
Piers that B(C6F5)3 serves to activate the silane in hydrosilylation reactions.86,106-111 Thus, 4-17
has been shown to activate both silane and olefin in independent experiments, which brings into
question the reaction pathway of the olefin hydrosilylation reactions. As mentioned previously,
in the presence of silane there was no evidence of olefin isomerization or polymerization. We
again turned to computational studies in order to elucidate which potential pathway was
occurring. The LUMO σ*-orbital of 4-17 was found to interact with both silane and olefin with
a ΔH of –15.2 kcal/mol and –8.1 kcal/mol respectively. These results are consistent with a
mechanism in which 4-17 activates the Si–H bond. This concept is further supported by the
observation that hydrosilylation does not occur with the sterically encumbered silane, iPr3SiH
(Table 5.2.2, Entry 13). The research groups of Oestreich87,112,113 and Gervorgyan114 have
unambiguously established an anti-1,2-addition of Si−H to methylcyclohexene using B(C6F5)3
and experimental results using 4-17 as the catalyst yield identical results (Table 5.2.2, Entry 14).
Thus, it appears that the mechanism of hydrosilylation using 4-17 is similar to that of B(C6F5)3
where addition of the olefin to the phosphonium activated silane generates a carbocation, to
which hydride is then delivered in an anti-fashion (Scheme 5.2.16).

166
Scheme 5.2.16 - Proposed hydrosilylation reaction mechanism with 4-17.
5.2.3.5 Phosphonium Catalyzed Dehydrocoupling Reactions
Further investigating reactions involving silanes led us to explore the potential of 4-17 to act as a
dehydrocoupling catalyst. Dehydrocoupling is a benign way of appending silyl protecting
groups to a heteroatom, with concurrent loss of H2. This offers certain advantages over classical
protection strategies involving chlorosilanes and strong bases.115 Another application of
dehydrocoupling reactions is the generation of H2 as a fuel source from ammonia-borane.116 To
that end, the research groups of Tilley and Manners have pioneered the development of transition
metal catalysts for this purpose.117,118 Furthermore, Hill et al.119 have described a
dehydrocoupling procedure involving Mg, Ca and Sr for the preparation of amino silanes while
Baba et al. have utilized InBr3 to dehydrocouple carboxylic acids with silanes.120
Dehydrocoupling reactions have also been examined from a metal-free perspective and B(C6F5)3
has been used as a catalyst as noted in the work of Piers,121 and this work has subsequently been
expanded by the groups of Oestreich,112 Brook,122 Rubinsztajn,123 and Paradies.124

167
Investigating the viability of 4-17 to act as a dehydrocoupling catalyst, 1.5 mol% of 4-17 was
used in an attempt to dehydrocouple Ph2NH and Et3SiH (Table 5.2.3, Entry 1). After 10 h the
reaction was complete and the dehydrocoupled product, Ph2NSiEt3, was obtained in quantitative
yield. 1H NMR analysis of the reaction showed the loss of both the NH resonance and the SiH
resonance, in addition to the generation of H2. The 29Si NMR spectrum was also indicative of
the formation of the dehydrocoupled product, with a chemical shift of 9.60 ppm. The silane used
can be altered in a similar fashion to the hydrosilylation reactions previously discussed. These
reactions (Table 5.2.3, Entries 2-5) show that the choice silane has an effect on the reaction time,
and these reactions are complete within 1 to 48 h. Again, no reaction occurs when the bulky
silane iPr3SiH was utilized, indicating that the dehydrocoupling mechanism is similar to that of
hydrosilylation. Di-p-tolylamine can also undergo dehydrocoupling with a number of silanes
(Table 5.2.3, Entries 6-8), albeit at a slower rate than diphenylamine. Increasing the basicity or
reducing the steric bulk around the nitrogen atom by using iPr2NH or PhNH2 resulted in no
product formation (Table 5.2.3, Entries 9 and 10). These Lewis bases are able to interact
strongly with 4-17, which consequently leads to its decomposition.
Expanding the scope of heteroatom-containing coupling partners, we focused our attention on
thiophenol derivatives (Table 5.2.3, Entries 11-15). The reaction of PhSH and Et3SiH was
complete within 1 h under the same conditions used for amine dehydrocoupling. The 1H NMR
spectrum showed the disappearance of the SH and SiH resonance with concurrent formation of
H2. The 29Si NMR spectrum showed a new resonance at 24 ppm, indicating the dehydrocoupled
product formation. Para-substituted thiophenols also underwent rapid dehydrocoupling,
resulting in quantitative formation of the product. The exception to this was C6F5SH, which
required nearly a week under ambient conditions to reach completion. This is likely due to the
decreased basicity at the S centre, which prevents attack at the activated silicon center. This was
overcome by heating the reaction mixture to 100 °C for 3 h, after which quantitative product
formation was observed. Phenol derivatives were also investigated, and like thiols, they also
underwent rapid dehydrocoupling (Table 5.2.3, Entries 16-20). It is interesting to note that the
reaction of 1 eq. of Et3SiH with p-MeO(C6F4)OH results in rapid dehydrocoupling of the silane
and alcohol to form the silyl ether. However, with two equivalents of silane, the methoxy-
functionality is replaced by another silylether with concurrent formation of CH4. This is again
evident in the 1H NMR spectrum with the disappearance of the methoxy resonance at 3.48 ppm.

168
This observation is similar to the hydrosilylation of p-methoxy-α-methylstyrene and has
subsequently been exploited for the degradation of lignin, which will be discussed briefly in the
next chapter. The carboxylic acid p-C8H17(C6H4)CO2H and Et3SiH also undergo rapid
dehydrocoupling to give the corresponding ester in less than 1 h (Table 5.2.3, Entry 21).

169
Table 5.2.3 - Catalytic dehydrocoupling reactions using 4-17.
Entry E-H Silane T (h) Yield (%)a
1 Ph2NH Et3SiH 10 h > 99
2 Ph2NH ClMe2SiH 1 h > 99
3 Ph2NH Ph3SiH 20 h > 99
4 Ph2NH PhMe2SiH 48 h > 99
5 Ph2NH iPr3SiH 4 d 0
6 p-Me(C6H4)2NH Et3SiH 30 h > 99
7 p-Me(C6H4)2NH ClMe2SiH 16 h > 99
8 p-Me(C6H4)2NH Ph3SiH 36 h 40
9 iPr 2NH Et3SiH 48 h 0
10 PhNH2 Et3SiH 48 h 0
11 PhSH Et3SiH <1 h > 99
12 p-Me(C6H4)SH Et3SiH <1 h > 99
13 p-Cl(C6H4)SH Et3SiH <1 h > 99
14 p-F(C6H4)SH Et3SiH <1 h > 99
15 C6F5SH Et3SiH 1 week > 99
16b PhOH Et3SiH 2 h > 99
17b o-(Me)2(C6H3)OH Et3SiH 2 h > 99
18b p-OMe(C6H4)OH Et3SiH 18 h > 99
19b p-Me(C6H4)OH Et3SiH 3 h > 99
20b C6F5OH Et3SiH 1 day > 99
21 p-C8H17(C6H4)CO2H Et3SiH 1h > 99 a Yields measured by 1H-NMR. b1.0 mol% of catalyst was used.

170
From these experimental results, it appeared likely that the dehydrocoupling reaction was
proceeding via a similar mechanism to that of the hydrosilylation reactions. The initial step
involves the activation of the Si–H bond by 4-17, followed by backside attack of the silicon
LUMO by the Lewis base (N, S, O). This generates a transient hypervalent silicon center, which
has both hydridic (Si-H) and protic (E-H, E = N, S, O) hydrogen atoms, prompting facile loss of
hydrogen gas (Scheme 5.2.17). The lack of reactivity with smaller amines suggests that the
interaction with 4-17 is stronger than with the silane. At the same time, sterically demanding
silanes do not undergo dehydrocoupling reactions, which is consistent with the hypervalent
silicon intermediate. Furthermore, using a large excess of silane (10 eq.) with respect to
p-tol2NH resulted in acceleration of the reaction, while using an excess of the amine dramatically
increases reaction time. These results support the view of competitive binding of the amine and
silane at 4-17 and the dehydrocoupling reaction proceeds via silane activation. The quest for
mechanistic insight prompted a gas-phase DFT study at the WB97XD/def2TZV125,126 level of
theory employing the cation of 4-17, Me3SiH and Ph2NH. Supporting the experimental results, it
was shown that both Me3SiH and Ph2NH interact with the fluorophosphonium center with
distances of 2.3 and 3.5 Å, respectively. Both interactions are exothermic by 15.19 kcal/mol
(ΔG = –1.8 kcal/mol) and 23.35 kcal/mol (ΔG = –6.3 kcal/mol), respectively. Coordination of
the amine to the phosphonium-bound silane generates a transient five-coordinate silicon
intermediate, analogous to the hydrosilylation mechanism. This interaction is also exothermic
with ΔH = –37.1 kcal/mol (ΔG = −23.7 kcal/mol). Subsequent generation of (C6F5)3P(F)H and
[Ph2N(H)SiMe3]+ is a slightly endothermic (ΔH = 2.8 kcal/mol), but exergonic (ΔG =
−9.3 kcal/mol) step. The subsequent reaction of these two species to yield the dehydrocoupling
product is driven by the liberation of H2, even though the reaction is somewhat endothermic and
endergonic (ΔH = 43.5 kcal/mol and ΔG = 34.1 kcal/mol). Overall, this catalytic cycle is
computed to be slightly exothermic (ΔH = −6.0 kcal/mol) with only a small overall ΔG of
−0.6 kcal/mol.

171
Scheme 5.2.17 - Proposed mechanistic pathways for dehydrocoupling of silane and amine with
4-17. Gibbs free energies and enthalpies in parentheses for every step are provided in kcal/mol.
5.2.3.6 Phosphonium Catalyzed Transfer Hydrogenations of Olefins
Hydrogenation reactions are of paramount importance to industry as a wide range of
petrochemicals, pharmaceuticals, commodity chemicals, materials, and foods rely on them. It
was nearly a century ago that Sabatier127 discovered the utility of amorphous Ni in hydrogenation
processes. Organometallic chemistry opened the door to homogeneous hydrogenation catalysts
and systems have been developed based on a number of precious metals.128-130 While these are
very effective, catalysts based on less toxic, cheaper “earth-abundant” elements are being
investigated. To that end, first row transition metals have been targeted and the groups of
Chirik,131,132 Morris,133 Beller134 and others135 have developed remarkably active and selective
catalysts based on Fe.
Transition metal-free strategies have been explored as alternatives to transition metal catalysts.
Examples of these include the use of Hantzsch esters,136 Birch reduction of arenes,137 or the use
of B or Al hydrides as effective substitutes, although these reagents are stoichiometric. Catalytic
examples for transfer hydrogenation have been developed. The Lewis acid B(C6F5)3 has been
shown to be an effective catalyst for the transfer hydrogenation of imines.138 Additionally, the

172
reversible P(III)/P(V) redox cycling of planar phosphorus compounds have been exploited for
the transfer hydrogenation of azobenzenes.139 As mentioned throughout this dissertation, the
advent of FLPs has led to the development of a number of metal free catalysts for the
hydrogenation of imines, enamines, silyl enol ethers, olefins and alkynes.140 We sought to
investigate the possibility of utilizing the H2 generated from the dehydrocoupling process in a
transfer hydrogenation reaction with the addition of an olefin.
1,1-Diphenylethylene was initially chosen as a test olefin, as it is known to be readily
hydrogenated using FLP catalysts.141 1,1-Diphenylethylene was added to a mixture of p-tol2NH,
Et3SiH or Ph3SiH with 1.5 mol% of 4-17 at elevated temperatures (100 °C). This reaction
resulted in quantitative dehydrocoupling and 1,1-diphenylethane formation in 60% and >99%
yield, respectively (Table 5.2.4, Entries 1 and 2). These reactions proceeded at room
temperature, however the reaction times and conversions were negatively affected. Substituted
thiophenols, phenols and carboxylic acids are significantly better proton sources, resulting in
complete reduction of the olefin in less than 1 h (Table 5.2.4, Entries 3-8). A number of other
olefins undergo hydrogenation using this process in relatively good yields (Table 5.2.4). Styrene
and stilbene derivatives can be reduced under these conditions in high yields (Table 5.2.4,
Entries 9-19). The reduction of t-butylethylene readily occurs with both Cl(C6H4)SH and p-
Cl(C6H4)OH, however it results in the formation of 2,3-dimethylbutane (Table 5.2.4, Entries 20
and 21). This rearrangement reaction is indicative of a carbocationic intermediate and gives us
some insight into the possible mechanism. Furthermore, methylenecyclohexane and
1-methylcyclohexene were successfully reduced in good yields (Table 5.2.4, Entries 22-26).
Ester functional groups were well tolerated under these conditions and dibutyl-2-
methylenesuccinate was hydrogenated in good yields (Table 5.2.4, Entries 27-29). An
intramolecular proton source can also be used, as evidenced by the combination of
2-methylenesuccinic acid and Et3SiH in the presence of a catalytic amount of 4-17, which results
in the reduction of the internal alkene. Subsequent hydrolysis of the silyl-ester afforded the
corresponding acid in 89% yield (Table 5.2.4, Entry 30). All of the aforementioned reactions do
not occur without the presence of a catalytic amount of 4-17, and no hydrosilylation of the olefin
substrates was observed. These results illustrate the first metal-free transfer hydrogenation
catalyst for olefins.

