Embed Size (px)
Citation preview

Glutamate is an excitatory neurotransmitter in the CNS thatplays a key role in long-term potentiation and cognitivefunctions such as learning and memory. However, prolongedexposure to excessive glutamate overactivates glutamatereceptors and initiates neurodegenerative processes (excito-toxicity) characterized by morphological changes in the axonand the cell body (Coleman 2005; Lau and Tymianski 2010).Excitotoxicity is linked to chronic neurological disorders,including Alzheimer’s disease (AD) and amyotrophic lateralsclerosis (ALS), and acute CNS insults, including ischemia.The mechanism underlying excitotoxicity is complex. Acti-vated NMDA receptors trigger calcium influx and induce theactivation of cysteine proteases, including calpain andcaspases (Brorson et al. 1995; Sattler et al. 1998; Tennetiet al. 1998), and the production of nitric oxide and freeradicals, both of which are detrimental to normal neuronalfunction (Lafon-cazal et al. 1993; Sattler et al. 1999).Despite these findings, pharmacological modulators ofexcitotoxicity have not provided significant neuroprotection
in clinical settings (Lau and Tymianski 2010). Thus,identifying alternate mechanisms that could modulate ex-citotoxic effects are important for therapeutic purposes and abetter understanding of neurodegenerative processes.
A key molecular feature of neurodegeneration is deficits inaxonal transport. Kinesins anterogradely transport cargosfrom the cell body to the distal axon, and the dynein-dynactincomplex retrogradely transports cargos from the distal axon
Received February 20, 2012; revised manuscript accepted March 26,2012; accepted March 27, 2012.Address correspondence and reprint requests to Takeshi Fujiwara,
PhD, Division of Biochemistry, Department of Biochemistry andMolecular Biology, Osaka University Graduate School of Medicine,Yamada-oka 1-3, Suita 565-0871, Osaka, Japan.E-mail: [email protected] used: AD, Alzheimer’s disease; ALS, amyotrophic
lateral sclerosis; a.a., amino acid; DIC, dynein intermediate chain; DIV,days in vitro; KD, knockdown; PI, propidium iodide; SDS, sodiumdodecyl sulfate; WT, wild-type.
*Department of Biochemistry and Molecular Biology, Osaka University Graduate School of Medicine,
Osaka, Japan
�Department of Pathology, Brain Research Institute, University of Niigata, Niigata, Japan
Abstract
Glutamate excitotoxicity causes neuronal dysfunction and
degeneration. It is implicated in chronic disorders, including
Alzheimer’s disease, and in acute CNS insults such as
ischemia. These disorders share prominent morphological
features, including axon degeneration and cell body death.
However, the molecular mechanism underlying excitotoxicity-
induced neurodegeneration remains poorly understood. A
key molecular feature of neurodegeneration is deficits
in microtubule-based cargo transport that plays a pivotal role
in maintaining the balance of survival and stress signaling in
the axon. We developed an excitotoxicity-induced neurode-
generation system in primary neuronal cultures. We find that
excitotoxicity generates a C-terminal truncated form of
p150Glued, a major component of the dynactin complex,
which exacerbates axon degeneration. This p150Glued
truncated form was identified in brain tissues of patients with
Alzheimer’s disease. Overexpression of wild-type (WT) dy-
nein intermediate chain (DIC), a dynein component that
interacts with p150Glued and links dynein and dynactin
complexes, DIC (S84D) mutant, and WT p150Glued sup-
pressed axon degeneration. These modulating effects of
p150Glued and DIC on excitotoxicity-induced axon degen-
eration are also observed in apoptosis and cell body death.
Thus, our findings identify retrograde transport proteins,
p150Glued and DIC, as novel modulators of neurodegener-
ation induced by glutamate excitotoxicity.
Keywords: apoptosis, dynactin, dynein, excitotoxicity, neu-
rodegeneration.
J. Neurochem. (2012) 10.1111/j.1471-4159.2012.07746.x
JOURNAL OF NEUROCHEMISTRY | 2012 doi: 10.1111/j.1471-4159.2012.07746.x
� 2012 The AuthorsJournal of Neurochemistry � 2012 International Society for Neurochemistry, J. Neurochem. (2012) 10.1111/j.1471-4159.2012.07746.x 1

to the cell body. These microtubule-based motors supplynewly synthesized materials to distal synapses, clear awaymisfolded/aggregated proteins, and deliver signals initiatedby changes in the environment from the axon to the cell body(Heerssen et al. 2004; Cosker et al. 2008; Perlson et al.2010). Identification of mutations in these motors fromhereditary neurodegenerative disease patients provided directevidence to implicate axonal transport defects to thepathogenesis of neurodegeneration (Zhao et al. 2001; Reidet al. 2002; Puls et al. 2003; Munch et al. 2004; Farrer et al.2009). In combination with findings from targeted disruptionof kinesin, transgenic or knock-in of autosomal dominantmutation in p150Glued dynactin, and disruption of dynactinby p50 dynamitin overexpression in mice confirmed thatdisruption of axonal transport system could induce neurode-generation (LaMonte et al. 2002; Lai et al. 2007; Laird et al.2008; Hirokawa et al. 2009). However, the role of axonaltransport in the context of excitotoxicity is poorly under-stood.
Here, we identify novel modulators of excitotoxicity-induced neurodegeneration using primary hippocampal cul-tures. We find that excitotoxicity generates a C-terminaltruncated form of p150Glued, a major component of thedynactin complex, which exacerbates excitotoxicity-inducedneurodegeneration and apoptosis. The p150Glued C-terminaltruncated form was detected in brain tissues from the frontalcortex of AD patients. We also demonstrate that wild-type(WT) dynein intermediate chain (DIC), a dynein componentthat interacts with p150Glued and links dynein and dynactincomplexes, p150Glued binding-defective DIC (S84D) mu-tant, and WT p150Glued suppress excitotoxicity-inducedneurodegeneration and apoptosis. These findings demon-strate p150Glued and DIC as novel modulators of excito-toxicity-induced neurodegeneration.
Materials and methods
cDNA cloning and expression vectorsp150Glued and DIC2 cDNAs were obtained by reverse transcrip-tion-PCR using RNeasy Plus Mini Kit (Qiagen, Valencia, CA,USA), GE Ready-To-Go You-Prime First Strand Beads (GEHealthcare, Little Chalfont, Buckinghamshire, UK), PrimeSTARMax DNA Polymerase (Takara Bio, Otsu, Shiga, Japan), and totalRNA from 10 days in vitro (DIV) hippocampal cultures. SalI-flanked forward (5¢-GCG GTC GAC ATG GCC CAG AGC AAGAGG CAC-3¢) and XhoI-flanked reverse (5¢-GCG CTC GAG TTAGGA GAT GAG ACG ACC GTG-3¢) primers were used to amplifyp150Glued, BglII-flanked forward (5¢-GCG AGA TCT ATG TCAGAC AAA AGT GAA TTA AAA G-3¢), and HindIII-flankedreverse (5¢-GCG AAG CTT CTA GGC AGG AAT CCG GG-3¢)primers were used to amplify DIC2 cDNAs, subcloned into pCR4Blunt TOPO cloning vector (Invitrogen, Carlsbad, CA, USA), andconfirmed by DNA sequencing (Applied Biosystems, Foster city,CA, USA). cDNAs were digested with SalI/XhoI for p150Glued,and BglII/HindIII for DIC2 and ligated into the pEGFP-C1
(Clontech) expression vector. pEGFP-C1-p150Glued WT plasmidwas digested with SmaI to remove its’ C-terminal portion and self-ligated to generate pEGFP-C1-p150Glued DC1. pEGFP-C1-p150Glued WT plasmid was digested with HindIII, and theresulting fragments were cloned into pEGFP-C1 to generatepEGFP-C1-p150Glued DC2. p150Glued DC3 fragment wasobtained by DraIII (blunted)/SalI digestion of p150Glued WT inpCR4 Blunt TOPO vector and cloned into SmaI/SalI digestedpEGFP-C1. To generate DIC2B (S84D) and -2C (S84D), PCR-based mutagenesis was performed using WT DIC2B and -2CcDNAs as templates. BglII-flanked forward and BsiHKAI-flankedinternal reverse (5¢-GCG GTG CTC ACC GAC TTG TCG G-3¢)primers were used to amplify the S84D mutated fragment. Underlineindicates the site of mutagenesis. The C-terminal fragments ofDIC2B and -2C were amplified by BsiHKAI-flanked internalforward (5¢-GCG GAG CAC GCC AAG TGA AGC-3¢) andHindIII-flanked reverse primers. DNA fragments were subclonedinto pCR4 Blunt TOPO vector, confirmed by DNA sequencing,digested with EcoRI/BglII for S84D mutated fragment and EcoRI/HindIII for C-terminal fragments. DNA fragments were furtherdigested with BsiHKAI, and cloned into BglII/HindIII site ofpEGFP-C1 to generate DIC2B (S84D) and -2C (S84D).
