Embed Size (px)
Citation preview

Einführung in dieBioinformatik
Kay NieseltIntegrative Transkriptomik
Zentrum für Bioinformatik Tü[email protected]
SS 20111. Was ist Bioinformatik?
2
Ablauf und Formales
• Ringvorlesung mit 5 Dozenten– Dr. Kay Nieselt (ZBIT/WSI)– Prof. Dr. Daniel Huson (ZBIT/WSI)– Prof. Dr. Oliver Kohlbacher (ZBIT/WSI)– Dr. Karsten Borgwardt (MPI)– Prof. Dr. Andrei Lupas (MPI)
• Webseite mit pdfs zu Vorlesungen,Übungen etc. unter:http://ab.inf.uni-tuebingen.de/teaching/ss2011/bioinf/
3
Modulbedingungen
• Um das Modul zu bestehen, muss eineam Ende des Semesters stattfindendeKlausur mitgeschrieben und bestandenwerden (Note ≥ 4,0).
• Am Ende jeder Vorlesung werdenÜbungsblätter zum Selbststudiumausgeteilt, deren Bearbeitung undAbgabe freiwillig ist.
• Antworten an Günter Jäger([email protected])schicken.

4
Themenübersicht
-Klausur21.7.
LupasDie Sprache der Proteine - Evolution konservierterProteinstrukturen
14.7.
KohlbacherImpfen gegen Krebs - Bioinformatik im Impfstoffentwurf7.7.
NieseltIt’s hip to Chip - von Microarrays zu personalisierter Medizin30.6.
NieseltGut vernetzt hält besser - Analyse biologischer Netzwerke9.6.
BorgwardtLernen mit Kernen - Maschinelles Lernen in der Bioinformatik26.5.
HusonAus einer Hand voll Erde - Metagenomik19.5.
KohlbacherMolekulare Maschinen - Proteinstrukturen und ihre Funktion12.5.
HusonDarwins Erben - Stammbäume auf Genomebene5.5.
KohlbacherDesignerdrogen - Wirkstoffe aus dem Rechner28.4.
HusonVon der DNA zur Datenbank - Sequenzierung, Assemblierung21.4.
NieseltWas ist Bioinformatik?14.4.
DozentThemaTermin
5
Was ist Bioinformatik?
Wir beginnen mit Nachschlagen:• Die Bioinformatik ist eine interdisziplinäre
Wissenschaft, die Probleme aus denLebenswissenschaften mit theoretischencomputergestützten Methoden löst. (Wikipedia)
• Bioinformatics is the field of science in which biology,computer science, and information technology mergeto form a single discipline. (NCBI)
• Bioinformatics deals with research, development, orapplication of computational tools and approaches forexpanding the use of biological, medical, behavioral orhealth data, including those to acquire, store,organize, archive, analyze, or visualize such data. (NIH)
6
Ursprung der Bioinformatik• Die Begründerin der
Bioinformatik ist MargaretDayhoff.
• Sie hat 1965 eine Sammlungvon Proteinsequenzen, auchbekannt als Atlas of ProteinSequence and Structureveröffentlicht.
• Zusammen mit Richard Eckhat sie den erstenphylogenetischen Baum mitmolekularen Sequenzenveröffentlicht.
M. Dayhoff, 1980, Quelle: Tochter

7
Themen der Bioinformatik
• DNA-Sequenzanalyse– Sequenzierung– Alignment von 2 oder mehreren Sequenzen– Verwandschaft von Sequenzen
• Suche nach Mustern / Motiven• Evolution von Arten• Genvorhersage incl. der Architektur (gene
finding / gene prediction)• Strukturen von Molekülen
– Protein-Strukturvorhersage– RNA-Strukturvorhersage
8
Themen der Bioinformatik
• Struktur-Funktionsverhältnisse• Metabolische und regulatorische
Stoffwechselwege• Genexpression
– Microarrays bzw. DNA-Chips
• Systeme: Zellen und Organismen, diesogenannten “omics”-Daten
• Datenaufbereitung und Speicherung inbiologischen Datenbanken– GenBank, SwissProt, viele weitere
• Mathematik, Statistik, künstliche Intelligenz
9
Berufsaussichten
• Stärke der Bioinformatik-Absolventen=> Interdisziplinarität
• Große Bandbreite an Arbeitsfeldern– Forschungsinstitute– Biotechnologische Firmen– Pharmafirmen– Softwareentwicklung– Chemische Entwicklung– Lebensmittelfirmen– Kliniken– Labore– Ämter– usw.