173
Table 5.2.4 - Transfer hydrogenation of olefins with concurrent dehydrocoupling catalysis using
4-17.
Entry E-H R3SiH T (°C) Time (h) Conv.a
1 p-Tol2NH Et3SiH 100 5 60
2 p-Tol2NH Ph3SiH 100 6 >99
3 p-TolSH Et3SiH 25 <1 >99
4 p-Cl(C6H4)SH Et3SiH 25 <1 >99 (94)
5 p-F(C6F4)SH Et3SiH 25 <1 >99 (96)
6 p-MeO(C6H4)OH Et3SiH 25 <1 >99 (93)
7 p-C8H17CO2H Et3SiH 25 1 >99
8 C6F5CO2H Et3SiH 25 1 >99 (94)
9 p-Tol2NH Et3SiH 100 3 56
10 p-TolSH Et3SiH 25 <1 75
11 p-TolSH Et3SiH 25 <1 67
12 p-F(C6F4)SH Et3SiH 25 <1 61
13 C6F5SH Et3SiH 100 3 >99 (98)
14 p-C8H17(C6H4)CO2H Et3SiH 25 1 55
15 p-C8H17(C6H4)CO2H Ph3SiH 25 3 90
16 C6F5CO2H Et3SiH 25 2 >99 (99)

174
17 C6F5CO2H Et3SiH 25 2 >99 (97)
18 p-Cl(C6H4)SH Et3SiH 25 1 76
19 p-Cl(C6H4)OH Et3SiH 25 3 69
20 p-Cl(C6H4)SH Et3SiH 25 2 60
21 p-Cl(C6H4)OH Et3SiH 25 3 77
22 p-Cl(C6H4)SH Et3SiH 25 12 50
23 p-Cl(C6H4)OH Et3SiH 25 1 65
24 C6F5CO2H Et3SiH 25 2 >99
25 p-Me(C6H4)SH Et3SiH 25 1 >99 (95)
26 p-Cl(C6H4)SH Et3SiH 25 1 89
27 p-F(C6H4)SH Et3SiH 25 1 98 (96)
28 - Et3SiH 25 20h 96 (89)b
a Yields determined by 1H NMR analysis, isolated yield in brackets. b Isolated yield of the
corresponding acid.
One can imagine that the mechanism of these transfer hydrogenation reactions proceeds by the
intervention of the olefin in the dehydrocoupling reaction pathway. As opposed to the

175
elimination of H2, the transient [Ar2N(H)SiR3]+ cation (or any other E element) protonates the
olefin, generating a carbocation which is subsequently attacked by the hydride from the
hydridofluorophosphorane, (C6F5)3PFH (Scheme 5.2.18), yielding the corresponding alkane and
dehydrocoupled product. This mechanism is reminiscent of FLP hydrogenations of olefins,
where the hydrogen-activated product is so acidic that it protonates the olefin, followed by
hydride delivery.142 This idea is supported by the observation that 1,1-disubstitued olefins are
the most reactive. In addition, the methyl migration observed with tert-butylethylene supports
this theory. Furthermore, when Et3SiD was used as the hydride source, it led to exclusive
deuteration of the more electrophilic position of the olefin. This is consistent with a
Markovnikov addition of a proton to the less hindered carbon, resulting in tertiary carbocation
formation, followed by hydride or deuteride delivery.
Once again, DFT calculations at the WB97XD/def2TZV125,126 level of theory were carried out to
support the possible mechanism. Looking at the 4-17 catalyzed transfer hydrogenation process
of 1,1-diphenylethylene with diphenylamine and trimethylsilane showed that protonation of the
olefin by the intermediate [Ph2N(H)SiMe3]+ is endothermic (ΔH = 14.1 (ΔG = 13.7) kcal/mol)
while the subsequent hydride delivery from (C6F5)3PFH which affords 1,1-diphenylethane and
regenerates the catalyst is almost thermoneutral (ΔH = 1.1 and ΔG = 0.0 kcal/mol). Overall,
tandem dehydrocoupling and transfer hydrogenation to an olefin is energetically favourable
compared to loss of H2 as it requires 28.3 kcal/mol less energy, and is lower in the Gibbs free
energy by 20.5 kcal/mol. This supports why the dehydrocoupling of p-Tol2NH with Et3SiH is
significantly slower than the analogous reaction in the presence of 1,1-diphenylethylene.

176
Scheme 5.2.18 - Proposed mechanistic pathways for dehydrocoupling of silane and amine and
transfer hydrogenation of olefins with 4-17. Gibbs free energies and enthalpies in parentheses for
every step are provided in kcal/mol.
5.2.3.7 Dihydrogen Activation with Fluorophosphonium Lewis Acids
One of the steps of the proposed mechanism in the 4-17 catalyzed transfer hydrogenation
reaction indicates the formation of a hydridofluorophosphorane species. This intermediate is the
product that would be formed from the FLP activation of H2 with a Lewis base and 4-17. This
observation indicates that 4-17 may have the ability to activate H2. While this is fairly
straightforward with borane based Lewis acids, it was not trivial using phosphonium Lewis
acids. This was due to the fact that 4-17 has a propensity to decompose in the presence of Lewis
bases which have been previously utilized to effect H2 activation.
The dehydrocoupling reactions have shown favourable reactivity between diarylamines and
4-17. Therefore, DFT calculations were undertaken at the WB97XD/def2TZV125,126 level of
theory to explore the possibility of H2 activation between diarylamines and 4-17. The interaction
between diphenylamine and 4-17 resulted in a complex which shows significant π-stacking
interactions between the two molecules and the nitrogen atom approaching the σ*-orbital of 4-17

177
(Scheme 5.2.19, A). This interaction is exothermic (ΔH = −6.3 kcal/mol and ΔG =
−23.4 kcal/mol) and shows a distance of 3.52 Å between the P and N atoms. The HOMO and
LUMO diagram indicate the FLP nature of this combination as there is no observable interaction
between the N lone pair and the P acceptor orbital (Scheme 5.2.19, B & C). Activation of H2
was calculated and was shown to be favourable with a ΔH = −22.6 kcal/mol and ΔG =
−31.8 kcal/mol.
Scheme 5.2.19 - Calculated interaction between 4-17 and Ph2NH (A). HOMO and LUMO
depictions of this combination (B & C).
These calculations prompted the investigation of the reactivity of diarylamines with 4-17. The
normally sharp resonances in the 1H NMR spectrum for di-p-tolylamine completely disappear in
a CD2Cl2 solution with one equivalent of 4-17, instead yielding a very broad signal at 7.10 ppm
with no observation of the p-methyl resonance. In addition, no decomposition of the
phosphonium cation is observed. These broad resonances indicate a rapid equilibrium between
the free species and an interaction between the Lewis acid and Lewis base (Scheme 5.2.20).

178
Scheme 5.2.20 - Exchange process between 4-17 and p-tol2NH.
To probe this interaction, a VT-NMR experiment was undertaken. Due to solubility reasons
[(C6F5)2PhPF] [B(C6F5)4] (4-16) was used and dissolved in a 1:1 ratio with di-p-tolylamine in
CD2Cl2. The 1H NMR spectra were obtained at several temperatures between 25 °C and –90 °C.
As the temperature decreases there is no discernable change in the one phenyl ring that belongs
to 4-16, however a drastic change can be seen in the resonances belonging to di-p-tolylamine. At
room temperature only a broad resonance at 7.10 ppm can be seen for the aromatic signals of the
amine and essentially no peak is observed corresponding to the p-methyl substituent. At –15 °C
the broad resonance at 7.10 ppm begins to develop a shoulder and a large, broad resonance
begins to appear around 2.20 ppm, the first evidence of a signal belonging to the p-methyl
substituents. At –90 °C the equilibrium process appears to slow and the peaks belonging to
di-p-tolylamine begin to resolve. Two resonances begin to appear at ~ 7 ppm, corresponding to
the aromatic protons of the tolyl substituent. Furthermore, a broad peak appears at 6 ppm which
can be attributed to the NH fragment, and the methyl resonance continues to sharpen at 2.20 ppm
(Figure 5.2.10). It has long been postulated that FLP reactivity occurs because an “encounter
complex” is formed in situ between the Lewis acid and base.143 These encounter complexes have
never been observed experimentally, and we sought to see whether or not we can observe one in
solution between 4-16 and p-tol2NH. However, performing low temperature DOSY and 1H/19F
HOESY NMR experiments gave no indication of an encounter complex in solution. Attempts to
co-crystallize the combination were also unsuccessful.

179
Figure 5.2.10 - Low temperature 1H VT-NMR study between 4-16 and p-tol2NH.
This rapid equilibrium interaction has also been observed with other FLP combinations. As
shown previously in Chapter 2, both diethyl ether and crown ethers show a rapid exchange
process with B(C6F5)3, but these combinations still allow effective H2 activation and further
hydrogenation of 1,1-diphenylethylene as well as anthracene.144 Seeking to develop a
hydrogenation catalyst based phosphonium Lewis acids, we subjected 1,1-diphenylethylene to
5 mol % of 4-17 and p-tol2NH under 4 atm of H2. However, even at 100 °C there was no
evidence of olefin hydrogenation. Subsequently, a mixture of 4-17 and p-tol2NH was
pressurized with 4 atm of HD and heated to 100 °C to determine if hydrogen activation could
occur. Indeed, after 18 h the 1H NMR spectrum shows the formation of H2, indicating that
reversible hydrogen activation is occurring (Figure 5.2.11).

180
Figure 5.2.11 - Observed HD scrambling after 18 h with p-tol2NH and 4-17 at 100 °C.
Aryl-substituted amines have been known to form radical cations145,146 so it was critical to
confirm that the H2 activation proceeds through a heterolytic FLP-type activation rather than a
homolytic process. Control experiments were performed in order to support this mechanism.
The first experiment was the stoichiometric mixture of 4-17 with p-tol3N, which when combined
in C6D5Br formed an intense dark blue solution, indicative of radical formation. After adding
HD to this mixture and heating to 100 °C no HD scrambling was observed, indicating that the H2
seen in the previous experiment is likely not generated homolytically. In order to prove a radical
was not being formed in the reaction between 4-17 and p-tol2NH, the two were subjected to the
same reaction conditions in the absence of HD, but in the presence of a radical trap,
1,8-dihydroanthracene. If a radical was being formed, H2 would be generated with concurrent
formation of anthracene, however no H2 is observed in this reaction, supporting the proposed
FLP type H2 activation with 4-17. These preliminary results show that phosphonium Lewis acids
are capable of activating H2 in an FLP fashion, and others in the group have begun to seek ways
of exploiting this reactivity.

181
5.3 Conclusions
This chapter has presented the first applications of highly electrophilic fluorophosphonium
cations. The addition of phosphine donors to 4-6 or 4-12 resulted in a nucleophilic aromatic
substitution reaction, similarly observed with B(C6F5)3. This reaction led to the generation of an
all-phosphorus FLP system with a free phosphine para-disposed to a phosphonium cation (4-2).
While this product did not possess sufficient Lewis acidity or basicity to activate small
molecules, one and two equivalents of CO2 were successfully activated in an FLP fashion using
an amidofluorophosphorane (5-4) or diamidophosphorane (5-8). The Lewis acid 4-17, which
was the most Lewis acidic phosphonium cation generated, has proven to have numerous
applications in catalysis. Initially, it was shown to be an extremely effective catalyst for the
hydrodefluorination of fluoroalkanes, with an expanded substrate scope compared to the
B(C6F5)3 catalyzed hydrodefluorination reactions. Mechanistic insight indicated that the process
was not simply initiated but indeed phosphonium catalyzed. Expanding the reaction scope,
hydrosilylation reactions catalyzed by 4-17 were investigated. Olefins were shown to interact
with 4-17 and 1-hexene was isomerized to 2-hexene. Olefins and alkynes were subsequently
shown to be effectively hydrosilylated with a catalytic amount of 4-17. Expanding upon this
reaction, 4-17 was also shown to be an effective catalyst for the dehydrocoupling of silanes with
a number of E–H containing functionalities (E = R2N, RS, RO). The H2 generated in this
reaction was subsequently utilized in a tandem transfer hydrogenation reaction with olefins,
resulting in the first metal-free transfer hydrogenation catalyst for olefins. Finally, we postulated
the ability of 4-17 to activate hydrogen since the proposed mechanism for the transfer
hydrogenation reaction proceeds through a hydridofluorophosphorane intermediate.
Calculations were undertaken to support this theory and 4-17 was indeed found to activate
hydrogen gas with the Lewis base, p-tol2NH.
5.4 Experimental Section
5.4.1 General Considerations
All preparations and manipulations were carried out under an anhydrous N2 atmosphere using
standard Schlenk and glovebox techniques. All glassware was oven-dried and cooled under
vacuum before use. Solvents were purified with a Grubbs-type column system manufactured by
Innovative Technologies and dispensed into thick-walled Schlenk glass flasks equipped with

182
Teflon valve stopcocks. All solvents were degassed prior to use via repeated freeze-pump-thaw
cycles. CD2Cl2 (Aldrich) was deoxygenated, distilled over CaH2, then stored over 4 Å molecular
sieves before use. C6D5Br (Aldrich) was deoxygenated and stored over 4 Å molecular sieves
before use. NMR spectra were obtained on a Bruker AvanceIII-400 MHz spectrometer or an
Agilent DD2-600 MHz spectrometer. In selective cases the resonance for ipso-carbons or C6F5
carbons were not located in the 13C{1H} NMR. DFT calculations were performed using Gaussian
09.147 Combustion analyses were performed in house at Analest, employing a Perkin Elmer 2400
Series II CHNS Analyzer. Ph2P(o-C6H4NHMe) and PhP(o-C6H4NHMe)2 were prepared
following literature methods.51 tBu3P, and Ph2PSiMe3 were purchased from Strem and used
without further purification. TMSOTf, Ph3P, tBuLi and Et3SiH were purchased from Sigma
Aldrich and used without further purification. XeF2 was purchased from Apollo Scientific and
used without further purification. All fluorocarbons were ordered from Sigma Aldrich or Alfa
Aesar and used without further purification. Finally, the olefin substrates used in hydrosilylation
and transfer hydrogenation and the amines, thiols, alcohols and carboxylic acids used in
dehydrocoupling were purchased from Sigma Aldrich and used without further purification.
5.4.2 Synthesis of Compounds
[Ph3P(C6F4)P(F)2Ph2] [FB(C6F5)3] (5-1)
A solution of Ph3P (12 mg, 46 μmol) in 2 mL of CH2Cl2 was added to a solution of 4-6 (43 mg,
47 μmol) in 2 mL of CH2Cl2 instantly producing a white precipitate. The mixture was heated to
40 °C for 15 min, over which time the precipitate gradually dissolved to form a colourless
solution. The solvent volume was reduced to approx. 0.5 mL in vacuo and 1 mL of Et2O was
added, followed by 1 mL of n-pentane. Slow evaporation of solvents from the solution produced
colourless, diffraction-quality crystals (53 mg, 45 μmol, 96%).
Anal. Calcd. for C54H25BF22P2: C, 55.66; H, 2.16%. Found: C, 55.16; H, 2.58%.
1H NMR (CD2Cl2, Me4Si, 400 MHz): δ 8.17 (m, 4H, C6H5), 7.92 (m, 3H, C6H5), 7.79 – 7.61
(14H, C6H5), 7.55 (m, 4H, C6H5).
11B NMR (CD2Cl2, 128 MHz, BF3·OEt2): δ –0.6 (d, 1JFB = 65 Hz, BF).
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –36.8 (dt, 1JPF = 699 Hz, 4JFF = 12 Hz, 2F, PF2), –123.5
(m, 2F, P(C6F4)), –128.5 (m, 2F, P(C6F4)), –136.6 (m/br, 6F, B(o-C6F5)), –163.5 (t, 3JFF = 20 Hz,
3F, B(p-C6F5)), –167.9 (m, 6F, B(m-C6F5)), –191.4 (m/br, 1F, BF).

