Embed Size (px)
Citation preview

Letter to the Editor
E-Cadherin Unlikely to Be a Common “LowPenetrance” Gene for Colorectal Cancer
To the Editor:
E-cadherin plays a key role in intercellular commu-nication for the control of cell growth, and loss of thisfunction may be associated with the evolution of ma-lignant change in epithelial tissues [Jawhari et al.,1997; Perl et al., 1998]. E-cadherin protein expressionis reduced in some colorectal cancers (CRC) [Agrez,1996], but apparently not as a consequence of alleleloss [Ilyas et al., 1997]. However, there is evidence forsomatic mutation in E-cadherin in another gastrointes-tinal cancer, namely sporadic gastric cancer [Becker etal., 1994; Oda et al., 1994]. Germline mutation at theE-cadherin locus has recently been identified as thebasis of familial dominantly inherited gastric cancer inthree New Zealand Maori families, albeit colorectalcancer was not a particular observation [Guilford et al.,1998]. E-cadherin mutations have been identified insporadic infiltrative lobular breast cancer [Berx et al.,1995] and in ovarian and endometrial cancer [Risingeret al., 1994]. Some similarity in the carcinogenetic pro-cesses of upper and lower gastrointestinal malignancyis suggested by the finding of somatic APC (adenoma-tous polyposis coli) mutations in a small number ofsporadic gastric cancers [Horii et al., 1992], and theintimate functional relationship that the APC proteinhas with E-cadherin [Jawhari et al., 1997] is also to benoted. Given these several observations, it is reason-able to postulate a possible role for E-cadherin in thegenesis of CRC, notwithstanding the negative findingsof Ilyas et al. noted above, and the lack of obvious as-sociation with CRC in the New Zealand E-cadheringastric cancer families. It is plausible that acquiredmutations could be contributory in some sporadic CRC,or that germline mutations could have a part to play insome nonmismatch repair hereditary nonpolyposis co-lorectal cancer (HNPCC) families.
Most familial CRC presents with a family historythat falls far short of typical dominant inheritance. Adominant gene could, nevertheless, be involved; but
such a gene would, presumably, have a low penetrance.The concept of low penetrance CRC genes has cur-rency; what remains is for the concept to achieve actu-ality with molecular proof that mutations at one ormore loci cosegregate with familial CRC. We havesought to determine whether E-cadherin might be alow penetrance CRC-predisposing gene.
MATERIALS AND METHODS
Cases were recruited through the Familial BowelCancer Service at The Royal Melbourne Hospital. Mostindividuals registered with the service have not them-selves had CRC, but came with a concern due to a rela-tive or relatives having had a diagnosis of CRC. Weidentified a subgroup who had a family history of CRCat age 50 or younger in a single first-degree relative, orof CRC in two or more first- or second-degree relativesat any age, with the (or an) affected member still living.Families with definite or suspected HNPCC were notincluded. The subject was contacted by letter explain-ing the basis of the study, and those willing to partici-pate were then sent a second letter which they wereasked to pass on to their affected relative. The affectedrelatives who were, in turn, willing to participate thensaw their own doctor, or a convenient pathology labo-ratory, and presented our letter giving instructions forpreparing a Guthrie card blood sample and a stampedaddressed envelope for return of the card. In mostcases, these diagnoses of CRC had been confirmed byour service through contact with the appropriate medi-cal authority.
DNA was extracted from the Guthrie cards, andSSCP mutation analysis done as described in Berx etal. [1995]. All 16 exons of the gene were screened. Thepolymerase chain reaction (PCR) products were elec-trophoresed at room temperature through a 6% nonde-naturing polyacrylamide gel without added glycerol.Products were detected by autoradiography. If anSSCP bandshift was observed, the Guthrie card DNAwas re-PCR amplified using the SSCP primers with a58 leader corresponding to the T3 sequencing primeradded to the sense primer. Direct sequencing was thencarried out using Thermosequenase (Amersham,Cleveland, OH) and an IR800 labelled (MWG Biotech,Ebersberg, Germany) T3 primer (3 pmoles/reaction).The products were analysed on a LiCor 4000L DNAsequencer.
Contract grant sponsor: New Zealand Health Research Coun-cil; Contract grant sponsor: Murdoch Institute.
