Embed Size (px)
Citation preview





List of Papers
This thesis is based on the following papers, which are referred to in the text by their Roman numerals.
I Ekerljung, L., Steffen, A-C., Carlsson, J., Lennartsson, J.
(2006) Effects of HER2-binding Affibody Molecules on Intra-cellular Signaling Pathways. Tumor Biology, 27:201–210
II Ekerljung, L., Lindborg, M., Gedda, L., Frejd, F., Carlsson, J., Lennartsson, J. (2008) Dimeric HER2-Specific Affibody Mole-cules Inhibit Proliferation of the SKBR-3 Breast Cancer Cell Line. Biochem Biophys Res Commun, 12;377(2):489–94
III Ekerljung, L., Lennartsson, J., Gedda, L. (2011) The HER2-Binding Affibody Molecule (ZHER2:342)2 Increases Radio-sensitivity in SKBR-3 cells. Manuscript
IV Nordberg, E., Ekerljung, L., Sahlberg, S.H., Carlsson, J., Len-nartsson, J., Glimelius, B. (2010) Effects of an EGFR-Binding Affibody Molecule on Intracellular Signaling Pathways. Int J Oncol, 36(4):967–72
V Ekerljung, L., Wållberg, H., Sohrabian, A., Andersson, K., Friedman, M., Frejd, F., Ståhl, S., Gedda, L. (2011) Generation and Evaluation of Bispecific Affibody Molecules for Simulta-neous Targeting of EGFR and HER2. Manuscript
Reprints were made with permission from the respective publishers.

Related Papers
Friedman, M., Lindström, S., Ekerljung, L., Andersson-Svahn, H., Carlsson, J., Brismar, H., Gedda, L., Frejd, F., Ståhl, S. (2009) Engi-neering and characterization of a bispecific HER2 × EGFR-binding affibody molecule. Biotechnol Appl Biochem, 21;54(2):121-31

Contents
1 Populärvetenskaplig sammanfattning (Summary in Swedish) .................. 11
2 Introduction ................................................................................................ 15 2.1 Cancer................................................................................................. 15
2.1.1 Detection and Treatment ............................................................. 16 2.1.2 Tumor Targeting ......................................................................... 16 2.1.3 Molecular Imaging and Radionuclide Therapy Today ............... 17
2.2 The EGFR/ErbB Family ..................................................................... 19 2.2.1 Structure ...................................................................................... 19 2.2.2 Receptor Dimerization ................................................................ 20 2.2.3 Receptor Activation .................................................................... 21 2.2.4 EGFR .......................................................................................... 22 2.2.5 HER2 .......................................................................................... 22 2.2.6 HER3 .......................................................................................... 23 2.2.7 HER4 .......................................................................................... 24
2.3 Cell Signaling ..................................................................................... 24 2.3.1 MAPK ......................................................................................... 26 2.3.2 PI3K / Akt ................................................................................... 26 2.3.3 PLC ........................................................................................... 27
2.4 Targeting Agents for the ErbB-Family .............................................. 27 2.4.1 Monoclonal Antibodies .............................................................. 27 2.4.2 Tyrosine Kinase Inhibitors ......................................................... 28
2.5 Affibody Molecules ............................................................................ 28 2.5.1 Affibody Molecules Used in the Present Study .......................... 30
3 The Present Study ...................................................................................... 31 3.1 Aim ..................................................................................................... 31 3.2 Summary of Included Papers ............................................................. 32
3.2.1 HER2-Targeting - Paper I-III ..................................................... 33 Intracellular Signaling ..................................................................... 33 Cell Growth ..................................................................................... 34 Migration ......................................................................................... 37 Survival ........................................................................................... 38 Dimerization ................................................................................... 40 Conclusions from Paper I-III .......................................................... 41

3.2.2 EGFR-Targeting - Paper IV ........................................................ 43 Intracellular Signaling ..................................................................... 43 Receptor Phosphorylation ............................................................... 43 Conclusions from Paper IV ............................................................. 45
3.2.3 Bispecific-Targeting - Paper V ................................................... 46 Production ....................................................................................... 46 Binding Properties ........................................................................... 47 Cellular Effects ............................................................................... 49 Conclusions from Paper V .............................................................. 52
4 Summary and Future Perspectives ............................................................. 53
5 Acknowledgments ...................................................................................... 55
6 References .................................................................................................. 57

Abbreviations
Ab Da ECD EGF EGFR ELISA Erk Gy HER2 IB IP KD mAb MAPK NRG PBS PI3K PLC
Antibody Dalton Extracellular domain Epidermal growth factor Epidermal growth factor receptor Enzyme-linked immunosorbent assay Extracellular signal regulated kinase Gray Human EGFR (ErbB2) Immunoblotting Immunoprecipitation Dissociation equilibrium constant Monoclonal antibody Mitogen-activated protein kinase Neuregulin Phosphate buffer saline Phosphatidylinositol-3-OH kinase Phospholipase
PTEN RTK SDS-PAGE
Phosphatase and tensin homolog deleted on chro-mosone 10 Receptor tyrosine kinase Sodium dodecyl sulfate polyacrylamide gel elec-trophoresis


11
1 Populärvetenskaplig sammanfattning (Summary in Swedish)
Avhandlingen beskriver olika ErbB-bindande affibody-molekylers inverkan på cancerceller. Här följer en kort introduktion till området samt en samman-fattning av de fem delarbeten som ingår i avhandlingen. Introduktion För att kunna vidta de åtgärder som är av vikt för cellens, och hela organ-ismens, överlevnad måste en cell kunna ta emot information (signaler) från omgivningen, överföra signalerna till cellens insida och där tolka dem. När en signal når cellens inre påverkas aktivering och uttryck av flera proteiner som styr exempelvis tillväxt, överlevnad och mobilitet. Ett exempel på en sådan signalkaskad förmedlas av tillväxtreceptorerna i ErbB-familjen. Re-ceptorerna sitter på ytan av celler och vidarebefodrar signaler till dess insida vid inbindning av olika naturliga ligander (bindare), så kallade tillväxtfak-torer. I ErbB-familjen finns fyra olika receptorer varav de mest studerade är EGFR och HER2. Dessa finns i ovanligt stor mängd på flertalet typer av cancerceller och är förknippade med särskilt aggressiva tumörer och en sämre överlevnadsprognos. Det gör receptorerna till attraktiva måltavlor för så kallade målsökande läkemedel. Med målsökande läkemedel eftersträvar man att behandla endast tumören och inte frisk vävnad. Detta skiljer från cytostatika som är vanligt vid cancerbehandling och som påverkar nästan alla celler i kroppen. Ett målsökande läkemedel kan ha en effekt i sig själv (genom att den påverkar det den binder till, t.ex. HER2), eller genom att man kopplar en toxisk substans till den. Ett annat användningsområde för måls-ökning är diagnostik. Genom att koppla en radioaktiv nuklid till den målsö-kande liganden kan man med specialla detektorer lokalisera tumörer och upptäcka om de har spridit sig och bildat dottertumörer.
Affibody-molekylerna är små ligander som är framtagna för att binda till exempelvis EGFR och HER2 med avsikten att de ska kunna användas kli-niskt för diagnostik och behandling av tumörer. I och med att affibody-molekylerna binder tillväxtreceptorer finns möjligheten att de har inverkan på cellen. Om dessa ligander ska användas i människokroppen är det viktigt att vara medveten om deras effekter eftersom de kan påverka den fortsatta tillväxten av tumören.

12
Arbete I-III I arbete I-III har tre stycken HER2-bindande affibody-molekyler undersökts. Dessa kallas (ZHER2:4)2, ZHER2:342 och (ZHER2:342)2. Medan monomeren ZHER2:342 bara består av en HER2-bindande del så består dimeren (ZHER2:342)2 av två sådana lika delar och har därmed den teoretiska möjligheten att samtidigt binda två HER2. Även (ZHER2:4)2 är en dimer. Skillnaden mellan (ZHER2:4)2 och (ZHER2:342)2 är att de har olika affinitet för receptorn, det vill säga de bin-der olika starkt till HER2.
Genom att behandla odlade cancerceller med de tre liganderna har jag un-dersökt vilken effekt de har med avseende på bland annat tillväxthastighet, cellsignallering och överlevnad efter bestrålning. Generellt sett hade den monomera liganden liten effekt medan dimererna hade större påverkan. Till exempel minskade både (ZHER2:4)2 och (ZHER2:342)2 tillväxten av bröstcancer-celler (se Figur 8, sida 35). (ZHER2:342)2 kunde även göra samma celler mer känsliga för bestrålning (se Figur 12 och 13, sida 39-40). Detta gör affibody-dimererna intressanta för cancerterapi. Genom att koppla en lämplig radioak-tiv nuklid till dimeren finns det en teoretisk möjlighet att målsöka tumörer med en enhet som skadar cancercellerna på två sätt, dels genom den radioak-tiva substansen och dels genom affibody-molekylens inverkan. Att den mo-nomera liganden inte har någon inverkan på celler är inte negativt utan gör den tvärtemot lämplig som målsökare där man inte vill ha någon effekt vid bindning till receptorn, till exempel vid diagnostik.
Arbete IV I studien har en EGFR-bindande affibody-molekyl, (ZEGFR:955)2, undersökts med avseende på två signalvägar som EGFR kan påverka. Det visade sig att när (ZEGFR:955)2 binder till EGFR aktiveras båda signalvägarna, som oftast är sammankopplade med ökad tillväxt och överlevnad. Eftersom cellsignalle-ring är en komplicerad process så är det biologiska utfallet svårt att förutse. I fortsatta studier skulle det därför vara intressant att undersöka den biologiska effekten av (ZEGFR:955)2, särskilt eftersom den uppvisar flera likheter med läkemedlet Erbitux som används vid behandling av vissa tumörer.
Arbete V Här presenteras sex nya så kallade bispecifika affibody-molekyler för att målsöka EGFR och HER2 samtidigt. Bispecifikerna består av två monome-rer, en EGFR-bindande och en HER2-bindande. Alla sex bispecifika ligan-der visade sig vara kapabla att samtidigt binda både HER2 och EGFR utan att ha en stor effekt på levande celler. Nämnvärt är att bindningsstyrkan på-verkades av den inbördes positioneringen av monomererna och även av sammansättningen av länken dem emellan.
Dubbelmålsökning är intressant ur flera aspekter. Dels för att det visat sig att när flera olika ErbB-receptorer (t.ex. EGFR och HER2) finns på cancer-

13
celler samtidigt så är det förknippat med en sämre patientöverlevnad. Dels för att cancerceller som behandlas med ErbB-målsökande läkemedel ofta uppvisar resistens efter en tids behandling genom att öka aktiveringen av andra ErbB-receptorer. Om en ligand kan binda två receptorer samtidigt och förhindra aktiveringen av båda två så kan man få dubbel effekt. En tredje aspekt är att det kan ge bättre målsökning eftersom tumörer är mer troliga än frisk vävnad att ha stora mängder av både EGFR och HER2. Med bättre målsökning kan man inom diagnostik lättare upptäcka tumörer och inom terapi få mindre skadliga effekter på frisk vävnad.

14

15
2 Introduction
This thesis is based on in vitro studies of EGFR- and HER2-binding affibody molecules, particularly focusing on their effect on cell growth and intracellu-lar signaling. The included papers will be presented in the section “the pre-sent study”.
2.1 Cancer Cancer is one of the most common diseases in the world, and approximately one in every three people will receive a cancer diagnosis during their life-time. There are about 200 different types of cancer, classified by the type of cell the tumor originates from, and the differences between two types can be very large. Most cancers occur in epithelial cells and are classified as carci-nomas. Common to all cancers is the disturbance of the normal balance be-tween proliferation and apoptosis, resulting in dysregulated cell growth. Another defining feature of malignant tumors is the ability to invade and spread from the site of origin. A benign tumor, which is not evidence of can-cer, can be large but does not infiltrate surrounding tissue or metastasize. Metastases are the major cause of death from cancer [1].
Cancer development is a multistep process through which normal cells evolve into invasive cancer. There are 8 hallmarks of cancers, which accord-ing to Weinberg and Hanahan, can be applicable to most cancers [2,3].
Sustained proliferative signaling Insensitivity to growth inhibitory
signals Invasion and metastasis Unlimited replication
Angiogenesis Evasion of apoptosis Deregulated cellular metabolism Evading the immune system
Each of these features is a potential target for cancer targeting therapeutics. Additionally, genome instability and inflammation are two characteristics that facilitate acquisition of the hallmarks [3].
Nevertheless, there are many factors that can cause cancer. Approximate-ly 30% of all cancers could be prevented by avoiding key risk factors, most importantly; smoking, obesity, physical inactivity, exposure to sunlight and

16
alcohol consumption [1]. Some genomic changes that might lead to cancer can also be inherited, however most cancers are not inheritable but acquired randomly. Cancer can affect people at any age, but the risk of acquiring can-cer increases with age, and more than 60% of people are above 65 years of age at the time of diagnosis [4].
The number of cancer cases diagnosed in Sweden has increased during the recent decades. The major reasons for this are the increasing age of the population together with improved diagnostic methods and more screening programs. There are about 50 000 new cases in Sweden each year, and can-cer is responsible for almost 25% of all deaths. Next to heart disease it is the most common cause of death. The most common types of cancer in Sweden are prostate- and breast cancer, while lung cancer causes the most deaths. During the last 30 years, the overall survival and, in particular the life expec-tancy of cancer patients has increased [4]. This indicates that treatment, as well as diagnosis, have improved.
2.1.1 Detection and Treatment An early diagnosis of cancer is very important for the potential survival of patients. If the tumor is detected at an early stage, i.e. before it has spread to other parts of the body, the effects of therapy and the prognosis of the dis-ease are improved. Detection can be done for example by biopsies, lab tests and different visualization techniques such as x-rays, computed tomography (CT), magnetic resonance imaging (MRI) and positron emission tomography (PET).
Surgery, chemotherapy and external radiation are the most common ther-apies used to treat cancer, and different combinations are often used. A drawback with surgery, and to some extent radiotherapy, is that small metas-tases are not removed or killed. Chemotherapy on the other hand, affects all dividing cells, which includes metastases but also healthy tissues. These three conventional treatment methods cure approximately 60% of cancer patient [5]. For many patients with metastases or disseminated tumors the best effect of chemotherapy is prolonged survival and/or a palliative effect [6]. Other complementary methods that can improve survival are; immuno-therapy, anti-angiogenesis and radionuclide therapy, among others.
2.1.2 Tumor Targeting The optimal way to treat or detect cancer is to target the cancer cells only, i.e. tumor targeting. This can be accomplished by molecular recognition of structures present on cancer cells only. However, in reality these structures are also present in normal tissue but often to a much lower extent than in tumors. In theory, tumor targeting is a good method to find and treat dissem-inated tumor cells and metastases, but in order for the targeting to work they