183
31P{1H} NMR (CD2Cl2, 162 MHz, H3PO4): δ 16.1 (t, 3JPF = 6 Hz, 1P, Ph3P), –58.4 (tm, 1JPF =
699 Hz, 1P, PF2).
13C{1H} NMR (CD2Cl2, 100 MHz, Me4Si): δ 136.8 (d, 4JPC = 3 Hz, P(p-C6H5)), 136.2 (dt, 2JPC
= 14 Hz, 3JFC = 11 Hz, P(o-C6H5)), 134.3 (d, 3JPC = 11 Hz, P(m-C6H5)), 133.8 (d, 4JPC = 4 Hz,
P(p-C6H5)), 131.2 (d, 2JPC = 14 Hz, P(o-C6H5)), 129.5 (dt, 3JPC = 18 Hz, 4JFC = 2 Hz, P(m-C6H5)),
129.4 (dt, 1JPC = 65 Hz, 2JFC = 16 Hz, P(i-C6H5)), 116.1 (d, 1JPC = 92 Hz, P(i-C6H5).
[Ph2P(C6F4)P(F)Ph2] [O3SCF3] (5-2)
A colourless solution of Ph2PSiMe3 (80 mg, 310 μmol) in 2 mL of C6H5Br was added to a flask
containing solid 4-12 (150 mg, 288 μmol). Agitation of the mixture for several minutes produced
a yellow solution containing trace particles, which was then filtered through a Kimwipe® plug in
a glass pipette. The solvent and volatile Me3SiF were removed in vacuo, resulting in a clear,
viscous, yellow oil. Et2O (10 mL) was then added, and the mixture was agitated thoroughly for
5 min. A yellow oil settled out from the colourless supernatant, which was then decanted, and the
residue was dried in vacuo. n-Pentane (10 mL) was then added, and the mixture was triturated to
a fine suspension before the precipitate was allowed to settle. The supernatant was decanted and
the solid was dried in vacuo yielding a light-yellow powder. X-Ray quality crystals were grown
from a mixture of CH2Cl2 and hexanes. (147 mg, 208 μmol, 72%).
Anal. Calcd. for C31H20F8O3P2S: C, 54.24; H, 2.94%. Found: C, 52.53; H, 3.05%. *Carbon was
consistently low over multiple experiments, even with crystalline samples.
1H NMR (CD2Cl2, Me4Si, 400 MHz): δ 8.18–8.03 (m, 6H, C6H5) 7.92 (m, 4H, C6H5), 7.63 (m,
4H, C6H5), 7.51–7.42 (m, 6H, C6H5).
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –78.9 (s, CF3, 3F), –123.1 (m, C6F4, 2F), –124.4 (dt,
1JPF = 1011 Hz, 4JFF = 14 Hz, PF, 1F), –125.9 (m, C6F4, 2F).
31P{1H} NMR (CD2Cl2, 162 MHz, H3PO4): δ 86.8 (dt, 1JPF = 1011 Hz, 3JPF = 7 Hz, PF), –15.0 (t,
3JPF = 23 Hz, Ph2P).
13C{1H} NMR (CD2Cl2, 100 MHz, Me4Si): δ 148.5 (dm, 1JFC = 260 Hz, C6F4), 147.7 (dm, 1JFC =
267 Hz, C6F4), 139.6 (dd, JPC = 3 Hz, JFC = 2 Hz, C6H5), 134.3 (d, JPC = 38 Hz, C6H5), 134.4 (s,
C6H5), 131.4 (d, JPC = 16 Hz, C6H5), 130.7 (s, C6H5), 129.4 (d, JPC = 8 Hz, C6H5), 121.0 (q, 1JFC
= 321 Hz, CF3), 116.4 (dm, 1JPC = 113 Hz, 2JFC = 12 Hz, i-C6H5), 115.2 (d, 1JPC = 14 Hz,
i-C6H5).

184
Ph2PF2(o-C6H4NHMe) (5-3)
In a glovebox, a 20 mL vial was charged with Ph2P(o-C6H4NHMe) (178 mg, 611 μmol), then
5 mL of CH2Cl2 were added, resulting in a colorless solution. A separate 20 mL vial was charged
with XeF2 (103 mg, 611 μmol), then 5 mL of CH2Cl2 were added, resulting in a colorless
solution. The XeF2 solution was cooled to –35 °C, and subsurface addition of the Ph2PAr
solution over 2 minutes resulted in immediate effervescence, producing a pale-yellow solution.
The solution was allowed to warm to ambient temperature and the solvent was completely
removed in vacuo yielding an off-white residue, which was triturated with 5 mL of n-pentane
resulting in a suspension. The solvent was completely removed in vacuo and the solid was
isolated in quantitative yield as an off-white powder. (201 mg, 610 μmol, 99%).
Anal. Calcd. for C19H18F2NP: C, 69.30; H, 5.51; N, 4.25%. Found: C, 68.82; H, 5.59; N, 4.16%.
1H NMR (400 MHz, CD2Cl2, Me4Si): δ 7.96 (m, 4H, Ph/Ar), 7.60 – 7.32 (8H, Ph/Ar), 6.70 (m,
2H, Ph/Ar), 4.68 (q/br, 3JHH = 5.0 Hz, 1H, NH), 2.67 (d, 3JHH = 5.0 Hz, 3H, CH3).
19F NMR (377 MHz, CD2Cl2, CFCl3): δ –35.9 (d, 1JPF = 625 Hz).
31P{1H} NMR (162 MHz, CD2Cl2, H3PO4): δ –45.6 (t, 1JPF = 625 Hz).
13C{1H} NMR (100 MHz, CD2Cl2, Me4Si): δ 150.6 (dt, JPC = 8 Hz, JFC = 2 Hz, Ph/Ar), 136.3
(dt, 1JPC = 176 Hz, 2JFC = 28 Hz, Ph/Ar), 134.1 (dt, JPC = 13 Hz, JFC = 9 Hz, Ph/Ar), 133.4 (dt,
JPC = 12 Hz, JFC = 6 Hz, Ph/Ar), 132.4 (dt, JPC = 3 Hz, JFC = 1 Hz, Ph/Ar), 131.9 (dt, JPC = 4 Hz,
JFC = 2 Hz, Ph/Ar), 128.8 (dt, JPC = 17 Hz, JFC = 1 Hz, Ph/Ar), 120.1 (dt, 1JPC = 180 Hz, 2JFC =
28 Hz, Ph/Ar), 116.2 (d, JPC = 17 Hz, Ph/Ar), 111.6 (d, JPC = 12 Hz, Ph/Ar), 30.8 (s, CH3).
[Ph2PF(o-C6H4NHMe)] [O3SCF3] (5-4)
In a glovebox, a 20 mL vial was charged with 5-3 (136 mg, 413 μmol) and 10 mL of CH2Cl2 was
added, producing a pale-yellow solution. A separate 20 mL vial was charged with Me3SiO3SCF3
(92 mg, 413 μmol), then cooled to –35 °C. The solution of 1 was added to the cold vial of
Me3SiO3SCF3 resulting in no visible change. The solution was allowed to warm to ambient
temperature, then solvent volume was reduced to approx. 1 mL in vacuo before adding 10 mL of
n-pentane, which resulted in a pale-yellow oil which was allowed to separate from the
supernatant. The supernatant was discarded and the oil was dried in vacuo producing an off-
white powder (123 mg, 268 μmol, 65%).

185
Anal. Calcd. for C20H18F4NO3PS: C, 52.29; H, 3.95; N, 3.05%. Found: C, 51.93; H, 4.12; N,
3.04%.
1H NMR (400 MHz, CD2Cl2, Me4Si): δ 8.01 (m, 2H, Ph/Ar), 7.84 – 7.73 (9H, Ph/Ar), 7.01 (2H,
Ph/Ar), 6.89 (m, 1H, Ph/Ar), 5.20 (q/br, 3JHH = 4.8 Hz, 1H, NH), 2.82 (d, 3JHH = 4.8 Hz, 3H,
CH3). 19F NMR (377 MHz, CD2Cl2, CFCl3): δ –79.9 (s, 3F, CF3), –125.3 (d, 1JPF = 980 Hz, 1F,
PF).
31P{1H} NMR (162 MHz, CD2Cl2, H3PO4): δ 94.4 (d, 1JPF = 980 Hz).
13C{1H} NMR (100 MHz, CD2Cl2, Me4Si): δ 154.9 (d, JPC = 5 Hz, JPF = 1 Hz, Ph/Ar), 140.7 (t,
JPC = 2 Hz, JFC = 2 Hz, Ph/Ar), 138.1 (dd, JPC = 3 Hz, JFC = 2 Hz, Ph/Ar), 136.3 (d, JPC = 16 Hz,
Ph/Ar), 134.1 (d, JPC = 13 Hz, Ph/Ar), 131.0 (dd, 1JPC = 265 Hz, 2JFC = 12 Hz, Ph/Ar), 131.0 (d,
JPC = 15 Hz, Ph/Ar), 121.2 (q, 1JFC = 322 Hz, CF3), 118.5 (d, JPC = 15 Hz, Ph/Ar), 117.4 (dd, 1JPC
= 110 Hz, 2JFC = 16 Hz, Ph/Ar), 114.3 (d, JPC = 9 Hz, Ph/Ar), 31.1 (s, CH3).
Ph2PF(o-C6H4NMe) (5-5)
In a glovebox, a flame-dried 50 mL Schlenk flask containing a stir bar was charged with
Ph2PF2(o-C6H4NHMe) (62 mg, 188 μmol) and 7 mL of THF were added, producing a pale-
yellow solution. The flask was sealed with a septum, transferred from the glovebox to a
dinitrogen/vacuum double manifold, and then cooled to –78 °C. While stirring, t-BuLi (118 μL,
1.7 M in n-pentane, 200 μmol) was added over 2 min via a microsyringe, instantly resulting in a
bright-yellow solution, which was allowed to warm to ambient temperature, then stirred for
5 min. The solvent was removed in vacuo, producing a yellow residue. The flask was transferred
to a glovebox where 3 mL of C7H8 was added, a small amount of an orange precipitate was
separated from the yellow solution by filtering through a Kimwipe® plug in a pipette into a 5 mL
vial. The solvent volume was reduced to approx. 1 mL in vacuo, then 1 mL of n-pentane was
added. Slow evaporation of the solvent yielded yellow, diffraction-quality crystals (52 mg,
168 μmol, 89%).
Anal. Calcd. for C19H17FNP: C, 73.78; H, 5.54; N, 4.53%. Found: C, 73.96; H, 5.80; N, 4.72%.
1H NMR (400 MHz, C7D8, Me4Si): δ 7.77 (m, 4H, Ph/Ar), 7.48 (m, 1H, Ph/Ar), 7.22 (m, 1H,
Ph/Ar), 7.13 – 7.01 (6H, Ph/Ar), 6.52 (m, 1H, Ph/Ar), 6.08, (m, 1H, Ph/Ar), 2.28 (dd, 3JPH =
4.4 Hz, 4JFH = 2.4 Hz, 3H, CH3).
19F NMR (377 MHz, C7D8, CFCl3): δ –44.4 (d, 1JPF = 679 Hz).
31P{1H} NMR (162 MHz, C7D8, H3PO4): δ –44.6 (d, 1JPF = 679 Hz).

186
13C{1H} NMR (100 MHz, C7D8, Me4Si): δ 162.2 (dd, JPC = 7 Hz, JFC = 6 Hz, Ph/Ar), 137.2
(dd, JPC = 3 Hz, JFC = 1 Hz, Ph/Ar), 133.8 (dd, 1JPC = 148 Hz, 2JFC = 30 Hz, Ph/Ar), 132.6 (dd,
JPC = 5 Hz, JFC = 3 Hz, Ph/Ar), 131.9 (dd, JPC = 12 Hz, JFC = 4 Hz, Ph/Ar), 130.8 (dd, JPC =
3 Hz, JFC = 1 Hz, Ph/Ar), 128.5 (dd, JPC = 16 Hz, JFC = 1 Hz, Ph/Ar), 127.8 (dd, 1JPC = 175 Hz,
2JFC = 50 Hz, Ph/Ar), 116.3 (d, JPC = 20 Hz, Ph/Ar), 103.7 (d, JPC = 18 Hz, Ph/Ar), 30.9 (dd, 2JPC
= 3 Hz, 3JFC = 2 Hz, CH3).
Ph2PF(o-C6H4N(Me)CO2) (5-6)
In a glovebox, a flame-dried 50 mL Schlenk flask containing a stir bar was charged with 5-5
(43 mg, 139 μmol) and 7 mL of THF were added, producing a bright-yellow solution. The flask
was sealed with a septum, transferred from the glovebox to a dinitrogen/vacuum double
manifold, cooled to –196 °C, evacuated, sealed, allowed to warm to ambient temperature, then
backfilled with approx. 1 atm of CO2, resulting in an instant color change to a pale-yellow. The
solvent was completely removed in vacuo, leaving an off-white residue. The flask was
transferred to a glovebox where 10 mL of Et2O were added, and the mixture was triturated,
yielding a suspension. The solid was allowed to settle and the supernatant was filtered through a
Kimwipe® plug into a 20 mL vial. Slow evaporation of the solvent yielded off-white, diffraction-
quality crystals (35 mg, 99 μmol, 71%).
Anal. Calcd. for C20H17FNO2P: C, 67.99; H, 4.85; N, 3.96%. Found: C, 67.72; H, 5.02; N,
3.82%.
IR νC=O: 1696 cm-1.
1H NMR (400 MHz, CD2Cl2, Me4Si): δ 8.02 (m, 1H, Ph/Ar), 7.82 (m, 4H, Ph/Ar), 7.63 (m, 1H,
Ph/Ar), 7.54 (m, 2H, Ph/Ar), 7.47 (m, 4H, Ph/Ar), 7.24 (m, 1H, Ph/Ar), 7.14 (m, 1H, Ph/Ar),
3.15 (s, 3H, CH3).
19F NMR (377 MHz, CD2Cl2, CFCl3): δ –34.2 (d, 1JPF = 664 Hz).
31P{1H} NMR (162 MHz, CD2Cl2, H3PO4): δ –57.0 (d, 1JPF = 664 Hz).
13C{1H} NMR (100 MHz, CD2Cl2, Me4Si): δ 153.1 (d, 2JPC = 8 Hz, CO2), 146.4 (dd, JPC = 8 Hz,
JFC = 5 Hz, Ph/Ar), 135.6 (dd, 1JPC = 177 Hz, 2JFC = 28 Hz, Ph/Ar), 135.2 (t, JPC = 10 Hz, JFC =
10 Hz, Ph/Ar), 134.1 (dd, JPC = 3 Hz, JFC = 2 Hz, Ph/Ar), 132.3 (dd, JPC = 13 Hz, JFC = 6 Hz,
Ph/Ar), 131.7 (dd, JPC = 4 Hz, JFC = 1 Hz, Ph/Ar), 128.7 (d, JPC = 17 Hz, Ph/Ar), 123.0 (d, JPC =
15 Hz, Ph/Ar), 119.8 (dd, 1JPC = 172 Hz, 2JFC = 31 Hz, Ph/Ar), 116.2 (dd, JPC = 11 Hz, JFC =
2 Hz, Ph/Ar), 32.8 (s, CH3).

