Embed Size (px)
Citation preview

Drug Discovery OverviewDrug Discovery Overview
Hits to the Clinic
1 Chatterjee – Lecture at TSRI Oct 29 2014

Outline
• Medicinal chemistry strategies– Physiochemical properties – General terms– On-target versus off-tissue effectsg– Target-based optimization case study
• Mdm-2/p53– Cell-based optimization case study
Leeson and SprinhthorpeNature Reviews Drug
• Malaria– Pharmacokinetic optimization
• Effects on cardiotoxicity (S1P1 agonists)
Discovery, 2007, 881-890.
• Take home messages– Principals for optimization
• Structure-based and physiochemical property based• Pharmacokinetic based (and interplay with toxicity)
– Understanding compound progression schemes• Lots of compounds so need clear benchmarks for success (attrition-based
strategy)
2 Chatterjee – Lecture at TSRI Oct 29 2014
strategy)• Close interplay between chemistry, biology, pharmacology, toxicology

S1P1 Receptor and Immune Diseases
Mechanism of Action• S1P Receptor agonists such as FTY720 (spingolipids) p g ( p g p )
leads to S1P1 down-regulation and prevents lymphocyte egress. Clin Neuropharmacol. 2010 ; 33(2): 91–101.
Key issuesKey issues• On-target bradychardia (slow heart rate defined as less
than 60 bpm) upon first dosingp ) p g– Likely a Cmax effect
• Truly on-target toxicity? S1P3 receptor• Can one modulate this effect my pushing out Tmax and
“flattening the PK curve”?
3 Chatterjee – Lecture at TSRI Oct 29 2014

Physicochemical properties for BBB permeability
Multiple-color band Radar chart, a tool for lead optimization. Here the combined shaded area represents the property ranges of 95% of approved CNS drugs The green bandapproved CNS drugs. The green band represents the most densely populated property range containing 50% of the approved CNS drugs. The red line represents the properties of the test molecule (amitriptyline, a tertiary amine tricyclic antidepressant, is structurally related to both the skeletal muscle relaxant cyclobenzaprine and the thioxanthene antipsychotics such as thiothixene). During lead optimization, it isthiothixene). During lead optimization, it is often found that even after ROF type rules have been satisfied the lead optimization process does not achieve the goal. In such cases, optimizing the physicochemical
ti t d th i hproperties toward the green region may have a better chance of success.
< ACS Chem Neurosci 2012 3 50−68 >
4 Chatterjee – Lecture at TSRI Oct 29 2014
< ACS Chem. Neurosci. 2012, 3, 50−68 >

Check points for the CNS drugs to Penetrate Blood Brain Barrier
Free Drug HypothesisFree Drug Hypothesis(total clearance adjusted for free fraction)
5 Chatterjee – Lecture at TSRI Oct 29 2014