17
need to express the target structure at sufficient levels. Examples of tumor targets are CEA, CD20 and the EGFR-family. Figure 1 shows the targeting principle.
The targeting agent can either have a therapeutic effect by itself, or an ef-fector substance can be coupled to the targeting agent. Depending on the desired effect, different effectors can be used. Examples of such are cytotox-ic drugs and radionuclides. The same concept is used for molecular imaging, which today is the most widely used application for the concept of targeting. It can be used for cancer diagnosis but also in other clinical areas like neu-rology and cardiology. Natural ligands, antibodies, antibody fragments and small peptides are examples of targeting agents.
Figure 1. Tumor targeting. The targeting agent recognizes cell surface proteins on the tumor cells, and by selection of effector molecule the desired effect can be achieved. Radionuclides as effectors can be used for both imaging (a) and therapy (b).
2.1.3 Molecular Imaging and Radionuclide Therapy Today Molecular imaging is used to visualize, characterize and measure biological processes at molecular and cellular level [7]. In oncology it can for example be used to locate tumors, identify targets for tumor-targeted therapy and to monitor response to therapy.
Since there are many types and subtypes of tumors it is natural that they respond differently to different treatments. This creates a need for personal-ized therapy, meaning the tailoring of each patient’s treatment according to the characteristics of his/her disease. Personalized medicine demands a lot of tools for diagnosis and the rapidly evolving field of molecular imaging has great potential in this area [8]. One great advantage is that it can be used to

18
identify and quantify tumor markers, e.g. HER2 (human epidermal growth factor receptor 2) or estrogen receptors in breast cancer, without the inva-siveness of a biopsy or surgery [9].
Radionuclide imaging techniques, which include single-photon emission computed tomography (SPECT) and positron emission tomography (PET), are powerful tools for detecting and staging tumors. Today, the most com-mon molecular imaging method in oncology is 18F-FDG-PET (fluoridediox-yglucose) [10].
For more than 50 years 131I has been used for therapy of thyroid cancer, but besides this few methods for targeted radionuclide therapy (TRT) are routinely used at the clinics. Two examples are Bexxar (131I-tositumomab) and Zevalin (90Y-ibritumomab tiuxetan) which are approved by the FDA (food and drug administration, USA) for clinical use in the treatment of CD20-positive non-Hodgkin’s lymphoma. Both compounds have shown impressive clinical outcomes with overall response rates of 80-100% and in some cases, no evidence of disease for several years [11]. So far TRT has not been very successful in treatment of solid tumors. Two reasons for this are the inability to deliver a sufficient radiation dose to tumor cells and the generally lower radiosensitivity of solid tumors [12]. During the last decade the field of TRT has expanded and the gradual shift towards treating patients with minimal residual disease instead of bulky tumors has given rise to sev-eral promising (phase I and II) studies [12,13,14].
TRT is a complex field that has to take many components into considera-tion. The choice of targeting agent and radionuclide is of course of great importance. Simplified, - emitters are often the best choice for solid tumors while -emitters may be better suited for micrometastases due to their short-er range. Also, the radiolabeling method is of importance since it can have large effects on for example binding affinity, biodistribution and retention of the radionuclide [15]. Other important factors to evaluate when choosing radionuclide for therapy is the half-life and the possibility for production [6].
When using tumor targeting, both for imaging and in particular for thera-py purposes where a toxic effector might be used, it is of great importance to be aware of the cellular events followed by binding of the target. The major goal in tumor targeting is either to ensure that the effector stays at the target for as long as possible, or that it relocates to the cell nucleus where the main target – the DNA, is situated. The targeting agent itself may also have a bio-logical effect on the cell, which could be both good and bad. A dream sce-nario would be that the targeting agent delivers the toxic substance to the cell while simultaneously sensitizing the cell to its lethal effects.

19
2.2 The EGFR/ErbB Family The epidermal growth factor receptor (EGFR) family consists of four mem-bers; EGFR, HER2, HER3 and HER4 (also known as ErbB1-4), from now on denoted as EGFR and HER2-4, while ErbB refers to all four receptors. The most extensively studied receptors are EGFR and HER2. During recent years HER3 has also come into focus, but less is known about HER4.
The ErbB receptors have evolved from a single ligand-receptor combina-tion found in C. elegans to a complex signaling network in humans. Upon ligand binding to the receptor a signal is brought from the outside of the cell to the inside, and eventually to the nucleus, where changes in gene expres-sion cause the cell to adjust to the environment. The many combinations in the ErbB-receptor network results in several regulatory levels and signal diversity, making it a robust system [16,17].
The receptors are expressed in most normal tissues, and the absence of one of the ErbBs is fatal during embryonic development. Abnormal expres-sion and signaling of the receptors is associated with the development and progression of several forms of cancer and in addition to enhanced invasive-ness and resistance to chemotherapy and radiation (see below for each recep-tor). Apart from cancer, the ErbB family has also been linked to other dis-eases like psoriasis, inflammation and Alzheimer’s diseases [18].
2.2.1 Structure The ErbBs are type I receptor tyrosine kinases and they share the same struc-tural basis. Each receptor contains an extracellular domain (ECD), a trans-membrane domain (TM) and an intracellular tyrosine kinase domain flanked by a juxtamembrane region and carboxyl tail. The ECD is composed of four different subdomains (I-IV). Natural ligands bind to domain I and III, and domain II is known as the dimerization domain. The receptors can be posed in a “closed” and an “open” position in which the dimerization arm is ex-posed (reviewed in [19]). It is generally accepted that binding of a ligand stabilizes the receptor in the open position, which enables dimerization and leads to activation , resulting in downstream signaling and subsequent cellu-lar processes such as proliferation, migration and apoptosis (reviewed in [20]). HER2 and HER3 are special since HER2 always has its dimerization arm exposed [21] and HER3 lacks a fully functional tyrosine kinase [22]. Figure 2 shows a simplified version of the four receptors and two receptors forming a heterodimer.
The receptors are controlled by at least 11 different EGF-related ligands, including EGF and neuregulin (NRG). Some ligands are receptor specific while others can bind more than one receptor. HER2 does not have any known ligand [23,24].

20
Figure 2. Receptor structures. While HER2 is always in an open state with the dimerization domain (II) exposed, the other receptors are posed in a “closed” position with the dimerization arm hidden. The intracellular kinase domain con-sists of an N-lobe and a C-lobe. The kinase domain of HER3 is dysfunctional. Binding of a natural ligand (L) stabilizes the open configuration and enables re-ceptor dimerization, leading to tyrosine phosphorylation (P).
2.2.2 Receptor Dimerization Receptor dimerization and activation is yet not fully understood. Crystallog-raphy studies have focused on the function of the extracellular domain in dimerization and have given a fairly good view of the ECD rearrangement required for ligand-induced dimerization. Unliganded receptors adopt an auto-inhibited configuration with the dimerization arm hidden [25]. Upon ligand binding, the receptor undergoes conformational changes that expose the dimerization arm [26,27]. The dimer interface is formed by domain II and a small part of domain IV [28]. It was earlier believed that one ligand could bind two receptors simultaneously and thus pull the receptors together, but it has been shown that two ligands are present in each EGFR homodimer [26,27]. Dimerization is thus entirely receptor-mediated since binding of a ligand merely stabilizes the receptor in the open position.
The steps between ligand binding and initiation of downstream signaling are more complex than previously believed. Seemingly, receptors can form both active and inactive dimers. Exposure of the dimerization arm is not sufficient for the formation of an active dimer but the intracellular part must also contribute. For the receptors to become active, asymmetric dimers must be formed in which the C-lobe of one kinase domain acts as an activator when binding to the N-lobe of the other receptor, resulting in kinase activa-tion (see Figure 2) [29].

21
Further complicating the understanding of dimerization is the documenta-tion of a variety of different dimers, with different requisitions for dimeriza-tion. Pre-formed dimers, even in the absence of ligand stimulation, have been reported by several groups [30,31], and some reports also speak of a higher order of oligomers, or receptor clusters [32,33,34]. Dimers formed by the kinase domain, and not the ECD, have also been studied. These so-called “quasi-dimers” are autoinhibited by the carboxyl-tail, which needs to unfold to enable dimerization [35,36]. Yet others have reported that for ligand-independent homodimerization, the extracellular domain is not needed and only the intracellular part is sufficient to form homodimers driven by over-expression [37,38].
The biological effect of inactive dimers and oligomers are to date not well understood. One possible explanation is that the inactive dimers are interme-diate states to activation with the function of increasing ligand affinity [39,40,41], and/or to prevent ligand-independent activation [36].
2.2.3 Receptor Activation Kinase activation leads to phosphorylation of tyrosine residues of the C-terminal tail. These residues serve as docking sites for signaling molecules containing Src homology 2 (SH2) and phosphotyrosine binding (PTB) do-mains [42], which in turn activates numerous signal transduction pathways. The effect on downstream signaling, and hence the biological outcome, de-pends on the composition of the receptor pair and the identity of the ligand [43,44,45]. Heterodimers are regarded as more signaling-potent than homo-dimers [46,47].
Depending on which phosphotyrosine sites are phosphorylated different signaling proteins are engaged. The number of docking sites for certain sig-naling proteins differs between the receptors, for example EGFR has several shc/grb2 binding sites to activate mitogen-activated protein kinase (MAPK) [48], while HER3 has more phosphatidylinositol-3-OH kinase (PI3K) bind-ing sites to initiate Akt signaling [49].
The ErbB receptors are capable of binding several docking proteins in-cluding; growth-factor-receptor-bound-2 (Grb2), Src-homology-2-contining (Shc), PI3K, STAT5 and Cbl, amongst others [48]. The focus of this thesis will be on the signaling pathways MAPK, PI3K/Akt and PLC , see the fol-lowing section “Cell Signaling”. To conclude, activation of the ErbBs is regulated at multiple levels by several auto-inhibitory mechanisms.

22
2.2.4 EGFR EGFR was the first receptor within the family to be identified, and it is also the most studied receptor, often used as a model of how the ErbB family works and functions. It was discovered by Stanley Cohen and co-workers in 1980 as the receptor to EGF [50]. Six years later, Cohen was awarded with the Nobel Prize in medicine for his discovery of growth factors.
EGFR can be bound by all the ligands in the family with the exception of the NRGs, and can form heterodimers with all three co-receptors. The lig-ands are EGF, transforming growth factor (TGF- ), betacellulin (BTC), heparin-binding EGF-like growth factor (HB-EGF), epiregulin (EPR), epigen (EPN), ampiregulin (AR) [51].
Ligand binding to EGFR results in rapid internalization [52]. The main internalization pathway is through clathrin-mediated endocytosis, in which the receptor-ligand complexes are clustered in clatherin coated pits and transported to early endosomes [53,54]. Depending on the pH-stability of the ligand, the receptor is then either ubiquitinated and subsequently degraded, or recycled to the cell surface [53]. The other receptors are considered as endocytosis-impaired [55], and are more or less inefficiently degraded [53]. Heterodimerization with HER2 decreases internalization, especially when HER2 is overexpressed [56].
EGFR in Cancer Deregulation of EGFR has been observed and is associated with poor prog-nosis in several cancers, for example head and neck, ovarian, breast and colorectal cancers [57]. But perhaps the best known example of EGFR over-expression is found in brain tumors where 40% of glioblastomas have gene amplifications [58]. Many cell types normally express EGFR, predominantly in squamous and glandular epithelium [59].
There are three main mechanisms by which EGFR can mediate tumor ini-tiation and progression: overexpression, mutation and autocrine signaling of ligands that results in constitutive activation [60,61]. The most common mutation, EGFRvIII, has for example been found in gliomas, lung- and breast cancers. This mutation results in a receptor that cannot bind any lig-and but is constitutively active [62].
2.2.5 HER2 HER2 is sometimes denoted as p185ErbB2/neu or neu. Unlike the other recep-tors in the family, HER2 does not have a natural ligand. Instead it has a con-stitutively open dimerization arm, similar to the configuration of the other receptors open state. This structure makes the receptor unable to bind EGF-related peptides because domains I and III are too close for a ligand to fit [24]. Instead HER2 functions as a co-receptor for the other members of the

23
family. It is actually considered as the preferred dimerization partner for the other receptors [63,64]. Heterodimerization with HER2 leads to more effec-tive signaling, for example by decreasing the rate of ligand dissociation and receptor down-regulation [56,65,66]. In addition, HER2 is capable of recruit-ing more signaling proteins than the other ErbBs, especially when overex-pressed [67].
HER2 is considered as an oncogene that can cause cell transformation even in the absence of a ligand [68]. It is generally thought that when HER2 is overexpressed (approximately >106 receptors/ cell) it can induce ligand-independent dimerization, both with itself and with other receptors of the ErbB family, resulting in constitutive activation [37,46,69,70]. Even though it is likely that HER2 homodimers exist, crystallography studies of the ECD have failed to detect such [24].
Interestingly, recent studies have proposed the existence of a membrane-bound ligand for HER2, more precisely the cell surface mucin Muc4 [71,72].
HER2 in Cancer The second member of the ErbB family was first found as an amplified gene (HER2) in breast cancer [73], and as a product of the neu oncogene in rat neuroblastomas [74]. Since then it has held its position as one of the most studied areas in human cancer and is a pursued target for therapy.
Increased expression and activity of HER2 induces cell transformation and tumorigenesis in several tumor types and model systems (reviewed in [75]). Overexpression due to gene amplification or transcriptional deregula-tion is seen in approximately 20-30% of breast cancer [76,77]. The HER2 status is often maintained during progression to invasive disease and breast cancer metastases generally overexpress HER2 to the same extent as the corresponding primary tumors [75,78]. Overexpression is also associated with poor patient prognosis, enhanced invasiveness and resistance to con-ventional therapies, e.g. chemotherapy and radiation [79,80]. Even though HER2 overexpression is not as common as for EGFR, it has been found in several other cancers, for example ovarian- [81], colorectal- [82] and gastric cancer [83]. Mutated HER2 has also been found in a subset of lung cancer [77]. The expression of HER2 is low to medium in normal tissue and gener-ally lower than the expression of EGFR [59].
2.2.6 HER3 HER3 lacks a fully functional kinase domain but is capable of activating other ErbB receptors when forming heterodimers [84]. In fact, HER2-HER3 heterodimers are considered as the most potent receptor couple for mitogenic signaling [67,85]. Moreover, HER3 has 6 binding sites for PI3K and is thus a powerful activator of the PI3K/Akt pathway which affects cell survival. EGFR and HER2 cannot bind PI3K directly [49].