187
PhPF2(o-C6H4NHMe)2 (5-7)
The compound was prepared by a method analogous to that of 5-3, using PhPAr2 (212 mg,
662 μmol), XeF2 (112 mg, 662 μmol), CH2Cl2 (5 mL), n-pentane (5 mL) and was isolated in
quantitative yield as an off-white powder (236 mg, 658 μmol, 99%).
Anal. Calcd. for C20H21F2N2P: C, 67.03; H, 5.91; N, 7.82%. Found: C, 66.72; H, 6.04; N,
7.75%.
1H NMR (400 MHz, CD2Cl2, Me4Si): δ 7.90 (m, 2H, Ph/Ar), 7.57 (m, 1H, Ph/Ar), 7.50 (m, 2H,
Ph/Ar), 7.41 – 7.32 (4H, Ph/Ar), 4.74 (s/br, 2H, NH), 2.70 (s, 6H, CH3).
19F NMR (377 MHz, CD2Cl2, CFCl3): δ –32.1 (d, 1JPF = 588 Hz).
31P{1H} NMR (162 MHz, CD2Cl2, H3PO4): δ –38.8 (t, 1JPF = 588 Hz).
13C{1H} NMR (100 MHz, CD2Cl2, Me4Si): δ 150.8 (d, JPC = 8 Hz, Ph/Ar), 134.4 (d, JPC = 2 Hz,
Ph/Ar), 134.1 (m/br, Ph/Ar), 133.6 (m/br, Ph/Ar), 133.1 (d, JPC = 11 Hz, Ph/Ar), 132.7 (d, JPC =
2 Hz, Ph/Ar), 132.0 (d, JPC = 4 Hz, Ph/Ar), 128.8 (d, JPC = 17 Hz, Ph/Ar), 116.3 (d, JPC = 17 Hz,
Ph/Ar), 111.8 (d, JPC = 12 Hz, Ph/Ar), 30.9 (s, CH3).
PhP(o-C6H4NMe)2 (5-8)
The compound was prepared by a method analogous to that of 5-5, using 5-7 (151 mg,
421 μmol), t-BuLi (496 μL, 1.7 M in n-pentane, 842 μmol), THF (15 mL), toluene (5 mL) and
was isolated as a yellow solid (101 mg, 317 μmol, 75%).
Anal. Calcd. for C20H19N2P: C, 75.46; H, 6.02; N, 8.80%. Found: C, 75.31; H, 6.15; N, 8.80%.
1H NMR (400 MHz, C7D8, Me4Si): δ 7.39 – 7.27 (m, 5H, Ph), 7.05 (m, 2H, Ar), 6.97 (m, 2H,
Ar), 6.61 (m, 2H, Ar), 6.24 (m, 2H, Ar), 2.55 (d, 3JPH = 6.8 Hz, 3H, CH3).
31P{1H} NMR (162 MHz, C7D8, H3PO4): δ –58.7 (s).
13C{1H} NMR (100 MHz, C7D8, Me4Si): δ 162.1 (d, 4JPC = 3 Hz, Ph/Ar), 136.5 (d, 3JPC = 3 Hz,
Ph/Ar), 133.1 (d, 1JPC = 121 Hz, Ph/Ar), 131.2 (d, 1JPC = 117 Hz, Ph/Ar), 131.2 (d, 3JPC = 4 Hz,
Ph/Ar), 129.8 (d, 4JPC = 3 Hz, Ph/Ar), 129.3 (d, 3JPC = 11 Hz, Ph/Ar), 128.6 (d, 2JPC = 14 Hz,
Ph/Ar), 116.8 (d, 2JPC = 20 Hz, Ph/Ar), 104.4 (d, 2JPC = 17 Hz, Ph/Ar), 31.2 (d, 2JPC = 4 Hz,
CH3).
PhP(o-C6H4N(Me)CO2)2 (5-9)
The compound was prepared by a method analogous to that of 5-6, using 5-8 (50 mg, 157 μmol),
except that after removal of THF in vacuo, CH2Cl2 (1 mL) was added to the off-white solid that

188
remained. The mixture was triturated, yielding a suspension, which was filtered through a
Kimwipe® plug into a 5 mL vial. Slow evaporation of the solvent yielded off-white, diffraction-
quality crystals (56 mg, 138 μmol, 87%).
Anal. Calcd. for C22H19N2O4P·0.5CH2Cl2: C, 60.21; H, 4.49; N, 6.24%. Found: C, 60.02; H,
4.45; N, 6.15%.
IR νC=O: 1697 cm-1.
1H NMR (400 MHz, CD2Cl2, Me4Si): δ 8.12 (2H, Ph/Ar), 7.72 – 7.12 (m, 11H, Ph/Ar), 3.04 (s,
6H, CH3).
31P{1H} NMR (162 MHz, CD2Cl2, H3PO4): δ –68.6 (s).
13C{1H} NMR (100 MHz, CD2Cl2, Me4Si): δ 152.8 (d, 2JPC = 8 Hz, CO2), 146.6 (d, 2JPC = 8 Hz,
Ph/Ar), 135.2 (d, 1JPC = 177 Hz, Ph/Ar), 135.1 (d, 2JPC = 11 Hz, Ph/Ar), 134.2 (d, 4JPC = 3 Hz,
Ph/Ar), 131.5 (d, 4JPC = 5 Hz, Ph/Ar), 130.4 (d, 3JPC = 13 Hz, Ph/Ar), 128.6 (d, 3JPC = 18 Hz,
Ph/Ar), 123.5 (d, 3JPC = 16 Hz, Ph/Ar), 118.4 (d, 1JPC = 170 Hz, Ph/Ar), 116.4 (d, 2JPC = 12 Hz,
Ph/Ar), 32.9 (s, CH3).
PhP(o-C6H4N(Me)CO2)2 (5-10)
The compound was prepared by a method analogous to that of 5-4, except using 5-5 (50 mg,
152 μmol), and Me3SiO3SCF3 (37 mg, 166 μmol), and was isolated as an off-white powder
(58 mg, 132 μmol, 87%, satisfactory elemental analysis not obtained)
1H NMR (400 MHz, CD2Cl2, Me4Si): δ 8.02 – 7.89 (m, 7H, Ph/Ar), 6.78 – 7.70 (m, 5H, Ph/Ar),
7.18 (m, 1H, Ph/Ar), 6.96 (m, 1H, Ph/Ar), 2.97 (d, 3JPH = 14 Hz, 3H, CH3).
19F NMR (377 MHz, CD2Cl2, CFCl3): δ –79.9 (s, 3F, CF3).
31P{1H} NMR (162 MHz, CD2Cl2, H3PO4): δ 76.8 (s).
13C{1H} NMR (100 MHz, CD2Cl2, Me4Si): δ 161.0 (d, JPC = 8 Hz, Ph/Ar), 139.0 (d, JPC =
3 Hz, Ph/Ar), 137.3 (d, JPC = 3 Hz, Ph/Ar), 134.7 (d, JPC = 13 Hz, Ph/Ar), 131.6 (d, JPC = 2 Hz,
Ph/Ar), 130.6 (d, JPC = 14 Hz, Ph/Ar), 123.9 (d, JPC = 15 Hz, Ph/Ar), 121.2 (q, 1JCF = 322 Hz,
CF3), 119.4 (d, 1JPC = 105 Hz, Ph/Ar), 117.1 (d, 1JPC = 87 Hz, Ph/Ar), 109.8 (d, JPC = 17 Hz,
Ph/Ar), 30.4 (d, 2JPC = 3 Hz, CH3).
Ph3CF65
A solution of tBu3P (100 mg, 494 μmol) in CH2Cl2 was added to XeF2 (84 mg, 494 μmol) and
the resulting mixture was stirred until cessation of effervescence. This solution was then added to
[Ph3C] [B(C6F5)4] (456 mg, 494 μmol) and allowed to stir for one hour. The solvent was

189
removed in vacuo and the product was extracted from the solid with hexanes (3 x 5 mL). The
solvent was removed from the extract in vacuo, yielding Ph3CF (121 mg, 93%). 19F NMR
analysis of the product in CD2Cl2 shows a signal at δF –126.6.
Reaction of 4-17 with Ph3CF
A solution of Ph3CF (4 mg, 15 μmol) in CD2Cl2 (3 mL) was added to 4-17 (20 mg, 16 μmol).
The solution immediately turned from colorless to orange, and 31P{1H} NMR and 19F NMR
spectra indicated that the phosphonium salt was completely consumed, having formed
(C6F5)3PF2, and that Ph3CF was no longer present in solution. The sample was analyzed by 1H
NMR, confirming complete conversion of Ph3CF to [Ph3C] [B(C6F5)4], and by 19F and 31P{1H}
NMR, indicating conversion of 4-17 to (C6F5)3PF2 (4-8).
1H NMR (CD2Cl2, 400 MHz, Me4Si): δ 8.27 (tt, 3JHH = 7.5 Hz, 4JHH = 1.3 Hz, 3H, p-C6H5), 7.88
(dd, 3JHH = 8.4 Hz, 4JHH = 7.5 Hz, 6H, m-C6H5), 7.66 (dd, 3JHH = 8.4 Hz, 4JHH = 1.4 Hz, 6H,
o-C6H5).
19F NMR (CD2Cl2, CFCl3, 377 MHz): δ 1.5 (dsept, 1JPF = 695 Hz, 4JFF = 16 Hz, 2F, PF2),
−133.12 (m, 8F, o-C6F5, B(C6F5)4), –132.5 (m, 6F, o-C6F5, F2P(C6F5)3), –146.3 (t, 3JFF = 20 Hz,
3F, p-C6F5, F2P(C6F5)3), –159.3 (m, 6F, m-C6F5, F2P(C6F5)3), –163.7 (t, 3JFF = 20 Hz, 4F, p-C6F5,
B(C6F5)4), −167.6 (t, 3JFF = 17 Hz, 8F, m-C6F5, B(C6F5)4).
31P{1H} NMR (CD2Cl2, 162 MHz, H3PO4): δ –48.0 (tsept, 1JPF = 695 Hz, 3JPF = 10 Hz, PF2).
Reaction of 4-17 with 1-fluoropentane
In a glove box, 4-17 (25 mg, 20 μmol) was weighed into a 5 mL vial. 1-Fluoropentane (0.5 mL)
was then added, resulting a yellow solution. The vial was capped and left undisturbed for 5 min,
after which point the solution color had turned red. After 30 min the cap was removed from the
vial and the 1-fluoropentane was allowed to evaporate slowly from the orange-red solution. The
product mixture was then dissolved in 1 mL of C6D5Br and submitted for 31P NMR, revealing
only the presence of the difluorophosphorane 4-8 with a triplet of multiplets at –48.0 with a 1JPF
= 697 Hz. The 19F, 11B and 1H NMR spectroscopic analyses indicate decomposition of the pentyl
cation and borate anion to produce a mixture.