AntibodiesAntibodies used are as follows: Primary antibodies are ratmonoclonal anti-GFP (Nacalai Tesque, Kyoto, Japan), rabbitpolyclonal anti-GFP (MBL, Nagoya, Japan), mouse monoclonalanti-p150Glued (BD Biosciences, San Jose, CA, USA; N-terminalrecognition), goat polyclonal anti-DCTN1 (Abcam, Cambridge,UK; C-terminal recognition), mouse monoclonal anti-Dyneinintermediate chains cytoplasmic (Millipore Corporation, Billerica,MA, USA), mouse monoclonal anti-p50 (BD Biosciences), mousemonoclonal anti-kinesin heavy chain (KHC, H2; Chemicon,Temecula, CA, USA), rabbit polyclonal anti-cleaved caspase 3(Asp175) (Cell Signaling Technology, Danvers, MA, USA), rabbitpolyclonal anti-a-tubulin, mouse monoclonal anti-bIII-tubulin, ratmonoclonal anti-tubulin (YL1/2; Abcam), mouse monoclonal anti-TAU-1 (Chemicon), rabbit polyclonal anti-MAP2 (Chemicon);secondary antibodies are mouse IgG TrueBlot Ultra, rabbit IgGTrueBlot (eBioscience, San Diego, CA, USA), horseradish perox-idase-conjugated anti-mouse or anti-rabbit IgG (Cell Signaling),horseradish peroxidase-conjugated anti-goat IgG (Santa CruzBiotechnology, Santa Cruz, CA, USA).
TransfectionPlasmid DNA transfection into 293 cells was performed usingTransIT-LT1 Reagent (Mirus, Madison, WI, USA). Cells wereseeded at a density of 7 · 105 cells/6 cm dish (Corning, NY, USA)in Dulbecco’s modified Eagle’s medium (Invitrogen)/10% fetalbovine serum. Each plasmid of 3–4 lg were used according to thesupplier’s instructions and harvested for further analysis afterincubation for 48 h at 37�C, 5% CO2. Transfection into rathippocampal neurons was performed using AMAXA Nucleofectortransfection system (Lonza, Cologne, Germany). For plasmid DNA,cells of 2–3 · 106 and 2.5–4 lg of plasmid were used for eachtransfection. For RNA interference, cells of 4 · 106 and 8 lM ofON-TARGETplus Non-targeting pool siRNA or ON-TARGETplusSMARTpool siRNA for rat DCTN1 (Thermo Fisher Scientific,Waltham, MA, USA) was used for each transfection.
Journal of Neurochemistry � 2012 International Society for Neurochemistry, J. Neurochem. (2012) 10.1111/j.1471-4159.2012.07746.x� 2012 The Authors
2 | T. Fujiwara et al.

Neuron culture and immunofluorescencePrimary cultures of hippocampal neurons were prepared aspreviously described (Brewer et al. 1993) with some modifications.Hippocampi were dissected from Wistar rat embryonic day 18embryos (Kiwa Laboratory Animals Co., Ltd., Wakayama, Japan)and dissociated cells were resuspended and plated in neurobasalmedium (Invitrogen) supplemented with 2% B27 (Invitrogen) and0.5 mM L-glutamine (Invitrogen). Cells were seeded on 13 mmdiameter glass cover slips (Matsunami, Osaka, Japan) or plasticculture dishes (Corning) pre-coated with 0.05 mg/mL poly-L-lysine(Sigma-Aldrich, St. Louis, MO, USA) and grown for 8 DIV at 37�C,5% CO2. Procedures were approved by the Osaka UniversityInstitutional Guidelines for the Care and Use of LaboratoryAnimals. Transfected cells by electroporation was mixed withuntransfected cells at the ratio of 1 : 5–1 : 6 and seeded for culture.For indirect immunofluorescence labeling of transfected neurons, amixture of transfected and untransfected cells was seeded as 2–3 · 104 cells/cover slip. Neurons were fixed in 2% paraformalde-hyde (PFA)/4% sucrose/phosphate-buffered saline (PBS; pH 7.2–7.4), permeabilized with 0.2% Triton X-100/PBS, blocked by 3%bovine serum albumin/0.05% Tween20/PBS. Samples were furtherincubated with primary antibodies and secondary antibodiesconjugated with Alexa Fluor� 488, 568, or 647 fluorescents(Invitrogen) diluted in blocking buffer. Nuclei and chromosomeswere labeled with DAPI (Molecular Probes, Eugene, OR, USA).Images were captured by confocal laser microscopy FV1000 system(Olympus, Tokyo, Japan) with 20· and 40· oil-immersion objectivelenses, with 2· or 3· zoom.
Cell lysis, western blot detection, and immunoprecipitationTo obtain cell lysates, cells were harvested on ice with ice-cold PBSand lysed for 1–2 h at 4�C by rotation in either RIPA buffercontaining 1% NP-40/0.74% Na-deoxycolate/0.1% sodium dodecylsulfate (SDS)/1 mM EDTA/150 mM NaCl/50 mM Tris–Cl (pH7.4–7.5), or lysis buffer containing 1% NP-40/1 mM Na-orthovana-date/10% glycerol/1 mM EDTA/150 mM NaCl/20 mM Tris–Cl(pH 8.0), added with protease inhibitor cocktail tablets (RocheDiagnostics, Mannheim, Germany). Lysates were cleared bycentrifugation at 17 800 g for 15 min at 4�C and quantified withBCA protein detection kit (Thermo Fisher Scientific). Samples forSDS–polyacrylamide gel electrophoresis were prepared in samplebuffer containing 3.2% SDS/5.3% glycerol/66 mM Tris–Cl (pH6.8)/bromophenol blue (BPB), and 2-mercaptoethanol, and boiledfor 5 min at 95�C. Samples were loaded onto 10% or 12%polyacrylamide gel and transferred to nitrocellulose membranes byiblot TM gel transfer system (Invitrogen). Membranes were blockedwith either 5% skim milk/0.05% Tween 20/Tris-buffered saline (pH7.4) or 5% bovine serum albumin/0.1% TritonX-100/PBS. Super-Signal West Femto or West Dura Substrate (Thermo FisherScientific) was used for detection. For reuse of blotted membranes,antibodies were stripped off from the membrane by WB StrippingSolution (Nacalai Tesque), and subjected to new rounds of probing.Densitometry was performed on western blots for quantification.
Immunoprecipitation of GFP-DIC2 was performed with 70 lg oftransfected lysates from 293 cells prepared by RIPA buffer withoutSDS and EDTA, and added with 4 lg of rabbit polyclonal anti-GFPantibody. Protein G Sepharose 4 Fast Flow Beads (GE Healthcare)was added to each sample and further rotated for 1 h at 4�C to
capture the antibody–antigen complexes. Untransfected lysates of260 lg from 293 cells were further added to each sample androtated for 2 h to detect p150Glued.
Preparation of human brain extractsFresh–frozen brain tissues were obtained from The Brain ResourceCenter, Brain Research Institute, University of Niigata. Brain tissuesfrom the frontal cortex of patients with AD (n = 3: male 79-, female86-, and female 89-year-old), and aged-matched control individuals(n = 3: male 76-, female 79-, and male 82-year-old), and from themotor cortex of patients with ALS (n = 3: female 61-, female 82-,and female 83-year-old), and its’ controls (n = 3: male 67-, male 76-, and female 82-year-old) were used. For each case, the postmorteminterval was shorter than 4.5 h. The tissues were homogenized in 5volumes of RIPA buffer-containing protease inhibitor cocktails.Homogenates were rotated for 3 h at 4�C and subsequentlycentrifuged at 17 800 g for 15 min. The resulting supernatantswere quantified and used for analysis. The families of all subjectsprovided written informed consent to use the brain tissue forscientific purposes, and the study was performed with the approvalof the Ethics Committees of the University of Niigata and the OsakaUniversity Graduate School of Frontier Biosciences.