10
Entwicklung
11
2001 – 201110 Jahre Das Menschliche Genom
Science (2001), 291 (5507)Nature (2001), 409 (6822)
12
(Ultimatives) Ziel der Bioinformatik
Ultimatives Ziel der Bioinformatik ist• dazu beizutragen, dass neue
biologischen Einsichten entdecktwerden;
• eine globale Sicht zu erzeugen, von deraus vereinende biologische Prinzipienabgeleitet werden können.

13
Aufgaben der Bioinformatik
• Entwicklung und Implementierung vonWerkzeugen, die den effizientenZugang, Gebrauch und das Managementvon biologischen Daten / Informationenermöglichen.
• Entwicklung neuer effizienterAlgorithmen, mathematischer Methodenund Statistiken.
14
Typische Fragen der Bioinformatik
• Wie sieht eigentlich ein Protein aus?• Wie vergleicht man die Erbinformation
verschiedener Organismen?• Wie entstehen Tumorzellen?
15
Effiziente Algorithmen
• “Ein Algorithmus ist eine aus endlichvielen Schritten bestehende eindeutigeHandlungsvorschrift zur Lösung einesProblems oder einer Klasse vonProblemen.” (Quelle: Wikipedia)
• Effizient bedeutet: „es darf nicht zulange dauern“, selbst wenn es sich umein großes Problem handelt.

16
Effiziente Algorithmen
• Klassisches Beispiel aus dem Alltag:Gegeben seien zwei biologische Sequenzen.Aufgabe: wie ähnlich sind sich die beidenSequenzen?
• Lösung:Optimal: Needleman-Wunsch-Algorithmusbasierend auf dynamischem ProgrammierenEffizient: BLAST
17
Grundlagen der Molekularbiologie
• Gregor Mendel 1866– Konzept des Gens
• Max Perutz 1941– Biologische Rolle der DNS (Desoxy-
Ribonukleinsäure)
• James Watson, Francis Crick 1953– Struktur der DNS– DNS als Träger der genetischen Information
in allen lebenden Organismen.
18
Grundlagen der Molekularbiologie
• DNS (Desoxyribonukleinsäure)– 4 Basen (A, G, C, T)
• Adenin• Guanin• Thymin• Cytosin
– Doppelhelix– Wasserstoffbrückenbindungen
• Bevorzugt: A-T oder G-C

19
Zentrales Dogma der Molekularbiologie
Der Informationsfluss verläuft immer vom Genom zum Proteom.
20
Replikation der DNS
• Während des Prozesses der DNS-Replikationwerden die beiden DNS-Stränge aufgetrenntund jeder der beiden Stränge dient alsMatrize zur Erzeugung eines neuen Strangs.
• Auf Grund der Basenkomplementaritätwird der Erhalt der genetischen Informationgarantiert.
• Prozess wirddurch dasEnzymDNA-Polymerasekatalysiert.
21
Biologische Objekte
Sequenzen
Strukturen
Netzwerke
taaccctaac cctaacccta accctaaccc taaccctaac taaccctaac cctaacccta accctaaccc taaccctaacaaccctaacc ctaaccctaa ccctaacccc taaccctaacaaccctaacc ctaaccctaa ccctaaccct aaccctaacctaaccctaaa ccctaaaccc taaccctaac cctaacccta cccaacccca accccaaccc caaccctaac ccctaaccctcctaacccta accctaaccc taaccctaac ccctaaccccaccctaaccc taaccctaac ccctaaccct aaccctaacc