*Correspondence to: Dr. R.J.M. Gardner, Victorian Clinical Ge-netics Services, Royal Children’s Hospital, Parkville, Melbourne,Vic. 3052, Australia.
Received 24 August 1998; Accepted 13 January 1999
American Journal of Medical Genetics 84:169–171 (1999)
© 1999 Wiley-Liss, Inc.
RESULTS
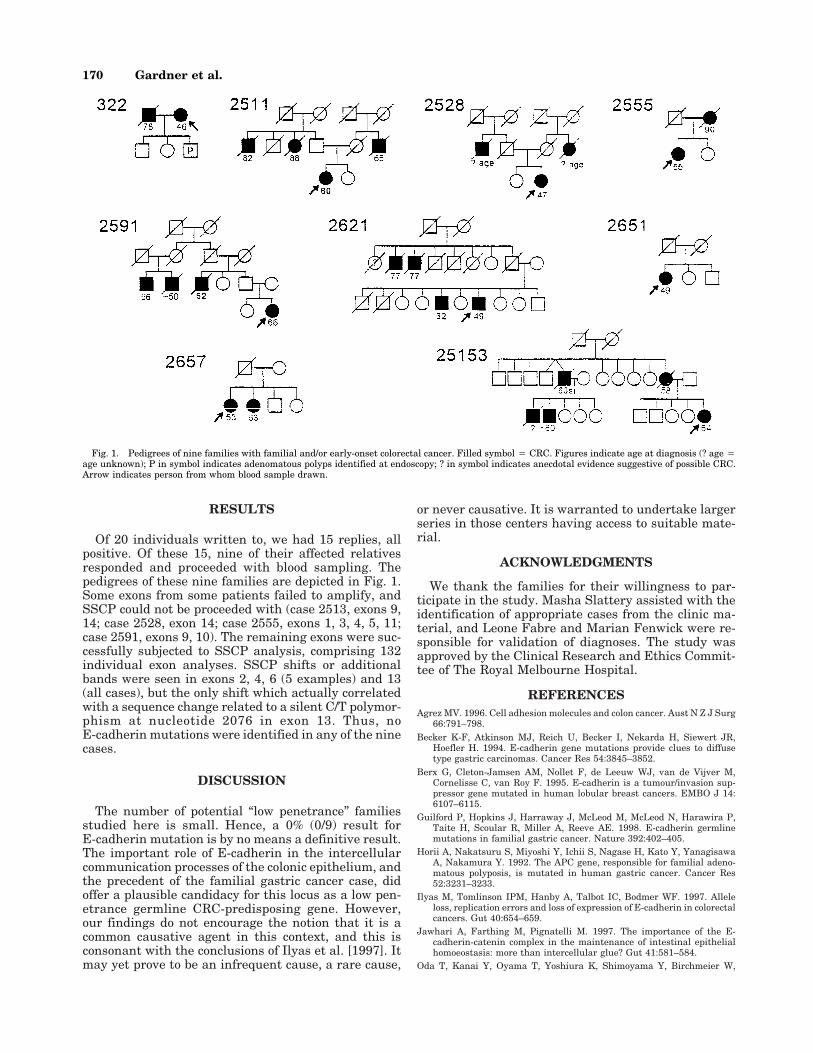
Of 20 individuals written to, we had 15 replies, allpositive. Of these 15, nine of their affected relativesresponded and proceeded with blood sampling. Thepedigrees of these nine families are depicted in Fig. 1.Some exons from some patients failed to amplify, andSSCP could not be proceeded with (case 2513, exons 9,14; case 2528, exon 14; case 2555, exons 1, 3, 4, 5, 11;case 2591, exons 9, 10). The remaining exons were suc-cessfully subjected to SSCP analysis, comprising 132individual exon analyses. SSCP shifts or additionalbands were seen in exons 2, 4, 6 (5 examples) and 13(all cases), but the only shift which actually correlatedwith a sequence change related to a silent C/T polymor-phism at nucleotide 2076 in exon 13. Thus, noE-cadherin mutations were identified in any of the ninecases.