Neurite beading, apoptosis, cell body death analysesA punctum was defined as follows: Using FV10-ASW1.7 software(Olympus), images were captured with the same exposure and thesignal intensity of a dot labeled with GFP and/or bIII-tubulin/tubulinwas ‘more than 1500’. When the signal intensity of the neurite shaftadjacent to the dot was ‘less than 250’ positive for GFP/bIII-tubulinor tubulin, ‘0’ for GFP only, the dot was defined as ‘a punctum’.Neurite of 100 lm was regarded as one segment and two segmentswere analyzed for each neuron treated with either 50 lM glutamateor vehicle. To define cleaved-caspase 3-positive neurons, culturedneurons were treated with 10 lM staurosporine (Enzo LifeSciences, Farmingdale, NY, USA) or dimethyl sulfoxide for 6 h.Quantification was achieved by measuring the ratio of GFP-positiveneurons containing beading neurites positive for cleaved caspase 3(Asp175) in the cell body and in neurites within GFP-positiveneurons. Neurons showing nuclei staining of cleaved caspase 3 wereconsidered negative. Cell body death was analyzed by propidiumiodide (PI) incorporation (Invitrogen). Neurons were incubated with5 lg/mL of PI for 30 min prior to fixation. Quantification wasachieved by measuring the ratio of PI-incorporated GFP-positiveneurons within DAPI-labeled GFP-positive neurons.
Statistical analysisStatistical analyses were performed by one-way ANOVA with eitherDunnett or Bonferroni/Dunn post-test, or chi-squared for indepen-dence test. In all instances, a value of p < 0.05 was consideredsignificant.
Results
Excitotoxicity triggers C-terminal truncation of p150GluedA focal bead-like swelling phenotype in neurites is an earlyneurodegenerative feature in acute and chronic neurologicaldisorders (Takeuchi et al. 2005). In our primary hippocampal
� 2012 The AuthorsJournal of Neurochemistry � 2012 International Society for Neurochemistry, J. Neurochem. (2012) 10.1111/j.1471-4159.2012.07746.x
Dynein and dynactin in excitotoxicity | 3

culture system at 8 DIV, 50 lM glutamate treatment induceda bead-like swelling phenotype in TAU-1-labeled axonsindicating degeneration (Fig. 1a). A large body of evidencehas demonstrated that axonal transport machinery is impairedduring neurodegeneration (Chevalier-Larsen and Holzbaur2006; De Vos et al. 2008). Thus, we first looked atbiochemical features of major transport proteins under50 lM glutamate excitotoxic condition. We found pro-nounced biochemical alterations in the components of thedynein-dynactin complex that transports cargos in a plus tominus end direction on microtubules (Fig. 1b). By detectionwith the N-terminal-recognizing antibody, p150Glued, acomponent of the dynactin complex, was cleaved and anapproximately 110K truncated form was generated underglutamate-treated conditions (Fig. 1b and c). Re-probing by aC-terminal-recognizing antibody of p150Glued did not detectthe 110K truncated form indicating that the 110K truncatedform is C-terminal truncated (Fig. 1d). The expression of
DIC, a component of the dynein complex that links dynein todynactin, was reduced approximately 35% by 6 h glutamateincubation (Fig. 1c and e). However, a dynactin componentp50 dynamitin, which binds the C-terminal region ofp150Glued, and kinesin heavy chain had no significantalteration (Fig. 1b and c). From these results, we hypothe-sized that p150Glued might function in the process ofexcitotoxicity-induced neurodegeneration, and that detectingthe p150Glued truncated form in vivo in the context ofneurological disorders would support our hypothesis. Weaddressed this question by using brain tissues from thefrontal and motor cortex of autopsied patients with AD andALS, respectively. The frontal cortex is a severely andconstantly affected region in patients with AD, wherepathological features including neuronal loss, b-amyloiddeposits, and neurofibrillary tangles are prominent (Leubaet al. 2009). In the frontal cortex of AD patients, thep150Glued truncated form corresponding to that generated in
150 K
100 K
150 K
100 K
150 K
100 K
GlutamateVehicle 1 h 3 h 6 h
p150Glued
DIC
p50
KHC
α-tubulin
Blots
0.0
0.2
0.4
0.6
0.8
1.0
1.2
DIC
rela
tive
inte
nsity
ver
sus
vehi
cle
trea
tmen
t
GlutamateVehicle 1 h 3 h 6 h
1.000.84 0.82
0.63
VehicleGlutamate
3 h
Blot:p150Glued mAb recognizingN-terminal region
150 K
100 K
Blot:p150Glued pAb recognizingC-terminal region
VehicleGlutamate
3 h
150 K
100 K
AD control AD
Blots
Hippocampalcultures
glutamate3 hN1 R8 N16 N30 N2N32
p150Glued
α-tubulin
ALS control ALS
N1 R6 N27 N25 N11N40 Blots
p150Glued
α-tubulin
Trun
cate
d/to
tal
p150
Glu
ed (%
)
0
20
40
60
80
100
69.8
30.5
9.8
N1 R8 N16 N30 N2N32AD control AD
N40 N1 N27 N25 N11R6
ALS control ALS
Trun
cate
d/to
tal
p150
Glu
ed (%
)
0
10
20
30
40
50
0.11 0.75 0.03
Vehi
cle
Glu
tam
ate
TAU-1 MAP2
TAU-1 MAP2
Microtubule– +
Cargo
DynactinDynein
p150Glued
p50
C-terminus
N-terminus
p150Glued
Dyneinintermediate
chain(DIC)
(a)
(c)
(f)
(g)
(d)(e)
(b)
Fig. 1 C-terminal truncation of p150Glued.
(a) Neurons incubated with 50 lM gluta-
mate or vehicle for 3 h labeled with anti-
bodies to TAU-1 and MAP2. Glutamate
induced a punctate pattern in TAU-1-posi-
tive axons (arrows). Bars: 40 lm. (b) A
concise illustration of the dynein–dynactin
complex (left), including p150Glued and
DIC (right). (c) Western blot detection of
transport proteins on a single sheet.
Detection of approximately 110K p150Glued
truncated form (arrow) by the N-terminal-
recognizing antibody using 20 lg of lysates
from cultures incubated with 50 lM gluta-
mate for indicated hours or vehicle for 6 h.
(d) Detection of p150Glued by the N-termi-
nal-recognizing or C-terminal-recognizing
antibody on a single sheet. The C-terminal-
recognizing antibody does not detect the
110K p150Glued truncated form (arrows).
(e) Quantified relative intensity of DIC nor-
malized by a-tubulin shown in panel (c). (f)
Using 20 lg lysates, the 110K p150Glued
truncated form is detected in AD patients
(asterisks) using the N-terminal-recognizing
antibody (n = 3/3, p < 0.05; chi-square test)
but not in ALS. p150Glued and a-tubulin
were detected on a single sheet. (g) Ratio
of truncated versus total p150Glued inten-
sity shown in panel (f).
Journal of Neurochemistry � 2012 International Society for Neurochemistry, J. Neurochem. (2012) 10.1111/j.1471-4159.2012.07746.x� 2012 The Authors
4 | T. Fujiwara et al.