190
Reaction of 4-17 with α,α,α-Trifluorotoluene
In a glove box, 4-17 (15 mg, 12 μmol) was weighed into a vial and dissolved in neat
trifluorotoluene. The colorless solution immediately began to turn yellow, eventually becoming
an orange-red color. Multinuclear NMR analysis indicated the incomplete conversion of 4-17 to
4-8 and numerous decomposition products.
11B NMR (CD2Cl2, 128 MHz, BF3·OEt2): δ 59.9 (s/br), 42.6 (s/br), 22.4 (s/br), –16.3 (s).
31P{1H} NMR (CD2Cl2, 162 MHz, H3PO4): δ 67.8 (d, 1JPF = 1048 Hz, PF), –48.0 (t, 1JPF =
738 Hz, PF2).
Reaction of 4-17 with [Et3Si·(C7H8)2] [B(C6F5)4] and Octafluorotoluene.
Compound 4-17 (20 mg, 35 μmol) was dissolved in 5 mL of C6H5Br and the resulting solution
was added to octafluorotoluene (8 mg, 34 μmol) at –35 °C. This cold solution was added to a
precooled sample of [Et3Si·(C7H8)2][B(C6F5)4] (32 mg, 33 μmol). As the [Et3Si·(C7H8)2]
[B(C6F5)4] dissolved, a white powder began to precipitate. The supernatant was decanted and the
solid was dried in vacuo. Isolated yield: (32 mg, 76 %). Multinuclear NMR analysis indicated
selective formation of 4-17.
11B NMR (CD2Cl2, 128 MHz, BF3·OEt2): δ –16.7 (s, B(C6F5)4).
19F NMR (CD2Cl2, 377 MHz, CFCl3): δ –119.5 (dsept, 1JPF = 1062 Hz, 4JFF = 14 Hz, 1F, PF),
−121.4 (m, 3F, P(p-C6F5)), –124.3 (ddd, 3JFF = 29 Hz, 4JFF = 14 Hz, 4JFF = 6 Hz, 6F, P(o-C6F5)),
–133.4 (m/br, 8F, B(o-C6F5)), –149.6 (m, 6F, P(m-C6F5)), –163.9 (t, 3JFF = 20 Hz, 4F,
B(p-C6F5)), –167.9 (m/br, 8F, B(m-C6F5)).
31P{1H} NMR (CD2Cl2, 162 MHz, H3PO4): δ 67.8 (dm, 1JPF = 1062 Hz, PF).
5.4.3 Catalytic Reaction Procedures
Hydrodefluorination Reactions
All reactions were carried out under identical conditions, although catalyst loadings vary. A
representative procedure for the reaction using 1-fluoropentane as the substrate follows: In a
glove box, 4-17 (1.0 mg, 0.80 μmol) was weighed directly into a J-Young NMR tube.
1-Fluoropentane (7 mg, 78 μmol) and Et3SiH (10 mg, 86 μmol) were then each weighed into
separate 5 mL vials. C6D5Br (0.5 mL) was added to the vial containing 1-fluoropentane, and the
resulting solution was transferred to the vial containing Et3SiH. Fluorobenzene (7.6 μL, 81 μmol)
was then added to this solution as an internal standard. The solution was transferred to the

191
J-Young NMR tube, which was then sealed, agitated and left at ambient temperature. The
reaction mixture was analyzed by 1H NMR and 19F spectroscopy after 2 h (Figure 5.4.1).
Figure 5.4.1 - 19F NMR spectrum of the reaction of 1-fluoropentane with Et3SiH in the presence
of 4-17 (1 mol%) after 2 h.
Hydrosilylation Reactions
All hydrosilylation reactions were carried out under similar conditions, so only one procedure is
described below:
In a glovebox, 1.5 mol% of 4-17 (7 mg, 5.7 μmol) was measured out into a vial and dissolved in
CD2Cl2 (or C6D5Br) and added to Et3SiH (52 mg, 450 μmol) measured out in a separate vial.
This mixture was then added to a vial containing 1-hexene (38 mg, 450 μmol) and then the
solution was added to an NMR tube and allowed to react. Monitoring the reaction by NMR
indicated the reaction was complete after 30 minutes, resulting in the formation of the
hydrosilylated product, triethylhexylsilane. A saturated solution of NaHCO3 was added and the
product was extracted with CH2Cl2. The mixture was filtered through a plug of silica and the
solvent removed in vacuo, yielding a colourless oil.
1H NMR (400 MHz, CDCl3, Me4Si): δ 1.30 (m, 8H), 0.94 (t, J = 7.9 Hz, 9H), 0.90 (t, J = 7.2 Hz,
3H), 0.51 (c, J = 8.3 Hz, 8H).
13C{1H} NMR (100 MHz, CDCl3, Me4Si): δ 33.7 (s), 31.7 (s), 23.9 (s), 22.7 (s), 14.1 (s), 11.4
(s), 7.5 (s), 3.4 (s).

192
29Si{1H} NMR (78.5 MHz, CD2Cl2, Me4Si): δ 6.5 (s).
1H NMR (400 MHz, CD2Cl2, Me4Si): δ 1.4-1.2 (m, 8H), 0.87 (t, 3JHH = 6.8 Hz, 3H), 0.81 (m,
2H), 0.38 (s, 6H).
13C{1H} NMR (100 MHz, CD2Cl2, Me4Si): δ 33.1 (s), 31.9 (s), 23.4 (s), 23.0 (s), 19.4 (s), 14.3
(s), 1.8 (s).
29Si{1H} NMR (78.5 MHz, CD2Cl2, Me4Si): δ 32.35 (s).
1H NMR (400 MHz, CD2Cl2, Me4Si): δ 7.6 (m, 6H), 7.4 (m, 9H), 1.6-1.1 (m, 10H), 0.90 (m,
3H), 0.81 (m, 2H).
13C{1H} NMR (100 MHz, CD2Cl2, Me4Si): δ 126.4 (s), 135.6 (s), 129.3 (s), 127.8 (s), 33.5 (s),
31.4 (s), 23.9 (s), 22.6 (s), 14.1 (s), 13.3 (s).
29Si{1H} NMR (78.5 MHz, CD2Cl2, Me4Si): δ –10.81 (s).
1H NMR (400 MHz, CDCl3, Me4Si): δ 7.18 (m, 8H), 7.05 (m, 2H), 3.99 (t, J = 7.9 Hz, 1H), 1.33
(d, J = 7.9 Hz, 2H), 0.75 (t, J = 8.1 Hz, 9H), 0.26 (c, J = 7.9 Hz, 6H).
13C{1H} NMR (100 MHz, CDCl3, Me4Si): δ 147.3 (s), 128.3 (s), 127.5 (s), 126.0 (s), 47.3 (s),
19.1 (s), 7.3 (s), 3.5 (s).
29Si{1H} NMR (87.5 MHz, CDCl3, Me4Si): δ 6.7 (s).
1H NMR (400 MHz, CDCl3, Me4Si): δ 7.17 (m, 4H), 7.08 (m, 1H), 2.79 (m, J = 7 Hz, 1H), 1.20
(d, 3JHH = 7 Hz, 3H), 0.92 (m, 1H), 0.83 (m, 1H), 0.80 (t, 3JHH = 8 Hz, 9H), 0.34 (m, 6H).

193
13C{1H} NMR (100 MHz, CDCl3, Me4Si): δ 150.2 (s), 128.2 (s), 126.6 (s), 125.7 (s), 35.2 (s),
26.5 (s), 21.5 (s), 7.4 (s), 3.7 (s).
29Si{1H} NMR (87.5 MHz, CDCl3, Me4Si): δ 6.2 (s)
1H NMR (400 MHz, CDCl3, Me4Si): δ 7.16 (d, J = 8.4 Hz, 2H), 7.08 (d, J = 8.3 Hz, 2H), 2.77
(sx., 3JHH = 7 Hz, 1H), 1.17 (d, 3JHH = 7 Hz, 3H), 0.92-0.80 (m, 2H), 0.80 (t, 3JHH = 8.0 Hz, 9H),
0.34 (m, 6H).
13C{1H} NMR (100 MHz, CDCl3, Me4Si): δ 148.6 (s), 131.2 (s), 128.3 (s), 127.9 (s), 35.7 (s),
26.5 (s), 21.5 (s), 7.4 (s), 3.7 (s).
29Si{1H} NMR (78.5 MHz, CDCl3, Me4Si): δ 6.2 (s).
1H NMR (400 MHz, CD2Cl2, Me4Si): δ 7.07 (d, 3JHH = 8 Hz, 2H), 6.72 (d, 3JHH = 8 Hz, 2H),
2.81 (sx, 3JHH = 7 Hz, 1H), 1.22 (d, 3JHH = 7 Hz, 3H), 1.00 (m, 2H), 0.93 (t, 3JHH = 7 Hz, 9H),
0.86 (t, 3JHH = 8 Hz, 9H), 0.53 (q, 3JHH = 8 Hz, 6H), 0.40 (m, 6H).
13C{1H} NMR (100 MHz, CD2Cl2, Me4Si), δ : 153.7 (s), 142.5 (s), 127.7 (s), 115.0 (s), 35.4 (s),
26.8 (s), 21.7 (s), 7.3 (s), 6.8 (s), 6.5 (s), 3.7 (s).
29Si{1H} NMR (87.5 MHz, CDCl3, Me4Si): δ 20.8 (s), 6.1 (s).
1H NMR (400 MHz, CDCl3, Me4Si): δ 1.68 (m, 5H), 1.18 (m, 5H), 0.90 (t, 3JHH = 8.0 Hz, 9H),
0.72 (m, 1H), 0.51 (q, 3JHH = 8 Hz, 6H).
13C{1H} NMR (100 MHz, CDCl3, Me4Si): δ 28.4 (s), 27.9 (s), 27.1 (s), 23.5 (s), 7.7 (), 1.9 (s).
29Si{1H} NMR (87.5 MHz, CDCl3, Me4Si): δ 5.8 (s).

194
1H NMR (400 MHz, CDCl3, Me4Si): δ 1.55-1.08 (m, 6H), 0.95 (t, 3JHH = 7.8 Hz, 9H), 0.93 (m,
3H), 0.90 (t, 3JHH = 7 Hz, 3H), 0.75 (m, 1H), 0.54 (q, 3JHH = 8 Hz, 6H).
13C{1H} NMR (100 MHz, CDCl3, Me4Si): δ 31.6 (s), 31.0 (s), 22.8 (s), 16.5 (s), 14.2 (s), 14.1
(s), 7.7 (s), 2.2 (s).
29Si{1H} NMR (87.5 MHz, CDCl3, Me4Si): δ 8.0 (s).
1H NMR (400 MHz, CDCl3, Me4Si): δ 6.38 (dt, 3JHH = 14, 3JHH = 7 Hz, 1H), 5.38 (d, 3JHH =
14 Hz, 1H), 2.09 (q, 3JHH = 7 Hz, 2H), 1.27 (m, 12H), 1.00-0.87 (m, 12H), 0.61 (q, 3JHH = 7 Hz,
6H).
13C{1H} NMR (100 MHz, CD2Cl2, Me4Si): δ 150.4 (s), 124.9 (s), 34.1 (s), 31.9 (s), 29.8 (s), 29.5
(s), 29.4 (s), 29.3 (s), 22.7 (s), 14.1 (s), 8.0 (s), 4.7 (s).
29Si{1H} NMR (87.5 MHz, CDCl3, Me4Si): δ –2.8.
1H NMR (400 MHz, CD2Cl2, Me4Si): δ 7.37-7.29 (m, 8H), 7.22 (m, 3H), 0.82 (t, 3JHH = 8 Hz,
9H), 0.43 (q, 3JHH = 8 Hz, 6H).
13C{1H} NMR (100 MHz, CD2Cl2, Me4Si): δ 148.3 (s), 146.8 (s), 145.8 (s), 140.6 (s), 128.9 (s),
128.4 (s), 128.3 (s), 128.0 (s), 127.8 (s), 126.1 (s), 7.9 (s), 5.3 (s).
The conformation was assigned by NMR comparison with the product from the analogous
reaction with B(C5F6)3.88
1H NMR (400 MHz, CDCl3, Me4Si): δ 2.00 (m, 1H), 1.70 (m, 1H), 1.55-1.39 (m, 6H), 1.21 (m,
1H), 0.98 (d, 3JHH = 7 Hz, 3H) 0.95 (t, 3JHH = 8 Hz, 9H), 0.95 (m, 1H), 0.55 (q, 3JHH = 8 Hz, 6H).

195
13C{1H} NMR (100 MHz, CDCl3, Me4Si): δ 35.3 (s), 29.2 (s), 28.7 (s), 28.1 (s), 22.4 (s), 21.1
(s), 16.5 (s), 7.9 (s), 3.1 (s).
29Si{1H} NMR (87.5 MHz, CDCl3, Me4Si): δ 6.2 (s).
Dehydrocoupling Reactions
All hydrosilylation reactions were carried out under similar conditions and the procedure is
described below:
To a solution of the catalyst 4-17 (1.0 – 1.5 mol%) in C6D5Br or CD2Cl2 (1.0 mL) was added the
corresponding silane (1.0-1.2 Eq) and RH (R= Ar2N, ArS, ArO, ArCO2,) (1.0 Eq) at rt. The
reaction was monitored by NMR analysis until the reaction was complete. Yield was determined
by 1H-NMR spectroscopy.
1H NMR (400 MHz, C6D5Br, Me4Si): δ 7.18 (t, 3JHH = 8 Hz, 4H), 6.92 (m, 6H), 0.86 (t, 3JHH =
8 Hz, 9H), 0.69 (q, 3JHH = 8 Hz, 6H).
13C{1H} NMR (100 MHz, C6D5Br, Me4Si): δ 147.9 (s), 128.6 (s), 123.9 (s), 121.7 (s), 6.8 (s), 4.9
(s).
29Si{1H} NMR (79.5 MHz, C6D5Br, Me4Si): δ 9.6 (s).
1H NMR (400 MHz, CD2Cl2, Me4Si): δ 7.63 (m, 6H), 7.40 (m, 3H), 7.33 (m, 6H), 7.07 (m, 4H),
6.95-6.87 (m, 6H).
13C{1H} NMR (100 MHz, CD2Cl2, Me4Si): δ 148.3 (s), 136.7 (s), 134.7 (s), 130.3 (s), 129.3 (s),
128.3 (s), 125.9 (s), 122.8 (s).
29Si{1H} NMR (79.5 MHz, CD2Cl2, Me4Si): δ –16.03 (s).

196
1H NMR (400 MHz, C6D5Br, Me4Si): δ 7.14 (m, 4H), 7.07 (m, 4H), 6.96 (t, 3JHH = 7 Hz, 2H),
0.40 (s, 6H).
13C{1H} NMR (100 MHz, C6D5Br, Me4Si): δ 146.4 (s), 128.7 (s), 125.2 (s), 123.5 (s), 2.9 (s).
29Si{1H} NMR (79.5 MHz, C6D5Br, Me4Si): δ 9.7 (s).
1H NMR (400 MHz, C6D5Br, Me4Si): δ 7.65 (m, 2H), 7.25 (m, 3H), 7.11 (t, 3JHH = 8 Hz, 4H),
6.91 (m, 6H), 0.33 (s, 6H).
13C{1H} NMR (100 MHz, C6D5Br, Me4Si): δ 147.5 (s), 138.1 (s), 133.1 (s), 128.9 (s), 128.6 (s),
127.7 (s), 124.2 (s), 121.9 (s), -0.3 (s).
29Si{1H} NMR (79.5 MHz, C6D5Br, Me4Si): δ –2.3 (s).
1H NMR (400 MHz, C6D5Br, Me4Si): δ 6.97 (d, 3JHH = 8 Hz, 2H), 6.87 (t, 3JHH = 8 Hz, 2H),
2.17 (s, 6H), 0.89 (t, 3JHH = 8 Hz, 9H), 0.71 (q, 3JHH = 8 Hz, 6H).
13C{1H} NMR (100 MHz, CD2Cl2, Me4Si): δ 145.1 (s), 130.3 (s), 128.8 (s), 123.5 (s), 19.8 (s),
6.4 (s), 4.5 (s).
29Si{1H} NMR (79.5 MHz, CD2Cl2, Me4Si): δ 9.5 (s).
1H NMR (400 MHz, CD2Cl2, Me4Si): δ 7.09 (d, 3JHH = 8 Hz, 2H), 7.00 (t, 3JHH = 8 Hz, 2H), 2.30
(s, 6H), 0.50 (s, 6H).