DISCUSSION
The number of potential “low penetrance” familiesstudied here is small. Hence, a 0% (0/9) result forE-cadherin mutation is by no means a definitive result.The important role of E-cadherin in the intercellularcommunication processes of the colonic epithelium, andthe precedent of the familial gastric cancer case, didoffer a plausible candidacy for this locus as a low pen-etrance germline CRC-predisposing gene. However,our findings do not encourage the notion that it is acommon causative agent in this context, and this isconsonant with the conclusions of Ilyas et al. [1997]. Itmay yet prove to be an infrequent cause, a rare cause,
or never causative. It is warranted to undertake largerseries in those centers having access to suitable mate-rial.
ACKNOWLEDGMENTS
We thank the families for their willingness to par-ticipate in the study. Masha Slattery assisted with theidentification of appropriate cases from the clinic ma-terial, and Leone Fabre and Marian Fenwick were re-sponsible for validation of diagnoses. The study wasapproved by the Clinical Research and Ethics Commit-tee of The Royal Melbourne Hospital.
REFERENCESAgrez MV. 1996. Cell adhesion molecules and colon cancer. Aust N Z J Surg
66:791–798.Becker K-F, Atkinson MJ, Reich U, Becker I, Nekarda H, Siewert JR,
Hoefler H. 1994. E-cadherin gene mutations provide clues to diffusetype gastric carcinomas. Cancer Res 54:3845–3852.
Berx G, Cleton-Jamsen AM, Nollet F, de Leeuw WJ, van de Vijver M,Cornelisse C, van Roy F. 1995. E-cadherin is a tumour/invasion sup-pressor gene mutated in human lobular breast cancers. EMBO J 14:6107–6115.
Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P,Taite H, Scoular R, Miller A, Reeve AE. 1998. E-cadherin germlinemutations in familial gastric cancer. Nature 392:402–405.
Horii A, Nakatsuru S, Miyoshi Y, Ichii S, Nagase H, Kato Y, YanagisawaA, Nakamura Y. 1992. The APC gene, responsible for familial adeno-matous polyposis, is mutated in human gastric cancer. Cancer Res52:3231–3233.
Ilyas M, Tomlinson IPM, Hanby A, Talbot IC, Bodmer WF. 1997. Alleleloss, replication errors and loss of expression of E-cadherin in colorectalcancers. Gut 40:654–659.
Jawhari A, Farthing M, Pignatelli M. 1997. The importance of the E-cadherin-catenin complex in the maintenance of intestinal epithelialhomoeostasis: more than intercellular glue? Gut 41:581–584.
Oda T, Kanai Y, Oyama T, Yoshiura K, Shimoyama Y, Birchmeier W,
Fig. 1. Pedigrees of nine families with familial and/or early-onset colorectal cancer. Filled symbol 4 CRC. Figures indicate age at diagnosis (? age 4age unknown); P in symbol indicates adenomatous polyps identified at endoscopy; ? in symbol indicates anecdotal evidence suggestive of possible CRC.Arrow indicates person from whom blood sample drawn.
170 Gardner et al.

Sugimura T, Hirohashi S. 1994. E-cadherin gene mutations in humangastric carcinoma cell lines. Proc Nat Acad Sci 91:1858–1862.
Perl A-K, Wilgenbus P, Dahl U, Semb H, Christofori G. 1998. A causal rolefor E-cadherin in the transition from adenoma to carcinoma. Nature392:190–193.
Risinger JI, Berchuck A, Kohler MF, Boyd J. 1994. Mutations of the E-cadherin gene in human gynaecologic cancers. Nature Genet 7:98–102.
R.J. McKinlay Gardner*Clara L. GaffVictorian Clinical Genetics ServicesRoyal Children’s HospitalMelbourne, Australia
Finlay A. MacraeD. James B. St. JohnDepartment of GastroenterologyThe Royal Melbourne HospitalMelbourne, Australia
Justin HopkinsParry J. GuilfordAnthony E. ReeveCancer Genetics LaboratoryDepartment of BiochemistryUniversity of OtagoDunedin, New Zealand
E-Cadherin and Colorectal Cancer 171




![CD146 mediates an E-cadherin-to-N-cadherin switch during TGF-β … · 2018. 7. 10. · expression [16–19]. N-cadherin is reported to be upregulated by TGF-β signaling [20,21]](https://img.dokumen.tips/doc/110x75/6126bb2c5b910b6f974c32bd/cd146-mediates-an-e-cadherin-to-n-cadherin-switch-during-tgf-2018-7-10-expression.jpg)
