glutamate-treated hippocampal cultures was observed (n = 3/3; Fig. 1f and g). The ratio of the truncated form relative tothe total amount of p150Glued indicated that all three braintissues of AD patients contained the truncated form rangingfrom approximately 70–10%, however, less than 1% in themotor cortex of ALS and controls, and in the frontal cortex ofcontrols for AD (Fig. 1f and g). This result indicates that thep150Glued truncated form exists in vivo in the context ofneurological disorders.
p150Glued modulates excitotoxicity-induced axondegenerationTo investigate the significance of the p150Glued C-terminaltruncated form on excitotoxicity-induced axon degeneration,we generated p150Glued WT and C-terminal truncatedmutants, DC1, DC2, and DC3 N-terminally fused to GFP,and confirmed the expression of each construct (Fig. 2a andb). Each C-terminal truncated mutant was designed toresemble the excitotoxicity-induced truncated form. Amongthem, GFP-p150Glued DC2 and DC3 mutants resembled themolecular weight of the excitotoxicity-induced GFP-p150Glued truncated form generated from GFP-p150GluedWT (Fig. 2c). Based on this result, we further analyzed thefunctional significance of the C-terminal truncatedp150Glued using DC2 and DC3 mutants. We examinedwhether overexpression of DC2 and DC3 mutants have animpact on axon degeneration by quantifying the neuritebeading phenotype. For this purpose, transfected neurons hadto be visually isolated. Transfection efficiency was between70 and 75% as evaluated by indirect immunofluorescenceusing anti-GFP antibody (T. Fujiwara and K. Morimoto,unpublished observation); therefore, analyses observing thetransfected population should represent the majority of theneuronal cultures. We defined bead-containing neurites asfollows: (i) a punctum was defined as described in Materialsand methods, (ii) transfected cells showing a punctate patternpositive for tubulin (Fig. 3a and d), double-positive for GFPand bIII-tubulin (Figs 2e–g, 5d and e), and positive for GFP(Figs 4a and b, 6a and b) were regarded as ‘beads’, (iii) aneurite of 100 lm was defined as one segment (Fig. 2d;yellow lines in bIII-tubulin panels), and (iv) two segmentswere randomly analyzed and when the total sum was morethan 10 beads, the neuron contained ‘bead-containingneurites’ (Fig. 2d). In most vehicle-treated samples, the totalsum of beads in two segments was 4 or less. Overexpressionof DC2 and DC3 mutants increased approximately 30%,whereas overexpression of WT suppressed approximately48% the number of neurons with beading neurites comparedwith that of control in 1 h glutamate incubation (Fig. 2e andf). In 3 h glutamate incubation, neurons overexpressingcontrol GFP, DC2, and DC3 mutants contained beadingneurites ranging from 42 to 45% showing no significantdifference among these transfected neurons (Fig. 2g). On thecontrary, neurons over-expressing WT showed an approxi-
mately 20% suppression of the number of neurons withbeading neurites compared with that of control (Fig. 2g).These results indicate that the C-terminal truncatedp150Glued exacerbates and that p150Glued WT suppressesexcitotoxicity-induced axon degeneration. In addition, theprotective effect of over-expressed p150Glued WT isreduced in 3 h glutamate incubation which supports the ideathat decreased amount of p150Glued WT allows axon todegenerate under excitotoxic condition. The effect of thep150Glued C-terminal truncated form could be a loss offunction or a toxic gain of function. To clarify the role ofp150Glued, p150Glued knockdown (KD) by siRNA wasperformed in cultured neurons (Fig. 3a–d). Expression ofDIC and p50 was not altered by p150Glued KD (Fig. 3b),and on average, approximately 80% of p150Glued KD wasachieved (Fig. 3c). p150Glued KD increased approximately2-fold the number of neurons with beading neuritescompared with that of control in 1 h glutamate incubation(Fig. 3d). These results indicate that the p150Glued truncatedform mimics a loss of function of p150Glued.
p150Glued modulates excitotoxicity-induced apoptosisand cell body deathExcitotoxicity can induce apoptosis depending on theduration and intensity of the excitotoxic insult (Ankarcronaet al. 1995; Bonfoco et al. 1995). NMDA and glutamatetrigger activation of caspase 3 and apoptosis in cerebrocor-tical and hippocampal neurons, respectively (Tenneti andLipton 2000; Brecht et al. 2001), indicating activatedcaspase 3 as a marker for neuronal apoptosis. By detectingactivated (cleaved) caspase 3, we assessed whetheroverexpression of p150Glued constructs modulate excito-toxicity-induced apoptosis. In glutamate-treated neurons thatcontained beading neurites, cleaved caspase 3 was observedin neurites and throughout the cell body, whereas in vehicle-treated control neurons, cleaved caspase 3 was observed inthe nuclei (Fig. 4a). As apoptotic neurons treated withstaurosporine show cleaved caspase 3 in neurites andthroughout the cell body, this pattern of labeling was definedas activated caspase 3 positive. In neurons containingbeading neurites, overexpression of DC2 and DC3 mutantsincreased the number of neurons with activated caspase 3approximately 50% in 1 h and 25% in 3 h glutamateincubation compared with that of control (Fig. 4b). Bycontrast, overexpression of WT suppressed the number ofneurons with activated caspase 3 approximately 50% in 1 hand 30% in 3 h glutamate incubation compared with that ofcontrol (Fig. 4b). This result indicates that p150Gluedmodulates the process of excitotoxicity-induced apoptosis.
In a number of neurodegenerative conditions, axondegeneration overlaps with degeneration of the cell body(Raff et al. 2002; Conforti et al. 2007). We assessed whetherp150Glued function is also involved in the process ofexcitotoxicity-induced cell body death by PI incorporation
� 2012 The AuthorsJournal of Neurochemistry � 2012 International Society for Neurochemistry, J. Neurochem. (2012) 10.1111/j.1471-4159.2012.07746.x
Dynein and dynactin in excitotoxicity | 5

CAP-Gly domain (microtubule binding)Basic serine rich regionCoiled-coil domain (dimerization)
WT1(a)
(c)
(e) (f)
(g)
(d)
(b)1280GFP
ΔC11 1083
GFP
ΔC21 944
GFP
ΔC31 931
GFP
200 K150 K100 K
75 K
50 K
37 K25 K
Blot: GFP
WT vectorΔC2ΔC3ΔC1
GFP
GFP-p150 ΔC2GFP-p150 WT
GFP
βIII-tubulin
Glutam
ateVehicle
βIII-tubulin
0
10
20
30
Cel
ls w
ith b
ead-
cont
aini
ng n
eurit
es (%
) ∗
∗
∗
Vector p150ΔC2
p150ΔC3
p150WT
Vector p150ΔC2
p150ΔC3
p150WT
Vehicle 1 h Glutamate 1 h
150 K
200 K
GFP-p150 WTglutamate
6 h ΔC2 ΔC1ΔC3
Blot: GFP
GFP Merge VehicleG
lutamate
GFP Merge
βIII-tubulin
βIII-tubulin
Cel
ls w
ith b
ead-
cont
aini
ng n
eurit
es (%
)
0
10
20
30
40
50
60∗
Vector p150ΔC2
p150ΔC3
p150WT
Vector p150ΔC2
p150ΔC3
p150WT
Vehicle 3 h Glutamate 3 h
Fig. 2 p150Glued modulates axon degeneration. (a) Schema of
p150Glued WT (1,280 a.a.), DC1 (1,083 a.a.), DC2 (944 a.a.), and
DC3 (931 a.a.), N-terminally fused to GFP. (b) Western blot detection
of GFP-p150Glued constructs in 293 cells using 30 lg of lysates. (c)
Western blot detection of glutamate-induced truncated form from
GFP-p150Glued WT, DC1, DC2, and DC3 constructs using 30 lg of
lysates. (d) Defining a ‘‘bead-containing neurite’’ in neurons. Shown
are vector-transfected neurons labeled for GFP (green) and bIII-
tubulin (red). Yellow lines in bIII-tubulin-labeled images represent
100 lm length. Bars: 20 lm. (e) Representative images of gluta-
mate- or vehicle-treated neurons transfected with WT and DC2, la-
beled for GFP and bIII-tubulin. Bars: 30 lm. (f, g) Quantification of
the number of neurons with beading neurites. In 1 h glutamate
incubation (> 824 cells for each condition), neurons transfected with
vector, WT, DC2, and DC3, were 18.3%, 9.4%, 23.7%, and 23.7%,
respectively (f, *p < 0.01). In 3 h (> 655 cells for each condition),
vector, WT, DC2, and DC3, were 42.0%, 33.5%, 43.8%, and 45.1%,
respectively (g, *p < 0.01). Statistics represent mean ± SEM of three
independent experiments and analyzed by one-way ANOVA with
Dunnett post-test.
Journal of Neurochemistry � 2012 International Society for Neurochemistry, J. Neurochem. (2012) 10.1111/j.1471-4159.2012.07746.x� 2012 The Authors
6 | T. Fujiwara et al.