197
13C{1H} NMR (100 MHz, CD2Cl2, Me4Si): δ 145.2 (s), 134.2 (s), 130.2 (s), 126.6 (s), 21.1 (s),
3.7 (s).
29Si{1H} NMR (79.5 MHz, CD2Cl2, Me4Si): δ 9.9 (s).
1H NMR (400 MHz, CD2Cl2, Me4Si): δ 7.43 (m, 2H), 7.07 (m, 3H), 0.93 (t, 3JHH = 8 Hz, 9H),
0.63 (q, 3JHH = 8 Hz, 6H).
13C{1H} NMR (100 MHz, CD2Cl2, Me4Si): δ 136.5 (s), 134.6 (s), 128.2 (s), 126.2 (s), 6.8 (s), 5.0
(s).
29Si{1H} NMR (79.5 MHz, CD2Cl2, Me4Si): δ 24.0 (s).
1H NMR (400 MHz, CD2Cl2, Me4Si): δ 7.32 (d, 3JHH = 8 Hz, 2H), 6.89 (d, 3JHH = 8 Hz, 2H),
2.11 (s, 3H), 0.94 (t, 3JHH = 8 Hz, 9H), 0.63 (q, 3JHH = 8 Hz, 6H).
13C{1H} NMR (100 MHz, CD2Cl2, Me4Si): δ 135.7 (s), 134.6 (s), 129.2 (s), 127.1 (s), 20.4 (s),
6.8 (s), 5.0 (s).
29Si{1H} NMR (79.5 MHz, CD2Cl2, Me4Si): δ 23.4 (s).
1H NMR (400 MHz, C6D5Br, Me4Si): δ 7.24 (d, 3JHH = 9 Hz, 2H), 7.02 (t, 3JHH = 9 Hz, 2H),
0.90 (t, 3JHH = 8 Hz, 9H), 0.59 (q, 3JHH = 8 Hz, 6H).
13C{1H} NMR (100 MHz, C6D5Br, Me4Si): δ 135.8 (s), 132.4 (s), 129.7 (s), 128.3 (s), 6.8 (s), 4.9
(s).
29Si{1H} NMR (79.5 MHz, C6D5Br, Me4Si): δ 24.5 (s).

198
1H NMR (400 MHz, C6D5Br, Me4Si): δ 7.24 (dd, 3JHH = 8 Hz, 3JHF = 5 Hz, 2H), 6.75 (t, 3JHH =
8 Hz, 2H), 0.91 (t, 3JHH = 8 Hz, 9H), 0.59 (q, 3JHH = 8 Hz, 6H).
13C{1H} NMR (100 MHz, C6D5Br, Me4Si): δ 161.6 (d, 1JCF = 244 Hz), 136.2 (d, 3JCF = 8 Hz),
132.4, 115.1 (d, 2JCF = 21 Hz), 6.8 (s), 4.8 (s).
29Si{1H} NMR (79.5 MHz, C6D5Br, Me4Si): δ 24.0 (s).
1H NMR (400 MHz, C6D5Br, Me4Si): δ 0.87 (t, 3JHH = 8 Hz, 9H), 0.57 (q, 3JHH = 8 Hz, 6H).
13C{1H} NMR (100 MHz, C6D5Br, Me4Si): δ 147.6 (d, 1JCF = 244 Hz), 139.7 (d, 1JCF = 250 Hz),
137.1 (d, 1JCF = 255 Hz) , 106.6 (td, 2JCF = 23 Hz, 3JCF = 5 Hz), 6.4 (s), 5.0 (s).
29Si{1H} NMR (79.5 MHz, C6D5Br, Me4Si): δ 28.3 (s).
1H NMR (400 MHz, C6D5Br, Me4Si): δ 7.15 (m, 2H), 6.87 (m, 3H), 0.95 (t, 3JHH = 8 Hz, 9H),
0.66 (q, 3JHH = 8 Hz, 6H).
13C{1H} NMR (100 MHz, C6D5Br, Me4Si): δ 155.7 (s), 129.5 (s), 121.4 (s), 120.0 (s), 6.9 (s), 5.2
(s).
29Si{1H} NMR (79.5 MHz, C6D5Br, Me4Si): δ 20.1 (s)
1H NMR (400 MHz, C6D5Br, Me4Si): δ 6.91 (d, 3J = 8 Hz, 2H), 6.76 (t, 3JHH = 7 Hz, 2H), 2.18
(s, 6H), 0.92 (t, J = 7.8 Hz, 9H), 0.68 (q, J = 7.9 Hz, 6H).
13C{1H} NMR (100 MHz, C6D5Br, Me4Si): δ 152.9 (s), 128.0 (s), 128.0 (s), 121.4 (s), 17.8 (s),
7.0 (s), 6.0 (s).

199
29Si{1H} NMR (79.5 MHz, C6D5Br, Me4Si): δ 18.2 (s)
1H NMR (400 MHz, C6D5Br, Me4Si): δ 6.79 (d, 3JHH = 9 Hz, 2H), 6.70 (d, 3JHH = 9 Hz, 2H),
3.48 (s, 3H), 0.97 (t, 3JHH = 8 Hz, 9H), 0.67 (q, 3JHH = 8 Hz, 6H).
13C{1H} NMR (100 MHz, C6D5Br, Me4Si): δ 154.3 (s), 149.3 (s), 120.5 (s), 114.6 (s), 55.1 (s),
6.9 (s), 5.2 (s).
29Si{1H} NMR (78.5 MHz, C6D5Br, Me4Si): δ 19.5 (s).
1H NMR (400 MHz, C6D5Br, Me4Si): δ 6.75 (s, 4H), 0.96 (t, 3JHH = 8 Hz, 18H), 0.66 (q, 3JHH =
8 Hz, 12 H).
13C{1H} NMR (100 MHz, C6D5Br, Me4Si): δ 149.84 (s), 120.53 (s), 6.88 (s), 5.19 (s).
29Si{1H} NMR (78.5 MHz, C6D5Br, Me4Si): δ 19.8 (s)
1H NMR (400 MHz, C6D5Br, Me4Si): δ 6.94 (d, 3JHH = 8 Hz, 2H), 6.79 (d, 3JHH = 9 Hz, 2H),
2.15 (s, 3H), 0.97 (t, 3JHH = 7 Hz, 9H), 0.67 (q, 3JHH = 8 Hz, 12H).
13C{1H} NMR (100 MHz, C6D5Br, Me4Si): δ 153.4 (s), 130.1 (s), 130.0 (s), 119.8 (s), 20.65 (s),
6.93 (s), 5.23 (s).
29Si{1H} NMR (78.5 MHz, C6D5Br, Me4Si): δ 19.7 (s).
1H NMR (400 MHz, C6D5Br, Me4Si): δ 0.92 (t, 3JHH = 8 Hz, 9H), 0.64 (q, 3JHH = 8 Hz, 12 H).
19F NMR (377 MHz, C6D5Br, Me4Si): δ –158.21 (m, 2F), –163.93 (m, 2F), –166.68 (tt, 3JFF =
23 Hz, 4JFF = 5 Hz, 1F)

200
13C{1H} NMR (100 MHz, C6D5Br, Me4Si): δ 140.8 (d, 1JCF = 244 Hz), 137.8 (d, 1JCF = 250 Hz),
135.7 (d, 1JCF = 247 Hz), 6.9 (s), 4.9 (s).
29Si{1H} NMR (78.5 MHz, C6D5Br, Me4Si): δ 29.5 (s).
1H NMR (400 MHz, C6D5Br, Me4Si): δ 8.10 (d, 3JHH = 8 Hz, 2H), 7.10 (d, 3JHH = 8 Hz, 2H),
2.50 (t, 3JHH = 8 Hz, 2H), 1.50 (m, 2H), 1.20 (m, 10H), 1.01 (t, 3JHH = 8 Hz, 9H), 0.85 (m, 9H).
13C{1H} NMR (100 MHz, C6D5Br, Me4Si): δ 165.8 (s), 147.9 (s), 129.9 (s), 128.4 (s), 127.9 (s),
35.5 (s), 31.5 (s), 30.7 (s), 29.1 (s), 28.9 (s), 28.9 (s), 22.4 (s), 13.8 (s), 6.3 (s), 4.4 (s).
29Si{1H} NMR (79.5 MHz, C6D5Br, Me4Si): δ 25.6 (s).
Transfer Hydrogenation Reactions
All transfer hydrogenation reactions were carried out under similar conditions, so only one
procedure is described below:
In a glovebox, Et3SiH (21 mg, 179 μmol), C6F5CO2H (34 mg, 163 μmol) and
1,1-diphenylethylene (30 mg, 163 μmol) were measured out into separate vials. They were
dissolved in C6D5Br (or CD2Cl2) and combined. This solution was then added to a catalytic
amount of 4-17 (1.5 mol%, 3 mg, 2.4 μmol) and allowed to react at rt. The reaction was
monitored by NMR until completion and yield was determined by 1H NMR spectroscopy (Figure
5.4.2).

201
Figure 5.4.2 - Transfer Hydrogenation of 1,1-diphenylethylene with tandem dehydrocoupling
between pentafluorophenyl carboxylic acid and Et3SiH using 4-17.
5.4.4 X-Ray Crystallography
5.4.4.1 X-Ray Collection and Reduction
Crystals were coated in Paratone-N oil in the glovebox, mounted on a MiTegen Micromount and
placed under an N2 stream, thus maintaining a dry, O2-free environment for each crystal. The
data were collected on a Bruker Kappa Apex II diffractometer. Data collection strategies were
determined using Bruker Apex 2 software and optimized to provide >99.5% complete data to a

202
2θ value of at least 55°. The data were collected at 150(±2) K for all. The data integration and
absorption correction were performed with the Bruker Apex 2 software package.148
5.4.4.2 X-Ray Solution and Refinement
Non-hydrogen atomic scattering factors were taken from the literature tabulations.149 The heavy
atom positions were determined using direct methods employing the SHELX-2013 direct
methods routine. The remaining non-hydrogen atoms were located from successive difference
Fourier map calculations. The refinements were carried out by using full-matrix least squares
techniques on F, minimizing the function ω (Fo-Fc)2 where the weight ω is defined as 4Fo2/2σ
(Fo2) and Fo and Fc are the observed and calculated structure factor amplitudes, respectively. In
the final cycles of each refinement, all non-hydrogen atoms were assigned anisotropic
temperature factors in the absence of disorder or insufficient data. In the latter cases atoms were
treated isotropically. C-H atom positions were calculated and allowed to ride on the carbon to
which they are bonded assuming a C-H bond length of 0.95 Å. H-atom temperature factors were
fixed at 1.20 times the isotropic temperature factor of the C-atom to which they are bonded. The
H-atom contributions were calculated, but not refined. The locations of the largest peaks in the
final difference Fourier map calculation as well as the magnitude of the residual electron
densities in each case were of no chemical significance.

203
Table 5.4.1 - Select Crystallographic Data for 5-1, 5-2, 5-5, 5-6 and 5-9.
5-1·0.25 C6H14 5-2 5-5
Formula C55.5H28.5BF15P2 C31H20F8O3P2S C19H19FNP
Formula weight 1186.03 686.47 311.32
Crystal System Triclinic Orthorhombic Monoclinic
Space group P-1 P212121 P21/c
a(Å) 10.7377(6) 8.2059(4) 12.2115(13)
b(Å) 13.3608(8) 14.1403(7) 10.8681(11)
c(Å) 18.5330(11) 25.1497(12) 11.6437(11)
α(deg) 85.615(3) 90 90
β(deg) 89.546(4) 90 95.523(5)
γ(deg) 71.427(3) 90 90
V(Å3) 2512.6(3) 2918.2(2) 1538.1(3)
Z 2 4 4
Temp. (K) 150 150 150
d(calc)gcm-1 1.568 1.562 1.344
Abs coeff,μ,mm-1 0.208 0.306 0.185
Data collected 12691 6690 2702
DataFO2
>3(FO2) 9213 5867 2233
Variables 739 406 219
R 0.0534 0.0346 0.0895
Rw 0.1673 0.0780 0.2225
GOF 1.029 1.024 1.122

204
5-6 5-9·0.5 CH2Cl2
Formula C20H17FNO2P C22.5H19.5ClN2O4P
Formula weight 353.32 448.32
Crystal System Monoclinic Monoclinic
Space group P21/c C2/c
a(Å) 17.0345(9) 28.4415(13)
b(Å) 11.3459(7) 9.3452(5)
c(Å) 18.3854(10) 15.5756(7)
α(deg) 90 90
β(deg) 107.910(2) 98.410(2)
γ(deg) 90 90
V(Å3) 3381.2(3) 4095.4(3)
Z 8 8
Temp. (K) 150 150
d(calc)gcm-1 1.388 1.454
Abs coeff,μ,mm-1 0.186 0.299
Data collected 7735 7801
DataFO2
>3(FO2) 5671 6006
Variables 451 281
R 0.0514 0.0384
Rw 0.1494 0.1057
GOF 1.040 1.027