24
The receptor has two known ligands; NRG 1 (also known as heregulin HRG and neuregulating factor NDF) and NRG2, which both have several splicing variants [51].
HER3 in Cancer High expression of HER3 has been reported in melanomas, colorectal and some lung cancers. Yet it is not considered as transforming when overex-pressed and no oncogenic mutations have so far been identified [86]. How-ever, HER3 is crucial for cell growth in tumors driven by HER2 overexpres-sion [87,88]. Increased HER3 signaling is also an escape route for resistance to ErbB-targeted tyrosine kinase inhibitors (TKI) [89]. This makes HER3 interesting for combinatorial therapies that target several ErbB receptors.
2.2.7 HER4 HER4 is the least studied member of the ErbB family. It has several ligands of which some are shared by EGFR (HB-EGF, BTC, EPR), some by HER3 (NRG1 and 2) and NRG3 and 4 that are exclusive for HER4 [51].
HER4 has four different isoforms caused by alternative splicing of mRNA. In addition, the JM-a isoform can be cleaved at two different sites. Cleavage results in a soluble intracellular domain (4ICD) that can translocate to the nucleus, where it affects transcription [90]. The other ErbB receptors have also been found in the nucleus [91].
The significance of HER4 in cancer, and especially breast cancer, is un-clear. Some reports describe HER4 as a tumor suppressor with pro-apoptotic effects, while others report that it can promote cell growth [92]. The contra-dicting evidence may be due to the different functions of the four isoforms [93].
2.3 Cell Signaling Important signaling pathways activated by the EGFR family are the mito-gen-activated protein kinase (MAPK), the phosphatidylinositol 3 kinase (PI3K)/Akt cascade and the phospholipas C (PLC ). Signaling through these pathways are important for proliferation, survival and migration [94,95,96]. These pathways are outlined in Figure 3 and will be further dis-cussed below.

25
Figure 3. Three major signaling pathways downstream of the ErbB receptors; PI3K/Akt, PLC and MAPK/Erk1/2 pathways. The PI3K/Akt pathway is asso-ciated with increased survival, PLC with migration, and MAPK signals are linked with an increase in proliferation.
Figure 3, which illustrates the pathways as more or less separate linear cas-cades, is a very simplified version of reality. Cellular signaling is a complex system with various crosstalks between a multitude of different players, both within the ErbB family, and to other signaling proteins in the surroundings. If one ErbB is blocked, signaling from other family members will be in-creased [89,97]. Co-activation of other receptor tyrosine kinases is believed to play an important role in tumor response to targeted therapies [98].
The strength and duration of signaling is regulated by several regulatory loops which give negative or positive feedback [17]. As in the case of MAPK signaling, the duration of the signal can affect the cellular outcome [99]. In general, endocytosis of receptors is considered as a way of down-regulating receptor-activated signaling. However, signaling can continue after internalization. It can even be initiated in the endosomes [54].
There are also several other signaling players affected by the ErbB recep-tors, for example STAT, catenin and Src [18]. Additionally, all ErbB recep-tors have also been reported in the nucleus where they may act as transcrip-tion factors [100].

26
2.3.1 MAPK There are at least four distinct MAPK cascades; extracellular signal-regulated kinases (Erk)1 and 2, Jun amino-terminal kinases (Jnk)1/2/3, p38-MAPK and Erk5. Even though all are regulated by growth factors, only Erk1/2 is regarded as growth factor controlled while the others are consid-ered more or less as stress-induced cascades [99]. All functional combina-tions of receptors and ligands are able to activate the Erk1/2 MAPK path-way, either directly through the SH2-binding domain or indirectly through binding of the Shc adaptor to the PTB-binding domain [20].
The MAPK signaling pathway is a three layer signaling cascade in which one MAPkinase phosphorylates the other (MAPKKK-MAPKK-MAPK). The Erk1/2 MAPK pathway starts with recruitment of Grb-2 to the phos-phorylated receptor (or the adaptor protein Shc). This leads to a linear kinase cascade that culminates in activation of Erk1/2 (see Figure 3). One im-portant regulator upstream of Erk1/2 is Ras. Erk1/2 phosphorylates several cytoplasmic proteins and is also translocated to the nucleus where it activates multiple transcription factors, for example regulators of cell cycle progres-sion [85,95].
The Erk1/2 cascade was initially identified as a growth-promoting path-way, but has also been found to contribute to cell migration, tumor invasion and cell survival [99]. A constitutively active form of MAPK-kinases can transform cells in culture [101]. Approximately 30% of human tumors con-tain oncogenic mutations in the Ras genes, causing constitutive activation of downstream effectors, e.g. Erk1/2 [102,103].
2.3.2 PI3K / Akt Activation of PI3K occurs through binding of its regulatory p85 subunit to a phosphotyrosine site on the receptor. This results in the recruitment of sever-al signaling effectors, e.g. Akt (also known as protein kinase B (PKB)), which is recruited to the plasma membrane and activated by phosphorylation (see Figure 3 for further details). Cytoplasmatic Akt, and Akt translocated to the nucleus, target regulators of apoptosis and cell survival, e.g. Bad and caspase 9 [104]. PI3K can also be activated by Ras in the MAPK pathway [95,105]. The Akt cascade is negatively regulated by PTEN (phosphatase and tensin homologue deleted on chromosome ten). Several cancers have mutations in PTEN, resulting in constitutive activation of Akt [106]. Signal-ing through the PI3K/Akt pathway has come forth as an important player in cancer tumorigenesis, especially with regards to resistance to ErbB-targeted therapy [107,108].

27
2.3.3 PLC PLC (PLC 1) is recruited to the membrane by activated EGFR and HER2, but can also be activated by PI3K products [20]. Activated PLC hydrolyzes lipids that generate second messengers. This results in calcium release and thereby activation of calcium/calmodulin-dependent kinases (see Figure 3) [109]. PKC can also activate Raf, thus implicating crosstalk to the MAPK pathways [95]. PLC promotes tumor invasion and is required for cell motil-ity induced by growth factors, e.g. through EGFR signaling [109]. The activ-ity of PLC has also been related to radiation and chemotherapy resistance [110].
2.4 Targeting Agents for the ErbB-Family Due to their overexpression and involvement in development of many can-cers, the ErbB receptors (particularly EGFR and HER2) have been pursued as therapeutic targets for several decades. So far, there are two types of drugs in clinical use that target the ErbB receptors: monoclonal antibodies (mAbs) and tyrosine kinase inhibitors (TKIs). Table 1 summarizes EGFR and HER2 targeting drugs.
Table 1. Examples of ErbB targeting agents.
Target Name Type Status Tumor type
EGFR Cetuximab/Erbitux® mAb appr. 2004 Colorectal, head and neck
EGFR Panitumumab/Vectibix® mAb appr. 2006 Colorectal
HER2 Trastuzumab/Herceptin® mAb appr. 1998 Breast
HER2 Pertuzumab/Omnitarg mAb ICT Prostate, breast, ovarian
EGFR Gefitinib/Iressa® TKI appr. 2004 NSCLC
EGFR Erlotinib/Tarceva® TKI appr. 2005 NSCLC, pancreatic
EGFR/
HER2
Lapatinib/Tykerb® TKI appr. 2007 Breast
Abbreviations: (mAb) monoclonal antibody, (TKI) tyrosine kinase inhibitor, (appr.) approved for clinical use by the FDA (Food and Drug Administration, USA), (ICT) in clinical trials, (NSCLC) non-small cell lung carcinoma.
2.4.1 Monoclonal Antibodies There are two monoclonal antibodies that target EGFR, cetuximab and pa-nitumumab. Both bind to the subdomain III of EGFR and prevent ligand binding [111]. Cetuximab has been shown to sensitize head and neck pa-tients to irradiation [112], but neither of the drugs seem to have an effect on patients with mutations in the ras genes [111]. The most common side effect of EGFR-targeted drugs is skin toxicity [113].

28
Trastuzumab is a monoclonal antibody that targets HER2. It binds to the extracellular domain IV [21] and effects downstream signaling. Trastuzumab has been reported to down-regulate the amount of HER2 at the cell surface and activate the human complement cascade [114]. Some of its effects are thought to be mediated by inhibition of the PI3K/Akt pathway, and this sig-naling cascade is also believed to play an important role in trastuzumab re-sistance [108]. Today, trastuzumab is used for treatment of HER2-positive metastatic breast cancer. Trastuzumab in combination with chemotherapy has given the best results and significantly improved overall survival [115]. Pertuzumab is another antibody that binds HER2. By binding domain II it prevents receptor dimerization [116]. Currently, pertuzumab is undergoing clinical trials.
2.4.2 Tyrosine Kinase Inhibitors TKIs target the kinase activity of the receptors. They are small molecule inhibitors that bind to the intracellular domain and prevent signal transduc-tion. Two TKIs that target EGFR have been approved for clinical use, ge-fitinib and erlotinib. However, only patients with mutated EGFR kinase domain, resulting in constitutively active receptor, respond to gefitinib treatment [117]. Lapatinib is a dual EGFR and HER2 tyrosine kinase in-hibitor that reversibly inhibits the ATP-binding site of their kinase domains. It has been proposed that lapatinib may be able to overcome trastuzumab resistance [118].
2.5 Affibody Molecules The affibody molecules are originally derived from domain B of the staphy-lococcal protein A. This so-called Z domain consists of 58 amino acids in a three-helix bundle. By randomizing 13 surface-exposed amino acids, librar-ies containing up to 1011 molecules with the potential to bind to as many different targets have been created [119,120], see Figure 4. Selection of new binders is most often done by phage display but other methods such as ribo-some display [120] and cell surface display [121] have also been used. If the affinity for the target is insufficient, affinity maturation can improve binding. One way to do this is by creating a new library where only some of the 13 amino acids are randomized [122]. Affibody molecules binding to a wide range of targets have been generated, for example insulin, amyloid beta pep-tide, CD28,TNF- , HER3 and IgE [123].
Even though the affibody molecules are small (about 6 kDa for monomers and 15 kDa for dimers), they have a binding area similar in size to that of an antibody. Multimeric constructs can be generated by head-to-tail genetic

29
fusion, which can be used to increase affinity [124] or, as shown in this the-sis, to generate a binder with different biological properties. The affibody molecules are stable at high temperature and in a wide pH-range [125]. By introduction of a terminal cysteine residue to the otherwise cysteine free scaffold, site specific labeling or immobilization on a solid surface can be performed. Another advantage is the possibility of synthetic production that gives the opportunity of adding conjugates like fluorophores or chelating groups for radiometal labeling [126,127].
Figure 4. The affibody molecules are based on the Z-domain scaffold derived from domain B of the Ig-binding region of protein A. Large libraries of potential binders can be generated by randomizing 13 amino acids on helices 1 and 2 (in-dicated by yellow). Printed with permission from Affibody AB.
Affibody molecules have been used for a wide variety of applications rang-ing from biotechnological applications (e.g. immunoprecipitation and affini-ty chromatography) to clinically intended tumor targeting (e.g. imaging and therapy) [123]. Ongoing clinical studies at Akademiska hospital in Uppsala are using 111In-labeled ABY-025 for SPECT/CT imaging in breast cancer patients. ABY-025 is a DOTA-conjugated variant of ZHER2:2891, which is a more stable variant of ZHER2:342 due to reengineering of the scaffold [128]. So far the results from the study are promising. HER2-expressing metastases in brain, axilla and adrenal gland were visible 24 hours post-injection, and the opportunity to visualize the HER2 status in metastases has been used to modify further treatment of patients (personal communication with Jörgen Carlsson).

30
Figure 5. A schematic presentation of the HER2-targeting affibody molecules used in this thesis. A) (ZHER2:4)2 B) ZHER2:342 C) (ZHER2:342)2. The grey dots indi-cate positions modified during affinity maturation.
2.5.1 Affibody Molecules Used in the Present Study In this thesis, several different affibody molecules have been used. The HER2-targeting (ZHER2:4)2 is a dimeric version of the first HER2-targeting affibody generated by Wikman et al. This first binder had an affinity for HER2 of 50 nM, but a fast off-rate [129]. The binding properties were im-proved by the dimeric (ZHER2:4)2 which has a KD of 3 nM [124]. In the affini-ty maturated version ZHER2:342, binding has been further enhanced and it has an apparent affinity for HER2 of 22 pM [122]. Schematic figures of the three HER2-binding ligands are presented in Figure 5.
The EGFR-targeting affibody molecule ZEGFR:955 has been generated and described by Friedman et al. [130]. In the present study, the dimeric (ZE-
GFR:955)2 and the affinity maturated ZEGFR:1907 [131] have been used. Accord-ing to Biacore analysis they bind EGFR with a KD of 50 nM and 5 nM, re-spectively [130,131].
A bispecific affibody molecule that simultaneously targets both EGFR and HER2 has previously been described by us and a group at KTH [132]. Paper V describes the generation of six new bispecific-targeting affibody molecules based on monomeric ligands.

31
3 The Present Study
3.1 Aim The aim of my Ph.D. work was to evaluate new ErbB-targeting affibody molecules regarding their cellular effects in vitro upon receptor binding. The affibody molecules are intended for tumor targeting with the prospect of clinical use in imaging or therapy. Ligand binding to an ErbB receptor may have an impact on the cell. When used in vivo, it is important to be aware of these effects as they may have consequences for the continued growth of the tumor.
Three types of affibody molecules have been studied;
HER2-targeting (paper I-III). EGFR-targeting (paper IV). Simultaneous targeting of both EGFR and HER2 (paper V).

32
3.2 Summary of Included Papers Studying the cellular effects of ligand binding has been achieved by studies of intracellular signaling (paper I-V), cell proliferation (paper I-II and V), migration and apoptosis (paper I), binding kinetics (paper II, III and V), sur-vival and receptor dimerization (III and V). Additionally, the generation of six bispecific affibody molecules is described in paper V.
Human cancer cells in cultivation were used as the model in which the ef-fects were studied. The cell lines used all overexpress at least one of the receptors in the ErbB family. Table 2 summarizes the cell lines and their expression levels of EGFR and HER2.
Table 2. Cell lines used in the present study. Additionally, HER3 is expressed in MCF-7 (~104) and SKBR-3 (~104).
Cell line EGFR* HER2* Tumor type origin Study
SKBR-3 104-105 2-6x106 Adenocarcinoma (breast) I-III, V SKOV-3 2x105 107 Adenocarcinoma (ovary) I-II MCF-7 5x103 2x104 Adenocarcinoma (breast) III A-431 2x106 2x105 Squamous-cell carcinoma
(epithelial vulva) IV-V
U-343 6x105 3x104 Glioblastoma (brain) IV * Approximate number of receptors per cell according to the literature [41,70,133].

33
3.2.1 HER2-Targeting - Paper I-III
Three different HER2-binding affibody molecules have been studied regard-ing their cellular effects. In paper I, (ZHER2:4)2 and ZHER2:342 were studied, while the focus of paper II and III is mainly on (ZHER2:342)2.The ligands are sometimes denoted as (Z4)2, Z342 and (Z342)2 to improve readability. The clinically used antibody trastuzumab was used as a reference substance. Three HER2-expressing cell lines have been used: SKBR-3, SKOV-3 and MCF-7.
Intracellular Signaling Activation of the downstream signaling pathways was studied by examining the phosphorylation status of Erk1/2, Akt, PLC and the receptor itself (p-HER2). This was done either by SDS-PAGE and western blot or by ELISA. To quantify the results from the western blot experiments, the AIDA or Image J softwares were used to measure the intensity of the immunoblot-ting bands. Figure 6 shows the phosphorylation levels of HER2, Erk1/2 and Akt in SKBR-3 cells, and Figure 7 shows the p-HER2 status in SKOV-3 and MCF-7 cells.
Figure 6. Phosphorylation and total levels of HER2 (A), Erk1/2 (B) and Akt (C) in SKBR-3 cells. A) Immunoprecipitation (IP) by HER2-specific antibody (Ab) was followed by SDS-PAGE and immunoblotting (Ib) with Abs specific for phosphotyrosine (pTyr) or total HER2. B, C) Immunoblotting was done with Abs specific for phosphorylated Erk1/2 (pErk), total Erk (Erk2), phosphorylated Akt (pAkt) or total Akt. Results from one representative experiment of three are shown.
Generally, the effects of the monomeric ZHER2:342 were modest in both SKBR-3 and SKOV-3 cells. The dimeric affibody molecules, on the other hand, had more pronounced effects. Both (ZHER2:4)2 and (ZHER2:342)2 activated the receptor, increased phosphorylation of Erk1/2 (although not in SKOV-3, since Erk1/2 is constitutively active in this cell line) and resulted in a ten-dency of increased phosphorylation levels of Akt.