Structure-based Design• p53/MDM-2 interaction blocker
– p53 is key tumor suppressor and 1st gen compound mimic MDM-2 peptide (though the interactions span ~120 residues and subsequent
tid d t h d l )peptides do not have good oral exposure)– HTS led to nutlins and subsequent optimization
• Top. Med Chem 2012, 8; 57-80.
6 Chatterjee – Lecture at TSRI Oct 29 2014
Flexible pocket
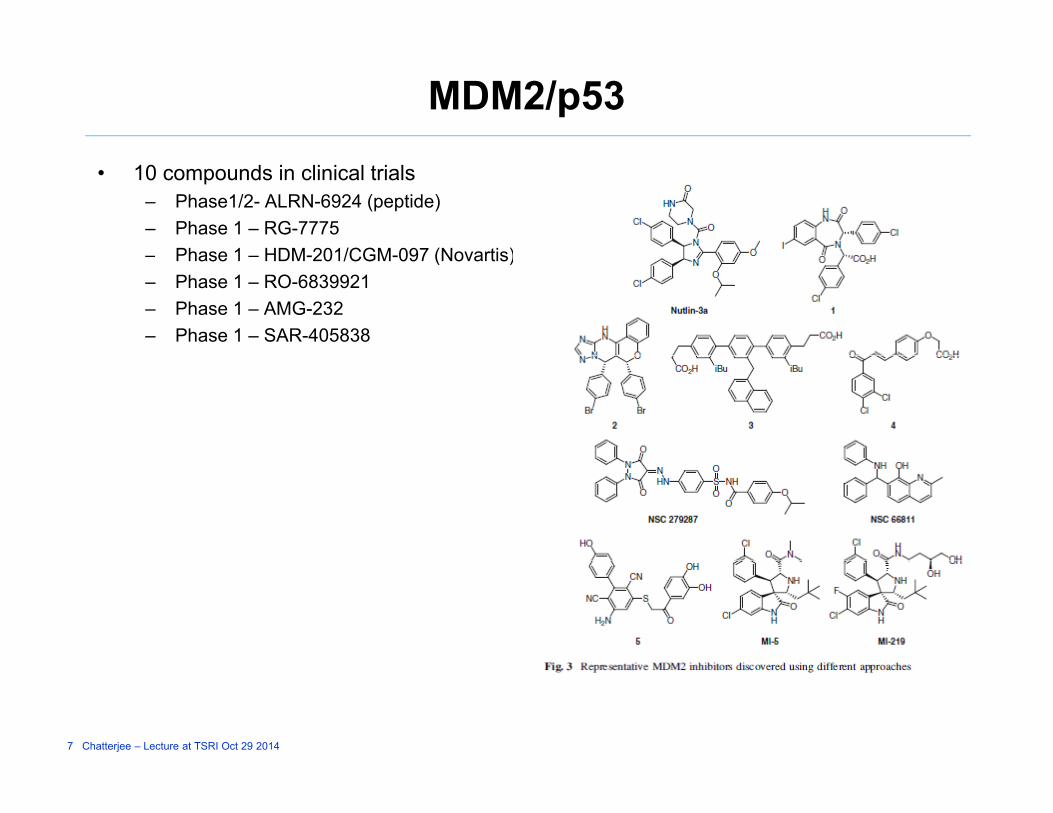
MDM2/p53
• 10 compounds in clinical trials– Phase1/2- ALRN-6924 (peptide)– Phase 1 – RG-7775
Phase 1 HDM 201/CGM 097 (Novartis)– Phase 1 – HDM-201/CGM-097 (Novartis)– Phase 1 – RO-6839921– Phase 1 – AMG-232– Phase 1 – SAR-405838
7 Chatterjee – Lecture at TSRI Oct 29 2014