22
Formalisieren der Daten
• Primärsequenzen werden alsZeichenketten über einem gegebenenAlphabet (z.B. dem Alphabet derNukleotide Σ={A,G,C,T} oder demAlphabet der AminosäurenΣ= {A,R,N,D,C,Q,...}) verwendet
• Beispiel NukleotidsequenzATGCAGGACGACGGATAACGATGACAG
• Beispiel AminosäuresequenzMQDDGQRGQTVVALKCCLLKCARDDG
23
Formalisieren biologischer Vorgänge
• Beispiel: Translation• Bei der Translation einer mRNA in eine
Proteinsequenz wird mittels desgenetischen Codes jedem Codon dermRNA seine zugehörige Aminosäurezugewiesen.
24
Formalisieren der Translation
• Formal:Sei ΣCodon das Alphabet der Codons(AAA,AAG,AAC,AAT,...,TTT). Sei ΣAA ={A,R,N,D,C,Q,...} das Alphabet derAminosäuren.
• Die Translation lässt sich als Funktionf: ΣCodon → ΣAA mRNA → Aminosäureseq.
beschreiben.

25
Mutationen
• Fehler während der Replikation• Lokale Veränderungen der
Primärsequenz der DNS– Substitutionen– Insertionen / Deletionen
• Umordnung ganzer Segmente entlangeines Chromosoms oder Austauschzwischen Chromosomen
26
Variabilität
• Mutationen sind die Quellephänotypischer Vielfalt, die dieGrundlage der natürlichen Selektiondarstellt.
• Veränderte/neue Spezies• Untersuchung von Mutationen führt zu
einem besseren Verständnis desevolutionären Prozesses.
• Ähnlichkeit von Genomen bestimmen
27
Sequenz-Ähnlichkeit
These:Ähnliche Gene / Proteine
haben auch ähnlicheFunktionen
Frage:Wie kann man feststellen
wie ähnlich sich zweiSequenzen sind?
Antwort: visuell
Aber kann man esauch berechnen?

28
Sequenz-AlignmentAlignment = Ausrichtung zweier Sequenzen
Seq 1 : t a t a - t a c g c t a g c aSeq 2 : t a t a a t a g g c t - g c a
Sind zwei ausgerichtete Nukleotide identisch ⇒ MatchSind zwei ausgerichtete Nukleotide nicht identisch ⇒ Mismatch
Daneben gibt es Lücken, die dadurch entstehen, dass indie Sequenzen Nukleotide eingefügt (Insertionen) oderentfernt werden (Deletionen).
Obiges Alignment hat 80% Identität, denn 12 von den 15ausgerichteten Positionen sind identisch.
29
Sequenz-Alignment
Offensichtlich gibt es viele Möglichkeiten zwei Sequenzenauszurichten (zu alignieren).
Folgendes Alignment hat nur 60% Identität.
Seq 1 : - t a t a t a c g c t a g c aSeq 2 : t a t a a t a g g c t - g c a
Das Alignment Problem:Gegeben seien zwei Nukleotid (Protein) Sequenzen.Gesucht ist das Alignment, dessen Wert für einegegebene Bewertungs-Funktion minimal ist.
Eine Generalisierung dieses Problems auf mehrereSequenzen führt zum multiplen Alignment Problem.
30
Bewertungsfunktion
• Wie bewerten man Mutationen, Deletionen /Insertionen?
• Annahme: alle Positionen evolvierenunabhängig voneinander → additive Funktion
• Benötigt wird Bewertung von Match,Mismatch, Gaps
• Beispiel: Match = +2, Mismatch = -1, Gap = -5
Seq 1 : - t a t a t a c g c t a g c aSeq 2 : t a t a a t a g g c t - g c a -5-1-1-1+2+2+2-1+2+2+2-5+2+2+2 = +4
Seq 1 : t a t a - t a c g c t a g c aSeq 2 : t a t a a t a g g c t - g c a +2+2+2+2-5+2+2-1+2+2+2-5+2+2+2 = +13

