Embed Size (px)
Citation preview

[CANCER RESEARCH 60, 3957–3964, July 15, 2000]
E1A Overcomes the Apoptosis Block in BCR-ABL1Leukemia Cells and RendersCells Susceptible to Induction of Apoptosis by Chemotherapeutic Agents1
Thorsten Stiewe,2 Keiarasch Parssanedjad,2 Helmut Esche, Bertram Opalka, and Brigitte M. Putzer3
Institute of Molecular Biology (Cancer Research) [T. S., K. P., H. E., B. M. P.] and Department of Internal Medicine, West German Cancer Center [B. O.], University of EssenMedical School, Hufelandstrasse 55,D-45122 Essen, Germany
ABSTRACT
A crucial function of the BCR-ABL chimeric gene in chronic myeloidleukemia is the prolongation of cell survival by inhibition of apoptosis.BCR-ABL expression confers cross-resistance to multiple genotoxic antican-cer drugs by inhibition of the apoptotic response to DNA damage in associ-ation with cell cycle arrest at the G2-M restriction point. Previous reportsindicated that BCR-ABL exerts its antiapoptotic effect against various apo-ptotic stimuli upstream to the cleavage and activity of caspase-3. Here weshow that the adenovirus E1A protein induces substantial apoptosis in BCR-ABL expressing K562 and LAMA-84 leukemia cells. This apoptotic activityof E1A is accompanied by processing of caspase-3 and cleavage of poly(ADP-ribose) polymerase and can be significantly blocked by z-VAD-fmk Z-Val-Ala-Asp(OCH3)-CH2F and the caspase-3-specific inhibitor Z-DEVD-FMKZ-Asp(OCH3)-Glu-Val-Asp(OCH3)-CH2F. Moreover, E1A renders K562cells, which are particularly resistant to cell death irrespective of the inducingagent, susceptible to induction of apoptosis by the chemotherapeutic agentsetoposide and daunorubicin. Counteracting the DNA damage-induced inac-tivation of cdc2 kinase, E1A reverses the drug-induced G2-M arrest. Theseresults indicate that solitary delivery of E1A significantly antagonizes BCR-ABL-induced antiapoptotic functions and circumvents the inherent resist-ance to DNA damage-induced apoptosis, supporting the use of E1A in com-bination with chemotherapeutic agents as a promising therapeutic strategyfor successful treatment of Philadelphia chromosome-positive leukemiainvivo.
INTRODUCTION
CML4 is a hematopoietic disorder that is characterized by thepresence of excessive numbers of mature myeloid cells in the periph-eral blood and bone marrow, considered to be a consequence of anexpanded population of progenitor cells (1). The cytogenetical hall-mark of CML is the presence of the Ph chromosome, which resultsfrom a mostly reciprocal translocation of c-ABL from chromosome 9to theBCRgene on chromosome 22, generating a chimeric BCR-ABLprotein that has elevated levels of ABL tyrosine kinase activity (2).
When theBCR-ABL gene is transduced into hematopoietic celllines, they become growth factor independent and have increasedproliferative capacity. Such studies have demonstrated that the mito-genic ability of BCR-ABL is mediated in part through activation of aRas (3) and phosphatidylinositol 39-kinase-dependent signaling path-ways (4), suggesting that the massive expansion of malignant cells inCML may be due to BCR-ABL-induced proliferation. However, thereis much support that the clonal expansion evident in this malignantdisorder is not a result of deregulated cellular proliferation (5, 6) but
rather occurs via prolongation of cell survival by prevention of apo-ptotic cell death (7, 8). Supporting the role of the BCR-ABL protein-tyrosine kinase as a negative regulator of apoptosis, deregulatedkinase activity confers cross-resistance to multiple anticancer agentsby inhibition of the apoptotic response to DNA damage (9, 10).BCR-ABL can protect growth factor-dependent hematopoietic celllines from apoptosis induced by factor withdrawal (11, 12) andFas-mediated apoptosis (13). In turn, recent observations indicatedrestoration of susceptibility to apoptosis and enhancement of survivalthrough inhibition of BCR-ABL expression by antisense oligonucleo-tides (9, 14), by the tyrosine kinase inhibitor CGP57148 (15), andFas-mediated down-modulation of BCR-ABL (16), confirming theanti-apoptotic function of the chimeric protein.
The importance of apoptosis in maintaining hematopoietic home-ostasis is evident from the consequences of its deregulation. Apoptosisoccurs under physiological conditions,e.g.,during T-cell maturationin the thymus, and is characterized by cell shrinkage, chromatincondensation, and DNA fragmentation (17). A variety of apoptoticstimuli cause the preapoptotic mitochondrial release of cytochromecinto the cytosol, which mediates activation of caspase-3 from aprecursor and cleavage of PARP (18), resulting in execution of thewhole program of apoptosis. However, recent findings indicated thatBCR-ABL expression blocks apoptosis upstream of procaspase-3activation (19) by preventing the cytosolic accumulation of cyto-chromec and other preapoptotic mitochondrial perturbations in, forexample, etoposide-treated K562 cells (20, 21).
One of the hallmarks of apoptosis is that it is genetically regulated.A number of products of tumor suppressor genes, proto-oncogenes,and some viral genes are known to regulate this process (reviewed inRefs. 22, 23), making it open to genetic manipulation and therebyraising the possibility of therapeutic intervention. The adenovirus 5E1A oncogene products interact with and perturb the function of keyregulators of cell proliferation, such as the RB protein (24). The resultof these interactions is induction of cellular DNA synthesis but alsoloss of cell viability and induction of apoptosis, which impedes boththe transformation of primary rodent cells and productive adenovirusinfection of human cells (25, 26). The ability of E1A to directapoptosis is thought to be related to its ability to cause the release ofthe transcription factor E2F-1 from RB binding (27). Enforced over-expression of E2F-1 has been shown to trigger apoptosis in quiescentfibroblasts and to suppress tumor growth in glioma cells (28, 29).
E1A produces two major mRNAs, encoding proteins of 289 and243 residues (289R and 243R), respectively, which differ only by the46-amino acid conserved region 3 in the 13S protein known toactivate expression of other early viral genes. The protein products ofthe E1A gene can induce apoptosis by both p53-dependent and-independent mechanisms. In the presence of wild-type p53, expres-sion of the E1A 12S transcript leads to an increase in the levels of p53,resulting in the deregulation of Bax and Bcl-2 (30), which correlateswith the induction of apoptosis (31–34). Within the viral context,however, the E1A 13S transcript can also induce apoptosis independ-ently of p53, which appears to be dependent on conserved region 3and the early region E4 (35, 36). Our group recently showed that bothE1A proteins are capable of inducing substantial apoptotic cell death
Received 11/22/99; accepted 5/16/00.The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby markedadvertisementin accordance with18 U.S.C. Section 1734 solely to indicate this fact.
1 This work was supported by the Interne Forschungsforderung Essen program of theMedical Faculty of the University of Essen.
2 These authors contributed equally to this work.3 To whom requests for reprints should be addressed, at Institute of Molecular Biology,
University of Essen Medical School, Hufelandstrasse 55, D-45122, Germany. Phone: (49)201-723-3158; Fax: (49) 201-723-5974; E-mail: [email protected].
4 The abbreviations used are: CML, chronic myelogenous leukemia; Ph, Philadelphia;RB, retinoblastoma; RSV, Rous sarcoma virus; PARP, poly(ADP-ribose) polymerase;GFP, green fluorescent protein; PI, propidium iodide; FACS, fluorescence-activated cellsorting; TK, thymidine kinase; CPP32, caspase-3.
3957
on July 18, 2018. © 2000 American Association for Cancer Research.cancerres.aacrjournals.org Downloaded from

in the absence of other adenoviral genes in cells lacking p53 (37).Previous reports demonstrated that E1A expression enhances thesensitivity to apoptosis by ionizing radiation and various cytotoxicagents in murine embryonic fibroblasts, murine keratinocytes, andhuman ovarian cancer cells (31, 38, 39).
In this study, we investigated the apoptotic activity of the E1Aprotein in the BCR-ABL-expressing leukemia cell lines K562 andLAMA-84, respectively. Our data indicate that E1A alone is capableof inducing substantial apoptosis by antagonizing the BCR-ABL-induced block within the apoptosis cascade of CML cells. E1Aovercomes the inherent resistance to DNA damage induced apoptosisby bypassing the drug-induced BCR-ABL-mediated G2-M arrest andrenders Ph-positive leukemia cells susceptible to induction of apo-ptosis by chemotherapeutic agents. Our results provide support for useof E1A in combination with chemotherapeutics as a promising ap-proach for elimination of Ph-positive leukemias in patients.