Protease inhibitors
Examples of efficient bond-constructionHCV NS3/4 protease inhibitor (MK-7009)p ( )
O OConvergent olefinON
NH
ON
NHHNConvergent olefin cross-metathesis
8 Chatterjee – Lecture at TSRI Oct 29 2014
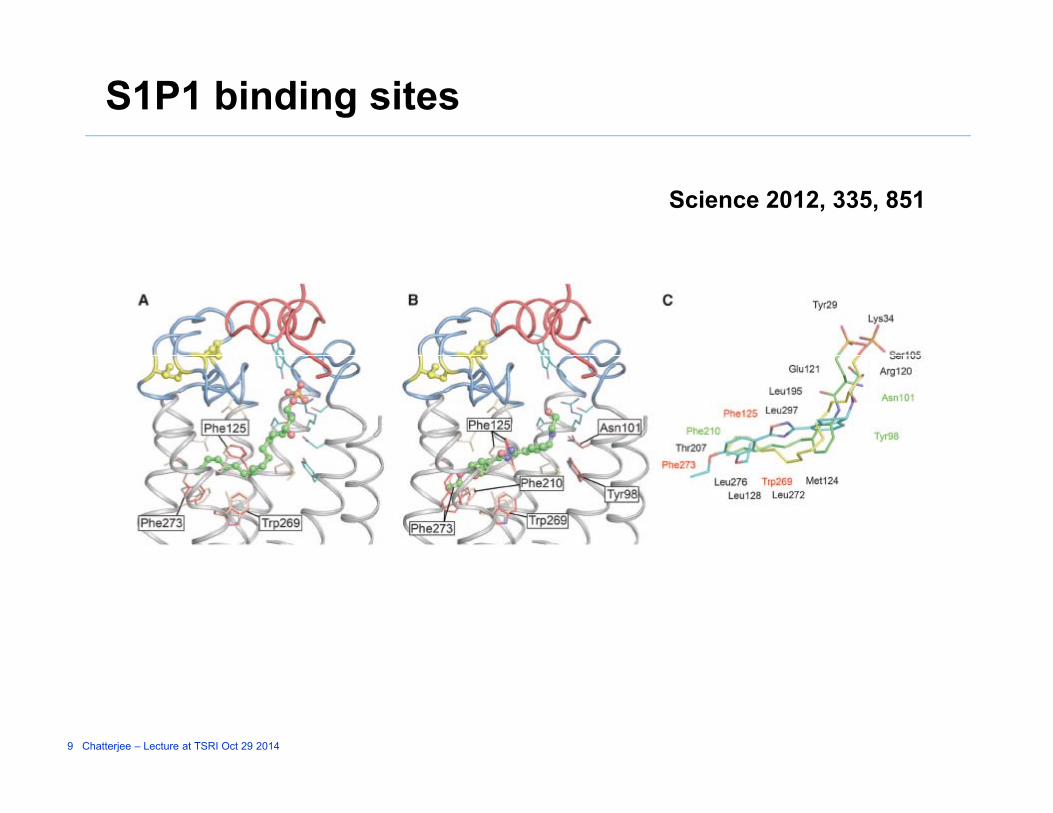
S1P1 binding sites
Science 2012, 335, 851
9 Chatterjee – Lecture at TSRI Oct 29 2014

Current Clinical Compounds FTY720
BAF312 (Phase 3 – Novartis)200 nM
Bradyarrhythmia at 10 mg/dayy y g y
SEW2871 (HTS hit J. Biol. Chem 2004, 13839.)
WO 2012/158550
20 pM20h human t1/2Tmax pushed out (1 7h versus 7h)WO 2012/158550WO 2011/060392 (1.7h versus 7h)
10 Chatterjee – Lecture at TSRI Oct 29 2014

Cell-based Optimization of Novel Anti-parasitics
11 Chatterjee – Lecture at TSRI Oct 29 2014

Drug Discovery Infrastructure
Clinic
Candidate Selection Point
D3D2D1D0
TargetValidation HTS
ExploratoryChemistry
Full LeadOptimization
Tox studies
Lead Discovery
D3D2D1D0
Medicinal & Analytical Chemistry Pharmacology/ADMETLead Discovery• Large, diverse compound library• Fully robotic uHTS system capable
of >2 million data points/day• Automated compound profiling
system
Medicinal & Analytical Chemistry• Automated library synthesis• Experienced medicinal chemistry
staff• High-throughput analytical and
purification technology
Pharmacology/ADMET• In vitro and in vivo ADMET
capabilities• State-of-the-art bioanalytical
technology• In vivo efficacy models insystem
• Screening informatics infrastructure (LDDB)
purification technology• Sophisticated compound
management
• In vivo efficacy models in metabolic disease, immunology and oncology
12 Chatterjee – Lecture at TSRI Oct 29 2014

Infectious Diseases of the Developing World
• Objective– Identification of new pharmacophore drug candidates
to improve treatment options for infectious diseases in low and middle income countries
Mosquito P.falciparum
low and middle income countries• Scientific Rationale
– Prioritized disease targets where current drugs are toxic/ ineffective and current drug discovery activities
Human M.tuberculosis
are low relative to medical burden– Deprioritized where other approaches are likely to be
more effective (vaccines, sanitation)– Programs focused on Malaria Tuberculosis
Sand fly L.donovani
Programs focused on Malaria, Tuberculosis, Kinetoplastid Diseases
• Approach– Reduction in burden of infectious agent will lead to
Kissing bug T.cruzi
improved clinical outcomes– Reliance on phenotypic screens for hit finding– Deconvolution of mechanism of resistance through
genetic selectionTsetse fly T.brucei
13 Chatterjee – Lecture at TSRI Oct 29 2014
genetic selection