34
It should be noted that trastuzumab induced phosphorylation of HER2 but decreased phosphorylation of Akt. This was seen in both cell lines. The liter-ature on phosphorylation status of HER2 upon trastuzumab treatment is somewhat contradicting. Most groups report that the antibody does not have any effect on the activation status [108,134], yet others confirm our results showing increased phosphorylation [135].
Figure 7. Phosphorylation levels of HER2 in (A) SKOV-3 cells, (B) MCF-7 cells. A) Immunoprecipitation was done with HER2-specific antibody prior to SDS-PAGE, followed by immunoblotting by antibodies specific for phosphoty-rosine (pTyr) or total HER2. Phosphorylation of HER2 is plotted as pTyr divid-ed by total HER2. Error bars represent standard deviation (n=3). B) Total cell lysates subjected to ELISA and detected by p-HER2-specific antibodies. Num-bers indicates minutes of stimulation with (ZHER2:342)2.
Cell Growth The effects on cell growth of the three different affibody molecules were tested in SKBR-3 and SKOV-3 cells. Cells were continuously treated with approximately 10 nM of each substance for a long period of time (33-56 days). The cell culture medium and treatment substances were changed three times a week. Once a week the cells were counted and a suitable amount of cells were reseeded to allow further proliferation. In the experi-ment described in paper II, the treatments were terminated after 56 days and the cells were further cultivated in complete cell culture medium without supplementing with the substances. Cell growth was monitored for an addi-tional 4-6 weeks. Control experiments that were performed in the HER2-negative cell line MDA-231 showed that (ZHER2:342)2 had no effect compared to untreated cells (data not shown).

35
Figure 8. Cell growth of SKBR-3 (A & B) and SKOV-3 (C & D) cells. The cells were continuously treated with the substances for 56 days after which the treatments were ended, indicated by interrupted lines in A and C. Cell growth after cessation of treatment is plotted in B and D. The number of cells is normal-ized to the starting value within each group, which in figure B and D is the total number of cells at day 56. The lines in A, C and D represent curve fits per-formed by non-linear regression of exponential growth. Error bars represent standard deviations (n=3). The experiment was repeated with similar results.
As can be seen in Figure 8, the substances have different effects and the outcomes of treatment are also different in the two cell lines. Generally, the effects of ZHER2:342 were minor. In SKOV-3 cells, both dimeric affibody mol-ecules had a small but significant growth-promoting effect. In SKBR-3 the consequences of treatment with the substances differed. Both (ZHER2:4)2 and (ZHER2:342)2 had a similarly suppressive effect on cell growth as the therapeu-tic drug trastuzumab. Interestingly, the cells treated by dimeric affibody molecules were further growth-suppressed when the treatments were ceased (Figure 8B). This phenomenon was again seen when the experiment was repeated (data not shown). Trastuzumab treated SKBR-3 cells immediately resumed to the same growth rate as untreated cells.
The differences between the cell lines could be explained by the fact that SKOV-3 is a cell line with low expression of PTEN [108] and lacks ARHI [136], which is a tumor suppressor protein sharing 60 % amino acid homo-

36
logy with Ras, leading to constitutively active Erk [108]. The low expression of PTEN is also a reason to why trastuzumab has no effect on SKOV-3, as the antibody is believed to recruit PTEN and thereby inhibit Akt signaling [134]. That SKOV-3 cells are resistant to trastuzumab treatment has been shown before [108].
The phosphorylation levels of Erk1/2 and Akt in SKBR-3 cells after pro-longed treatment were studied, in an attempt to learn more about the molecu-lar mechanism behind the growth inhibitory effects of the affibody mole-cules and trastuzumab seen in the proliferation study (Figure 8). This was done by reseeding some of the cells after counting, and after sufficient time for attachment the cells were lysed and subjected to SDS-PAGE and western blot. Figure 9 shows the levels of phosphorylated Erk1/2 and Akt in SKBR-3 cells at week 3, 8, 9 and 12. No deviations from the untreated con-trol could be detected in SKOV-3 cells (data not shown).
Figure 9. Phosphorylation levels of Erk1/2 (A) and Akt (B) in SKBR-3 cells from the proliferation study described in Figure 8. The phosphorylation levels are the intensity of the western blot bands quantified by the Image J software and normalized to the untreated control, which is indicated as a dotted line. Mean values and standard deviations from three measurements are shown.
Surprisingly considering the decrease in cell growth, both (ZHER2:4)2 and (ZHER2:342)2 strongly increased the levels of phosphorylated Erk1/2 after eight weeks of treatment (Figure 9A). It has been reported that prolonged hyper-activation of the Erk-pathway promotes cell cycle arrest and cell senescence [137,138]. A drastic drop in p-Erk1/2 levels were seen the week after, when the treatment had ended. Apart from this, the effects of all substances on phosphorylated Erk1/2 and Akt were modest. Nevertheless, it can be noted that trastuzumab decreased phosphorylation of Akt during treatment, which was also noted in the short time study (Figure 6). This further supports the general theory that some of trastuzumabs effects are mediated through atten-uation of the PI3K/Akt pathway [108,134]. The phosphorylation level of Akt immediately resumed to untreated levels, as did the growth rate, upon the cessation of treatment. In contrast, cells treated by (ZHER2:4)2 or (ZHER2:342)2 had decreased levels of p-Akt at weeks nine and twelve (Figure 9B). These

37
differences in signaling patterns indicate that even though trastuzumab and the dimeric affibody molecules target the same receptor and induce similar growth inhibitory effects on SKBR-3 cells, the effects are mediated through different molecular mechanisms.
Migration A so-called wound-healing assay was used to study the effects on migration, as previously described [139]. In this experiment “wounds” were introduced into a confluent layer of SKOV-3 cells, which were treated with (ZHER2:4)2, ZHER2:342, trastuzumab or left untreated. Cell migration to close the wounds was followed for 72h by measuring the width of the wound and taking phase-contrast microscopic pictures. The experiment was performed in se-rum-free cell culture medium to minimize proliferation. The result is pre-sented by photographs in Figure 10 and as a graph in Figure 11.
Figure 10. Photographs of the wound-healing assay taken in a phase-contrast microscope. Cell migration to close the wounds in confluent SKOV-3 cells was followed for 72h. Representative photographs from three experiments are shown.

38
Figure 11. Quantification of the migration assay. “Wounds” were introduced in confluent SKOV-3 cells that were treated with (ZHER2:4)2, ZHER2:342, trastuzumab or left untreated. Three wounds were introduced in each group. The widths of the wounds were measured in a microscope using a scale bar. The width of the wounds at time zero were set as 0% wound healing. Serum-free cell culture me-dium was used to minimize proliferation.
The cells treated with (ZHER2:4)2 closed the wound faster than the untreated cells. Trastuzumab decreased the rate of healing while ZHER2:342 had no ef-fect. However, proliferation may also have some effect on the outcome of this assay, even though serum-free cell culture medium was used. Increased migration has the risk of causing enhanced metastasizing in a clinical setting.
Survival The radiosensitizing effect of the affibody molecules, in particular (ZHER2:342)2, were evaluated in SKBR-3, MCF-7 and SKOV-3 cells. As shown in Figure 12A, treatment with (ZHER2:342)2 decreased SKBR-3 survival 4-fold after irradiation with 8 Gy compared to an irradiated control (S8Gy 0.006 and 0.023, respectively, P<0.01). After 4 Gy of radiation, the two groups did not significantly differ. For MCF-7 cells, treatment with (ZHER2:342)2 did not alter the survival, either at 4 or 8 Gy (Figure 12B).
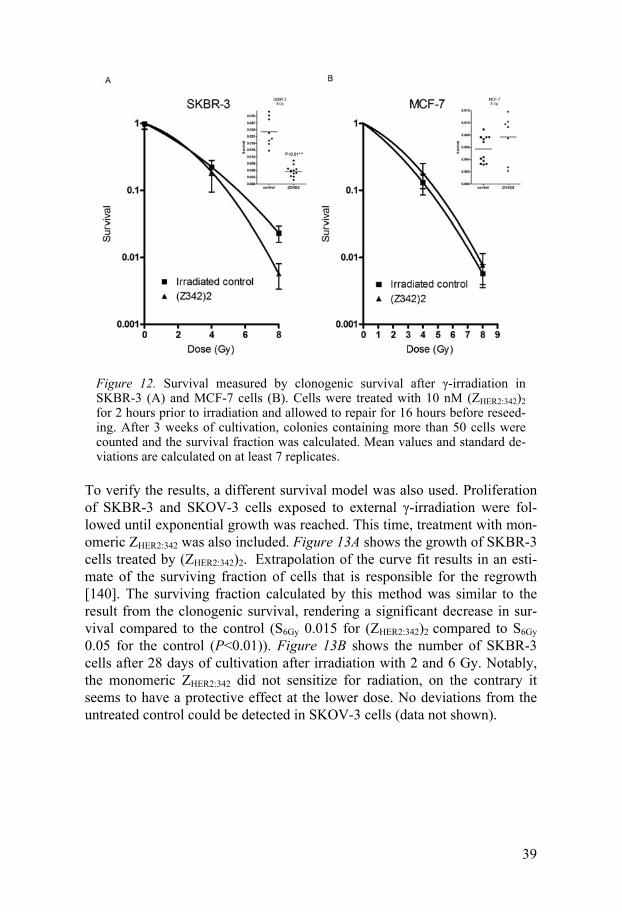
39
Figure 12. Survival measured by clonogenic survival after -irradiation in SKBR-3 (A) and MCF-7 cells (B). Cells were treated with 10 nM (ZHER2:342)2 for 2 hours prior to irradiation and allowed to repair for 16 hours before reseed-ing. After 3 weeks of cultivation, colonies containing more than 50 cells were counted and the survival fraction was calculated. Mean values and standard de-viations are calculated on at least 7 replicates.
To verify the results, a different survival model was also used. Proliferation of SKBR-3 and SKOV-3 cells exposed to external -irradiation were fol-lowed until exponential growth was reached. This time, treatment with mon-omeric ZHER2:342 was also included. Figure 13A shows the growth of SKBR-3 cells treated by (ZHER2:342)2. Extrapolation of the curve fit results in an esti-mate of the surviving fraction of cells that is responsible for the regrowth [140]. The surviving fraction calculated by this method was similar to the result from the clonogenic survival, rendering a significant decrease in sur-vival compared to the control (S6Gy 0.015 for (ZHER2:342)2 compared to S6Gy 0.05 for the control (P<0.01)). Figure 13B shows the number of SKBR-3 cells after 28 days of cultivation after irradiation with 2 and 6 Gy. Notably, the monomeric ZHER2:342 did not sensitize for radiation, on the contrary it seems to have a protective effect at the lower dose. No deviations from the untreated control could be detected in SKOV-3 cells (data not shown).

40
Figure 13. Cell growth of SKBR-3 cells after exposure to external -irradiation. A) Cells treated by 17 nM (ZHER2:342)2 for 2 hours prior to irradiation. Curve fits are based on the data points where exponential growth has been reached. B) Number of cells after 4 weeks of cultivation after irradiation. Mean values and standard deviations of 3 values are shown.
It should be noted that MCF-7 cells are more radiosensitive than SKBR-3 cells, mean survival after 8 Gy (S8Gy) was 6‰ compared to 23‰, respective-ly. SKOV-3 was the least sensitive cell line and had a calculated S8Gy of 3% according to the growth extrapolation method. It has been reported that in-creased levels of HER2 is corresponding to increased radioresistance [141], which is in agreement with this study. However, also other differences be-tween the cell lines can have an effect. In the case of SKOV-3 it is likely that the low expression of PTEN is contributing to its radioresistance.
Dimerization The membrane impermeable crosslinker bis[sulfosuccinimidyl] suberate (BS3) was used to examine receptor dimerization. After stimulation by the affibody molecules and natural ligands, the cells were treated with BS3 prior to cell lysis. This 11 Å long reagent crosslinks extracellular proteins through the amino groups. Monomeric and dimeric receptors were then separated by SDS-PAGE and detected by western blot.
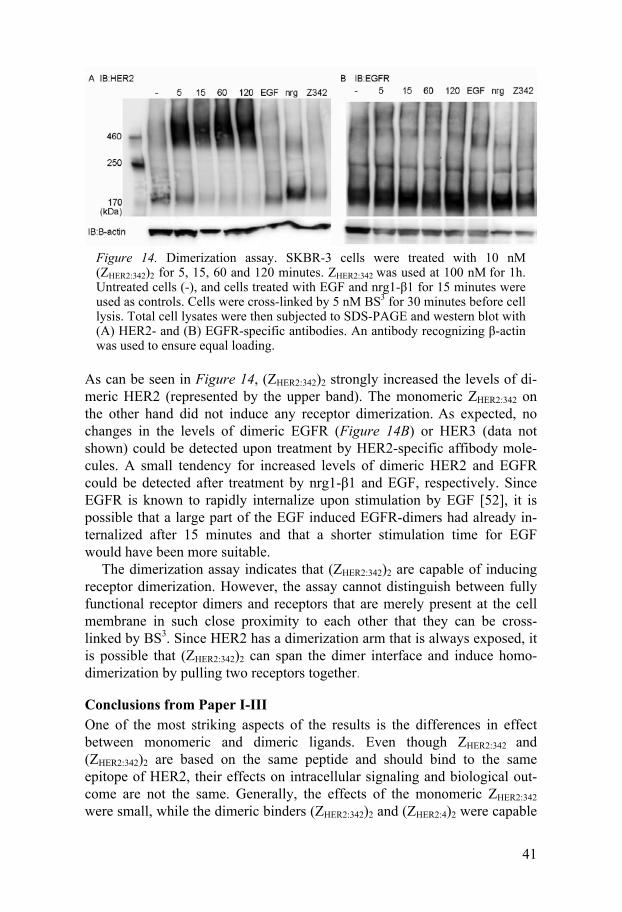
41
Figure 14. Dimerization assay. SKBR-3 cells were treated with 10 nM (ZHER2:342)2 for 5, 15, 60 and 120 minutes. ZHER2:342 was used at 100 nM for 1h. Untreated cells (-), and cells treated with EGF and nrg1- 1 for 15 minutes were used as controls. Cells were cross-linked by 5 nM BS3 for 30 minutes before cell lysis. Total cell lysates were then subjected to SDS-PAGE and western blot with (A) HER2- and (B) EGFR-specific antibodies. An antibody recognizing -actin was used to ensure equal loading.
As can be seen in Figure 14, (ZHER2:342)2 strongly increased the levels of di-meric HER2 (represented by the upper band). The monomeric ZHER2:342 on the other hand did not induce any receptor dimerization. As expected, no changes in the levels of dimeric EGFR (Figure 14B) or HER3 (data not shown) could be detected upon treatment by HER2-specific affibody mole-cules. A small tendency for increased levels of dimeric HER2 and EGFR could be detected after treatment by nrg1- 1 and EGF, respectively. Since EGFR is known to rapidly internalize upon stimulation by EGF [52], it is possible that a large part of the EGF induced EGFR-dimers had already in-ternalized after 15 minutes and that a shorter stimulation time for EGF would have been more suitable.
The dimerization assay indicates that (ZHER2:342)2 are capable of inducing receptor dimerization. However, the assay cannot distinguish between fully functional receptor dimers and receptors that are merely present at the cell membrane in such close proximity to each other that they can be cross-linked by BS3. Since HER2 has a dimerization arm that is always exposed, it is possible that (ZHER2:342)2 can span the dimer interface and induce homo-dimerization by pulling two receptors together.
Conclusions from Paper I-III One of the most striking aspects of the results is the differences in effect between monomeric and dimeric ligands. Even though ZHER2:342 and (ZHER2:342)2 are based on the same peptide and should bind to the same epitope of HER2, their effects on intracellular signaling and biological out-come are not the same. Generally, the effects of the monomeric ZHER2:342 were small, while the dimeric binders (ZHER2:342)2 and (ZHER2:4)2 were capable