205
5.5 References
(1) Dilworth, J. R.; Wheatley, N. Coordination Chemistry Reviews 2000, 199, 89-158.
(2) Tolman, C. A. Chemical Reviews 1977, 77, 313-348.
(3) Stephan, D. W.; Erker, G. Angewandte Chemie International Edition 2010, 49, 46-76.
(4) Ménard, G.; Stephan, D. W. Angewandte Chemie International Edition 2011, 50, 8396-
8399.
(5) Schäfer, A.; Reißmann, M.; Schäfer, A.; Saak, W.; Haase, D.; Müller, T. Angewandte
Chemie International Edition 2011, 50, 12636-12638.
(6) Spikes, G. H.; Fettinger, J. C.; Power, P. P. Journal of the American Chemical Society
2005, 127, 12232-12233.
(7) Harrison, G. C.; Diehl, H. Organic Syntheses 1943, 23, 32.
(8) Dillon, K. B.; Mathey, F.; Nixon, J. F. Phosphorus: The Carbon Copy: From
Organophosphorus to Phospha-organic Chemistry; Wiley, 1998.
(9) Carmalt, C. J.; Lomeli, V.; McBurnett, B. G.; Cowley, A. H. Chemical Communications
1997, 2095-2096.
(10) Denk, M. K.; Gupta, S.; Lough, A. J. European Journal of Inorganic Chemistry 1999,
1999, 41-49.
(11) Gudat, D.; Haghverdi, A.; Hupfer, H.; Nieger, M. Chemistry – A European Journal 2000,
6, 3414-3425.
(12) Burck, S.; Forster, D.; Gudat, D. Chemical Communications 2006, 2810-2812.
(13) Gudat, D. Accounts of Chemical Research 2010, 43, 1307-1316.
(14) Brazeau, A. L.; Caputo, C. A.; Martin, C. D.; Jones, N. D.; Ragogna, P. J. Dalton
Transactions 2010, 39, 11069-11073.
(15) Burford, N.; Ragogna, P. J. Journal of the Chemical Society, Dalton Transactions 2002,
4307-4315.
(16) Dyker, C. A.; Burford, N. Chemistry – An Asian Journal 2008, 3, 28-36.
(17) Hardman, N. J.; Abrams, M. B.; Pribisko, M. A.; Gilbert, T. M.; Martin, R. L.; Kubas, G.
J.; Baker, R. T. Angewandte Chemie International Edition 2004, 43, 1955-1958.
(18) Slattery, J. M.; Hussein, S. Dalton Transactions 2012, 41, 1808-1815.
(19) He, R.; Maruoka, K. Synthesis 2009, 2009, 2289-2292.
(20) Clark, J. H.; Tavener, S. J.; Barlow, S. J. Chemical Communications 1996, 2429-2430.
(21) Tindale, J. J.; Ragogna, P. J. Chemical Communications 2009, 1831-1833.

206
(22) Lall-Ramnarine, S. I.; Mukhlall, J. A.; Wishart, J. F.; Engel, R. R.; Romeo, A. R.; Gohdo,
M.; Ramati, S.; Berman, M.; Suarez, S. N. Beilstein Journal of Organic Chemistry 2014, 10,
271-275.
(23) Musher, J. I. Angewandte Chemie International Edition 1969, 8, 54-68.
(24) Gabbaï, F. P. Science 2013, 341, 1348-1349.
(25) Terada, M.; Kouchi, M. Tetrahedron 2006, 62, 401-409.
(26) Matsukawa, S.; Kajiyama, K.; Kojima, S.; Furuta, S.-y.; Yamamoto, Y.; Akiba, K.-y.
Angewandte Chemie International Edition 2002, 41, 4718-4722.
(27) Wittig, G.; Schöllkopf, U. Chemische Berichte 1954, 87, 1318-1330.
(28) Staudinger, H.; Meyer, J. Helvetica Chimica Acta 1919, 2, 635-646.
(29) Mukaiyama, T.; Matsui, S.; Kashiwagi, K. Chemistry Letters 1989, 18, 993-996.
(30) Mukaiyama, T.; Kashiwagi, K.; Matsui, S. Chemistry Letters 1989, 18, 1397-1400.
(31) McNulty, J.; Dyck, J.; Larichev, V.; Capretta, A.; Robertson, A. J. Letters in Organic
Chemistry 2004, 1, 137-139.
(32) Córdoba, R.; Plumet, J. N. Tetrahedron Letters 2003, 44, 6157-6159.
(33) Wang, X.; Tian, S.-K. Tetrahedron Letters 2007, 48, 6010-6013.
(34) Hudnall, T. W.; Chiu, C.-W.; Gabbaï, F. P. Accounts of Chemical Research 2009, 42,
388-397.
(35) Sole, S.; Gabbai, F. P. Chemical Communications 2004, 1284-1285.
(36) Zhao, H.; Gabbaı, F. P. Organometallics 2012, 31, 2327-2335.
(37) Zhao, H.; Leamer, L. A.; Gabbai, F. P. Dalton Transactions 2013, 42, 8164-8178.
(38) Wade, C. R.; Broomsgrove, A. E. J.; Aldridge, S.; Gabbaï, F. P. Chemical Reviews 2010,
110, 3958-3984.
(39) Kim, Y.; Gabbaï, F. P. Journal of the American Chemical Society 2009, 131, 3363-3369.
(40) Hudnall, T. W.; Kim, Y.-M.; Bebbington, M. W. P.; Bourissou, D.; Gabbai, F. o. P.
Journal of the American Chemical Society 2008, 130, 10890-10891.
(41) Hirai, M.; Gabbai, F. P. Chemical Science 2014, 5, 1886-1893.
(42) Pan, B.; Gabbaï, F. P. Journal of the American Chemical Society 2014, 136, 9564-9567.
(43) Welch, G. C.; Cabrera, L.; Chase, P. A.; Hollink, E.; Masuda, J. D.; Wei, P.; Stephan, D.
W. Dalton Transactions 2007, 3407-3414.
(44) Welch, G. C.; Prieto, R.; Dureen, M. A.; Lough, A. J.; Labeodan, O. A.; Holtrichter-
Rossmann, T.; Stephan, D. W. Dalton Transactions 2009, 1559-1570.

207
(45) Cabrera, L.; Welch, G. C.; Masuda, J. D.; Wei, P.; Stephan, D. W. Inorganica Chimica
Acta 2006, 359, 3066-3071.
(46) Hounjet, L. J.; Caputo, C. B.; Stephan, D. W. Angew. Chem. 2012, 51, 4714-4717.
(47) Sheldrick, G. M. Acta Crystallographica 1975, B31, 1776-1778.
(48) Welch, G. C.; Juan, R. R. S.; Masuda, J. D.; Stephan, D. W. Science 2006, 314, 1124-
1126.
(49) Brisdon, A. K.; Herbert, C. J. Chemical Communications 2009, 6658-6660.
(50) Reis, A.; Dehe, D.; Farsadpour, S.; Munstein, I.; Sun, Y.; Thiel, W. R. New Journal of
Chemistry 2011, 35, 2488-2495.
(51) Hounjet, L. J.; Bierenstiel, M.; Ferguson, M. J.; McDonald, R.; Cowie, M. Dalton
Transactions 2009, 4213-4226.
(52) Light, R. W.; Hutchins, L. D.; Paine, R. T.; Campana, C. F. Inorganic Chemistry 1980,
19, 3597-3604.
(53) Cavell, R. G.; The, K. I.; Griend, L. V. Inorganic Chemistry 1981, 20, 3813-3818.
(54) Cavell, R. G.; Vande-Griend, L. Inorganic Chemistry 1986, 25, 4699-4704.
(55) Cavell, R. G.; Vande-Griend, L. Inorganic Chemistry 1983, 22, 2066-2070.
(56) Hounjet, L. J.; Ferguson, M. J.; Cowie, M. Organometallics 2011, 30, 4108-4114.
(57) Hounjet, L. J.; McDonald, R.; Ferguson, M. J.; Cowie, M. Inorganic Chemistry 2011, 50,
5361-5378.
(58) Hounjet, L. J.; Bierenstiel, M.; Ferguson, M. J.; McDonald, R.; Cowie, M. Inorganic
Chemistry 2010, 49, 4288-4300.
(59) Mömming, C. M.; Otten, E.; Kehr, G.; Fröhlich, R.; Grimme, S.; Stephan, D. W.; Erker,
G. Angewandte Chemie International Edition 2009, 48, 6643-6646.
(60) Spies, P.; Erker, G.; Kehr, G.; Bergander, K.; Fröhlich, R.; Grimme, S.; Stephan, D. W.
Chemical Communications 2007, 5072-5074.
(61) Dureen, M. A.; Stephan, D. W. Journal of the American Chemical Society 2010, 132,
13559-13568.
(62) Zhu, J.; An, K. Chemistry – An Asian Journal 2013, 8, 3147-3151.
(63) Lemal, D. M. The Journal of Organic Chemistry 2004, 69, 1-11.
(64) Shine, K. P.; Sturges, W. T. Science 2007, 315, 1804 -1805.
(65) Weigert, F. J. Journal of Organic Chemistry 1980, 45, 3476-3483.
(66) Douvris, C.; Ozerov, O. V. Science 2008, 321, 1188-1190.

208
(67) Muetterties, E. L.; Mahler, W.; Schmutzler, R. Inorganic Chemistry 1963, 2, 613-618.
(68) Caputo, C. B.; Stephan, D. W. Organometallics 2012, 31, 27-30.
(69) Alcarazo, M.; Gomez, C.; Holle, S.; Goddard, R. Angewandte Chemie International
Edition 2010, 49, 5788-5791.
(70) Gu, W.; Haneline, M. R.; Douvris, C.; Ozerov, O. V. Journal of the American Chemical
Society 2009, 131, 11203-11212.
(71) Schleyer, P. v. R.; Fort, R. C.; Watts, W. E.; Comisarow, M. B.; Olah, G. A. Journal of
the American Chemical Society 1964, 86, 4195-4197.
(72) Scott, V. J.; Celenligil-Cetin, R.; Ozerov, O. V. Journal of the American Chemical
Society 2005, 127, 2852-2853.
(73) Bruce, M. I. Journal of the Chemical Society A: Inorganic, Physical, Theoretical 1968,
1459-1464.
(74) Aizenberg, M.; Milstein, D. Science 1994, 265, 359-361.
(75) Johnson, S. A.; Huff, C. W.; Mustafa, F.; Saliba, M. Journal of the American Chemical
Society 2008, 130, 17278-17280.
(76) Doster, M. E.; Johnson, S. A. Angewandte Chemie International Edition 2009, 48, 2185-
2187.
(77) Kraft, B. M.; Lachicotte, R. J.; Jones, W. D. Journal of the American Chemical Society
2000, 122, 8559-8560.
(78) Rieth, R. D.; Brennessel, W. W.; Jones, W. D. European Journal of Inorganic Chemistry
2007, 2007, 2839-2847.
(79) Chapman, A. M.; Haddow, M. F.; Wass, D. F. Journal of the American Chemical Society
2011, 133, 18463-18478.
(80) Allemann, O.; Duttwyler, S.; Romanato, P.; Baldridge, K. K.; Siegel, J. S. Science 2011,
332, 574-577.
(81) Duttwyler, S.; Douvris, C.; Fackler, N. L. P.; Tham, F. S.; Reed, C. A.; Baldridge, K. K.;
Siegel, J. S. Angewandte Chemie International Edition 2010, 49, 7519-7522.
(82) Hatnean, J. A.; Johnson, S. A. Organometallics 2012, 31, 1361-1373.
(83) Clot, E.; Eisenstein, O.; Jasim, N.; Macgregor, S. A.; McGrady, J. E.; Perutz, R. N.
Accounts of Chemical Research 2011, 44, 333-348.
(84) Blanksby, S. J.; Ellison, G. B. Accounts of Chemical Research 2003, 36, 255-263.

209
(85) Hong, A. C.; Young, C. J.; Hurley, M. D.; Wallington, T. J.; Mabury, S. A. Geophysical
Research Letters 2013, 40, 6010-6015.
(86) Parks, D. J.; Blackwell, J. M.; Piers, W. E. The Journal of Organic Chemistry 2000, 65,
3090-3098.
(87) Rendler, S.; Oestreich, M. Angewandte Chemie, International Edition 2008, 47, 5997-
6000.
(88) Rubin, M.; Schwier, T.; Gevorgyan, V. The Journal of Organic Chemistry 2002, 67,
1936-1940.
(89) Troegel, D.; Stohrer, J. Coordination Chemistry Reviews 2011, 255, 1440-1459.
(90) Karstedt, B.; Google Patents: 1973.
(91) Speier, J. L. In Advances in Organometallic Chemistry; Stone, F. G. A., Robert, W., Eds.;
Academic Press: 1979; Vol. Volume 17, p 407-447.
(92) Markó, I. E.; Stérin, S.; Buisine, O.; Mignani, G.; Branlard, P.; Tinant, B.; Declercq, J.-P.
Science 2002, 298, 204-206.
(93) Holwell, A. J. Platinum Metals Review 2008, 52, 243-246.
(94) Oestreich, M.; Rendler, S. Angewandte Chemie International Edition 2005, 44, 1661-
1664.
(95) Glaser, P. B.; Tilley, T. D. Journal of the American Chemical Society 2003, 125, 13640-
13641.
(96) Königs, C. D. F.; Klare, H. F. T.; Oestreich, M. Angewandte Chemie International
Edition 2013, 52, 10076-10079.
(97) Tondreau, A. M.; Atienza, C. C. H.; Weller, K. J.; Nye, S. A.; Lewis, K. M.; Delis, J. G.
P.; Chirik, P. J. Science 2012, 335, 567-570.
(98) Oertle, K.; Wetter, H. Tetrahedron Letters 1985, 26, 5511-5514.
(99) Asao, N.; Sudo, T.; Yamamoto, Y. The Journal of Organic Chemistry 1996, 61, 7654-
7655.
(100) Lambert, J. B.; Zhao, Y.; Wu, H. The Journal of Organic Chemistry 1999, 64, 2729-
2736.
(101) Zhao, X.; Stephan, D. W. Chemical Science 2012, 3, 2123-2132.
(102) Zhao, X.; Stephan, D. W. Journal of the American Chemical Society 2011, 133, 12448-
12450.
(103) Evans, A. G.; Owen, E. D. Journal of the Chemical Society 1959, 4123-4125.