MATERIALS AND METHODS
Cell Culture and Drug Treatment. K562 and LAMA-84 Ph1cell lines,derived from patients during the blast crisis phase of CML, were obtained fromthe German Collection of Microorganisms and Cell Cultures (Braunschweig,Germany). K562 and LAMA-84 cells both contain b3-a2 P210bcr-abl (40).Cells were cultured in RPMI 1640 supplemented with 10% FCS, 2 mM
L-glutamine, 100mg/ml penicillin, and 100 units/ml streptomycin in a 37°Cincubator containing 5% CO2. Culture media and supplements were obtainedfrom Life Technologies (Karlsruhe, Germany). Etoposide and daunorubicinwere obtained from Sigma-Aldrich (Deisenhofen, Germany) and dissolved inacidified ethanol or sodium chloride solution, respectively. For treatment withchemotherapeutic agents, 73 106 cells were treated by continuous exposure to0.2mg/ml daunorubicin or etoposide at the indicated concentrations for 2 days.Drug treatment of transfected cultures was started 24 h after transfection.
Plasmids and DNA Transfection. The adenovirus 5 E1A cDNAs werecloned into pRc/RSV (Invitrogen, Groningen, the Netherlands) for expressionfrom the RSV promoter. A 498-bp fragment from the human cdc2 promoter wasamplified by PCR using the following oligonucleotides as primers: 59-TTAGGT-CACTGAAATGTGCTCCTTG-39 (forward, bp2466 to2441) and 59-CAATT-TCCAAGAGCCAGCTTTGAAG-39 (reverse, bp18 to 133). The fragment wassubsequently cloned blunt-ended into theSmaI site of pGL3basic (Promega,Mannheim, Germany). Plasmid DNA was prepared by the alkaline lysis methodand purified by CsCl-ethidium bromide density gradient centrifugation. Transfec-tions were performed as described by using the electroporation method (41).
Western Blotting. Cell lysates were prepared after transfection, and pro-tein levels were analyzed by Western blot essentially as described (27). Theantibodies used were directed against E1A (M73; Calbiochem, Bad Soden,Germany), CPP32 17-kDa subunit (E-8; Santa Cruz Biotechnology, Heidel-berg, Germany), PARP 85 kDa (7D3–6; PharMingen, San Diego, CA), cdc2(9112; New England Biolabs, Schwalbach, Germany), phospho-cdc2 (Tyr-15;9111; New England Biolabs), and PKAa cat, thea catalytic subunit of proteinkinase A (sc-903; Santa Cruz Biotechnology). CPP32 or PARP cleavageproducts, cdc2, phospho-cdc2, and PKAa cat were detected by using celllysate from GFP-positive, transfected cells sorted out by flow cytometryanalysis. Immune complexes were visualized by enhanced chemiluminescence(Amersham Pharmacia Biotech, Braunschweig, Germany).
Clonogenic Assay.The ability to grow in soft agarose was determined asdescribed previously (42). Briefly, 10mg of plasmid DNA expressing E1A orcontrol vector were transfected together with the puromycin-N-acetyltrans-ferase-expressing plasmid into 73 106 K562 cells by electroporation. Twenty-four hours after transfection, cells were washed and plated in a six-well platein culture medium containing puromycin (1mg/ml) and 0.35% agarose over-lying a 0.7% agarose layer. The cells were incubated at 37°C for 3 weeks, afterwhich puromycin-resistant colonies were counted under light microscopy.
Flow Cytometry Analysis. Ten micrograms of plasmid DNA expressingE1A were transfected into 73 106 cells. Where required, the peptide caspase-inhibitors z-VAD-fmk or Z-DEVD-FMK (Calbiochem) were added simulta-neously with the apoptotic-triggering signal at a final concentration of 50mM. Tomeasure the transfection efficiency, 2mg of GFP reporter plasmid encoding the
membrane-localized enhanced GFP were cotransfected to ensure optimal fluores-cence intensity in combination with ethanol fixation (43). To quantitate apoptosisby flow cytometry, floating and adherent cells were harvested 72 h after transfec-tion, fixed in ethanol, and stained for DNA content with PI. Cells were measuredfor green fluorescence intensity (channel FL-1) and PI fluorescence (channel FL-3)in a fluorescence-activated cell sorter (FACSVantage; Becton Dickinson, Moun-tain View, CA) using CELLquest software (Becton Dickinson). The cells that didnot express GFP were used to set the baseline to allow the gating of the GFP-positive cells. The percentage of apoptotic cells seen in the population by electro-poration alone (typically 2–6%) was subtracted.
Luciferase Assay.K562 cells were cotransfected by electroporationwith 1 mg of the pGL3-basic (Promega) or pGL3-cdc2 firefly luciferasereporter plasmid and 2mg of the E1A expression plasmid or the pRc/RSVcontrol vector plasmid (Invitrogen), respectively. In all transfections 1mgof pRL-TK (Promega) encoding for Renilla luciferase under the control ofthe herpes simplex virus TK promoter region was cotransfected to accountfor differences in transfection efficiency. Treatment with 5mM etoposidewas initiated 24 h after transfection. Cells were collected 48 h aftertransfection in passive lysis buffer (Promega). Firefly and Renilla lucifer-ase activities were determined by a premanufactured dual luciferase re-porter assay system (Promega). To account for differences in transfectionefficiencies, firefly luciferase activity was normalized to Renilla luciferaseactivity. Error bars represent the SD within a representative experiment.Each experiment was repeated at least three times.
RESULTS
E1A Induces Substantial Cytotoxicity and Apoptosis in Ph-positive Chronic Myeloid Leukemia Cells. BCR-ABL has beenshown to contribute to the protection of hematopoetic cells from theinduction of apoptosis by cytokine withdrawal (14), Fas ligation (13), andtreatment with cytotoxic drugs (9). By contrast, E1A expression has beenshown previously to induce apoptosis and enhancein vitro cytotoxicity toionizing radiation and chemotherapeutic agents (38, 44). The ability ofE1A to mediate cytotoxicity in BCR-ABL-positive K562 erythroleuke-mia cells was analyzed by clonogenic survival in soft agarose. As shownin Fig. 1A, the numbers of formed colonies were markedly decreased inthe E1A 13S-transfected cells compared with mock-transfected cells. Toinvestigate whether the observed loss of viability in Ph-positive leukemiacells on overexpression of E1A protein is due to apoptosis, we analyzedK562 (Fig. 1B, I–III) and a second BCR-ABL positive cell line, LA-MA-84 (Fig. 1B, I, IV, andV), transiently transfected with the E1A 13ScDNA using FACS analysis of PI-stained cells. This flow cytometricassay measures the apoptotic rate at the time of harvesting rather thancumulative apoptosis (45). Transfected cells were gated on the basis ofthe expression of GFP, which was cotransfected as a transfection marker(cotransfection rate of;95%), and apoptosis was measured by theaccumulation of cells with a sub-G1 DNA content 72 h after transfection.Quantification of sub-G1 cells revealed a significant,;3–5-fold increasein apoptotic cells in both E1A-transfected BCR-ABL-positive leukemiacell lines (Fig. 1B, I, III, andV) compared with cells transfected with thecontrol vector (Fig. 1B, I, II, andIV). E1A expression in these cellsproduced typical apoptotic features with striking changes in the nuclearmorphology, characterized by intense staining of condensed chromatinand nuclear fragmentation as analyzed by fluorescence microscopy (datanot shown). Because K562 cells are negative for p53 expression (46, 47),the observed apoptotic response in the p53-negative cell system is con-sistent with our data, demonstrating apoptosis induction by solitary de-livery of E1A in the absence of functional p53 as well as other adenoviralgene products (37). As an indication of the transfection efficiency, E1Aexpression in K562 cells was monitored by Western blot analysis(Fig. 1C).