42
of promoting phosphorylation of HER2 and inducing changes in signal transduction and biological outcome. For example, treatment with (ZHER2:342)2 decreased cell growth and increased radiosensitivity in SKBR-3 cells. A possible explanation for the observed differences was found in the results of the dimerization study. The dimeric (ZHER2:342)2 is capable of induc-ing HER2 dimerization, while the monomeric ZHER2:342 is not. However, the assay cannot distinguish between functional receptor dimers and receptors that simply are close enough to be cross-linked. Taken together with all the results from paper I-III it is likely that the dimeric affibody molecules are capable of inducing effective HER2 dimerization.
It is intriguing that increased HER2-dimerization, which is generally con-sidered as mediating oncogenic effects, would result in decreased cell growth and increased radiosensitivity. High expression of HER2, as in the SKBR-3 cell line, is believed to result in ligand-independent dimerization, both with itself and with other receptors of the ErbB family [37,142]. It is possible that the (ZHER2:342)2-induced dimers differ from these “naturally” induced receptor dimers. A possible way to examine this would be to study the phosphorylation of specific tyrosine residues in HER2. Another potential explanation is that (ZHER2:342)2 might prevent ligand-induced formation of the more signaling potent heterodimers by locking HER2 in homodimers. How-ever, this must be studied in a cell line with a more balanced expression level of the different receptors than in SKBR-3. Additionally, also other bivalent binders that target the ErbB-family have been shown to sensitize to irradia-tion, for example trastuzumab and cetuximab [112,141].
The effects of the ligands are also dependent on the cell line. While (ZHER2:342)2 has a slightly growth promoting effect in SKOV-3 cells, it de-creases growth of SKBR-3 cells. This finding could provide clues as to how the ligand induces its biological effects. Since SKOV-3 has constitutively active Erk1/2 and Akt, it is likely that at least part of the effects induced by (ZHER2:342)2 are mediated by these pathways.
The dimeric (ZHER2:342)2, and perhaps also (ZHER2:4)2, show promising characteristics. It sensitizes SKBR-3 cells to irradiation and decreases cell growth to the same extent as the clinically approved mAb trastuzumab. This makes (ZHER2:342)2 interesting for therapy purposes, while ZHER2:342 might be better suited for imaging since it does not seem to induce any large effects on cells.

43
3.2.2 EGFR-Targeting - Paper IV
In this article, the intracellular signaling effects of the EGFR-binding af-fibody molecule (ZEGFR:955)2 are examined. The clinically approved antibody cetuximab (Erbitux®) and the natural ligand EGF were included as reference molecules. The studies were performed in the EGFR-expressing cell lines A-431 and U-343.
Intracellular Signaling The effect on the downstream signaling pathways MAPK/Erk and PI3K/Akt were analyzed by studying the phosphorylation levels of Erk1/2 and Akt, see Figure 15. All three substances were able to induce phosphorylation of Erk1/2 in both cell lines. The low background levels of activated Erk1/2 were rapidly increased upon stimulation. While the phosphorylation caused by EGF was still visible after 2 hours, signals caused by (ZEGFR:955)2 and ce-tuximab were more transient. The substances were also able to increase phosphorylation of Akt in A-431 cells, but not in the U-343 cells. This was due to the high background level of activated Akt since the U-343 cell line has mutations in PTEN which result in constitutive activation of Akt [143].
Figure 15. Phosphorylated Erk1/2 (A) and Akt (B) in A-431 (upper panel) and U-343 (lower panel) cells. Treatment time is indicated in minutes. Untreated cells were used as negative controls (-), and cells incubated with EGF for 15 minutes as positive control (+).
Receptor Phosphorylation The EGFR receptor was phosphorylated by EGF and cetuximab in both cell lines. Treatment by EGF resulted in strong phosphorylation as early as 5 minutes after stimulation and lasted for at least 2 hours in A-431 cells while it was more transient in U-343 cells. Phosphorylation induced by ce-tuximab was weaker. (ZEGFR:955)2, on the other hand, did not induce phos-phorylation of EGFR. This was very surprising since the affibody molecule can activate downstream signaling. Phosphorylated EGFR is shown in Fig-ure 16.

44
Figure 16. Phosphorylated EGFR in A-431 (A) and U-343 (B) cells. Phosphory-lation levels are shown to the left and total receptor amount to the right. Treat-ment time is indicated in minutes. Untreated cells were used as negative controls (-), and cells incubated with EGF for 15 minutes as positive control (+).
To see if the downstream signaling was caused by phosphorylation of one of the other receptors in the family, HER2, HER3 and HER4 were also studied. However, no phosphorylation after treatment by (ZEGFR:955)2 could be detect-ed in either of these receptors, see Figure 17. Cetuximab, on the other hand, phosphorylated all receptors. Treatment with EGF resulted in activation of HER2 and HER4 in A-431 but not HER3.
Figure 17. Phosphorylated receptors in A-431 (A) and U-343 cells (B). Phos-phorylation levels are shown to the left and total receptor amount to the right. Cells were treated with approximately 5-10 nM of substances for 15 minutes. Untreated cells were used as controls (-).
One explanation for the lack of detectable receptor phosphorylation despite activation of Erk1/2 and Akt by (ZEGFR:955)2 could be due to the limitations of the western blot method and the antibodies used. The levels of phosphory-lated receptor could be so low that we are not able to detect them, yet still

45
sufficient for activation of the signaling cascades. The receptors are probably present at the cell membrane in the “closed” configuration in equilibrium with the “open” configuration (further discussed in section 2.2). It is thus possible that the affibody ligand can bind to a receptor in the open state with its dimerization arm exposed. If one ligand happens to catch two open recep-tors, dimerization might occur and the signaling cascades are activated. Since the probability for this should be low, it could explain the very low levels of phosphorylated receptor (which are too low for detection). Phos-phorylation of Erk1/2 without notable activation of HER2 was observed in a concentration study of the HER2-binding affibody (ZHER2:4)2 on SKBR-3 cells (data not shown). Phosphorylation of Erk1/2 was noted after stimula-tion by such low amounts as 0.2 nM of (ZHER2:4)2 while phosphorylation of the receptor was visible first at 10 nM.
Another explanation could be that (ZEGFR:955)2 binds to EGFR in such a way that EGFR interacts with other protein tyrosine kinases. EGFR is for example known to be activated by G protein-coupled receptor (GPCR) and growth hormone activation of Jak2 (Janus kinase) [100].
Conclusions from Paper IV A surprising observation from this study was that (ZEGFR:955)2 could induce activation of the downstream signaling proteins Erk1/2 and Akt without any obvious phosphorylation of EGFR. The most likely explanation for this is that the level of phosphorylated receptors (EGFR or other members of the ErbB family) is too low to be detected by our method.
The fact that the induced signaling pattern of (ZEGFR:955)2 is similar to that induced by cetuximab makes the affibody molecule a potentially interesting candidate for EGFR-targeting therapy, especially since Cetuximab has been shown to sensitize to irradiation [112]. Cetuxumab and (ZEGFR:955)2 have also been shown to compete for the same binding site and are internalized in a similar manner in A-431 cells [144].

46
3.2.3 Bispecific-Targeting - Paper V
A bispecific EGFR- and HER2-targeting affibody molecule has previously been generated and described by us and collaborators at KTH [132]. A prob-lem with this ligand was that it could not simultaneously bind both EGFR and HER2 if the HER2-binding part was allowed to bind first. Only if EGFR was targeted first, binding could occur to both receptors, see Figure 18. This ligand was denoted (ZHER2:342)2-(G4S)3-(ZEGFR:1907)2, and was thus constructed of a dimeric HER2-binder, (ZHER2:342)2, and a dimeric EGFR-binder, (ZEGFR:1907)2. A probable explanation for the domination of the HER2-binding part could be the differences in affinity to the receptors. (ZHER2:342)2 has a higher association rate and a more than 100-fold better affinity for HER2 than (ZEGFR:1907)2 has for EGFR [122,131]. It is therefore likely that binding to HER2 takes place first. The lack of subsequent detectable binding to EGFR could be caused by inaccessibility of the EGFR-binding epitope in the bispecific affibody molecule due to steric interference or rigidity. Other possible explanations are methodological limitations and/or insufficient con-centrations to allow detection of the binding with lower affinity.
Figure 18. A) The binding of 14C-labelled A-431 cells to SKBR-3 cells was monitored in real-time by LigandTracer White. B) The opposite set-up, 14C-labelled SKBR-3 cells were allowed to bind A-431 cells. The 14C-labelled cell suspension was pre-incubated with bispecific affibody molecule, and then added to a cell dish with SKBR-3 or A-431 cells and the uptake of 14C was measured. 14C-labelled cells without bispecific affibody molecule were used as negative controls. (cps) counts/s.
Production The second generation of bispecific affibody molecules was based on mon-omeric ligands. For the EGFR-binding part ZEGFR:1907, was used. This is an affinity maturated version of ZEGFR:955 which is evaluated in its dimeric form in paper IV. ZEGFR:1907 is reported to bind to EGFR with an affinity of 5 nM [131]. Two different HER2-binding ligands were used, ZHER2:4 which binds

47
to HER2 with an affinity of 50 nM [129], and the affinity maturated binder ZHER2:342, which has an affinity of 22 pM to HER2 [122]. The affinities to-wards the receptors are thus more equal in some of these constructs than in the former bispecific ligand [132].
The two binding domains, directed towards EGFR and HER2, were as-sembled head-to-tail and separated by a spacer sequence to facilitate flexibil-ity and separation of the two binding arms. This so-called linker was a 20 amino acid long glycine or serine based sequence, (G4S)3 and (S4G)3. Fur-thermore, the influence of the order of the affibody molecules constituting the bispecific constructs was investigated by alternating the placement of the EGFR- and HER2-binding domains either at the N- or the C-terminus. The six different constructs are listed in Table 3. The constructs are herewith denoted by their roman numerals (I-VI).
Table 3. Constructs of six bispecific binders. To improve readability, the constructs are herewith named by roman numerals.
I ZHER2:4-(S4G)3-ZEGFR:1907 II ZEGFR:1907-(S4G)3- ZHER2:4 III ZHER2:342-(S4G)3- ZEGFR:1907 IV ZEGFR:1907-(S4G)3- ZHER2:342 V ZHER2:4-(G4S)3- ZEGFR:1907 VI ZEGFR:1907-(G4S)3- ZHER2:4
Binding Properties The binding properties of the six constructs were examined both in Biacore and in a cellular setting in LigandTracer. The kinetic data are presented in Table 4 and Table 5, respectively. Both methods give similar results, show-ing that all constructs are able to bind both receptors. It is also evident that binding of all three receptor targeting ligands are benefited by placement at the N-terminus of the construct. This is especially notable in the Biacore set-up, while it is most evident for constructs V and VI in the LigandTracer experiment. In SKBR-3 cells, in which the binding predominately originates from the HER2-binding part, construct V has higher affinity than VI. The opposite is shown in A-431 cells where binding is predominantly towards EGFR, here construct VI has higher affinity than V (see Table 5). According to the Biacore analysis, constructs III and IV are the most promising candi-dates. Especially construct IV shows avidity when allowed to bind to both targets (Table 4).

48
Table 4. Kinetic data from the Biacore assay. Bispecific affibody molecules (I-VI) were injected over immobilized EGFR, HER2 and a mixed surface containing both EGFR and HER2. Kinetic parameters were estimated using TraceDrawer, using a 1:1-binding model.
EGFR HER2 EGFRxHER2
ka (M-1s-1) kd (s
-1) KD (nM) ka (M-1s-1) kd (s
-1) KD (nM) KD (nM)
I 1.1 x 104 5.3 x 10-4 47 9.6 x 104 1.1 x 10-2 115 NA
II 1.7 x 104 5.0 x 10-4 29 9.8 x 104 1.7 x 10-2 172 NA
III 6.8 x 103 5.9 x 10-4 86 5.3 x 104 3.2 x 10-4 6 4
IV 1.1 x 104 6.1 x 10-4 55 4.2 x 104 4.1 x 10-4 10 0.3
V 1.3 x 104 5.6 x 10-4 43 1.1 x 105 1.3 x 10-2 118 NA
VI 1.5 x 104 5.2 x 10-4 35 8.7 x 104 1.9 x 10-2 219 NA
NA = not available.
Interestingly, bispecific constructs containing the novel (S4G)3 linker (I and II) displayed a higher affinity in cell binding, as compared to constructs containing the (G4S)3 linker (V and VI), see Table 5. An avidity effect of constructs I and II were seen in SKBR-3 cells in the LigandTracer experi-ment. Blocking only one of the receptors resulted in weak but visible bind-ing, which was notable during the association phase but had a very quick off-rate. Without any pre-blocking a clear binding rate could be seen both during the binding- and retention phase, clearly indicating avidity. In A-431 cells, constructs II-V showed increased binding when both receptors were present compared to when only EGFR was available. When EGFR was blocked, no binding could be detected in this cell line (Table 5).
Table 5. Kinetic data from binding assays in LigandTracer. Kinetic parameters were estimated using TraceDrawer, using a 1:1-binding model.
Binding to SKBR-3 cells KD (nM)
Binding to A-431 cells KD (nM)
No block Pre-block ZHER2:4/ ZHER2:342
Pre-block ZEGFR:1907
No block Pre-block ZHER2:4/ ZHER2:342
Pre-block ZEGFR:1907
I 14 W W 7 9 ND
II 24 W W 8 16 ND
III 5 ND 6.3 64 ND ND
IV 2 ND 2 0.3 2.7 ND
V 370 ND W 120 W ND
VI 1000 ND ND 28 17 ND
W= weak binding, no kinetic parameters could be calculated. ND= not detectable (no signal of binding was obtained).