Scope and Scientific Approaches
• Whole pathogen cell-based screening approach– Majority of anti-infectives discovered through cell-based screening
• Facilitates simultaneous interrogation of multiple nodes/ pathways• Consideration of compound permeability and metabolism in-built
– Target-based approaches have been less successful, including internal programs– HTS on >2 million compounds for all pathogens, with broad compound profiling in secondary
assays– Closely coordinated chemistry, pharmacology and biology teams
• Target identification methods– Lab-evolved strains resistant to compound treatment followed by genetic analysisab e o ed s a s es s a o co pou d ea e o o ed by ge e c a a ys s– Promoter signature profiling– Compound affinity pull-downs
• Ensures no shared in vitro resistance mechanism with current drugs • Target identification enables biochemical optimization approach for follow-on compounds• Target identification enables biochemical optimization approach for follow-on compounds
Presentation will highlight work in Malaria, Leishmania and Chagas Disease
14 Chatterjee – Lecture at TSRI Oct 29 2014
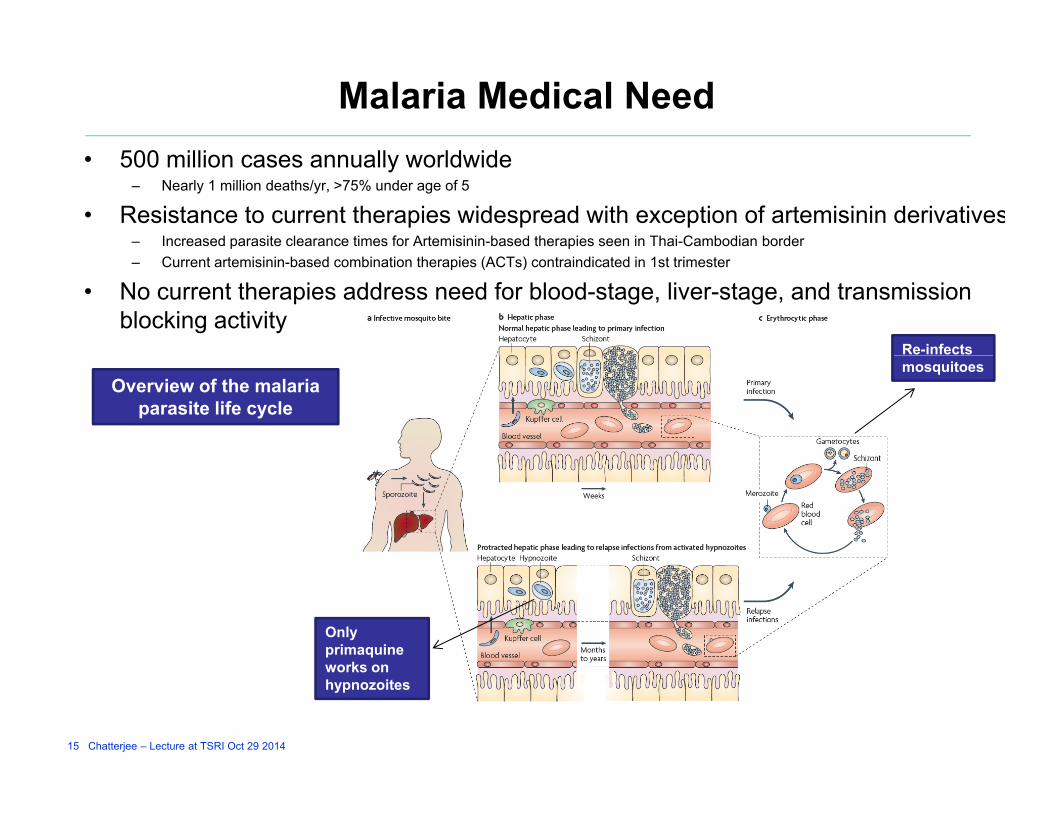
Malaria Medical Need00 illi ll ld id• 500 million cases annually worldwide– Nearly 1 million deaths/yr, >75% under age of 5
• Resistance to current therapies widespread with exception of artemisinin derivatives– Increased parasite clearance times for Artemisinin-based therapies seen in Thai-Cambodian border– Current artemisinin-based combination therapies (ACTs) contraindicated in 1st trimester
• No current therapies address need for blood-stage, liver-stage, and transmission blocking activity
Re-infects
Overview of the malaria parasite life cycle
mosquitoes
Only primaquine
k
15 Chatterjee – Lecture at TSRI Oct 29 2014
works on hypnozoites