Induction of Apoptosis by E1A Is Mediated by Caspase-3 Ac-tivation, Which Is Specifically Blocked in BCR-ABL-expressingLeukemia Cells. Previous studies have indicated that caspase acti-vation plays a critical role in the initiation of the active phase of
3958
APOPTOSIS BY E1A IN BCR-ABL1 LEUKEMIA CELLS
on July 18, 2018. © 2000 American Association for Cancer Research.cancerres.aacrjournals.org Downloaded from

apoptosis (48–50). In addition, it has been suggested that the blockageof cytochromec release and caspase-3 activation is a mechanism bywhich the deregulated BCR-ABL tyrosine kinase prevents apoptoticcell death (19, 21, 51). These observations prompted us to investigatethe effect of E1A treatment on processing of procaspase-3 (pro-CPP32) into the active 17- and 12-kDa subunits. To detect particularlyshort-lived proteins in a relatively small number of E1A-transfectedcells in front of the untransfected background, the effect of E1Aexpression in K562 cells was determined by Western blot analysisusing whole-cell extracts prepared from the GFP-positive, transfectedpopulation sorted out by FACS. In these cells, stimulation of apo-ptosis by E1A triggered processing of procaspase-3, as revealed by theappearance of an;17-kDa product, which corresponds to the 17-kDa
subunit of activated caspase-3 (Fig. 2A). As shown in Fig. 2A, induc-tion of apoptosis by E1A was also accompanied by cleavage of the116-kDa intact form of PARP to the 85-kDa fragment intimatelylinked to the induction of apoptosis in other systems. Interestingly, theapoptotic activity of E1A in K562 leukemia cells, measured by flowcytometry analysis at 72 h after transfection, was significantly antag-onized by the wide-spectrum caspase inhibitor z-VAD-fmk, resultingin an;50% reduction of relative apoptosis (Fig. 2B). In addition, thesame inhibitory effect on E1A-induced apoptosis was evident inE1A-transfected cells treated with the caspase-3-specific inhibitorZ-DEVD-fmk. These observations indicated that transiently expressedE1A is sufficient to abolish the antiapoptotic function of BCR-ABLby initiating the caspase cascade.
Fig. 1. Ectopic expression of E1A triggers apoptosis inPh-positive chronic myeloid leukemia cells.A, clonogenicassay of E1A 13S cDNA- and control plasmid-transfected(mock) K562 cells after selection for stable transfectants withpuromycin for 3 weeks. The average number of resultingcolonies is shown in theleft panel; representative phase-contrast micrographs are shown on theright. B, FACS anal-ysis of E1A 13S cDNA-transfected (I, III, andV) and controlplasmid-transfected (mock;I, II, and IV) PI-stained K562(I–III) and LAMA-84 (I, IV, and V) cells, respectively. Thepercentage of apoptotic cells by electroporation alone (EP) isas indicated (I). Transfected cells were gated on the basis ofGFP expression. Apoptosis was measured by the accumula-tion of cells with a sub-G1 DNA content 72 h after transfec-tion. Thediagram represents the mean of three independentexperiments.C, Western blot analysis of E1A expression intransfected K562. 293 cells are shown as a positive control.
3959
APOPTOSIS BY E1A IN BCR-ABL1 LEUKEMIA CELLS
on July 18, 2018. © 2000 American Association for Cancer Research.cancerres.aacrjournals.org Downloaded from

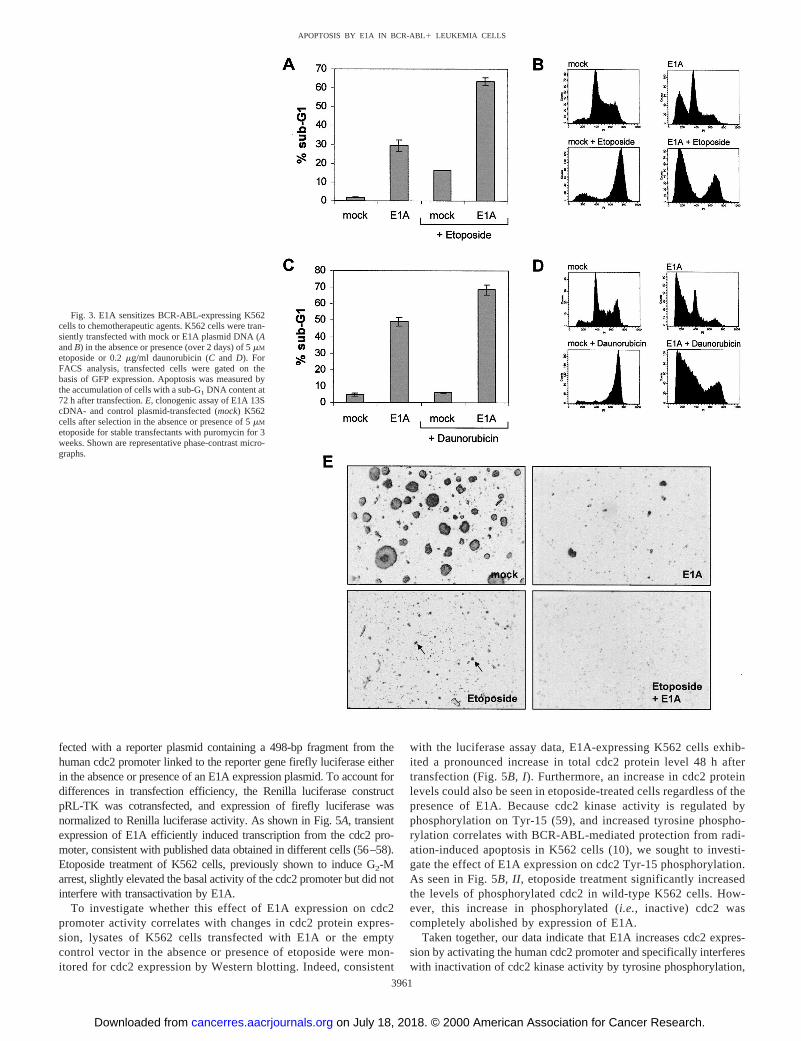
E1A Converts the BCR-ABL-mediated G2 Arrest after Treat-ment with Chemotherapeutic Agents to Induction of Apoptosis.Previous studies have shown that Ph-positive human chronic myeloidleukemia cells such as K562 are particularly resistant to cell death viaapoptosis, irrespective of the inducing agent used (10, 51). BCR-ABLexpression prevents the apoptotic deletion of damaged cells by pro-longation of cell cycle arrest in the G2-M phase (52). The importanceof G2 delay as a critical determinant of radio- or chemosensitivity wasrecently acknowledged by data indicating that abrogation of G2-Marrest in Ph-positive cells with caffeine neutralized the protectiveeffect of BCR-ABL kinase (10). Thus, to achieve maximal eradicationof CML cells in vivo, the use of proteins able to reduce G2-M arrestafter genotoxic damage may be beneficial. Based on our resultsdescribed above, indicating that E1A is a potent inducer of apoptosisin K562 cells, we sought to investigate whether apoptosis induction byE1A is also correlated with increased susceptibility to chemothera-peutic drugs. If G2-M arrest is tightly linked to the apoptotic protec-tion against DNA-damaging agents afforded by BCR-ABL kinaseexpression, this effect should be abolished by E1A. Mock- and E1A13S cDNA-transfected K562 cells were treated by continuous expo-sure with 5mM etoposide and 0.2mg/ml daunorubicin, respectively,over 2 days, and flow cytometry analysis was performed 72 h aftertransfection. Treatment with either drug alone at the particular dose aswell as higher doses (up to 68mM etoposide or 1mg/ml daunorubicin)did not result in the induction of significant amounts of apoptosis,which is consistent with previous reports (20, 53). In contrast, com-pared with E1A-transfected cells in the absence of chemotherapeuticagents, the population of sub-G1 cells significantly increased aftertreatment of E1A-expressing cells with either of these agents (Fig. 3,A–D). At 72 h after transfection;70% of K562 cells treated with E1Aplus etoposide showed a sub-G1 DNA content, compared with only
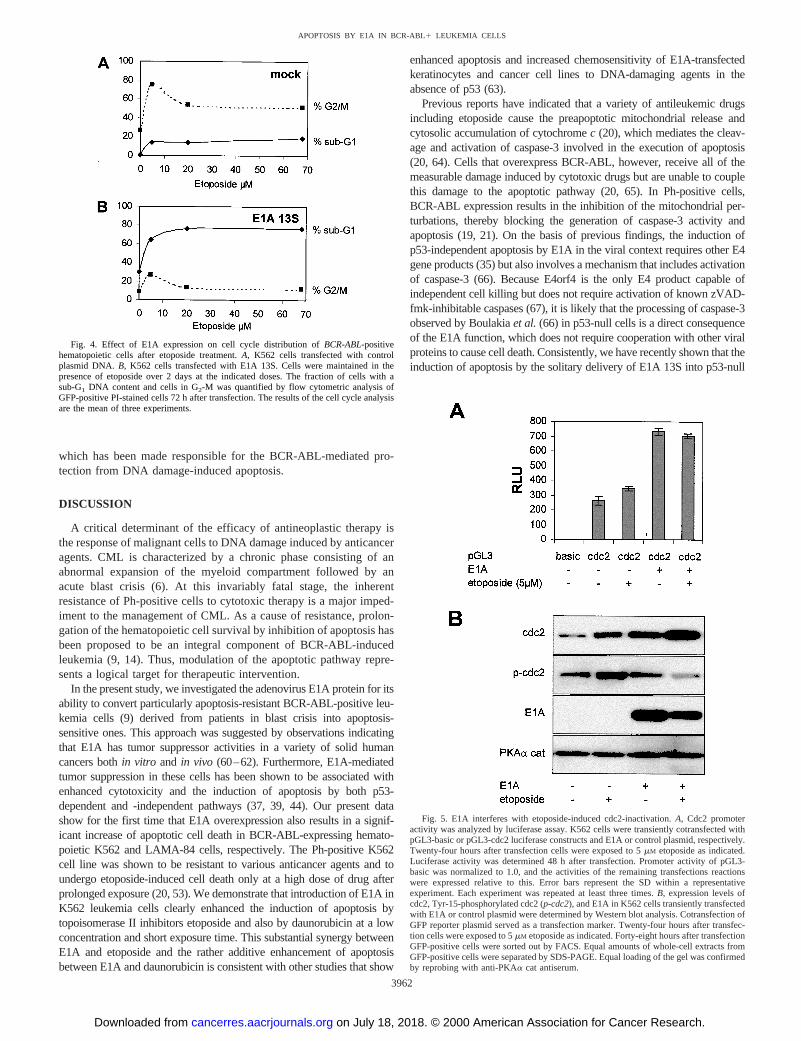