31
AA-Substitutionsmatrizen
Am häufigsten werden in der PraxisPAM- und BLOSUM-Matrizen verwendet.
PAM:PAM steht für Percent Accepted Mutation; die Matrizenwurden in den 70er Jahren von Margaret Dayhoffentwickelt.
BLOSUM (Blocks Substitution Matrix):1992 von Jorja und Steven Henikoff aufgestellt.
Spezielle mathematische Methoden zur Bewertung vonder Substitution einer Aminosäure durch eine andere Aminosäure → Substitutionsmatrix
32
BLOSUM62
33
Globales vs. lokales Alignment

34
Sequenz-Alignment - OptimalOptimale Lösung:
• Global: Needleman-Wunsch Algorithmus• Lokal: Smith-Waterman Algorithmus
• Beides sind dynamische Programmierverfahren, eineTechnik für das Lösen kombinatorischerOptimierungsprobleme mit den folgendenEigenschaften:• Rekursiv definierter exponentieller Suchraum,• Wiederauftretende Matrizen,• Zusammensetzung der optimalen Lösung aus den
„optimalen“ Lösungen von Teilproblemen
• DPs spielen eine große Rolle in der Bioinformatik
35
BLASTBasic Local Alignment Search Tool - kurz BLAST• Ist das häufigst benutzte Programm zur Suche einer
Sequenz (Anfrage - query) in einer Sequenz-Datenbank(subjects, targets)
• Es ist ein lokales Alignment-Verfahren• Es ist eine Heuristik: schnell ohne Garantie der
Optimalität• Basiert auf der Seed-Extend-Strategie: kurze sehr gute
übereinstimmende Teilsequenzen zwischen query undsubject werden gesucht, die dann nach links und rechts solange ausgedehnt werden, bis ein vorgegebener Wertüberschritten ist.
• Ausgabe ist ein high scoring segment pair, kurz HSP, dasAlignment
• Berechnet Signifikanz eines Treffers in der Datenbank: derE-Wert (e-value)
36
Die BLAST-Familie
37
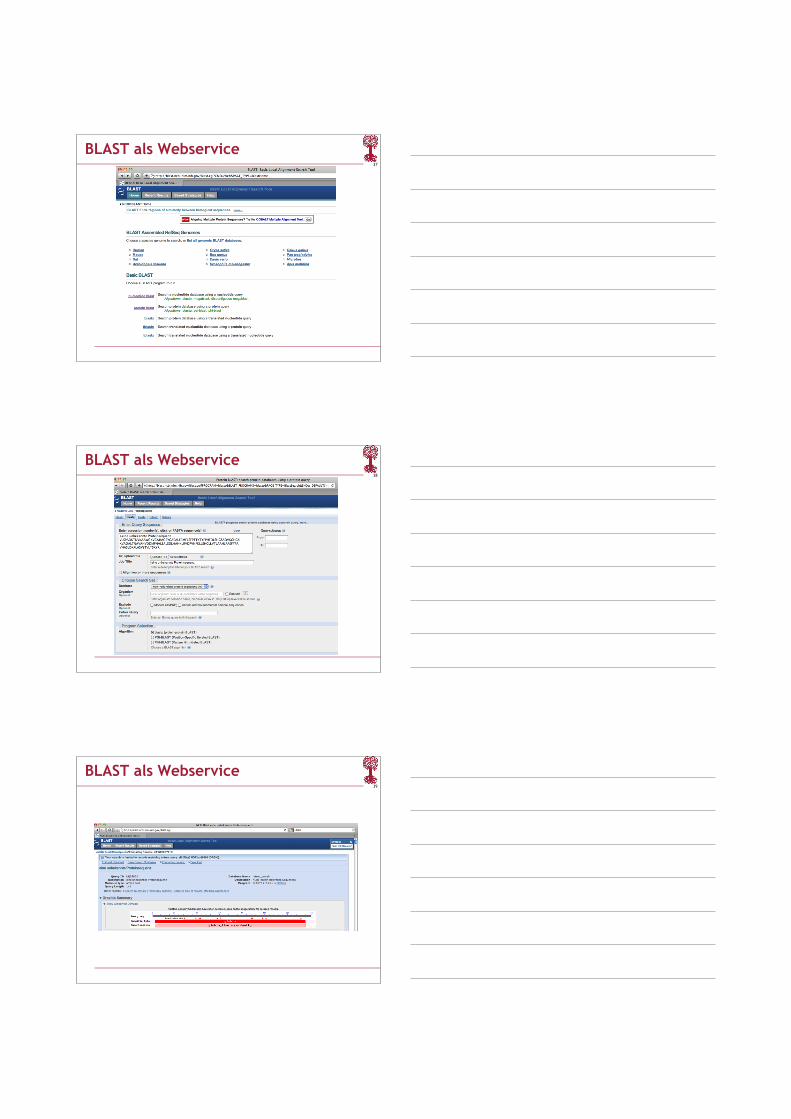
BLAST als Webservice
38
BLAST als Webservice
39
BLAST als Webservice