The NGBS consortium
• Novartis Institute for Tropical Diseases (NITD), Singapore– Team leaders: Bryan Yeung, Zou Bin, Christophe Bodenreider
• Genomic Institute of the Novartis Research Foundation (GNF)– Team leaders: Arnab Chatterjee, Kelli Kuhen and Elizabeth Winzeler
• Biomedical Primates Research Center (BPRC), Rijswijk (NL)– Team leader: Clemens Kocken
• Swiss Tropical Institute (STI), Basel (CH)– Team leader: Matthias Rottmann
• The NGBS (NITD GNF BPRC STI) consortium• The NGBS (NITD, GNF, BPRC, STI) consortium – Program Head: Thierry Diagana (NITD)
• NGBS is funded by:
16 Chatterjee – Lecture at TSRI Oct 29 2014

GNF Malaria Cell-based Screening Summary
• > 2M compounds screened at 1.25 M (3d7)
• 4851 hits < 1.2 M EC50 vs W2 and/or 3d7 – “Malaria Box” public resource for
l i h itmalaria research community– https://www.ebi.ac.uk/chembldb/index.p
hp/compound
• 1,256 < 200 nM EC50 vs 3d7
• >200 scaffolds represented in the reconfirmed compound set
• A much smaller subset active on liver-stages gof infection ~270 cmpds
How to best prioritize h i t ?chemistry resources?
Bl d t HTS t d i Pl ff d k PNAS 2008 9059
17 Chatterjee – Lecture at TSRI Oct 29 2014
Blood-stage HTS reported in: Plouffe and co-workers PNAS 2008, 9059.
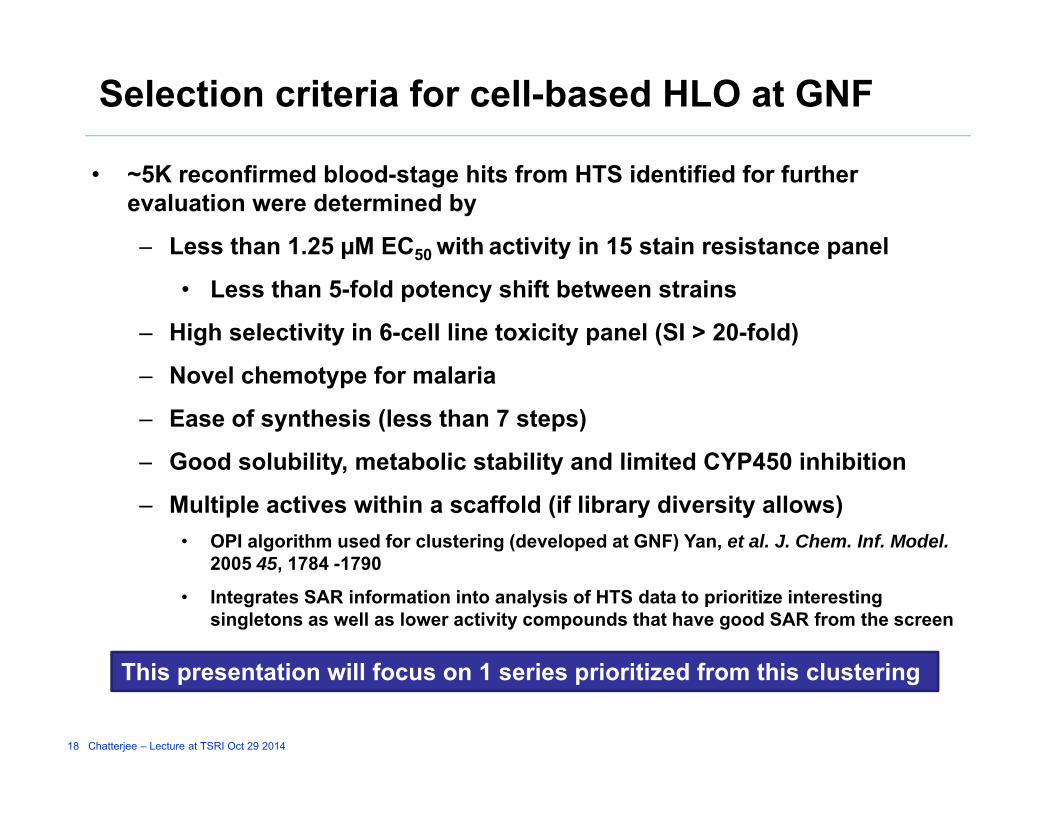
Selection criteria for cell-based HLO at GNF
• ~5K reconfirmed blood-stage hits from HTS identified for further evaluation were determined by