30% (Fig. 3, A and B) of cells introduced with E1A alone. Thissensitization to etoposide by E1A could also be observed in LA-MA-84 leukemia cells (data not shown), indicating that E1A rendersBCR-ABL-positive cells susceptible to apoptotic cell death by che-motherapeutic agents. A less pronounced but significant effect wasapparent in E1A-transfected cells treated with daunorubicin (Fig. 3,Cand D). As shown by Lock and Ross (54), our results also demon-strated that the majority of BCR-ABL kinase-expressing cells arrest inG2-M after etoposide treatment of mock-transfected cells (Figs. 3B,left bottom panel, and 4A). The same effect was evident after treat-ment with daunorubicin (Fig. 3D, left bottom panel). In contrast, indrug-treated BCR-ABL-positive cells, which express E1A, inductionof apoptosis was associated with a marked decrease of cells in G2-M(Fig. 3, B and D, right bottom panel). A maximum in apoptosisinduction by E1A with most of K562 cells in the sub-G1 fraction(76.6%)versusonly a very small fraction of cells arresting in G2-M(13.2%) was already achieved at low etoposide doses between 5 and20 mM (Fig. 4B). Together, these data suggest that temporary expres-sion of E1A is sufficient to convert preexisting resistance to genotoxicdamage by BCR-ABL-mediated cell cycle block to induction ofapoptosis.
To assess the combined effect of E1A expression and treatment withDNA-damaging agents on the long-term proliferative capacity ofBCR-ABL-positive cells, we analyzed clonogenic survival of E1A or mock-transfected cells in the absence or presence of 5mM etoposide (Fig. 3E).Whereas mock transfection gave rise to numerous, highly proliferatingcolonies, the number of viable colonies was significantly reduced by E1Aexpression alone. We note that E1A expressing colonies are variable insize, most likely due to differences in E1A expression levels. The nu-merous colonies surviving etoposide treatment alone were barely visibleand comprised only 10–20 cells on average. This antiproliferative effectof etoposide is consistent with the observed cell cycle arrest at the G2-Mtransition. Importantly, no viable, proliferating colonies could be ob-served on combination treatment with E1A and etoposide, indicating thatonly the combination of E1A expression and etoposide treatment isefficient to completely inhibit clonogenic survival.
E1A Interferes with Etoposide-induced cdc2 Inactivation.Toinvestigate the mechanism of E1A-induced reversal of BCR-ABL-mediated inhibition of DNA damage-induced apoptosis, we firstsought to analyze the effect of E1A expression on the tyrosine kinaseactivity of BCR-ABL. We therefore transfected K562 cells in theabsence or presence of etoposide with an E1A-expressing plasmid andmonitored the pattern of phosphotyrosine proteins by Western blottingusing a phosphotyrosine-specific antibody. There were certainchanges in the phosphorylation pattern with an E1A-induced increaseor decrease in the phosphorylation levels of different protein species(data not shown). However, the changes did not correlate with thedifferences observed when K562 cells were treated with a BCR-ABL-selective tyrosine kinase inhibitor (data not shown), suggesting thatthese changes are not specific for BCR-ABL tyrosine kinase activity.
Although E1A does not seem to directly inhibit BCR-ABL kinaseactivity, E1A might interfere with downstream functions responsible forthe protection from DNA damage-induced apoptosis. G2-M arrest in-duced by various stimuli including irradiation and etoposide has beenshown to be linked to the inhibition of p34cdc2 (cdc2) kinase (54). Inaddition, BCR-ABL-mediated resistance to radiation-induced apoptosiscorrelates with increased tyrosine phosphorylation (i.e., inhibition) ofcdc2 (10). E1A has been shown to induce cdc2 expression and kinaseactivity by transactivation of the human cdc2 promoter (55, 56). ThusE1A might specifically interfere with the BCR-ABL-mediated delay ofG2-M transition after DNA damage by activating cdc2. We thereforeanalyzed the effect of E1A expression on cdc2 promoter activity inBCR/ABL-expressing leukemia cells. K562 cells were transiently trans-
Fig. 2. Apoptosis induction by E1A in BCR-ABL-positive K562 cells involves acti-vation of caspase-3.A, activation of caspase-3 (CPP32) and PARP cleavage in cellstransiently transfected with E1A or control plasmid (mock) was analyzed by Western blot.Equal amounts of whole-cell extracts from GFP-positive cells sorted out by FACS wereseparated by SDS-PAGE. Full-length caspase-3 and the cleaved 17-kDa subunit as wellas PARP (116 kDa) and the 85-kDa proteolytic cleavage product are indicated byarrows.B, the inhibition of E1A-mediated apoptosis by z-VAD-fmk and the caspase-3-specificinhibitor Z-DEVD-fmk (50 mM) was quantitated by flow cytometry. Relative apoptosis72 h after transfection is as indicated. Apoptosis (as determined by cells with a sub-G1
DNA content) was calculated by subtraction of the percentage of apoptotic cells seen inthe population by electroporation alone. Apoptosis in mock-transfected cells (4.226 1%)was set as 1. Thegraph represents the mean of two independent experiments.
3960
APOPTOSIS BY E1A IN BCR-ABL1 LEUKEMIA CELLS
on July 18, 2018. © 2000 American Association for Cancer Research.cancerres.aacrjournals.org Downloaded from
fected with a reporter plasmid containing a 498-bp fragment from thehuman cdc2 promoter linked to the reporter gene firefly luciferase eitherin the absence or presence of an E1A expression plasmid. To account fordifferences in transfection efficiency, the Renilla luciferase constructpRL-TK was cotransfected, and expression of firefly luciferase wasnormalized to Renilla luciferase activity. As shown in Fig. 5A, transientexpression of E1A efficiently induced transcription from the cdc2 pro-moter, consistent with published data obtained in different cells (56–58).Etoposide treatment of K562 cells, previously shown to induce G2-Marrest, slightly elevated the basal activity of the cdc2 promoter but did notinterfere with transactivation by E1A.