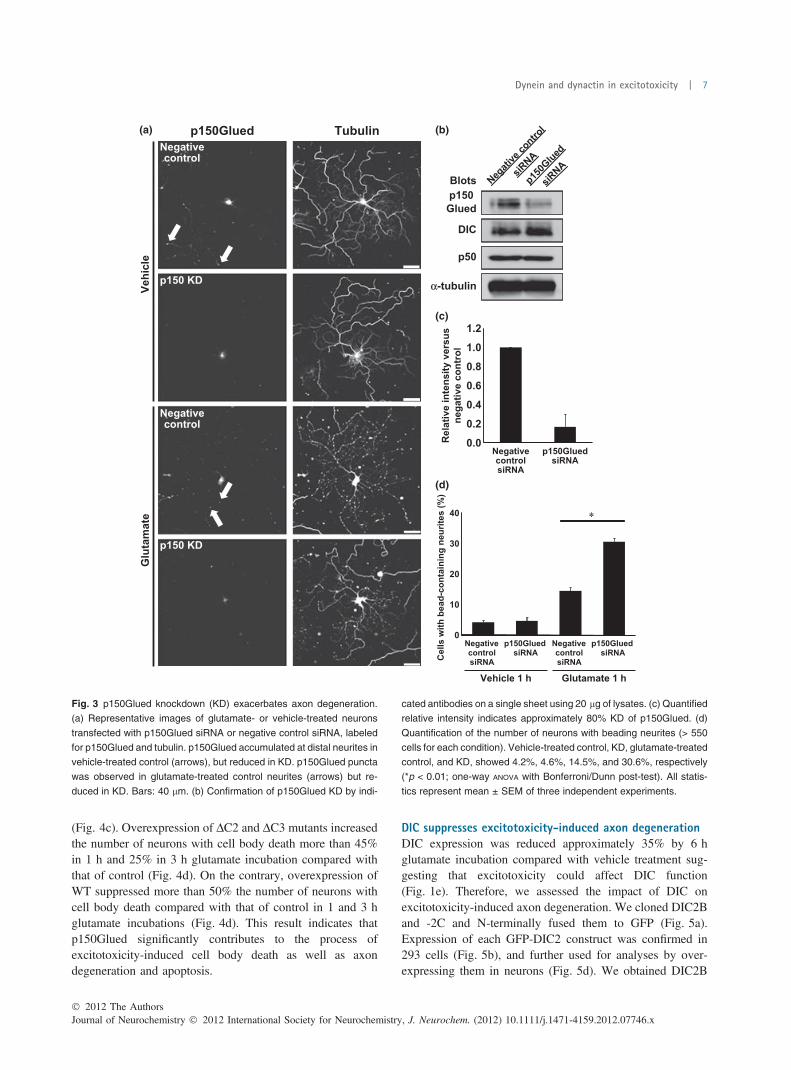
(Fig. 4c). Overexpression of DC2 and DC3 mutants increasedthe number of neurons with cell body death more than 45%in 1 h and 25% in 3 h glutamate incubation compared withthat of control (Fig. 4d). On the contrary, overexpression ofWT suppressed more than 50% the number of neurons withcell body death compared with that of control in 1 and 3 hglutamate incubations (Fig. 4d). This result indicates thatp150Glued significantly contributes to the process ofexcitotoxicity-induced cell body death as well as axondegeneration and apoptosis.
DIC suppresses excitotoxicity-induced axon degenerationDIC expression was reduced approximately 35% by 6 hglutamate incubation compared with vehicle treatment sug-gesting that excitotoxicity could affect DIC function(Fig. 1e). Therefore, we assessed the impact of DIC onexcitotoxicity-induced axon degeneration. We cloned DIC2Band -2C and N-terminally fused them to GFP (Fig. 5a).Expression of each GFP-DIC2 construct was confirmed in293 cells (Fig. 5b), and further used for analyses by over-expressing them in neurons (Fig. 5d). We obtained DIC2B
0.0
0.2
0.4
0.6
0.8
1.0
1.2
Rel
ativ
e in
tens
ity v
ersu
sne
gativ
e co
ntro
lNegativecontrolsiRNA
p150GluedsiRNA
0
10
20
30
40 ∗
Cel
ls w
ith b
ead-
cont
aini
ng n
eurit
es (%
)
NegativecontrolsiRNA
p150GluedsiRNA
p150GluedsiRNA
NegativecontrolsiRNA
Vehicle 1 h Glutamate 1 h
p150Glued TubulinG
luta
mat
eVe
hicl
e
p150 KD
p150 KD
Negativecontrol
Negativecontrol
Blotsp150Glued
DIC
p50
α-tubulin
Negati
ve co
ntrol
siRNA
p150G
lued
siRNA
(a) (b)
(c)
(d)
Fig. 3 p150Glued knockdown (KD) exacerbates axon degeneration.
(a) Representative images of glutamate- or vehicle-treated neurons
transfected with p150Glued siRNA or negative control siRNA, labeled
for p150Glued and tubulin. p150Glued accumulated at distal neurites in
vehicle-treated control (arrows), but reduced in KD. p150Glued puncta
was observed in glutamate-treated control neurites (arrows) but re-
duced in KD. Bars: 40 lm. (b) Confirmation of p150Glued KD by indi-
cated antibodies on a single sheet using 20 lg of lysates. (c) Quantified
relative intensity indicates approximately 80% KD of p150Glued. (d)
Quantification of the number of neurons with beading neurites (> 550
cells for each condition). Vehicle-treated control, KD, glutamate-treated
control, and KD, showed 4.2%, 4.6%, 14.5%, and 30.6%, respectively
(*p < 0.01; one-way ANOVA with Bonferroni/Dunn post-test). All statis-
tics represent mean ± SEM of three independent experiments.
� 2012 The AuthorsJournal of Neurochemistry � 2012 International Society for Neurochemistry, J. Neurochem. (2012) 10.1111/j.1471-4159.2012.07746.x
Dynein and dynactin in excitotoxicity | 7

GFP/PI
PI/DAPI
Vehicle Glutamate
GFP Cleaved caspase 3
Vehicle
(a)
(b)
(c) (d)
Glutamate
MergeC
leav
ed c
aspa
se 3
inbe
adin
g ne
uron
s/ne
uron
s (%
)
0
10
20
∗ ∗∗ ∗
∗ ∗ ∗∗
Glutamate 1 hVehicle 1 h
Vector p150WT
p150ΔC2
p150ΔC3
Vector p150WT
p150ΔC2
p150ΔC3
Glutamate 3 hVehicle 3 h
Cle
aved
cas
pase
3 in
bead
ing
neur
ons/
neur
ons
(%)
0
10
20
30∗ ∗ ∗
∗ ∗
∗ ∗∗ ∗
Vector p150WT
p150ΔC2
p150ΔC3
Vector p150WT
p150ΔC2
p150ΔC3
Vehicle 3 h Glutamate 1 h Glutamate 3 h
0
10
20
30
PI-in
corp
orat
ed c
ells
(%)
∗∗ ∗ ∗
∗
∗ ∗ ∗∗
∗ ∗ ∗
Vector p150WT
p150ΔC2
p150ΔC3
Vector p150WT
p150ΔC2
p150ΔC3
Vector p150WT
p150ΔC2
p150ΔC3
Fig. 4 p150Glued modulates apoptosis and cell body death. (a) Rep-
resentative images of glutamate- or vehicle-treated neurons trans-
fected with DC3 labeled for GFP (green) and cleaved caspase 3 (red).
Bars: 30 lm. (b) Quantification of the number of neurons with cleaved
caspase 3-positive beading neurites. In 1 h glutamate incubation
(> 450 cells for each condition), neurons transfected with vector, WT,
DC2, and DC3, showed 9.5%, 4.5%, 14.9%, and 14.8%, respectively
(*p < 0.01, **p < 0.05). In 3 h (> 581 cells for each condition), vector,
WT, DC2, and DC3, showed 16.7%, 11.6%, 21.4%, and 21.3%,
respectively (**p < 0.05). (c) Representative images of PI-incorporated
neurons transfected with DC2 labeled for GFP (green), PI (red), and
DAPI (blue). Neurons were incubated with glutamate for 1 and 3 h or
vehicle for 3 h. Bars: 30 lm. (d) Quantification of the number of neu-
rons with cell body death (> 509 cells for each condition). In 1 h glu-
tamate incubation, neurons transfected with vector, WT, DC2, and
DC3, showed 9.0%, 3.6%, 13.3%, and 13.2%, respectively (*p < 0.01,
**p < 0.05). In 3 h, vector, WT, DC2, and DC3, showed 16.5%, 7.8%,
21.1%, and 20.9%, respectively (*p < 0.01, **p < 0.05). All statistics
represent mean ± SEM of three (d) or four (b) independent experi-
ments and analyzed by one-way ANOVA with Dunnett post-test.