– Less than 1.25 µM EC50 with activity in 15 stain resistance panelLess than 1.25 µM EC50 with activity in 15 stain resistance panel
• Less than 5-fold potency shift between strains
– High selectivity in 6-cell line toxicity panel (SI > 20-fold)
– Novel chemotype for malaria
– Ease of synthesis (less than 7 steps)
Good solubility metabolic stability and limited CYP450 inhibition– Good solubility, metabolic stability and limited CYP450 inhibition
– Multiple actives within a scaffold (if library diversity allows)• OPI algorithm used for clustering (developed at GNF) Yan, et al. J. Chem. Inf. Model.
2005 45 1784 17902005 45, 1784 -1790
• Integrates SAR information into analysis of HTS data to prioritize interesting singletons as well as lower activity compounds that have good SAR from the screen
Thi i ill f 1 i i i i d f hi l i
18 Chatterjee – Lecture at TSRI Oct 29 2014
This presentation will focus on 1 series prioritized from this clustering

Imidazolopiperazines Hit series
GNF-Pf-5069 GNF-Pf-5179 GNF-Pf-5466
EBI ID HTS W2 EC50
HTS 3D7EC50
HTS Huh7EC50
Powder W2EC50
Powder 3D7EC50
Powder Huh7 EC50
(µM) (µM) (µM) (µM) (µM) (µM)GNF-Pf-5069
0.097 0.063 >10 0.198, 0.473
0.161,0.46
>100
GNF-Pf-5179
0.271 0.235 >10 0.122 0.119 >79
GNF-Pf-5466
0.119 0.116 >10 0.029 0.030 42.82
19 Chatterjee – Lecture at TSRI Oct 29 2014
5466