To investigate whether this effect of E1A expression on cdc2promoter activity correlates with changes in cdc2 protein expres-sion, lysates of K562 cells transfected with E1A or the emptycontrol vector in the absence or presence of etoposide were mon-itored for cdc2 expression by Western blotting. Indeed, consistent
with the luciferase assay data, E1A-expressing K562 cells exhib-ited a pronounced increase in total cdc2 protein level 48 h aftertransfection (Fig. 5B, I). Furthermore, an increase in cdc2 proteinlevels could also be seen in etoposide-treated cells regardless of thepresence of E1A. Because cdc2 kinase activity is regulated byphosphorylation on Tyr-15 (59), and increased tyrosine phospho-rylation correlates with BCR-ABL-mediated protection from radi-ation-induced apoptosis in K562 cells (10), we sought to investi-gate the effect of E1A expression on cdc2 Tyr-15 phosphorylation.As seen in Fig. 5B, II, etoposide treatment significantly increasedthe levels of phosphorylated cdc2 in wild-type K562 cells. How-ever, this increase in phosphorylated (i.e., inactive) cdc2 wascompletely abolished by expression of E1A.
Taken together, our data indicate that E1A increases cdc2 expres-sion by activating the human cdc2 promoter and specifically interfereswith inactivation of cdc2 kinase activity by tyrosine phosphorylation,
Fig. 3. E1A sensitizes BCR-ABL-expressing K562cells to chemotherapeutic agents. K562 cells were tran-siently transfected with mock or E1A plasmid DNA (AandB) in the absence or presence (over 2 days) of 5mM
etoposide or 0.2mg/ml daunorubicin (Cand D). ForFACS analysis, transfected cells were gated on thebasis of GFP expression. Apoptosis was measured bythe accumulation of cells with a sub-G1 DNA content at72 h after transfection.E, clonogenic assay of E1A 13ScDNA- and control plasmid-transfected (mock) K562cells after selection in the absence or presence of 5mM
etoposide for stable transfectants with puromycin for 3weeks. Shown are representative phase-contrast micro-graphs.
3961
APOPTOSIS BY E1A IN BCR-ABL1 LEUKEMIA CELLS
on July 18, 2018. © 2000 American Association for Cancer Research.cancerres.aacrjournals.org Downloaded from
which has been made responsible for the BCR-ABL-mediated pro-tection from DNA damage-induced apoptosis.
DISCUSSION
A critical determinant of the efficacy of antineoplastic therapy isthe response of malignant cells to DNA damage induced by anticanceragents. CML is characterized by a chronic phase consisting of anabnormal expansion of the myeloid compartment followed by anacute blast crisis (6). At this invariably fatal stage, the inherentresistance of Ph-positive cells to cytotoxic therapy is a major imped-iment to the management of CML. As a cause of resistance, prolon-gation of the hematopoietic cell survival by inhibition of apoptosis hasbeen proposed to be an integral component of BCR-ABL-inducedleukemia (9, 14). Thus, modulation of the apoptotic pathway repre-sents a logical target for therapeutic intervention.
In the present study, we investigated the adenovirus E1A protein for itsability to convert particularly apoptosis-resistant BCR-ABL-positive leu-kemia cells (9) derived from patients in blast crisis into apoptosis-sensitive ones. This approach was suggested by observations indicatingthat E1A has tumor suppressor activities in a variety of solid humancancers bothin vitro and in vivo (60–62). Furthermore, E1A-mediatedtumor suppression in these cells has been shown to be associated withenhanced cytotoxicity and the induction of apoptosis by both p53-dependent and -independent pathways (37, 39, 44). Our present datashow for the first time that E1A overexpression also results in a signif-icant increase of apoptotic cell death in BCR-ABL-expressing hemato-poietic K562 and LAMA-84 cells, respectively. The Ph-positive K562cell line was shown to be resistant to various anticancer agents and toundergo etoposide-induced cell death only at a high dose of drug afterprolonged exposure (20, 53). We demonstrate that introduction of E1A inK562 leukemia cells clearly enhanced the induction of apoptosis bytopoisomerase II inhibitors etoposide and also by daunorubicin at a lowconcentration and short exposure time. This substantial synergy betweenE1A and etoposide and the rather additive enhancement of apoptosisbetween E1A and daunorubicin is consistent with other studies that show
enhanced apoptosis and increased chemosensitivity of E1A-transfectedkeratinocytes and cancer cell lines to DNA-damaging agents in theabsence of p53 (63).
Previous reports have indicated that a variety of antileukemic drugsincluding etoposide cause the preapoptotic mitochondrial release andcytosolic accumulation of cytochromec (20), which mediates the cleav-age and activation of caspase-3 involved in the execution of apoptosis(20, 64). Cells that overexpress BCR-ABL, however, receive all of themeasurable damage induced by cytotoxic drugs but are unable to couplethis damage to the apoptotic pathway (20, 65). In Ph-positive cells,BCR-ABL expression results in the inhibition of the mitochondrial per-turbations, thereby blocking the generation of caspase-3 activity andapoptosis (19, 21). On the basis of previous findings, the induction ofp53-independent apoptosis by E1A in the viral context requires other E4gene products (35) but also involves a mechanism that includes activationof caspase-3 (66). Because E4orf4 is the only E4 product capable ofindependent cell killing but does not require activation of known zVAD-fmk-inhibitable caspases (67), it is likely that the processing of caspase-3observed by Boulakiaet al.(66) in p53-null cells is a direct consequenceof the E1A function, which does not require cooperation with other viralproteins to cause cell death. Consistently, we have recently shown that theinduction of apoptosis by the solitary delivery of E1A 13S into p53-null
Fig. 4. Effect of E1A expression on cell cycle distribution ofBCR-ABL-positivehematopoietic cells after etoposide treatment.A, K562 cells transfected with controlplasmid DNA. B, K562 cells transfected with E1A 13S. Cells were maintained in thepresence of etoposide over 2 days at the indicated doses. The fraction of cells with asub-G1 DNA content and cells in G2-M was quantified by flow cytometric analysis ofGFP-positive PI-stained cells 72 h after transfection. The results of the cell cycle analysisare the mean of three experiments.
Fig. 5. E1A interferes with etoposide-induced cdc2-inactivation.A, Cdc2 promoteractivity was analyzed by luciferase assay. K562 cells were transiently cotransfected withpGL3-basic or pGL3-cdc2 luciferase constructs and E1A or control plasmid, respectively.Twenty-four hours after transfection cells were exposed to 5mM etoposide as indicated.Luciferase activity was determined 48 h after transfection. Promoter activity of pGL3-basic was normalized to 1.0, and the activities of the remaining transfections reactionswere expressed relative to this. Error bars represent the SD within a representativeexperiment. Each experiment was repeated at least three times.B, expression levels ofcdc2, Tyr-15-phosphorylated cdc2 (p-cdc2), and E1A in K562 cells transiently transfectedwith E1A or control plasmid were determined by Western blot analysis. Cotransfection ofGFP reporter plasmid served as a transfection marker. Twenty-four hours after transfec-tion cells were exposed to 5mM etoposide as indicated. Forty-eight hours after transfectionGFP-positive cells were sorted out by FACS. Equal amounts of whole-cell extracts fromGFP-positive cells were separated by SDS-PAGE. Equal loading of the gel was confirmedby reprobing with anti-PKAa cat antiserum.
3962
APOPTOSIS BY E1A IN BCR-ABL1 LEUKEMIA CELLS
on July 18, 2018. © 2000 American Association for Cancer Research.cancerres.aacrjournals.org Downloaded from

human tumor cells can be linked to caspase activity (37). Our present datademonstrate that the apoptotic activity of E1A in hematopoietic K562cells is accompanied by processing of caspase-3, and cleavage of PARPand can be significantly blocked by z-VAD-fmk and the caspase-3-specific inhibitor Z-DEVD-fmk. Thus, transient expression of the E1Aprotein is apparently sufficient to antagonize the BCR-ABL-inducedblock in the apoptosis signaling pathway triggered by DNA-damagingagents.