Journal of Neurochemistry � 2012 International Society for Neurochemistry, J. Neurochem. (2012) 10.1111/j.1471-4159.2012.07746.x� 2012 The Authors
8 | T. Fujiwara et al.

25 K
100 K75 K
50 K
37 K
0
10
20
30
40
∗ ∗ ∗∗
Cel
ls w
ith b
ead-
cont
aini
ng n
eurit
es (%
)
Vector DIC2B DIC2CDIC2BS84D
DIC2CS84D
Vector DIC2B DIC2CDIC2BS84D
DIC2CS84D
Glutamate 3 hVehicle 3 h
Blot:GFP
25 K
100 K75 K
50 K
37 K
DIC2B DIC2C
VectorWTWT S84D S84D
DIC2A(a)
(b)
(d)
(e)
(c)
p150Glued-bindingdomain
1 638
TcTex-1-binding domai nLC8-binding domain
Roadblock-binding domainWD repeats
DIC2B WT1 632
GFP
DIC2C WTGFP1 612
S84D1 632S84D
GFP
S84D61 12
S84D
GFP
Input IP: Anti-GFP2BWT
2CWT
2CS84D
2BS84D
2BWT
2CWT
2CS84D
2BS84D VectorVector Blots
GFP
p150Glued
GlutamateGFP/DAPI βIII-tubuin/DAPIGFP/DAPI βIII-tubuin/DAPI
Vehicle
Vector Vector
GFP-DIC2B
GFP-DIC2B
Fig. 5 DIC suppresses axon degeneration. (a) Schema of DIC2B,
-2C, -2B (S84D), and -2C (S84D), N-terminally fused to GFP. DIC2B
(76–81 a.a. deletion of DIC2A) and -2C (76–81, 113–132 a.a. deletion
of DIC2A) were cloned and an S84D mutation was introduced. (b)
Western blot detection of GFP-DIC2 constructs using 40 lg of lysates.
(c) GFP-DIC2 WT and S84D immunoprecipitated by GFP antibody
were detected with GFP and p150Glued antibodies on a single sheet.
(d) Representative images of glutamate- or vehicle-treated neurons
transfected with DIC2B WT and vector labeled for GFP (green), bIII-
tubulin (red), and DAPI (blue). Bars: 50 lm. (e) Quantification of the
number of neurons with beading neurites (> 490 cells for each con-
dition). In 3 h glutamate incubation, vector, DIC2B, -2B (S84D), -2C,
and -2C (S84D), showed 36.5%, 7.0%, 6.2%, 6.5%, and 6.5%,
respectively (*p < 0.01). Statistics represent mean ± SEM of three
independent experiments and analyzed by one-way ANOVA with
Dunnett post-test.
� 2012 The AuthorsJournal of Neurochemistry � 2012 International Society for Neurochemistry, J. Neurochem. (2012) 10.1111/j.1471-4159.2012.07746.x
Dynein and dynactin in excitotoxicity | 9

and -2C but not -2A from total RNA prepared from our 10DIV hippocampal cultures. A recent report described thatDIC2B and -2C are embryonically expressed whereasDIC2A is expressed in adult rat and mice (Kuta et al.2010), explaining why we were able to isolate only DIC2Band -2C cDNAs. DIC2-over-expressing neurons showedmore than 80% suppression of the number of neurons withbeading neurites compared with that of control in 3 hglutamate incubation (Fig. 5e), showing a protective effect ofDIC against excitotoxicity. As a similar result was obtainedwith p150Glued WT, we speculated that p150Glued-DICinteraction could be vital for this protective effect. Previ-ously, a nucleotide substitution at serine 84 to aspartate(S84D) of DIC has been shown to significantly reduce theinteraction with p150Glued (Vaughan et al. 2001). Wegenerated DIC2B (S84D) and -2C (S84D) mutants andN-terminally fused them to GFP (Fig. 5a and b). p150Gluedco-immunoprecipitated with GFP-DIC2 WT but not with theGFP-DIC2 (S84D) mutants confirming that the generatedDIC2 (S84D) mutants were defective in the interaction withp150Glued (Fig. 5c). DIC2 (S84D) overexpression resultedin more than 80% suppression of the number of neurons withbeading neurites compared with that of control in 3 hglutamate incubation (Fig. 5e). These results indicate thatoverexpression of DIC suppresses excitotoxicity-inducedaxon degeneration irrespective of the interaction withp150Glued.
DIC suppresses excitotoxicity-induced apoptosis and cellbody deathWe also assessed the significance of DIC on excitotoxicity-induced apoptosis and cell body death by over-expressingDIC2B and -2C in neurons (Fig. 6a and c). In neuronscontaining beading neurites, overexpression of DIC2 WTand S84D mutants suppressed more than 45% and 65% thenumber of neurons with activated caspase 3 compared withthat of control in 1 and 3 h glutamate incubations, respec-tively (Fig. 6b). When the effect on cell body death wasanalyzed (Fig. 6c), overexpression of DIC2 WT and S84Dmutants suppressed more than 50% the number of neuronswith cell body death compared with that of control in 3 hglutamate incubation (Fig. 6d). These results indicate thatDIC suppresses excitotoxicity-induced apoptosis and cellbody death irrespective of the interaction with the dynactincomplex.
Discussion
Here, we identify p150Glued and DIC as novel modulatorsof neurodegeneration induced by excitotoxicity. The signif-icance of our findings includes the identification of a C-terminal truncated p150Glued that contributes to the processof neurodegeneration. Importantly, identification of thep150Glued truncated form in the frontal cortex of patients
with AD implies a contribution of the p150Glued truncatedform in the pathological process of human neurologicaldisorders.
The frontal cortex is important for executive functioninfluencing memory. Patients with AD show executivedeficits in the early to middle stages of the clinical course,and during the long disease duration, pathological featuresincluding neuronal loss, b-amyloid deposits, and neurofibril-lary tangles are manifested gradually in the frontal cortex(Leuba et al. 2009). The temporal lobe including theparahippocampal gyrus and the hippocampus in patientswith advanced stages of AD shows severe loss of neurons,therefore, it is sometimes difficult to obtain protein extractsfrom this region. In this study, we extracted proteins from thefrontal cortex of AD patients and identified the C-terminaltruncated form of p150Glued (Fig. 1f and g). The findingallows us to further investigate the frontal cortex/whitematter of patients with AD to test whether p150Glued andits’ related transport proteins could be a pathological featureof AD.
We expected to observe the truncated form of p150Gluedin the motor cortex of ALS patients because glutamateexcitotoxicity is considered to be a hallmark of ALS. Recentfindings show that ubiquitin-immunoreactive cytoplasmicinclusions that are positive for TAR DNA-binding protein 43in lower motor neurons represent a characteristic patholog-ical feature of ALS and that the TAR DNA-binding protein43 positivity was absent in those with SOD1 mutations (Araiet al. 2006; Neumann et al. 2006; Mackenzie et al. 2007),indicating that proteinopathy is also a hallmark of ALS andcould be distinguished from excitotoxicity. Thus, the expla-nation to why the p150Glued truncated form was notobserved is that the cause of ALS of our analyzed braintissues could be proteinopathy or those that do not relate toexcitotoxicity.
In our cellular studies of excitotoxicity, the ratio of GFP-positive 8 DIV neurons with bead-containing neuritesreached the saturation point at approximately 40–50% by3–6 h glutamate incubation (T. Fujiwara and K. Morimoto,unpublished observation). Ionotropic glutamate receptorscluster in dendrites starting from 3 DIV and increase thenumber of clusters as well as synapses at least until 17 DIV(Washbourne et al. 2002). At 8 DIV, there might be only 40–50% of neurons that cluster glutamate receptors at synapsesthat are sufficient to respond to excessive glutamate.