49
A possible explanation for the discrepancy between the two methods (as between the two cell lines) is the variances in receptor numbers. In the Biacore set-up, the EGFR and the HER2 receptor are present in equal num-bers, while in LigandTracer it depends on the cell line. SKBR-3 cells have approximately 6x106 HER2 receptors but only 4x105 EGFR receptors [41]. Furthermore, in Biacore the binding is measured to recombinant proteins in an artificial environment, while the binding in LigandTracer is measured to functional receptors in living cells.
Apparently, the balance in receptor number as well as the differences in affinity for the two targets have large impact on the behavior of a bispecific binder. This can be exemplified by construct IV: In SKBR-3 cells, binding is predominantly originating from the high affinity HER2-binder ZHER2:342, and no apparent contribution is made by the EGFR-binding part. On the contrary, in A-431 cells and to the mixed receptor surface in the Biacore assay, the construct shows avidity.
Cellular Effects The dimerization study presented in Figure 19 clearly shows that all six bispecific affibody molecules induce dimeric EGFR in SKBR-3 cells, but no dimeric receptors could be detected in A-431 cells that mostly express high numbers of EGFR. However, the assay cannot distinguish between EGFR-containing heterodimers and homodimers. Immunoblotting with an antibody specific for HER2 rendered ambiguous results. This was probably due to the high background level of dimerized HER2 which conceals the small changes induced by the bispecific affibody molecules (data not shown).
Figure 19. Dimerization assay of the bispecific binders. Monomeric affibody molecules and EGF were used as controls. After stimulation by 100 nM of each substance for 1 h at 37°C, the chemical crosslinker BS3 was added. EGF-treatment lasted only 5 minutes to minimize internalization. After cell lysis, the samples were subjected to SDS-PAGE, followed by transfer and western blot. A) SKBR-3 cells. B) A-431 cells.
The phosphorylation studies, which were performed both by ELISA and western blot, show a small but significant increase in phosphorylated EGFR after treatment with bispecific binders. Figure 20 shows the phosphorylation levels of EGFR measured by ELISA.

50
Figure 20. Phosphorylation study of the EGFR receptor by ELISA. SKBR-3 cells (A) or A-431 cells (B) were stimulated with 100 nM of each substance for 1 h at 37°C. EGF-treatment was only 5 minutes in order to minimize internaliza-tion. To enable comparison between different experiments, the obtained values were normalized to their respective untreated control. The graphs show mean values from 4 experiments with the standard deviations. In A) construct I-VI and EGF deviate significantly from the untreated control (unpaired t-test, P<0.05).
Both the dimerization and the phosphorylation studies indicate that the bispecific affibody molecules are able to dimerize the receptors when both HER2 and EGFR are present. However, the assay cannot distinguish be-tween fully functional receptor dimers and receptors that are merely present at the cell membrane in such proximity of each other that they can be cross-linked by BS3. The increase in EGFR phosphorylation indicates activation of the receptor and not only a close proximity of the receptors. It is also diffi-cult to say if we are detecting hetero- or homodimers, but heterodimers are more likely as the monomeric ligands do not seem to be able to induce di-merization and activation of the receptors (Figure 19 and Figure 20). Since the configuration of EGFR needs to change for the receptors to dimerize, and binding of the monomeric ligand does not seem capable of inducing this change, a possibility is that the ZEGFR:1907 part of the bispecific ligand catches an EGFR in its open state. The HER2-binding part binds to HER2 and the receptors are thus pulled together by the bispecific ligand. If the EGFR that ZEGFR:1907 binds to is in the “closed” configuration, no dimerization and phosphorylation of the receptor is possible. This could explain the low levels of phosphorylated EGFR observed.
Why no significant HER2 dimerization and phosphorylation could be measured in SKBR-3 cells might be explained by the high background level of activated HER2, together with the known differences in receptor number of EGFR and HER2. There are more than ten times as many HER2 as EGFR on SKBR-3 cells [41,70]. Probably the small increase of heterodimeric

51
HER2 is concealed by the large amount of HER2 dimers already existing and can therefore not be quantified by our methods.
Figure 21. Clonogenic survival of SKBR-3 cells after irradiation by 8 Gy. Cells were treated with construct IV, ZHER2:342, (ZHER2:342)2-(G4S)3-(ZEGFR:1907)2 (denot-ed as X) or (ZHER2:342)2 for 2 hours prior to irradiation and allowed to repair for 16 hours before reseeding. After 3 weeks of cultivation, colonies containing more than 50 cells were counted and the survival fraction was calculated. (ZHER2:342)2 and the bispecific (ZHER2:342)2-(G4S)3-(ZEGFR:1907)2 significantly de-creased survival (P<0.001, un-paired t-test), while treatment with ZHER2:342 or construct IV did not differ from the irradiated control. Mean values and standard deviations are calculated on at least 3 replicates.
Clonogenic survival after exposure to external -irradiation was performed in SKBR-3 cells. Construct IV was compared to the previously generated bispecific binder, (ZHER2:342)2-(G4S)3-(ZEGFR:1907)2 [132], and their respective HER2-binding ligands, ZHER2:342 and (ZHER2:342)2. As can be seen in Figure 21, construct IV had no effect on survival, similar to that of its HER2-binding part ZHER2:342. (ZHER2:342)2-(G4S)3-(ZEGFR:1907)2 on the other hand (de-noted as X in the figure) had a similar radiosensitizing effect as (ZHER2:342)2. These results suggest that (ZHER2:342)2-(G4S)3-(ZEGFR:1907)2 primarily binds to cells by its HER2-binding part.
Furthermore, cell proliferation in SKBR-3 and NCI-N87 cells was also studied. For this assay, construct II was chosen since it was the most promis-ing candidate according to the result from the binding studies in living cells. No differences compared to untreated cells were observed (data not shown).

52
Conclusions from Paper V All six bispecific ligands were able to simultaneously bind both EGFR and HER2. The use of monomeric EGFR and HER2 binders, rather than their dimeric equivalents, is more successful in terms of making functional bispecific affinity proteins. It can also be concluded that an N-terminal posi-tioning was favorable for the affinity to the respective targets. Furthermore, the serine linker (S4G)3 renders higher affinity of the bispecific binders com-pared to (G4S)3. None of the six bispecific constructs were able to induce any large effects on cellular outcome. Seemingly, they show more resemblance to the monomeric than the dimeric affibody molecules (evaluated in paper I-IV). The observation that the balance in receptor number as well as the dif-ferences in affinity for the two targets have large impact on the binding char-acteristics of a bispecific binder can be used when generating other new bispecific binders. Tuned bispecific binders can potentially be used to im-plicitly target receptors that are expressed also in normal tissue. By making a bispecific binder that binds moderately good to two different receptors, of which one is expressed also in normal tissue, the avidity effect will keep the bispecific binder bound mainly to cells expressing both receptors. The more a receptor is abundantly expressed in normal tissue, the weaker the respec-tive binder should be selected for the bispecific construct. This opens up the possibility to target EGFR positive tumors without excessive uptake in EGFR-expressing healthy tissues.
The expanding knowledge of the ErbB family suggests that heterodimers might be particularly oncogenic. For many years it has been known that het-erodimers are more signaling-potent than homodimers [46,47]. It has also been shown that suppressing one receptor may result in upregulation and increased signaling of another ErbB receptor [89,145]. Additionally, tumors that co-express several ErbB receptors are often more aggressive and associ-ated with a worse prognosis [146,147,148]. This suggests that for cancer prognosis and prediction of response to targeted therapies, the total ErbB expression pattern might be more important than the expression level of one single receptor. Bispecific ligands could thus be used as imaging agents with prognostic value. Another aspect of dual targeting is the possibility of in-creased tumor specificity since tumors are more likely than healthy tissue to express high amounts of two receptors.

53
4 Summary and Future Perspectives
Three types of targeting agents have been studied in this thesis, HER2-specific, EGFR-specific and dual EGFR- and HER2-specific affibody mole-cules. Studies in this thesis have shown that when these ligands bind to the receptors it can mediate intracellular signals and influence biological out-come. The affibody molecules are primarily intended as targeting agents for imaging and/or therapy of ErbB-expressing human cancers. Knowledge of how they interact and effect cells is a key step towards this goal.
Studies of the HER2-binding ligands showed that the valence of the ligand is of importance and can influence the effects. Treating SKBR-3 cells with the dimeric (ZHER2:342)2 induced large effects on intracellular signaling, in vitro proliferation and survival, while its monomeric equivalent ZHER2:342 did not. Dimerization studies showed that (ZHER2:342)2 was able to induce western blot bands corresponding to dimeric HER2. Thus, it is possible that (ZHER2:342)2 induces HER2 homodimerization. Why HER2-dimerization, which is gener-ally considered as mediating oncogenic effects, would result in decreased proliferation and increased radiosensitivity is somewhat of a mystery. Possi-ble ways to examine this is to see if the (ZHER2:342)2-induced dimers are dif-ferent from the homodimers induced by overexpression, and if (ZHER2:342)2 can prevent ligand-induced activation of HER2 by heterodimerization. The HER2-targeting (ZHER2:342)2 sensitizes SKBR-3 cells to -irradiation, which makes it interesting for therapy purposes. By labeling (ZHER2:342)2 with a suit-able radionuclide it could be possible to deliver the toxic effect to the tumors while at the same time sensitizing the tumor cells to irradiation. Since the monomeric ZHER2:342 did not have any large effects on neither of the cell lines, it could be better suited for imaging.
The EGFR-binding (ZEGFR:955)2 was found to activate the downstream sig-naling pathways MAPK/Erk1/2 and PI3K/Akt, without any detectable phos-phorylation of EGFR. This is probably due to the detection limitations of the western blot method and the antibody used. It would be interesting to see if the increased signaling could induce changes in terms of biological outcome. Since ZEGFR:955 has been shown to partially block the binding of EGF to the receptor [144], it would also be interesting to study if the affibody molecule could inhibit ligand-induced activation of EGFR.

54
It should be noted that the effects of the same affibody molecule differed in different cell lines. Ideally, similar studies should be performed on addi-tional cell lines to verify that effects that are not cell-line dependent. It could also be of interest to perform studies in a more controlled system, e.g. with transfected receptors and without signaling alterations in the cells.
The bispecific ligands generated and evaluated in paper V were all shown to be able to simultaneously bind to both EGFR and HER2. Their effects on cultured cells were minor. Furthermore, it was shown that N-terminal posi-tioning of the monomeric affibody molecules was favorable to promote the binding to respective target and that the linker (S4G)3 renders higher affinity of the bispecific binders compared to (G4S)3. Dual targeting of ErbB recep-tors is interesting from several points of view. First, heterodimerization is considered as more signaling-potent than homodimerization [20]. Second, suppression of one receptor may result in upregulation and increased signal-ing of another ErbB receptor [89]. Clinically, it has been implicated that coexpression of EGFR and HER2 is correlated with poor patient prognosis for breast cancer patients [146,149]. Bispecific ligands could thus be used as imaging agents with prognostic value. Another aspect of dual targeting is the possibility of increased tumor specificity, since tumors are more likely than healthy tissue to express high amounts of two receptors. It would also be interesting to see if the properties of a bispecific ligand can be changed by varying the length and flexibility of the linker.
Furthermore, HER2-overexpressing tumor cells are dependent on HER3 for proliferation [87], and HER3 has been found as an escape route for can-cer cells treated by ErbB-targeting TKIs [89]. A bispecific ligand that targets both these receptors has the opportunity to abrogate the potent signaling of HER2-HER3 dimers. This makes dual targeting of HER2 and HER3 a high-ly interesting research area.

55
5 Acknowledgments
This work has mainly been carried out at the Division of Biomedical Radia-tion Sciences (BMS) at Uppsala University with financial support from the Swedish Cancer Society and VINNOVA. Some of the experiments have also been performed at the Ludwig Institute for Cancer Research in Uppsala. Many people have contributed to my work on this thesis and I would like to express my gratitude to them. Especially, I would like to thank:
Mina handledare Lars Gedda och Johan Lennartsson som under olika peri-oder delat på handledarskapet. Tack för att ni alltid tagit er tid att lära mig nya experiment, förklara komplicerade saker och läsa igenom det jag skrivit. Ni har lärt mig ”allt” jag kan!
Min första handledare och numera pensionerade professor Jörgen Carlsson. Tack för att du inspirerar genom ditt stora kunnande på de mest oväntade områden.
Min handledare för examensarbetet till civilingenjörsutbildningen, Ann-Charlott Steffen. Utan dig och din trevliga introduktion av enheten hade det inte blivit något exjobb på BMS och än mindre någon avhandling!
Alla sammarbetspartner på KTH och Affibody AB, speciellt: Fredrik Frejd, tack för tillgången till alla affibody molekyler och din positiva energi på alla möten. Stefan Ståhl, Mikaela Friedman och Helena Wållberg, tack för bra samarbete och spännande bispecifika bindare.
Alla på Ludwiginstitutet som delat med sig av reagenser och bänkyta när jag kommit dit och labbat.
All mina rumskamrater; Ylva Almqvist, Marika Nestor, Erika Nordberg, Sara Ahlgren, Lovisa Göstring och Jennie Malmberg. Tack för alla trevliga pratstunder! Ni har gjort det till ett nöje att komma till jobbet.

56
Alla gamla och nya BMSare samt Ridgeview gänget. AmelieFondell, tack för att du har synkat ditt liv så bra med mitt! Vem skulle jag annars ha kun-nat diskutera graviditetskrämpor, barnprat och avhandlingsarbete? Marika, Mikael Vikström, Åsa Sundberg och Shirin Abbasinejad, tack för alla un-derhållande historier! Karl Andersson, tack för ”snurran”, uppfriskande syn-punkter på receptor-ligand interaktioner och för att du tagit dig tid att svara på alla mina kinetikfrågor. Hanna Björkelund, tack för givande dimerise-rings-diskussioner och fotosession. Sara Sahlberg, Diana Spiegelberg och Ann-Sofie Gustafsson, vad roligt att några fler har följt med mig ner i wes-tern blot träsket. Det var trevligt att inte längre vara helt själv på labbet. Samt alla icke nämnda kollegor, tack för ert sällskap vid fikabordet, stora engegemang vid filminspelningar och framförallt för att jag alltid kunnat be om er hjälp vid laborativa och vetenskapliga frågor.
Henry Ekerljung och Alvar Eriksson. Morfar, tack för att du fortsatte vara nyfiken och lära dig nya saker livet ut. Farfar, tack för att du alltid är intres-serad och ställer massor av frågor.
Lina Andersson Helin, den enda syster jag har. Tack för att du alltid föregått med gott exempel, särskilt i valet att plugga till civilingenjör här i Uppsala.
Siv och Kim Larsson. Tack för att ni är bästa tänkbara farmor och faster till mina barn!
Alla mina föräldrar. Tack för att ni lärt mig att det allra mesta är möjligt om man själv vill. Utan er inställning att kunskap är värdefullt hade det inte bli-vit någon avhandling.
Alva och Emma. Ni har snarare stjälpt än hjälpt när det gäller avhandlingen, men utan er skulle livet vara tämligen förutsägbart och långtråkigt. Tack för att ni får mig att glömma allt som har med jobbet att göra när jag är hemma.
Patrik. I tio år har det varit du och jag. Jag ser fram emot nästa tio år, och dom tio efter det.