The delay in the apoptosis signaling cascade in BCR-ABL-express-ing K562 cells treated with etoposide is known to be associated withthe induction of cell cycle arrest in the G2-M phase, shown in thepresent paper and by Lock and Ross (54), which leads to apoptosisprotection. In contrast, etoposide treatment of E1A-expressing K562cells correlates with the inhibition of G2-M arrest and substantiallyincreased chemosensitivity, resulting in increased apoptosis ofBCR-ABL-positive cells and significant reduction of long-term clonogenicsurvival. As our data indicate, E1A does not seem to interfere directlywith the tyrosine kinase activity of BCR-ABL, because the changesobserved in the pattern of phosphotyrosine proteins do not correlatewith the changes induced by a BCR-ABL-selective tyrosine kinaseinhibitor. However, E1A might interfere with downstream functionsresponsible for the protection from DNA damage-induced apoptosis.G2-M arrest induced by various stimuli including etoposide has beenshown to be linked to the inhibition of p34cdc2 (cdc2) kinase (54). Inmammalian cells, G2-M transition is controlled by cdc2 kinase activ-ity (68), which is normally regulated by phosphorylation of cdc2protein by the inhibitory kinase Wee1 and dephosphorylation by theactivating phosphatase cdc25C. In some systems, increased phospho-rylation of cdc2 in cells expressing Wee1 kinase occurred in associ-ation with protection from apoptosis (69). On the other hand, induc-tion of apoptosis is associated with premature activation (bydephosphorylation) of cdc2 kinase (70). Previous studies have re-vealed that the human cdc2 promoter is transcriptionally activated byE1A proteins in cycling cells, which is mediated through two CCAATbox binding motifs (56–58). With regard to this aspect, we detectedincreased cdc2 promoter activity and cdc2 protein levels in E1A-transfected K562 cells irrespective of the presence of etoposide. ThusE1A-induced up-regulation of cdc2 expression is apparently respon-sible for overriding the etoposide-associated inhibition of cdc2 kinase.In addition, etoposide-induced Tyr-15 phosphorylation (i.e., inactiva-tion) of cdc2 was completely abolished in the presence of E1A. Thismay result at least in part from a rapid cleavage of Wee1 (71), whichhas been shown to be a substrate of the caspase-3-like protease duringFas-induced apoptosis (71). On the other hand, inhibition of etoposideinduced cdc2 phosphorylation may also be due to activation of thephosphatase cdc25C. Indeed, CCAAT box motifs responsible for theE1A-mediated transactivation of the cdc2 promoter are also present inthe cdc25C promoter, and nuclear extracts from human E1A-immor-talized 293 cells bind the CCAAT elements of the cdc25C promoterto form specific DNA-protein complexes (57). Together, these datastrongly suggest that E1A overrides BCR-ABL-mediated DNA dam-age induced G2-M arrest by inhibition of cdc2 inactivation as apotential mechanism for E1A-induced cell death in CML.
The ability of E1A gene products to induce apoptosis in the presenceof p53 is well established (32). E1A interactions with RB and p300 havebeen linked to perturbation of cell cycle and apoptosis induction (34, 72)resulting in up-regulation of the ARF gene product (73) and stabilizationof p53. In this context, ectopic expression of E2F1 as a critical down-stream effector of RB, and E1A activity directly correlates with increasedexpression of ARF (74). Our results in CML cells and those of otherstudies in cancer cells from solid tumors demonstrate that enhancedapoptosis and chemosensitivity by E1A can be unrelated to the amount ofp53 present (63). Investigation of E1A targets in the cell cycle regulatory
pathway revealed that E1A-induced apoptosis was preceded not only bya rise in p53 but also by a precipitous drop in topoisomerase IIa,suggesting that E1A can activate or induce components responsible fordegrading topoisomerase IIa (75). Previous findings demonstrated thatenforced E2F1 expression in myeloid progenitor cells confers preferentialsensitivity to p53-independent apoptosis mediated by the topoisomeraseII inhibitors etoposide and doxorubicin (76, 77). On the basis of those andour data, we can speculate that E2F1 may be a potential target inE1A-expressing CML cells to sensitize for chemotherapeutic agents suchas etoposide and daunorubicin to undergo p53-independent apoptosis.
Even when it is likely that multiple pathways are involved in theregulation of cell growth and apoptosis and also the response tocytotoxic agents in BCR-ABL-positive tumors, by targeting the finalcommon pathway we may be able to create a potent therapeuticstrategy to improve survival of CML patients. From our data, E1A-based therapy may represent a potent new approach by itself and whenadministered in conjunction with conventional cytotoxic drugs for thetreatment of CML in the final stage of blast crisis.
ACKNOWLEDGMENTS
We thank S. Zimmermann and B. Pollmeier for technical assistance and K.Lennarz for support in flow cytometry analysis.
REFERENCES
1. Champlin, R. E., and Golde, D. Chronic myelogenous leukemia: recent advances.Blood, 65: 1039–1047, 1981.
2. Sawyers, C. L., Denny, C. T., and Witte, O. N. Leukemia and the disruption of normalhematopoiesis. Cell,64: 337–350, 1991.
3. Pendergast, A. M., Quilliam, L. A., Cripe, L. D., Bassing, C. H., Dai, Z., Li, N.,Batzer, A., Rabun, K. M., Der, C. J., and Schlessinger, J. BCR-Able-induced onco-genesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptorprotein. Cell,75: 175–185, 1993.
4. Skorski, T., Kanakaraj, P., Nieborowska-Skorska, M., Ratajczak, M. Z., Wen, S. C.,Zon, G., Gewirtz, A. M., Perussia, B., and Calabretta, B. Phosphatidylinositol-3kinase activity is regulated by BCR-ABL and is required for the growth of Philadel-phia chromosome-positive cells. Blood,86: 726–736, 1995.
5. Koeffler, P. H., and Golde, D. Chronic myelogenous leukemia-new concepts.N. Engl. J. Med.,319: 1201–1209, 1981.
6. Strife, A., and Clarkson, B. Biology of chronic myelogenous leukemia: is discordantmaturation the primary effect. Semin. Hematol.,25: 1–19, 1988.
7. Cotter, T. G. Bcr-Abl. An anti-apoptosis gene in chronic myelogenous leukemia.Leuk. Lymphoma,18: 231–236, 1995.
8. Samali, A., Gorman, A. M., and Cotter, T. G. Apoptosis—the story so far. Experi-entia,52: 933–941, 1996.
9. McGahon, A., Bissonnette, R., Schmitt, M., Cotter, K. M., Green, D. R., and Cotter,T. G. Bcr-abl maintains resistance of chronic myelogenous leukemia cells to apoptoticcell death. Blood,83: 1179–1187, 1994.
10. Nishii, K., Kabarowski, J. H. S., Gibbons, D. L., Griffiths, S. D., Titley, I., Wiedemann,L. M., and Greaves, M. F. ts BCR-ABL kinase activation confers increased resistance togenotoxic damage via cell cycle block. Oncogene,13: 2225–2234, 1996.
11. Evans, C. A., Owen-Lynch, P. J., Whetton, A. D., and Dive, C. Activation of theAbelson tyrosine kinase activity is associated with suppression of apoptosis inhemopoietic cells. Cancer Res.,53: 1735–1738, 1993.
12. Cambier, N., Chopra, R., Strasser, A., Metcalf, D., and Elefanty, A. G. Bcr-abl activatespathways mediating cytokine independence and protection against apoptosis in murinehematopoietic cells in a dose-dependent manner. Oncogene,16: 335–348, 1998.
13. McGahon, A., Nishioka, W. K., Martin, S. J., Mahboubi, A., Cotter, T. G., and Green,D. R. Regulation of the Fas apoptotic cell death pathway by abl. J. Biol. Chem.,270:22625–22631, 1995.
14. Bedi, A., Zehnbauer, B. A., Barber, J. P., Sharkis, S. J., and Jones, R. J. Inhibition ofapoptosis by bcr-abl in chronic myeloid leukemia. Blood,83: 2038–2044, 1994.
15. Dan, S., Naito, M., and Tsuruo, T. Selective induction of apoptosis in Philadelphiachromosome-positive chronic myelogenous leukemia cells by an inhibitor of bcr-abltyrosine kinase, CGP 57148. Cell Death Differ.,5: 710–715, 1998.
16. Selleri, C., Maciejewski, J. P., Pane, F., Luciano, L., Raiola, A. M., Mostarda, I.,Salvatore, F., and Rotoli, B. Fas-mediated modulation of bcr-abl in chronic myelog-enous leukemia results in differential effects on apoptosis. Blood,92: 981–989, 1998.
17. Kerr, J. F., Winterford, C. M., and Harmon, B. V. Apoptosis. Its significance in cancerand cancer therapy. Cancer (Phila.),73: 2013–2026, 1994.
18. Ibrado, A. M., Huang, Y., Fang, G., and Bhalla, K. Bcl-xL overexpression inhibitsTaxol-induced Yama protease activity and apoptosis. Cell Growth Differ.,7: 1087–1094, 1996.
19. Dubrez, L., Eymin, B., Sordet, O., Droin, N., Turhan, A. G., and Solary, E. BCR-ABLdelays apoptosis upstream of procaspase-3 activation. Blood,91: 2415–2422, 1998.