Both C-terminal truncated p150Glued DC2 and DC3mutants exacerbated excitotoxicity-induced axon degenera-tion, apoptosis, and cell body death. A 13 amino aciddifference between DC2 and DC3 mutants did not causesignificant difference in their functions. Notably, over-expression of DC2 and DC3 mutants were not sufficient toinduce neurodegeneration. As the C-terminal truncated formis produced by excitotoxic conditions and is rarely detectablein normal cultures, it is likely that the C-terminal truncated
Journal of Neurochemistry � 2012 International Society for Neurochemistry, J. Neurochem. (2012) 10.1111/j.1471-4159.2012.07746.x� 2012 The Authors
10 | T. Fujiwara et al.

form acts synergistically with known or unknown molecularpathways and contributes to excitotoxicity-induced degener-ative processes.
Overexpression of p150Glued WT, DIC2 WT, and DIC2S84D mutants protects neurons from excitotoxicity-inducedneurodegeneration and apoptosis. These results indicate that
GFP(a)
(b)
(c) (d)
Cleaved caspase 3
Vehicle
Glutamate
Merge
0
10
20
30
Cle
aved
cas
pase
3 in
bead
ing
neur
ons/
neur
ons
(%)
∗ ∗ ∗∗∗ ∗ ∗ ∗
Vector DIC2B DIC2CDIC2BS84D
DIC2CS84D
Vector DIC2B DIC2CDIC2BS84D
DIC2CS84D
Vector DIC2B DIC2CDIC2BS84D
DIC2CS84D
Glutamate 3 hGlutamate 1 hVehicle 3 h
Vehicle Glutamate
GFP/PI
PI/DAPI
0
5
10
15
∗ ∗ ∗∗
PI-in
corp
orat
ed c
ells
(%)
Vector DIC2B DIC2CDIC2BS84D
DIC2CS84D
Vector DIC2B DIC2CDIC2BS84D
DIC2CS84D
Glutamate 3 hVehicle 3 h
Fig. 6 DIC suppresses apoptosis and cell body death. (a) Repre-
sentative images of glutamate- or vehicle-treated neurons transfected
with DIC2B WT labeled for GFP (green) and cleaved caspase 3 (red).
Bars: 30 lm. (b) Quantification of the number of neurons with cleaved
caspase 3-positive beading neurites. In 1 h glutamate incubation
(> 443 cells for each condition), vector, DIC2B, -2B (S84D), -2C, and -
2C (S84D), showed 8.6%, 4.9%, 4.6%, 4.9%, and 4.6%, respectively
(*p < 0.01). In 3 h (> 451 cells for each condition), vector, DIC2B, -2B
(S84D), -2C, and -2C (S84D), showed 20.5%, 6.2%, 6.4%, 6.1%, and
6.1%, respectively (*p < 0.01). (c) Representative images of gluta-
mate- or vehicle-treated PI-incorporated neurons transfected with
DIC2B WT labeled for GFP (green), PI (red), and DAPI (blue). Bars:
30 lm. (d) Quantification of the number of neurons with cell body
death (> 614 cells for each condition). In 3 h glutamate incubation,
vector, DIC2B, -2B (S84D), -2C, and -2C (S84D), showed 10.2%,
4.3%, 4.2%, 4.3%, and 3.4%, respectively (*p < 0.01). All statistics
represent mean ± SEM of three (d) or four (b) independent experi-
ments and analyzed by one-way ANOVA with Dunnett post-test.
� 2012 The AuthorsJournal of Neurochemistry � 2012 International Society for Neurochemistry, J. Neurochem. (2012) 10.1111/j.1471-4159.2012.07746.x
Dynein and dynactin in excitotoxicity | 11

reduced interaction with p150Glued does not abrogate DICfunction. If the protective effect of DIC downstream ofexcitotoxicity requires DIC–p150Glued interaction, oneexplanation would be that low-rate-binding of p150Glued issufficient to facilitate retrograde cargo transport by dynein inneurons. In this case, p150Glued might localize and loadcargos onto dynein at microtubule plus-ends, or holding orrecruiting dynein to positions where ready-to-be transportedcargos are accumulated, for example, in distal neurites(Vaughan et al. 2002; Lansbergen et al. 2004; Lomakinet al. 2009). It is also possible that p150Glued and DIC exerttheir function through mutually exclusive signaling cascades.In this case, identification of interacting proteins or cargos fordynactin and dynein would help to understand p150Glued- orDIC-dependent regulation of neurodegenerative processes.Microtubule stabilization by taxol protects against excitotox-icity-induced neurodegeneration (Furukawa and Mattson1995). Thus, p150Glued and DIC could function through amicrotubule stabilization pathway. Taxol does not suppressthe generation of the p150Glued truncated form (T. Fujiwaraand K. Morimoto, unpublished observation), suggesting thatthe downstream pathways of p150Glued and taxol forprotection of neurons from degeneration are mutually exclu-sive or that the pathways activated by taxol lie downstream ofp150Glued. Concerning the latter case, we did not observealtered microtubule organization within neurites by over-expressing DC2 and DC3 mutants (Fig. 2e), therefore, it is notlikely that the effect of taxol lies downstream of p150Glued.
Glutamate or NMDA induces excitotoxicity via theactivation of ionotropic glutamate receptors. Followingexcitotoxicity, caspase 3 is activated from early time pointsin cultured hippocampal and cerebrocortical neurons toinduce apoptosis (Tenneti and Lipton 2000; Brecht et al.2001). It is clear that p150Glued and DIC play a role intransport processes, yet also regulate excitotoxicity-inducedcaspase 3-dependent apoptosis. Recent studies using dorsalroot ganglia cultured explants, as well as in mice and flieshave identified a NAD+-sensitive pathway as a parallelpathway of caspases to regulate nerve growth factor deple-tion-induced axon degeneration (Schoenmann et al. 2010).The NAD+-sensitive pathway plays a major role in axondegeneration induced by injury and trophic deprivation, anddendritic pruning during development (Schoenmann et al.2010). Thus, the NAD+-sensitive pathway could be onecandidate that operates synergistic with, or independent ofcaspase 3-dependent apoptosis in the context of excitotox-icity-induced axon degeneration, and whether p150Gluedand DIC functions are involved remains to be clarified.
Acknowledgements
We thank Drs Y. Yoneda and M. Silverman for valuable comments,Drs Y. Tsujimoto, Y. Matsuoka, T. Yamashita, Y. Shima, and D.
Pellman for their support. This work was supported in part by TheOsaka University Global COE Program, Grant-in-Aid for challeng-ing Exploratory Research from JSPS, The Collaborative ResearchProject (2011-2306) of the Brain Institute, Niigata University, TheKurata Memorial Hitachi Science and Technology Foundation,Takeda Science Foundation, and The Japan Health Foundation. Theauthors declare no conflicts of interest.
References
Ankarcrona M., Dypbukt J. M., Bonfoco E., Zhivotovsky B., OrreniusS., Lipton S. A. and Nicotera P. (1995) Glutamate-induced neu-ronal death – a succession of necrosis or apoptosis depending onmitochondrial-function. Neuron 15, 961–973.
Arai T., Hasegawa M., Akiyama H. et al. (2006) TDP-43 is a componentof ubiquitin-positive tau-negative inclusions in frontotemporallobar degeneration and amyotrophic lateral sclerosis. Biochem.Biophys. Res. Commun. 351, 602–611.
Bonfoco E., Krainc D., Ankarcrona M., Nicotera P. and Lipton S. A.(1995) Apoptosis and necrosis – 2 distinct events induced,respectively, by mild and intense insults with N-methyl-D-aspar-tate or nitric-oxide superoxide in cortical cell-cultures. Proc. NatlAcad. Sci. USA 92, 7162–7166.
Brecht S., Gelderblom M., Srinivasan A., Mielke K., Dityateva G. andHerdegen T. (2001) Caspase-3 activation and DNA fragmentationin primary hippocampal neurons following glutamate excitotoxic-ity. Mol. Brain Res. 94, 25–34.
Brewer G. J., Torricelli J. R., Evege E. K. and Price P. J. (1993) Opti-mized survival of hippocampal neurons in B27-supplementedNeurobasal, a new serum-free medium combination. J. Neurosci.Res. 35, 567–576.