210
(104) Hounjet, L. J.; Bannwarth, C.; Garon, C. N.; Caputo, C. B.; Grimme, S.; Stephan, D. W.
Angewandte Chemie International Edition 2013, 52, 7492-7495.
(105) Hasegawa, M.; Usui, I.; Konno, S.; Murakami, M. Organic & Biomolecular Chemistry
2010, 8, 4169-4175.
(106) Piers, W. E.; Marwitz, A. J. V.; Mercier, L. G. Inorganic Chemistry 2011, 50, 12252-
12262.
(107) Piers, W. E.; Marwitz, A. J.; Mercier, L. G. Inorganic Chemistry 2011, 50, 12252-12262.
(108) Berkefeld, A.; Piers, W. E.; Parvez, M. Journal of the American Chemical Society 2010,
132, 10660-10661.
(109) Roesler, R.; Har, B. J. N.; Piers, W. E. Organometallics 2002, 21, 4300-4302.
(110) Blackwell, J. M.; Morrison, D. J.; Piers, W. E. Tetrahedron 2002, 58, 8247-8254.
(111) Parks, D. J.; Blackwell, J. M.; Piers, W. E. J. Org. Chem. 2000, 65, 3090-3098.
(112) Hermeke, J.; Mewald, M.; Oestreich, M. Journal of the American Chemical Society 2013,
135, 17537-17546.
(113) Simonneau, A.; Oestreich, M. Angewandte Chemie International Edition 2013, 52,
11905-11907.
(114) Rubin, M.; Schwier, T.; Gevorgyan, V. Journal of Organic Chemistry 2002, 67, 1936-
1940.
(115) Djuric, S.; Venit, J.; Magnus, P. Tetrahedron Letters 1981, 22, 1787-1790.
(116) Staubitz, A.; Sloan, M. E.; Robertson, A. P. M.; Friedrich, A.; Schneider, S.; Gates, P. J.;
Günne, J. S. a. d.; Manners, I. Journal of the American Chemical Society 2010, 132, 13332-
13345.
(117) Sadow, A. D.; Tilley, T. D. Journal of the American Chemical Society 2005, 127, 643-
656.
(118) Waterman, R. Chemical Society Reviews 2013, 42, 5629-5641.
(119) Hill, M. S.; Liptrot, D. J.; MacDougall, D. J.; Mahon, M. F.; Robinson, T. P. Chemical
Science 2013, 4, 4212-4222.
(120) Nishimoto, Y.; Okita, A.; Yasuda, M.; Baba, A. Angewandte Chemie International
Edition 2011, 50, 8623-8625.
(121) Blackwell, J. M.; Foster, K. L.; Beck, V. H.; Piers, W. E. Journal of Organic Chemistry
1999, 64, 4887-4892.

211
(122) Brook, M. A.; Grande, J. B.; Ganachaud, F. Advances in Polymer Science 2011, 235,
161-183.
(123) Cella, J.; Rubinsztajn, S. Macromolecules 2008, 41, 6965-6971.
(124) Greb, L.; Tamke, S.; Paradies, J. Chemical Communications 2014, 50, 2318-2320.
(125) Grimme, S. Journal of Computational Chemistry 2006, 27, 1787-1799.
(126) Chai, J. D.; Head-Gordon, M. Physical Chemistry Chemical Physics 2008, 10, 6615-
6620.
(127) Sabatier, P. Industrial and Engineering Chemistry 1926, 18, 1005-1008.
(128) Hallman, P. S.; Evans, D.; Osborn, J. A.; Wilkinson, G. Chemical Communications 1967,
305-306.
(129) Boerner, A.; Holz, J. Transition Metals for Organic Synthesis (2nd Edition) 2004, 2, 3-
13.
(130) Pettinari, C.; Marchetti, F.; Martini, D. Comprehensive Coordination Chemistry II 2004,
9, 75-139.
(131) Friedfeld, M. R.; Shevlin, M.; Hoyt, J. M.; Krska, S. W.; Tudge, M. T.; Chirik, P. J.
Science 2013, 342, 1076-1080.
(132) Trovitch, R. J.; Lobkovsky, E.; Bill, E.; Chirik, P. J. Organometallics 2008, 27, 1470-
1478.
(133) Zuo, W. W.; Lough, A. J.; Li, Y. F.; Morris, R. H. Science 2013, 342, 1080-1083.
(134) Jagadeesh, R. V.; Surkus, A. E.; Junge, H.; Pohl, M. M.; Radnik, J.; Rabeah, J.; Huan, H.
M.; Schunemann, V.; Bruckner, A.; Beller, M. Science 2013, 342, 1073-1076.
(135) Bullock, R. M. Science 2013, 342, 1054-1055.
(136) Hantzsch, A. Chemische Berichte 1881, 14, 1637-1638.
(137) Birch, A. J. Journal of the Chemical Society 1944, 314-315.
(138) Farrell, J. M.; Heiden, Z. M.; Stephan, D. W. Organometallics 2011, 30, 4497-4500.
(139) Dunn, N.; Ha, M.; Radosevich, A. Journal of the American Chemical Society 2012, 134,
11330-11333.
(140) Stephan, D. W.; Erker, G. Frustrated Lewis Pairs I: Uncovering and Understanding
2013, 332, 85-110.
(141) Greb, L.; Oña-Burgos, P.; Schirmer, B.; Grimme, S.; Stephan, D. W.; Paradies, J.
Angewandte Chemie International Edition 2012, 51, 10164-10168.

212
(142) Greb, L.; Tussing, S.; Schirmer, B.; Ona-Burgos, P.; Kaupmees, K.; Lokov, M.; Leito, I.;
Grimme, S.; Paradies, J. Chemical Science 2013, 4, 2788-2796.
(143) Rokob, T. A.; Hamza, A.; Stirling, A.; Soós, T.; Pápai, I. Angewandte Chemie
International Edition 2008, 47, 2435-2438.
(144) Hounjet, L. J.; Bannwarth, C.; Garon, C. N.; Caputo, C. B.; Grimme, S.; Stephan, D. W.
Angewandte Chemie International Edition 2013, 52, 7492-7495.
(145) Lam, K.; Geiger, W. E. The Journal of Organic Chemistry 2013, 78, 8020-8027.
(146) O'Connor, A. R.; Nataro, C.; Golen, J. A.; Rheingold, A. L. Journal of Organometallic
Chemistry 2004, 689, 2411-2414.
(147) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman,
J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li,
X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara,
M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.;
Nakai, H.; Vreven, T.; Montgomery, J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.;
Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.;
Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene,
M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.
E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.;
Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.;
Daniels, A. D.; Farkas; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Wallingford CT,
2009.
(148) Bruker AXS Inc. : 2013.
(149) D. T. Cromer, J. T. W. Int. Tables X-Ray Crystallography, 1974; Vol. 4.

213
Chapter 6 Conclusion
6
6.1 Thesis Summary
The work presented in this thesis explored the reactivity of boron based frustrated Lewis pairs
with novel substrates and unprecedented Lewis bases. The focus then shifted to uncovering the
reactivity of highly electrophilic fluorophosphonium cations (EPCs). The work described herein
illustrates the first reports of EPC chemistry, which is proving to be an emerging field of
chemistry. The main conclusions are summarized below:
First, mechanically interlocked molecules can function as frustrated Lewis bases. Sterically
unencumbered amines were shown to form irreversible adducts with B(C6F5)3. When these
amines were incorporated into a [2]rotaxane, H2 activation was found to occur in the presence of
B(C6F5)3. The crown ether is able to provide sufficient steric bulk without the need for covalent
modification of the Lewis base. Mechanistic studies on this system indicated that the crown
ether oxygen atoms are capable of acting as a Lewis base to allow for H2 activation with
B(C6F5)3. This concept was subsequently exploited and the first oxygen/boron FLP
hydrogenation catalyst was developed based on crown ethers and B(C6F5)3.
Second, traditional P/B FLP systems were shown to activate inert C–F bonds in fluoroalkanes,
resulting in the formation of phosphonium fluoroborate salts. These reactions were shown to be
chemoselective, in which trifluoromethyl moieties remained intact throughout the activation of
primary fluoroalkanes. Furthermore, catalytic hydrodefluorination could be achieved using
5 mol% of B(C6F5)3 with Et3SiH. This was the first demonstration of selective metal-free
hydrodefluorination.
Third, xenon difluoride cleanly reacts with a number of P/B FLPs to yield fluorophosphonium
fluoroborate salts of the form [R3PF] [FB(C6F5)3]. The introduction of electron withdrawing
pentafluorophenyl substituents on the phosphorus center altered the reactivity, to the extent that
B(C6F5)3 was no longer a strong enough Lewis acid to abstract a fluoride from the corresponding
difluorophosphoranes. This indicated that the fluorophosphonium cations generated from those

214
systems should be more Lewis acidic than B(C6F5)3. Indeed, highly electrophilic
fluorophosphonium cations are generated by using cationic silylium Lewis acids to abstract a
fluoride. The product [(C6F5)3PF] [B(C6F5)4] (4-17) was shown both experimentally and
computationally to be more Lewis acidic than B(C6F5)3.
Lastly, these EPCs have been shown to exhibit remarkable reactivity. They have been exploited
to activate small molecules such as silanes, CO2 and fluoroalkanes. The scope of fluoroalkane
activation was expanded using [(C6F5)3PF] [B(C6F5)4] compared to that of B(C6F5)3. Catalytic
hydrodefluorination was achieved with this fluorophosphonium cation, as well as catalytic
hydrosilylation. Furthermore, it was discovered to be the first metal-free catalyst to effect the
transfer hydrogenation of olefins via a dehydrocoupling reaction. Finally, preliminary results
indicate that [(C6F5)3PF] [B(C6F5)4] can activate hydrogen in a FLP manner with p-tol2NH.
6.2 Future Work
While the use of ether Lewis bases in FLP chemistry was not a major focus of my research,
others have expanded upon and showed the utility of this work. Recently, two reports were
published using etheral solvents as a Lewis base, one from our lab,1 and the other from the
Ashley lab2 describing the first catalytic reduction of carbonyl functionalities using FLPs.
Further expansion of substrates for these B/O FLP systems should be investigated, including
those substrates which were previously thought to be incompatible with FLPs.
The field of electrophilic fluorophosphonium cations is still in its infancy; there remains a
plethora of unexplored chemistry and now nearly a dozen members of the lab are investigating
this topic. A significant number of these chemists are studying the potential use of 4-17 and
other EPCs as hydrogenation catalysis. Additionally, we have begun to explore the use of 4-17
as a catalyst for the degradation of lignin. Lignin is a complex biopolymer consisting of
aromatic ethers and alcohols, commonly derived from wood. Recently, research groups have
focused attention on the degradation of lignin with the goals of generating value added
products.3-5 As was noted in Chapter 5, 4-17 is able to catalyze the cleavage of aryl ether bonds
in the presence of silanes. This led us to investigate the reactivity of 4-17 with lignin model
compounds. Preliminary results indicate that 4-17 is very effective at degrading the β-O4

215
linkages in model compounds and 4-17 has shown encouraging results with samples of the
biomass itself (Scheme 6.2.1). Work on this project is currently ongoing.
Scheme 6.2.1 - Proposed lignin degradation using 4-17.
An obvious extension of the reported hydrosilylation chemistry is to look at the reactivity of
ketones, imines and other polar unsaturated compounds with 4-17 and silanes. B(C6F5)3
catalyzed hydrosilylation of these substrates is well known6,7 and these reactions will likely
proceed using 4-17. Furthermore, it would be interesting to see if it is possible to develop a
chiral transfer hydrogenation catalyst based on 4-17. One could imagine the use of a chiral
proton source (such as a chiral phosphoric acid) or a chiral silane in an attempt to generate a
chiral alkane. If these pathways proved to be unsuccessful, chiral EPCs can be developed using
a chiral phosphine. Subsequent oxidation using xenon difluoride and fluoride ion abstraction
may lead to an active chiral catalyst.
New derivatives of EPCs should be developed. Others in our group have transitioned away from
pentafluorophenyl rings and have developed dicationic fluorophosphonium salts with pendant
NHCs (Scheme 6.2.2, top).8 This compound was shown to be an effective catalyst for the
hydrodefluorination of fluoroalkanes as well as the hydrosilylation of olefins and alkynes. The
P–F moiety in the developed EPCs are crucial for the reactivity, however it is also a source of

216
instability. In most catalytic reactions, the deactivation product is free phosphine, indicating that
P–F bond cleavage occurs. The inherent electronegativity that fluorine possesses unlocks the
low lying acceptor orbital which gives the EPCs their reactivity. Other electronegative
substituents, such as P–O or P–N moieties, should be investigated as possible replacements. A
specific example would be the use of a Martin type ligand on phosphorus to form a chelating
P−O bond to potentially unlock Lewis acidic behaviour (Scheme 6.2.2, bottom).9-11
Scheme 6.2.2 - Dicationic fluorophosphonium salt (top) and a potential P–O EPC with a Martin
type ligand (bottom).
6.3 References
(1) Mahdi, T.; Stephan, D. W. Journal of the American Chemical Society 2014, 136, 15809-
15812.
(2) Scott, D. J.; Fuchter, M. J.; Ashley, A. E. Journal of the American Chemical Society
2014, 136, 15813-15816.
(3) Zhang, J.; Chen, Y.; Brook, M. A. ACS Sustainable Chemistry & Engineering 2014, 2,
1983-1991.
(4) Rahimi, A.; Ulbrich, A.; Coon, J. J.; Stahl, S. S. Nature 2014, 515, 249-252.
(5) vom Stein, T.; Weigand, T.; Merkens, C.; Klankermayer, J.; Leitner, W. ChemCatChem
2013, 5, 439-441.
(6) Blackwell, J. M.; Sonmor, E. R.; Scoccitti, T.; Piers, W. E. Organic Letters 2000, 2,
3921-3923.

217
(7) Parks, D. J.; Piers, W. E. Journal of the American Chemical Society 1996, 118, 9440-
9441.
(8) Holthausen, M. H.; Mehta, M.; Stephan, D. W. Angewandte Chemie International
Edition 2014, 53, 6538-6541.
(9) Perozzi, E. F.; Martin, J. C. Journal of the American Chemical Society 1979, 101, 1591-
1593.
(10) Matsukawa, S.; Yamamoto, Y.; Akiba, K.-y. Phosphorus, Sulfur, and Silicon and the
Related Elements 2009, 184, 928-935.
(11) Ross, M. R.; Martin, J. C. Journal of the American Chemical Society 1981, 103, 1234-
1235.