3963
APOPTOSIS BY E1A IN BCR-ABL1 LEUKEMIA CELLS
on July 18, 2018. © 2000 American Association for Cancer Research.cancerres.aacrjournals.org Downloaded from

20. Martins, L. M., Mesner, P. W., Kottke, T. J., Basi, G. S., Sinha, S., Tung, J. S., Svingen,P. A., Madden, B. J., Takahashi, A., McCormick, D. J., Earnshaw, W. C., and Kaufmann,S. H. Comparison of caspase activation and subcellular localization in HL-60 and K562cells undergoing etoposide-induced apoptosis. Blood,90: 4283–4296, 1997.
21. Amarante-Mendes, G. P., Kim, C. N., Liu, L., Huang, Y., Perkins, C. L., Green, D. R.,and Bhalla, K. Bcr-Abl exerts its antiapoptotic effect against diverse apoptotic stimulithrough blockage of mitochondrial release of cytochrome c and activation ofcaspase-3. Bood,91: 1700–1705, 1998.
22. Samali, A., Gorman, A. M., and Cotter, T. G. Role of bcr-abl kinase in resistance toapoptosis. Adv. Pharmacol.,41: 533–552, 1997.
23. McKenna, S. L., and Cotter, T. G. Functional aspects of apoptosis in hematopoiesisand consequences of failure. Adv. Cancer Res.,71: 121–164, 1997.
24. Whyte, P., Ruley, H. E., and Harlow, E. Two regions of the adenovirus early region1A proteins are required for transformation. J. Virol.,62: 257–265, 1988.
25. Rao, I., Debbas, M., Sabbatini, P., Hockenbery, D., Korsmeyer, S., and White, E. Theadenovirus E1A proteins induce apoptosis, which is inhibited by the E1B 19.kDa andBcl-2 proteins. Proc. Natl. Acad. Sci. USA,89: 7742–7746, 1992.
26. White, E., Chiou, S. K., Rao, L., Sabbatini, P., and Lin, H. J. Control of p53-dependent apoptosis by E1B, Bcl-2. Ha-ras proteins. Cold Spring Harb. Symp. Quant.Biol., 59: 395–402, 1994.
27. Hsieh, J. K., Fredersdorf, S., Kouzarides, T., Martin, K., and Lu, X. E2F1-inducedapoptosis requires DNA binding but not transactivation and is inhibited by theretinoblastoma protein through direct interaction. Genes Dev.,11: 1840–1852, 1997.
28. Qin, X. Q., Livingston, D. M., Kaelin, W. G., and Adams, P. D. Deregulated E2F-1expression leads to S-phase entry and p53-mediated apoptosis. Proc. Natl. Acad. Sci.USA, 91: 10918–10922, 1994.
29. Fueyo, J., Gomez-Manzano, C., Yung, W. K. A., Liu, T. J., Alemany, R., McDonnell,T. J., Shi, X., Rao, J. S., Levin, V. A., and Kyritsis, A. P. Overexpression of E2F-1in glioma triggers apoptosis and suppresses tumor growth in vitro and in vivo. Nat.Med., 4: 685–690, 1998.
30. White, E. Regulation of apoptosis by the transforming genes of the DNA tumor virusadenovirus. Proc. Soc. Exp. Biol. Med.,204: 30–39, 1993.
31. Lowe,S. W., and Ruley, H. E. Stabilization of the p53 tumor suppressor is inducedby adenovirus-5 E1A and accompanies apoptosis. Genes Dev.,7: 535–545, 1993.
32. Debbas, M., and White, E. Wild-type p53 mediates apoptosis by E1A, which isinhibited by E1B. Genes Dev.,7: 546–554, 1993.
33. Teodoro, J. G., Shore, G. C., and Branton, P. E. Adenovirus E1A proteins induceapoptosis by both p53-dependent and p53-independent mechanisms. Oncogene,11:467–474, 1995.
34. Querido, E., Teodoro, J. G., and Branton, P. E. Accumulation of p53 induced by theadenovirus E1A protein requires regions involved in the stimulation of DNA synthe-sis. J. Virol.,71: 3526–3533, 1997.
35. Marcellus, R. C., Teodoro, J. G., Wu, T., Brough, D. E., Ketner, G., Shore, G. C., andBranton, P. E. Adenovirus type 5 early region 4 is responsible for E1A-inducedp53-independent apoptosis. J. Virol.,70: 6207–6215, 1996.
36. Marcellus, R. C., Lavoie, J. N., Boivin, D., Shore, G. C., Ketner, G., and Branton,P. E. The early region 4 orf4 protein of human adenovirus type 5 induces p53-independent cell death by apoptosis. J. Virol.,72: 7144–7153, 1998.
37. Putzer, B. M., Stiewe, T., Parssanedjad, K., Rega, S., and Esche, H. E1A is sufficientby itself to induce apoptosis independent of p53 and other adenoviral gene products.Cell Death Differ.,7: 177–188, 2000.
38. Sanchez-Prieto, R., Lleonart, M., and Ramon y Cajal, S. Lack of correlation betweenp53 protein level and sensitivity to DNA-damaging agents in keratinocytes carryingadenovirus E1A mutants. Oncogene,11: 675–682, 1995.
39. Brader, K. R., Wolf, J. K., Hung, M. C., Yu, D., Crispens, M. A., van Golen, K. L.,and Price, J. E. Adenovirus E1A expression enhances the sensitivity of an ovariancancer cell line to multiple cytotoxic agents trough an apoptotic mechanism. Clin.Cancer Res.,3: 2017–2024, 1997.
40. Van Denderen, J., Hermans, A., Meeuwsen, T., Troelstra, C., Zegers, N., Borsma, W.,Grosveld, G., and Van Ewijk, W. Antibody recognition of the tumor-specific bcr-abljoining region in chronic myeloid leukemia. J. Exp. Med.,169: 87–98, 1989.
41. van den Hoff, M. J. B., Moorman, A. F. M., and Lamers, W. H. Electroporationintracellular buffer increases cell survival. Nucleic Acids Res.,20: 2902, 1992.
42. Yu, D., Wolf, J. K., Scalon, M., Price, J. E., and Hung, M. C. Enhanced c-erbB-2/neuexpression in human ovarian cancer cells correlates with more severe malignancy thatcan be suppressed by E1A. Cancer Res.,53: 891–898, 1993.
43. Kalejta, R. F., Shenk, T., and Beavis, A. J. Use of a membrane-localized greenfluorescent protein allows simultaneous identification of transfected cells and cellcycle analysis by flow cytometry. Cytometry,29: 286–291, 1997.
44. Deng, J., Xia, W., and Hung, M. C. Adenovirus 5 E1A-mediated tumor suppressorassociated with E1A-mediated apoptosis in vivo. Oncogene,17: 2167–2175, 1998.
45. Rowan, S., Ludwig, R. L., Haupt, Y., Bates, S., Lu, X., Oren, M., and Vousden, K. H.Specific loss of apoptotic but not cell cycle arrest function in a human tumour derivedp53 mutant. EMBO J.,15: 827–838, 1996.
46. Lubbert, M., Miller, C. W., Crawford, K., and Koeffler, H. P. p53 in chronicmyelogenous leukemia. J. Exp. Med.,167: 873–886, 1988.
47. Law, J. C., Ritke, M. K., Yalowich, J. C., Leder, G. H., and Ferrel, R. E. Mutationalinactivation of the p53 gene in the human erythroid leukemic K562 cell line.Leukemia Res.,17: 1045–1050, 1993.
48. Casciola, R. L., Nicholson, D. W., Chong, T., Rowan, K. R., Thornberry, N. A., Miller,D. K., and Rosen, A. Apopain/CPP32 cleaves protein that are essential for cellular repair:a fundamental principle of apoptotic death. J. Exp. Med.,183: 1957–1964, 1964.
49. Liu, X., Zou, H., Slaughter, C., and Wang, X. DFF, a heterodimeric protein thatfunctions downstream of caspase-3 to trigger DNA fragmentation during apoptosis.Cell, 89: 175–184, 1997.
50. Miller, D. K. The role of the caspase family of cysteine proteases in apoptosis. Semin.Immunol.,9: 35–49, 1997.
51. Martin, S. J., Lennon, S. V., Bonham, A. M., and Cotter, T. G. Induction of apoptosis(programmed cell death) in human leukemic HL-60 cells by inhibition of RNA andprotein synthesis. J. Immunol.,145: 1859–1867, 1990.