Imidazolopiperazines Hit Series
• Strengths– Unique scaffold for malaria, no biological activity
annotation with structureannotation with structure– 4 step synthesis– Very good solubility; no activity on Cyp450sy g y; y yp– Not identified in >100 HTS screens performed at GNF
• Issues– Moderate in vitro potency – In vivo snapshot PK: poor oral exposure
• Likely due to catechol and glycinamideLikely due to catechol and glycinamide– hERG inhibition (35% at 10 M, patch clamp)
20 Chatterjee – Lecture at TSRI Oct 29 2014
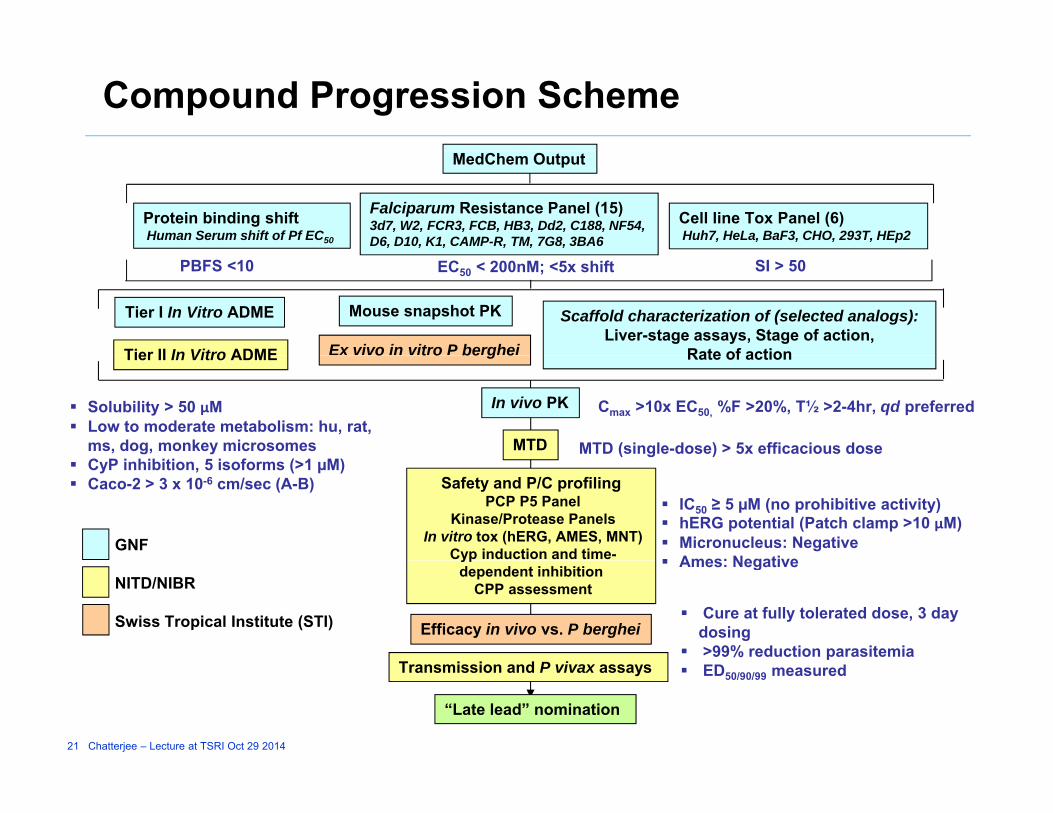
Compound Progression Scheme
Cell line Tox Panel (6) Huh7, HeLa, BaF3, CHO, 293T, HEp2
Protein binding shift Human Serum shift of Pf EC50
Falciparum Resistance Panel (15)3d7, W2, FCR3, FCB, HB3, Dd2, C188, NF54, D6, D10, K1, CAMP-R, TM, 7G8, 3BA6
MedChem Output
EC50 < 200nM; <5x shiftPBFS <10 SI > 50
Ex vivo in vitro P berghei
Tier I In Vitro ADME
Tier II In Vitro ADME
Mouse snapshot PK Scaffold characterization of (selected analogs):Liver-stage assays, Stage of action,
Rate of action
Solubility > 50 µM Low to moderate metabolism: hu, rat,
ms, dog, monkey microsomes MTD (single-dose) > 5x efficacious dose
Cmax >10x EC50, %F >20%, T½ >2-4hr, qd preferred
MTD
In vivo PK
o t o be g eTier II In Vitro ADME Rate of action
CyP inhibition, 5 isoforms (>1 µM) Caco-2 > 3 x 10-6 cm/sec (A-B)
IC50 ≥ 5 µM (no prohibitive activity) hERG potential (Patch clamp >10 µM) Micronucleus: Negative Ames: Negative
GNF
Safety and P/C profilingPCP P5 Panel
Kinase/Protease PanelsIn vitro tox (hERG, AMES, MNT)
Cyp induction and time-
Efficacy in vivo vs. P berghei Cure at fully tolerated dose, 3 day
dosing >99% reduction parasitemia
Ames: Negative NITD/NIBR
Swiss Tropical Institute (STI)
ypdependent inhibition
CPP assessment
T i i d P i
21 Chatterjee – Lecture at TSRI Oct 29 2014
“Late lead” nomination
ED50/90/99 measuredTransmission and P vivax assays