57
6 References
1. World Health Organization (2011) Cancer, fact sheet no 297. 2. Hanahan D, Weinberg RA (2000) The Hallmarks of Cancer. Cell
100: 57-70. 3. Hanahan D, Weinberg Robert A (2011) Hallmarks of Cancer: The
Next Generation. Cell 144: 646-674. 4. Cancerfonden (2011) Cancerfondsrapporten 2011. 5. Ringborg U, Dalianis, T., Henriksson, R. (2008) Onkologi
[Oncology]. 6. Stigbrand T, Carlsson, J., and Adams, GP. (2008) Targeted
Radionuclide Tumor Therapy - Biological Aspects. 7. Mankoff DA (2007) A definition of molecular imaging. Journal of
nuclear medicine : official publication, Society of Nuclear Medicine 48.
8. McLarty K, Reilly RM (2007) Molecular imaging as a tool for personalized and targeted anticancer therapy. Clinical Pharmacology and Therapeutics 81: 420-424.
9. Pysz MA, Gambhir SS, Willmann JK (2010) Molecular imaging: current status and emerging strategies. Clinical Radiology 65: 500-516.
10. Peterson TE, Manning HC (2009) Molecular imaging: 18F-FDG PET and a whole lot more. Journal of Nuclear Medicine Technology 37: 151-161.
11. Goldsmith SJ (2010) Radioimmunotherapy of Lymphoma: Bexxar and Zevalin. Seminars in Nuclear Medicine 40: 122-135.
12. Oyen WJG, Bodei L, Giammarile F, Maecke HR, Tennvall J, et al. (2007) Targeted therapy in nuclear medicine - Current status and future prospects. Annals of Oncology 18: 1782-1792.
13. Nestor MV (2010) Targeted radionuclide therapy in head and neck cancer. Head and Neck 32: 666-678.
14. Goldenberg DM, Ablin RJ, Gold P (2007) Radiolabelled monoclonal antibodies in the treatment of metastatic cancer. Current Oncology 14: 39-42.
15. Tolmachev V, Orlova A (2010) Influence of labelling methods on biodistribution and imaging properties of radiolabelled peptides for visualisation of molecular therapeutic targets. Current Medicinal Chemistry 17: 2636-2655.
16. Holbro T, Hynes NE (2004) ErbB Receptors: Directing Key Signaling Networks Throughout Life. pp. 195-217.

58
17. Citri A, Yarden Y (2006) EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol 7: 505-516.
18. Burgess AW (2008) EGFR family: Structure physiology signalling and therapeutic target. Growth Factors 26: 263-274.
19. Burgess AW, Cho HS, Eigenbrot C, Ferguson KM, Garrett TPJ, et al. (2003) An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Molecular Cell 12: 541-552.
20. Marmor MD, Skaria KB, Yarden Y (2004) Signal transduction and oncogenesis by ErbB/HER receptors. International Journal of Radiation Oncology Biology Physics 58: 903-913.
21. Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, et al. (2003) Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 421: 756-760.
22. Guy PM, Platko JV, Cantley LC, Cerione RA, Carraway Iii KL (1994) Insect cell-expressed p180(erbB3) possesses an impaired tyrosine kinase activity. Proceedings of the National Academy of Sciences of the United States of America 91: 8132-8136.
23. Klapper LN, Glathe S, Vaisman N, Hynes NE, Andrews GC, et al. (1999) The ErbB-2/HER2 oncoprotein of human carcinomas may function solely as a shared coreceptor for multiple stroma-derived growth factors. Proceedings of the National Academy of Sciences 96: 4995-5000.
24. Garrett TPJ, McKern NM, Lou M, Elleman TC, Adams TE, et al. (2003) The Crystal Structure of a Truncated ErbB2 Ectodomain Reveals an Active Conformation, Poised to Interact with Other ErbB Receptors. Molecular Cell 11: 495-505.
25. Ferguson KM, Berger MB, Mendrola JM, Cho H-S, Leahy DJ, et al. (2003) EGF Activates Its Receptor by Removing Interactions that Autoinhibit Ectodomain Dimerization. Molecular Cell 11: 507-517.
26. Garrett TPJ, McKern NM, Lou M, Elleman TC, Adams TE, et al. (2002) Crystal Structure of a Truncated Epidermal Growth Factor Receptor Extracellular Domain Bound to Transforming Growth Factor alpha. Cell 110: 763-773.
27. Ogiso H, Ishitani R, Nureki O, Fukai S, Yamanaka M, et al. (2002) Crystal Structure of the Complex of Human Epidermal Growth Factor and Receptor Extracellular Domains. Cell 110: 775-787.
28. Dawson JP, Berger MB, Lin C-C, Schlessinger J, Lemmon MA, et al. (2005) Epidermal Growth Factor Receptor Dimerization and Activation Require Ligand-Induced Conformational Changes in the Dimer Interface. Mol Cell Biol 25: 7734-7742.
29. Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J (2006) An Allosteric Mechanism for Activation of the Kinase Domain of Epidermal Growth Factor Receptor. Cell 125: 1137-1149.
30. Tao R-H, Maruyama IN (2008) All EGF(ErbB) receptors have preformed homo- and heterodimeric structures in living cells. Journal of Cell Science 121: 3207-3217.

59
31. Liu P, Sudhaharan T, Koh RML, Hwang LC, Ahmed S, et al. (2007) Investigation of the dimerization of proteins from the epidermal growth factor receptor family by single wavelength fluorescence cross-correlation spectroscopy. Biophysical Journal 93: 684-698.
32. Saffarian S, Li Y, Elson EL, Pike LJ (2007) Oligomerization of the EGF Receptor Investigated by Live Cell Fluorescence Intensity Distribution Analysis. Biophysical Journal 93: 1021-1031.
33. Clayton AHA, Tavarnesi ML, Johns TG (2007) Unligated epidermal growth factor receptor forms higher order oligomers within microclusters on A431 cells that are sensitive to tyrosine kinase inhibitor binding. Biochemistry 46: 4589-4597.
34. Ariotti N, Liang H, Xu Y, Zhang Y, Yonekubo Y, et al. (2010) Epidermal growth factor receptor activation remodels the plasma membrane lipid environment to induce nanocluster formation. Molecular and Cellular Biology 30: 3795-3804.
35. Bublil EM, Pines G, Patel G, Fruhwirth G, Ng T, et al. (2010) Kinase-mediated quasi-dimers of EGFR. The FASEB Journal 24: 4744-4755.
36. Jura N, Endres NF, Engel K, Deindl S, Das R, et al. (2009) Mechanism for Activation of the EGF Receptor Catalytic Domain by the Juxtamembrane Segment. Cell 137: 1293-1307.
37. Penuel E, Akita RW, Sliwkowski MX (2002) Identification of a region within the ErbB2/HER2 intracellular domain that is necessary for ligand-independent association. Journal of Biological Chemistry 277: 28468-28473.
38. Yu X, Sharma KD, Takahashi T, Iwamoto R, Mekada E (2002) Ligand-independent Dimer Formation of Epidermal Growth Factor Receptor (EGFR) Is a Step Separable from Ligand-induced EGFR Signaling. Molecular Biology of the Cell 13: 2547-2557.
39. Özcan F, Klein P, Lemmon MA, Lax I, Schlessinger J (2006) On the nature of low- and high-affinity EGF receptors on living cells. Proceedings of the National Academy of Sciences of the United States of America 103: 5735-5740.
40. Endres NF, Engel K, Das R, Kovacs E, Kuriyan J (2011) Regulation of the catalytic activity of the EGF receptor. Current Opinion in Structural Biology.
41. Björkelund H, Gedda L, Barta P, Malmqvist M, Andersson K (2011) Gefitinib Induces Epidermal Growth Factor Receptor Dimers Which Alters the Interaction Characteristics with 125I-EGF. PLoS ONE 6: e24739.
42. Schlessinger J, Lemmon MA (2003) SH2 and PTB domains in tyrosine kinase signaling. Sci STKE 2003: RE12.
43. Olayioye MA, Graus-Porta D, Beerli RR, Rohrer J, Gay B, et al. (1998) ErbB-1 and ErbB-2 Acquire Distinct Signaling Properties Dependent upon Their Dimerization Partner. Mol Cell Biol 18: 5042-5051.

60
44. Sweeney C, Lai C, Riese DJ, Diamonti AJ, Cantley LC, et al. (2000) Ligand Discrimination in Signaling through an ErbB4 Receptor Homodimer. Journal of Biological Chemistry 275: 19803-19807.
45. Weiß FU, Wallasch C, Campiglio M, Issing W, Ullrich A (1997) Distinct characteristics of heregulin signals mediated by HER3 or HER4. Journal of Cellular Physiology 173: 187-195.
46. Wallasch C, Weiss FU, Niederfellner G, Jallal B, Issing W, et al. (1995) Heregulin-dependent regulation of HER2/neu oncogenic signaling by heterodimerization with HER3. EMBO Journal 14: 4267-4275.
47. Pinkas-Kramarski R, Soussan L, Waterman H, Levkowitz G, Alroy I, et al. (1996) Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO Journal 15: 2452-2467.
48. Schulze WX, Deng L, Mann M (2005) Phosphotyrosine interactome of the ErbB-receptor kinase family. Molecular systems biology [electronic resource] 1.
49. Soltoff SP, Carraway Iii KL, Prigent SA, Gullick WG, Cantley LC (1994) ErbB3 is involved in activation of phosphatidylinositol 3-kinase by epidermal growth factor. Molecular and Cellular Biology 14: 3550-3558.
50. Cohen S, Carpenter G, King Jr L (1980) Epidermal growth factor-receptor-protein kinase interactions. Co-purification of receptor and epidermal growth factor-enhanced phosphorylation activity. Journal of Biological Chemistry 255: 4834-4842.
51. Riese DJ, Stern DF (1998) Specificity within the EGF family/ErbB receptor family signaling network. BioEssays 20: 41-48.
52. Wiley HS (2003) Trafficking of the ErbB receptors and its influence on signaling. Experimental Cell Research 284: 78-88.
53. Sorkin A, Goh LK (2009) Endocytosis and intracellular trafficking of ErbBs. Experimental Cell Research 315: 683-696.
54. Warren CM, Landgraf R (2006) Signaling through ERBB receptors: Multiple layers of diversity and control. Cellular Signalling 18: 923-933.
55. Baulida J, Kraus MH, Alimandi M, Fiore PPD, Carpenter G (1996) All ErbB Receptors Other Than the Epidermal Growth Factor Receptor Are Endocytosis Impaired. Journal of Biological Chemistry 271: 5251-5257.
56. Worthylake R, Opresko LK, Wiley HS (1999) ErbB-2 Amplification Inhibits Down-regulation and Induces Constitutive Activation of Both ErbB-2 and Epidermal Growth Factor Receptors. Journal of Biological Chemistry 274: 8865-8874.
57. Nicholson RI, Gee JMW, Harper ME (2001) EGFR and cancer prognosis. European Journal of Cancer 37, Supplement 4: 9-15.
58. Hatanpaa KJ, Burma S, Zhao D, Habib AA (2010) Epidermal growth factor receptor in glioma: Signal transduction,

61
neuropathology, imaging, and radioresistance1. Neoplasia 12: 675-684.
59. www.proteinatlas.org. 60. Sebastian S, Settleman J, Reshkin SJ, Azzariti A, Bellizzi A, et al.
(2006) The complexity of targeting EGFR signalling in cancer: from expression to turnover. Biochim Biophys Acta 1766: 120-139.
61. Stern HM (2008) EGFR family heterodimers in cancer pathogenesis and treatment. In: Gullick WJ, Haley JD, editors. EGFR Signaling Networks in Cancer Therapy: Humana Press. pp. 14-29.
62. Wikstrand CJ, Sampson JH, Bigner DD (2000) EGFRvIII: An oncogene deletion mutant cell surface receptor target expressed by multiple tumour types. Expert Opinion on Therapeutic Targets 4: 497-514.
63. Graus-Porta D, Beerli RR, Daly JM, Hynes NE (1997) ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO Journal 16: 1647-1655.
64. Tzahar E, Waterman H, Chen X, Levkowitz G, Karunagaran D, et al. (1996) A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol Cell Biol 16: 5276-5287.
65. Lenferink AEG, Pinkas-Kramarski R, Van De Poll MLM, Van Vugt MJH, Klapper LN, et al. (1998) Differential endocytic routing of homo- and hetero-dimeric ErbB tyrosine kinases confers signaling superiority to receptor heterodimers. EMBO Journal 17: 3385-3397.
66. Karunagaran D, Tzahar E, Beerli RR, Chen X, Graus-Porta D, et al. (1996) ErbB-2 is a common auxiliary subunit of NDF and EGF receptors: Implications for breast cancer. EMBO Journal 15: 254-264.
67. Jones RB, Gordus A, Krall JA, MacBeath G (2006) A quantitative protein interaction network for the ErbB receptors using protein microarrays. Nature 439: 168-174.
68. Di Fiore PP, Pierce JH, Kraus MH, Segatto O, King CR, et al. (1987) erbB-2 is a potent oncogene when overexpressed in NIH/3T3 cells. Science 237: 178-182.
69. Samanta A, LeVea CM, Dougall WC, Qian X, Greene MI (1994) Ligand and p185c-neu density govern receptor interactions and tyrosine kinase activation. Proceedings of the National Academy of Sciences 91: 1711-1715.
70. Yang S, Raymond-Stintz MA, Ying W, Zhang J, Lidke DS, et al. (2007) Mapping ErbB receptors on breast cancer cell membranes during signal transduction. Journal of Cell Science 120: 2763-2773.
71. Alvarado D, Klein DE, Lemmon MA (2009) ErbB2 resembles an autoinhibited invertebrate epidermal growth factor receptor. Nature 461: 287-291.
72. Kozloski GA, Carraway CAC, Carraway KL (2010) Mechanistic and signaling analysis of Muc4–ErbB2 signaling module: New