52. Bedi, A., Barber, J. P., Bedi, G. C., el-Deiry, W. S., Sidransky, D., Vala, M. S.,Akhtar, A. J., Hilton, J., and Jones, R. J. Bcr-Abl-mediated inhibition of apoptosiswith delay of G2/M transition after DNA damage: a mechanism of resistance tomultiple anticancer agents. Blood,86: 1148–1158, 1995.
53. Riordan, F. A., Bravery, C. A., Mengubas, K., Ray, N., Borthwick, N. J., Akbar,A. N., Hart, S. M., Hoffbrand, A. V., Mehta, A. B., and Wickremasinghe, R. G.Herbimycin A accelerates the induction of apoptosis following etoposide treatment org-irradiation of bcr/abl-positive leukaemia cells. Oncogene,16: 1533–1542, 1998.
54. Lock, R. B., and Ross, W. E. Inhibition of p34cdc2 kinase activity by etoposide orirradiation as a mechanism of G2 arrest in Chinese hamster ovary cells. Cancer Res.,50: 3761–3771, 1990.
55. Wang, H. G., Draetta, G., and Moran, E. E1A induces phosphorylation of theretinoblastoma protein independently of direct physical association between the E1Aand retinoblastoma products. Mol. Cell. Biol.,11: 4253–4265, 1991.
56. Kao, C.-Y., Tanimoto, A., Arima, N., Sasaguri, Y., and Padmanabhan, R. Transac-tivation of the human cdc2 promoter by adenovirus E1A. J. Biol. Chem.,274:23043–23051, 1999.
57. Tanimoto, A., Kao, C.-Y., Chang, C.-C., Sasaguri, Y., and Padmanabhan, R. Deregulationof cdc2 gene expression correlates with overexpression of a 110 kDa CCAAT boxbinding factor in transformed cells. Carcinogenesis (Lond.),19: 1735–1741, 1998.
58. Tanimoto, A., Chen, H., Kao, C. Y., Moran, E., Sasaguri, Y., and Padmanabhan, R.Transactivation of the human cdc2 promoter by adenovirus E1A in cycling cells ismediated by the induction of a 110-kDa CCAAT-box-binding factor. Oncogene,17:3103–3114, 1998.
59. Gould, K. L., Moreno, S., Tonks, N. K., and Nurse, P. Complementation of themitotic activator, p80cdc25, by a human protein-tyrosine phosphatase. Science(Washington DC),250: 1573–1576, 1990.
60. Frisch, S. M. Antioncogenic effect of adenovirus E1A in human tumor cells. Proc.Natl. Acad. Sci. USA,88: 9077–9081, 1991.
61. Chang, J. Y., Xia, W., Shao, R., and Hung, M. C. Inhibition of intratracheal lungcancer development by systemic delivery of E1A. Oncogene,13: 1405–1412, 1996.
62. Chang, J. Y., Xia, W., Shao, R., Sorgi, F., Hortobagyi, G. N., Huang, L., and Hung,M. C. The tumor suppression activity of E1A in HER-2/neu-overexpressing breastcancer. Oncogene,14: 561–568, 1997.
63. Sanchez-Prieto, R., Quintanilla, M., Cano, A. R., Lleonart, M., Martin, P., Anaya, A.,and Cajal, S. R. Carcinoma cell lines become sensitive to DNA-damaging agents bythe expression of the adenovirus E1A gene. Oncogene,13: 1083–1092, 1996.
64. Kim, C. N., Wang, X., Huang, Y., Ibrado, A. M., Liu, L., Fang, G., and Bhalla, K.Overexpression of Bcl-xL inhibits Ara-C-induced mitochondrial loss of cytochrome cand other perturbations that activate the molecular cascade of apoptosis. Cancer Res.,57: 3115–3120, 1997.
65. Dubrez, L., Goldwasser, F., Genne, P., Pommier, Y., and Solary, E. The role of cellcycle regulation and apoptosis triggering in determining the sensitivity of leukemiccells to topoisomerase I and II inhibitors. Leukemia,9: 1013–1024, 1995.
66. Boulakia, C. A., Chen, G., Ng, F. W., Teodoro, J. G., Branton, P. E., Nicholson,D. W., Poirier, G. G., and Shore, G. C. Bcl-2 and adenovirus 19KDA protein preventE1A-induced processing of CPP32 and cleavage of poly(ADP-ribose) polymerase.Oncogene,12: 529–535, 1996.
67. Lavoie, J. N., Nguyen, M., Marcellus, R. C., Branton, P. E., and Shore, G. C. E4orf4,a novel adenovirus death factor that induces p53-independent apoptosis by a pathwaythat is not inhibited by zVAD-fmk. J. Cell Biol.,140: 637–645, 1997.
68. Fang, F., and Newport, J. W. Evidence that the G1-S G2-M transitions are controlledby different cdc2 proteins in higher eukaryotes. Cell,66: 731–742, 1991.
69. Chen, G., Shi, L., Litchfield, D. W., and Greenberg, A. H. Rescue from granzymeB-induced apoptosis by Wee1 kinase. J. Exp. Med.,181: 2295–2300, 1995.
70. Shi, L., Nishioka, W. K., Th’ng, J., Bradbury, E. M., Litchfield, D. W., and Greeberg,A. H. Premature p34cdc2 activation required for apoptosis. Science (WashingtonDC), 263: 1143–1145, 1994.
71. Zhou, B. B., Li, H., Yuan, J., and Kirschner, M. W. Caspase-dependent activation ofcyclin-dependent kinases during Fas-induced apoptosis in Jurkat cells. Proc. Natl.Acad. Sci. USA,95: 6785–6790, 1998.
72. Mymryk, J. S., Shire, K., and Bayley, S. T. Induction of apoptosis by adenovirus type5 E1A in rat cells requires a proliferation block. Oncogene,9: 1187–1193, 1994.
73. deStanchina, E., McCurrach, M. E., Zindy, F., Shieh, S. Y., Ferbeyre, G., Samuelson,A. V., Prives, C., Roussel, M. F., Sherr, C. J., and Lowe, S. W. E1A signaling to p53involves the p19ARF tumor suppressor. Genes Dev.,12: 2434–2442, 1998.
74. Bates, S., Phillips, A. C., Clarke, P., Scott, F., Peters, G., Ludwig, R. L., and Vousden,K. H. p14ARF links the tumor suppressors RB and p53. Nature (Lond.),395:124–125, 1998.
75. Nakajima, T., Morita, K., Ohi, N., Arai, T., Nozaki, N., Kikuchi, A., Osaka, F.,Yamao, F., and Oda, K. Degradation of topoisomerase IIa during adenovirus E1A-induced apoptosis is mediated by the activation of the ubiquitin proteolysis system.J. Biol. Chem.,271: 24842–24849, 1996.
76. Nip, J., Strom, D. K., Fee, B. E., Zambetti, G., Cleveland, J. L., and Hiebert, S. W.E2F-1 cooperates with topoisomerase II inhibition and DNA damage to selectivelyaugment p53-independent apoptosis. Mol. Cell. Biol.,17: 1049–1056, 1997.
77. Meng, R. D., Phillips, P., and El-Deiry, W. S. p53-independent increase in E2F-1expression enhances the cytotoxic effects of etoposide and of Adriamycin. Int. J.Oncol.,14: 5–14, 1999.
3964
APOPTOSIS BY E1A IN BCR-ABL1 LEUKEMIA CELLS
on July 18, 2018. © 2000 American Association for Cancer Research.cancerres.aacrjournals.org Downloaded from

2000;60:3957-3964. Cancer Res Thorsten Stiewe, Keiarasch Parssanedjad, Helmut Esche, et al. by Chemotherapeutic AgentsCells and Renders Cells Susceptible to Induction of Apoptosis E1A Overcomes the Apoptosis Block in BCR-ABL+ Leukemia
Updated version
http://cancerres.aacrjournals.org/content/60/14/3957
Access the most recent version of this article at:
Cited articles
http://cancerres.aacrjournals.org/content/60/14/3957.full#ref-list-1
This article cites 72 articles, 37 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/60/14/3957.full#related-urls
This article has been cited by 9 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/60/14/3957To request permission to re-use all or part of this article, use this link
on July 18, 2018. © 2000 American Association for Cancer Research.cancerres.aacrjournals.org Downloaded from





