40
BLAST als Webservice
41
BLAST als Webservice
42
Woher bekommt man Sequenzen?
• Primäre Sequenzdatenbanken– GenBank am National Center for
Biotechnology Information (NCBI), USA;http://www.ncbi.nlm.nih.gov/,Suchmaschine ENTREZ
– European Molecular Biology Laboratory(EMBL) am European BioinformaticsInstitute (EBI) in Hinxton, England;http://www.ebi.ac.uk/
– UniProt (Universal Protein Resource);http://www.uniprot.org/
– many many more

43
FASTA Format
• erste Zeile: beginnt mit >, gefolgt vomSequenznamen und evtl. Beschreibungder Sequenz.
• zweite Zeile: eigentliche Sequenz.
>emb|AL096836| Pyrococcus abyssi complete genomeGGGCTTTAGCCTCCTTCACCGCTTCCACGATTTTCTGCCTGTCAAAGTGGTTTTTAATTAAAAATTCAAGGTGGAGTAAAAAGGGATGTTTTTAAATTCTCAAATAGCTCGTCGTAAACCCCTTCATCTATTTCTCTCTGAACTCGGTAACTCCCATGCTTAAAGCCGTTCCAATGACTTCCTTGGCGGCAGCTGGTTTCTCTTCATCTTAGCTATCTTGATAACTTGCTCCATCGTTAAGGCTCACCGCTGCCCTTCTCGAGCCCTAGTTCCTTCTTTATCAACTGCCTATCTCGAACTGCTTGGTTACTGGATCTACGATGATCTTCACTGGGA
44
Fazit• Während der letzten 3 Jahrzehnte ist bedingt
durch bedeutende Fortschritte auf dem Gebietder Molekularbiologie, verbunden mit denFortschritten in genomischen Technologien, einenormer Wachstum der durch diewissenschaftliche Gemeinschaft erzeugtenbiologischen Information / Daten zu beobachten.
• Diese Explosion genomischer Daten hat damitnotwendigerweise Bereitstellung vonDatenbanken für das Aufbewahren, Organisierenund Indizieren sowie speziell entwickeltenWerkzeugen zur Ansicht und Analyse der Datengeführt.
45
Referenzen
• Altschul SF, Gish W, Miller W, Myers EW,Lipman DJ. J Mol Biol. 1990 Oct 5;215(3):403-10.
• Dayhoff, Eck, Chang, and Sochard. Atlas ofProtein Sequence and Structure, 1965
• Eck RV, Dayhoff MO.Science. 1966 Apr15;152(3720):363-6.
• Henikoff S, Henikoff JG. Proc Natl Acad SciUSA. 1992 Nov 15;89(22):10915-9.
• Needleman SB, Wunsch CD. J Mol Biol. 1970Mar;48(3):443-53.