Brorson J. R., Marcuccilli C. J. and Miller R. J. (1995) Delayedantagonism of calpain reduces excitotoxicity in cultured neurons.Stroke 26, 1259–1266.
Chevalier-Larsen E. and Holzbaur E. L. F. (2006) Axonal transport andneurodegenerative disease. Biochim. Biophys. Acta 1762, 1094–1108.
Coleman M. (2005) Axon degeneration mechanisms: commonality amiddiversity. Nat. Rev. Neurosci. 6, 889–898.
Conforti L., Adalbert R. and Coleman M. P. (2007) Neuronal death:where does the end begin? Trends Neurosci. 30, 159–166.
Cosker K. E., Courchesne S. L. and Segal R. A. (2008) Action in theaxon: generation and transport of signaling endosomes. Curr.Opin. Neurobiol. 18, 270–275.
De Vos K. J., Grierson A. J., Ackerley S. and Miller C. C. J. (2008) Roleof axonal transport in neurodegenerative diseases. Annu. Rev.Neurosci. 31, 151–173.
Farrer M. J., Hulihan M. M., Kachergus J. M. et al. (2009) DCTN1mutations in Perry syndrome. Nat. Genet. 41, 163–165.
Furukawa K. and Mattson M. P. (1995) Taxol stabilizes [Ca2+]i andprotects hippocampal neurons against excitotoxicity. Brain Res.689, 141–146.
Heerssen H. M., Pazyra M. F. and Segal R. A. (2004) Dynein motorstransport activated Trks to promote survival of target-dependentneurons. Nat. Neurosci. 7, 596–604.
Hirokawa N., Noda Y., Tanaka Y. and Niwa S. (2009) Kinesin super-family motor proteins and intracellular transport. Nat. Rev. Mol.Cell Biol. 10, 682–696.
Kuta A., Deng W. H., El-Kadi A. M., Banks G. T., Hafezparast M.,Pfister K. K. and Fisher E. M. C. (2010) Mouse cytoplasmic dyneinintermediate chains: identification of new isoforms, alternativesplicing and tissue distribution of transcripts. PLoS ONE 5,e11682.
Journal of Neurochemistry � 2012 International Society for Neurochemistry, J. Neurochem. (2012) 10.1111/j.1471-4159.2012.07746.x� 2012 The Authors
12 | T. Fujiwara et al.

Lafon-cazal M., Pietri S., Culcasi M. and Bockaert J. (1993) NMDA-dependent superoxide production and neurotoxicity. Nature 364,535–537.
Lai C., Lin X., Chandran J., Shim H., Yang W. J. and Cai H. B. (2007)The G59S mutation in p150(glued) causes dysfunction of dynactinin mice. J. Neurosci. 27, 13982–13990.
Laird F. M., Farah M. H., Ackerley S. et al. (2008) Motor neurondisease occurring in a mutant dynactin mouse model is charac-terized by defects in vesicular trafficking. J. Neurosci. 28, 1997–2005.
LaMonte B. H., Wallace K. E., Holloway B. A., Shelly S. S., Ascano J.,Tokito M., Van Winkle T., Howland D. S. and Holzbaur E. L. F.(2002) Disruption of dynein/dynactin inhibits axonal transport inmotor neurons causing late-onset progressive degeneration. Neuron34, 715–727.
Lansbergen G., Komarova Y., Modesti M. et al. (2004) Conformationalchanges in CLIP-170 regulate its binding to microtubules anddynactin localization. J. Cell Biol. 166, 1003–1014.
Lau A. and Tymianski M. (2010) Glutamate receptors, neurotoxicityand neurodegeneration. Pflugers. Arch-Eur. J. Physiol. 460, 525–542.
Leuba G., Vernay A., Zimmermann V., Saini K., Kraftsik R. and SaviozA. (2009) Differential damage in the frontal cortex with aging,sporadic and familial Alzheimer’s disease. Brain Res. Bull. 80,196–202.
Lomakin A. J., Semenova I., Zaliapin I., Kraikivski P., Nadezhdina E.,Slepchenko B. M., Akhmanova A. and Rodionov V. (2009) CLIP-170-dependent capture of membrane organelles by microtubulesinitiates minus-end directed transport. Dev. Cell 17, 323–333.
Mackenzie I. R. A., Bigio E. H., Ince P. G. et al. (2007) PathologicalTDP-43 distinguishes sporadic amyotrophic lateral sclerosis fromamyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol.61, 427–434.
Munch C., Sedlmeier R., Meyer T. et al. (2004) Point mutations of thep150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63,724–726.
Neumann M., Sampathu D. M., Kwong L. K. et al. (2006) UbiquitinatedTDP-43 in frontotemporal lobar degeneration and amyotrophiclateral sclerosis. Science 314, 130–133.
Perlson E., Maday S., Fu M. M., Moughamian A. J. and Holzbaur E. L.F. (2010) Retrograde axonal transport: pathways to cell death?Trends Neurosci. 33, 335–344.
Puls I., Jonnakuty C., LaMonte B. H. et al. (2003) Mutant dynactin inmotor neuron disease. Nat. Genet. 33, 455–456.
Raff M. C., Whitmore A. V. and Finn J. T. (2002) Neuroscience –axonal self-destruction and neurodegeneration. Science 296, 868–871.
Reid E., Kloos M., Ashley-Koch A. et al. (2002) A kinesin heavy chain(KIF5A) mutation in hereditary spastic paraplegia (SPG10). Am. J.Hum. Genet. 71, 1189–1194.
Sattler R., Charlton M. P., Hafner M. and Tymianski M. (1998) Distinctinflux pathways, not calcium load, determine neuronal vulnera-bility to calcium neurotoxicity. J. Neurochem. 71, 2349–2364.
Sattler R., Xiang Z. G., Lu W. Y., Hafner M., MacDonald J. F. andTymianski M. (1999) Specific coupling of NMDA receptor acti-vation to nitric oxide neurotoxicity by PSD-95 protein. Science284, 1845–1848.
Schoenmann Z., Assa-Kunik E., Tiomny S., Minis A., Haklai-Topper L.,Arama E. and Yaron A. (2010) Axonal degeneration is regulatedby the apoptotic machinery or a NAD(+)-sensitive pathway ininsects and mammals. J. Neurosci. 30, 6375–6386.
Takeuchi H., Mizuno T., Zhang G. Q., Wang J. Y., Kawanokuchi J.,Kuno R. and Suzumura A. (2005) Neuritic beading induced byactivated microglia is an early feature of neuronal dysfunctiontoward neuronal death by inhibition of mitochondrial respirationand axonal transport. J. Biol. Chem. 280, 10444–10454.
Tenneti L. and Lipton S. A. (2000) Involvement of activated caspase-3-like proteases in N-methyl-D-aspartate-induced apoptosis in cere-brocortical neurons. J. Neurochem. 74, 134–142.
Tenneti L., D’Emilia D. M., Troy C. M. and Lipton S. A. (1998) Role ofcaspases in N-methyl-D-aspartate-induced apoptosis in cerebro-cortical neurons. J. Neurochem. 71, 946–959.
Vaughan P. S., Leszyk J. D. and Vaughan K. T. (2001) Cytoplasmicdynein intermediate chain phosphorylation regulates binding todynactin. J. Biol. Chem. 276, 26171–26179.
Vaughan P. S., Miura P., Henderson M., Byrne B. and Vaughan K. T.(2002) A role for regulated binding of p150(Glued) to microtubuleplus ends in organelle transport. J. Cell Biol. 158, 305–319.
Washbourne P., Bennett J. E. and McAllister A. K. (2002) Rapidrecruitment of NMDA receptor transport packets to nascent syn-apses. Nat. Neurosci. 5, 751–759.
Zhao C., Takita J., Tanaka Y. et al. (2001) Charcot-Marie-Tooth diseasetype 2A caused by mutation in a microtubule motor KIF1B beta.Cell 105, 587–597.
� 2012 The AuthorsJournal of Neurochemistry � 2012 International Society for Neurochemistry, J. Neurochem. (2012) 10.1111/j.1471-4159.2012.07746.x
Dynein and dynactin in excitotoxicity | 13





