62
insights into the mechanism of ligand-independent ErbB2 activity. Journal of Cellular Physiology 224: 649-657.
73. King CR, Kraus MH, Aaronson SA (1985) Amplification of a novel v-erbB-related gene in a human mammary carcinoma. Science 229: 974-976.
74. Stern DF, Heffernan PA, Weinberg RA (1986) P185, a product of the neu proto-oncogene, is a receptorlike protein associated with tyrosine kinase activity. Molecular and Cellular Biology 6: 1729-1740.
75. Moasser MM (2007) The oncogene HER2: Its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 26: 6469-6487.
76. Slamon DJ, Clark GM, Wong SG (1987) Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235: 177-182.
77. Baselga J, Swain SM (2009) Novel anticancer targets: Revisiting ERBB2 and discovering ERBB3. Nature Reviews Cancer 9: 463-475.
78. Carlsson J, Nordgren H, Sjöström J, Wester K, Villman K, et al. (2004) HER2 expression in breast cancer primary tumours and corresponding metastases. Original data and literature review. British Journal of Cancer 90: 2344-2348.
79. Chen JS, Lan K, Hung MC (2003) Strategies to target HER2/neu overexpression for cancer therapy. Drug Resistance Updates 6: 129-136.
80. Ross JS, Fletcher JA, Linette GP, Stec J, Clark E, et al. (2003) The HER-2/neu gene and protein in breast cancer 2003: Biomarker and target of therapy. Oncologist 8: 307-325.
81. Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, et al. (1989) Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 244: 707-712.
82. Kavanagh DO, Chambers G, O'Grady L, Barry KM, Waldron RP, et al. (2009) Is overexpression of HER-2 a predictor of prognosis in colorectal cancer? BMC Cancer 9.
83. Gravalos C, Jimeno A (2008) HER2 in gastric cancer: a new prognostic factor and a novel therapeutic target. Annals of Oncology 19: 1523-1529.
84. Shi F, Telesco SE, Liu Y, Radhakrishnan R, Lemmon MA (2010) ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proceedings of the National Academy of Sciences 107: 7692-7697.
85. Citri A, Skaria KB, Yarden Y (2003) The deaf and the dumb: The biology of ErbB-2 and ErbB-3. Experimental Cell Research 284: 54-65.
86. Amin DN, Campbell MR, Moasser MM (2010) The role of HER3, the unpretentious member of the HER family, in cancer biology and

63
cancer therapeutics. Seminars in Cell & Developmental Biology 21: 944-950.
87. Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas Iii CF, et al. (2003) The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proceedings of the National Academy of Sciences of the United States of America 100: 8933-8938.
88. Lee-Hoeflich ST, Crocker L, Yao E, Pham T, Munroe X, et al. (2008) A Central Role for HER3 in HER2-Amplified Breast Cancer: Implications for Targeted Therapy. Cancer Research 68: 5878-5887.
89. Sergina NV, Rausch M, Wang D, Blair J, Hann B, et al. (2007) Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature 445: 437-441.
90. Koutras AK, Fountzilas G, Kalogeras KT, Starakis I, Iconomou G, et al. (2010) The upgraded role of HER3 and HER4 receptors in breast cancer. Critical Reviews in Oncology/Hematology 74: 73-78.
91. Wang YN, Yamaguchi H, Hsu JM, Hung MC (2010) Nuclear trafficking of the epidermal growth factor receptor family membrane proteins. Oncogene 29: 3997-4006.
92. Sundvall M, Iljin K, Kilpinen S, Sara H, Kallioniemi OP, et al. (2008) Role of ErbB4 in breast cancer. Journal of Mammary Gland Biology and Neoplasia 13: 259-268.
93. Sundvall M, Veikkolainen V, Kurppa K, Salah Z, Tvorogov D, et al. (2010) Cell Death or Survival Promoted by Alternative Isoforms of ErbB4. Molecular Biology of the Cell 21: 4275-4286.
94. Datta SR, Brunet A, Greenberg ME (1999) Cellular survival: a play in three Akts. Genes Dev 13: 2905-2927.
95. Zhang W, Liu HT (2002) MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res 12: 9-18.
96. Thomas SM, Coppelli FM, Wells A, Gooding WE, Song J, et al. (2003) Epidermal growth factor receptor-stimulated activation of phospholipase Cgamma-1 promotes invasion of head and neck squamous cell carcinoma. Cancer Res 63: 5629-5635.
97. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, et al. (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316: 1039-1043.
98. Xu AM, Huang PH (2010) Receptor Tyrosine Kinase Coactivation Networks in Cancer. Cancer Research 70: 3857-3860.
99. Katz M, Amit I, Yarden Y (2007) Regulation of MAPKs by growth factors and receptor tyrosine kinases. Biochimica et Biophysica Acta - Molecular Cell Research 1773: 1161-1176.
100. Wieduwilt MJ, Moasser MM (2008) The epidermal growth factor receptor family: Biology driving targeted therapeutics. Cellular and Molecular Life Sciences 65: 1566-1584.
101. Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S, et al. (1994) Transformation of mammalian cells by constitutively active MAP kinase kinase. Science 265: 966-970.

64
102. Thompson N, Lyons J (2005) Recent progress in targeting the Raf/MEK/ERK pathway with inhibitors in cancer drug discovery. Current Opinion in Pharmacology 5: 350-356.
103. Roberts PJ, Der CJ (2007) Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26: 3291-3310.
104. Downward J (2004) PI 3-kinase, Akt and cell survival. Seminars in Cell and Developmental Biology 15: 177-182.
105. Franke TF, Kaplan DR, Cantley LC (1997) PI3K: Downstream AKTion blocks apoptosis. Cell 88: 435-437.
106. Li L, Ross AH (2007) Why is PTEN an important tumor suppressor? Journal of Cellular Biochemistry 102: 1368-1374.
107. Kruser TJ, Wheeler DL (2010) Mechanisms of resistance to HER family targeting antibodies. Experimental Cell Research 316: 1083-1100.
108. Longva KE, Pedersen NM, Haslekås C, Stang E, Madshus IH (2005) Herceptin-induced inhibition of ErbB2 signaling involves reduced phosphorylation of Akt but not endocytic down-regulation of ErbB2. International Journal of Cancer 116: 359-367.
109. Wells A, Grandis JR (2003) Phospholipase C-gamma1 in tumor progression. Clin Exp Metastasis 20: 285-290.
110. Maddens S, Charruyer A, Plo I, Dubreuil P, Berger S, et al. (2002) Kit signaling inhibits the sphingomyelin-ceramide pathway through PLC 1: Implication in stem cell factor radioprotective effect. Blood 100: 1294-1301.
111. Duffy MJ, O’Donovan N, Crown J (2011) Use of molecular markers for predicting therapy response in cancer patients. Cancer Treatment Reviews 37: 151-159.
112. Bonner JA, Harari PM, Giralt J, Cohen RB, Jones CU, et al. (2010) Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. The Lancet Oncology 11: 21-28.
113. Potthoff K, Hofheinz R, Hassel JC, Volkenandt M, Lordick F, et al. (2011) Interdisciplinary management of EGFR-inhibitor-induced skin reactions: a German expert opinion. Annals of Oncology 22: 524-535.
114. Albanell J, Codony J, Rovira A, Mellado B, Gascón P (2003) Mechanism of action of anti-HER2 monoclonal antibodies: Scientific update on trastuzumab and 2C4. pp. 253-268.
115. Baselga J, Perez EA, Pienkowski T, Bell R (2006) Adjuvant trastuzumab: A milestone in the treatment of HER-2-positive early breast cancer. Oncologist 11: 4-12.
116. Franklin MC, Carey KD, Vajdos FF, Leahy DJ, de Vos AM, et al. (2004) Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell 5: 317-328.

65
117. Gazdar AF, Shigematsu H, Herz J, Minna JD (2004) Mutations and addiction to EGFR: the Achilles ‘heal’ of lung cancers? Trends in Molecular Medicine 10: 481-486.
118. Untch M, Lück HJ (2010) Lapatinib - Member of a new generation of ErbB-Targeting drugs. Breast Care 5: 8-12.
119. Nord K, Nilsson J, Nilsson B, Uhlen M, Nygren PA (1995) A combinatorial library of an -helical bacterial receptor domain. Protein Engineering 8: 601-608.
120. Grimm S, Yu F, Nygren P-Å (2011) Ribosome Display Selection of a Murine IgG1 Fab Binding Affibody Molecule Allowing Species Selective Recovery Of Monoclonal Antibodies. Molecular Biotechnology 48: 263-276.
121. Kronqvist N, Löfblom J, Jonsson A, Wernérus H, Ståhl S (2008) A novel affinity protein selection system based on staphylococcal cell surface display and flow cytometry. Protein Engineering, Design and Selection 21: 247-255.
122. Orlova A, Magnusson M, Eriksson TLJ, Nilsson M, Larsson B, et al. (2006) Tumor imaging using a picomolar affinity HER2 binding Affibody molecule. Cancer Research 66: 4339-4348.
123. Löfblom J, Feldwisch J, Tolmachev V, Carlsson J, Ståhl S, et al. (2010) Affibody molecules: Engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Letters 584: 2670-2680.
124. Steffen AC, Wikman M, Tolmachev V, Adams GP, Nilsson FY, et al. (2005) In vitro characterization of a bivalent anti-HER-2 affibody with potential for radionuclide-based diagnostics. Cancer Biotherapy and Radiopharmaceuticals 20: 239-248.
125. Friedman M, Ståhl S (2009) Engineered affinity proteins for tumour-targeting applications. Biotechnology and Applied Biochemistry 53: 1-29.
126. Engfeldt T, Renberg B, Brumer H, Nygren PA, Karlström AE (2005) Chemical synthesis of triple-labelled three-helix bundle binding proteins for specific fluorescent detection of unlabelled protein. ChemBioChem 6: 1043-1050.
127. Orlova A, Tolmachev V, Pehrson R, Lindborg M, Tran T, et al. (2007) Synthetic affibody molecules: A novel class of affinity ligands for molecular imaging of HER2-expressing malignant tumors. Cancer Research 67: 2178-2186.
128. Feldwisch J, Tolmachev V, Lendel C, Herne N, Sjöberg A, et al. (2010) Design of an Optimized Scaffold for Affibody Molecules. Journal of Molecular Biology 398: 232-247.
129. Wikman M, Steffen AC, Gunneriusson E, Tolmachev V, Adams GP, et al. (2004) Selection and characterization of HER2/neu-binding affibody ligands. Protein Engineering, Design and Selection 17: 455-462.
130. Friedman M, Nordberg E, Höidén-Guthenberg I, Brismar H, Adams GP, et al. (2007) Phage display selection of Affibody molecules with

66
specific binding to the extracellular domain of the epidermal growth factor receptor. Protein Engineering Design and Selection 20: 189-199.
131. Friedman M, Orlova A, Johansson E, Eriksson TLJ, Höidén-Guthenberg I, et al. (2008) Directed Evolution to Low Nanomolar Affinity of a Tumor-Targeting Epidermal Growth Factor Receptor-Binding Affibody Molecule. Journal of Molecular Biology 376: 1388-1402.
132. Friedman M, Lindström S, Ekerljung L, Andersson-Svahn H, Carlsson J, et al. (2009) Engineering and characterization of a bispecific HER2 × EGFR-binding affibody molecule. Biotechnology and Applied Biochemistry 54: 121-131.
133. Aguilar Z, Akita RW, Finn RS, Ramos BL, Pegram MD, et al. (1999) Biologic effects of heregulin/neu differentiation factor on normal and malignant human breast and ovarian epithelial cells. Oncogene 18: 6050-6062.
134. Nagata Y, Lan K-H, Zhou X, Tan M, Esteva FJ, et al. (2004) PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 6: 117-127.
135. Gijsen M, King P, Perera T, Parker PJ, Harris AL, et al. (2010) HER2 phosphorylation is maintained by a PKB negative feedback loop in response to anti-HER2 herceptin in breast cancer. PLoS Biology 8.
136. Bao JJ, Le XF, Wang RY, Yuan J, Wang L, et al. (2002) Reexpression of the tumor suppressor gene ARHI induces apoptosis in ovarian and breast cancer cells through a caspase-independent calpain-dependent pathway. Cancer Research 62: 7264-7272.
137. Pumiglia KM, Decker SJ (1997) Cell cycle arrest mediated by the MEK/mitogen-activated protein kinase pathway. Proceedings of the National Academy of Sciences 94: 448-452.
138. Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M, et al. (1998) Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes & Development 12: 3008-3019.
139. Rodriguez LG, Wu X, Guan J-L (2005) Wound-Healing Assay. In: Guan J-L, editor. Cell Migration: Humana Press. pp. 23-29.
140. Johansson L, Nilsson K, Carlsson J (1981) Radiation effects on cultured human lymphoid cells. Analysis using the growth extrapolation method. Acta Radiologica Oncology Radiation Therapy Physics and Biology 20: 51-59.
141. Liang K, Lu Y, Jin W, Ang KK, Milas L, et al. (2003) Sensitization of breast cancer cells to radiation by trastuzumab. Molecular Cancer Therapeutics 2: 1113-1120.
142. Ignatoski KMW, LaPointe AJ, Radany EH, Ethier SP (1999) erbB-2 Overexpression in Human Mammary Epithelial Cells Confers Growth Factor Independence. Endocrinology 140: 3615-3622.

67
143. Sauvageot CM-E, Weatherbee JL, Kesari S, Winters SE, Barnes J, et al. (2009) Efficacy of the HSP90 inhibitor 17-AAG in human glioma cell lines and tumorigenic glioma stem cells. Neuro-Oncology 11: 109-121.
144. Nordberg E, Friedman M, Gostring L, Adams GP, Brismar H, et al. (2007) Cellular studies of binding, internalization and retention of a radiolabeled EGFR-binding affibody molecule. Nucl Med Biol 34: 609-618.
145. Kong A, Calleja V, Leboucher P, Harris A, Parker PJ, et al. (2008) HER2 oncogenic function escapes EGFR tyrosine kinase inhibitors via activation of alternative HER receptors in breast cancer cells. PLoS ONE 3.
146. Tsutsui S, Ohno S, Murakami S, Kataoka A, Kinoshita J, et al. (2003) Prognostic value of the combination of epidermal growth factor receptor and c-erbB-2 in breast cancer. Surgery 133: 219-221.
147. Hirsch FR, Varella-Garcia M, Cappuzzo F (2009) Predictive value of EGFR and HER2 overexpression in advanced non-small-cell lung cancer. Oncogene 28: S32-S37.
148. Xia W, Lau Y-K, Zhang H-Z, Xiao F-Y, Johnston DA, et al. (1999) Combination of EGFR, HER-2/neu, and HER-3 Is a Stronger Predictor for the Outcome of Oral Squamous Cell Carcinoma Than Any Individual Family Members. Clinical Cancer Research 5: 4164-4174.
149. DiGiovanna MP, Stern DF, Edgerton SM, Whalen SG, Moore Ii D, et al. (2005) Relationship of epidermal growth factor receptor expression to ErbB-2 signaling activity and prognosis in breast cancer patients. Journal of Clinical Oncology 23: 1152-1160.
















-labeled](https://img.dokumen.tips/doc/110x75/60079f9e83133342bc389bd0/affibody-molecules-for-pet-imaging-844633-hahaha-hehehe-hihihi-or-hkhkhk.jpg)










![HAHAHA, HEHEHE, HIHIHI, or HKHKHK: Influence of Position and Composition of Histidine Containing Tags on Biodistribution of [ 99mTc(CO)3] + -Labeled Affibody Molecules](https://img.dokumen.tips/doc/110x75/55cf8d015503462b139129b1/hahaha-hehehe-hihihi-or-hkhkhk-influence-of-position-and-composition-of.jpg)