Hit-to-lead optimization Summary
H2N
O NN
N
NH
O O
• GNF-Pf-5069– Pf 3D7 (EC50): 460 nM– hERG (binding) IC50 = 19 µM
• Compound GNF776– Pf 3D7 (EC50): 20 nM– hERG (binding) IC50 = 3.95 µM
– Mouse metabolic stability (ER): 0.24– Solubility (pH 6.8): > 175 µM– PO Cmax (D.N.) = 16 nM / (mg/kg)– PO AUC0 5h (D.N.) = 48.6 h*nM / (mg/kg)
– CL = 60 mL/min/kg; Vss = 23.7 L/kg; T½ = 6.2 h – PO Cmax (D.N.) = 60 nM / (mg/kg) – PO AUC0-inf (D.N.) = 596.5 h*nM / (mg/kg)– F (%) = 87
0-5h ( ) ( g g)Poor exposure limited any further work – Efficacy achieved in p berghei mouse model
(*D.N.: dose normalized) Wu, et.al. J. Med. Chem. 2011, 5116.
22 Chatterjee – Lecture at TSRI Oct 29 2014
Still need to improve hERG and potency

Groebke-Blackburn Cyclization Route
• 4 steps and 53% overall yield• No column chromatography
P falciparum 3D7 EC50 = 30 nM
23 Chatterjee – Lecture at TSRI Oct 29 2014

Imidazolopiperazine Core Optimization8,8-dimethyl substitution significantly improves profile
• GNF268– Pf W2 (EC50): 9 nM– hERG (binding) IC50 = 6.6 µM
CL 1179 L/ i /k V 106 76 L/k
• GNF179– Pf W2 (EC50): 6 nM– hERG (binding) IC50 = 7.2 µM
CL 21 92 L/ i /k V 11 8 L/k– CL = 1179 mL/min/kg; Vss = 106.76 L/kg– Oral T½ = 2.2 h– PO Cmax (D.N.) = 3.3 nM / (mg/kg)– PO AUCinf (D.N.) = 30 hr*nM
– CL = 21.92 mL/min/kg; Vss = 11.8 L/kg– Oral T½ = 8.4 h– PO Cmax (D.N.) = 60.5 nM / (mg/kg)– PO AUCinf (D.N.) = 1035 hr*nM (mg/kg)
– F (%) = 17– 30x cross-resistance to GNF-Pf-5069 lab
evolved resistance strain
– F (%) = 58– 7x cross-resistance to GNF-Pf-5069 lab
evolved resistance strain(*D.N.: dose normalized)
24 Chatterjee – Lecture at TSRI Oct 29 2014
gem-Dimethyl group on piperazine improves potency and PK in mice
J. Med. Chem. 2012, in press.

SAR Summary• Optimization of the Imidazolopiperazine scaffold:
– Introduction of 8,8-dimethylgroup on piperazine improves in vitro and in vivopotency by 10-fold
– Required glycine functionality is primary cause for hERG activity– Required glycine functionality is primary cause for hERG activity
• Aryl required• Para and meta substituted aromatic preferred
Amino acid SAR•NH2 preferred; but also can use NHR, NR2, OH and alkyl groups
H2N
O NN
NF
• Ortho substituted aromatic not preferred• Strong electron-withdrawing group not preferred
• 1-2 carbon spacer to NH2 preferred•Dimethylglycine improves in vivo PK relative to glycine (mouse and rat)• Glycine least active on hERG (2-3 fold), while all other amino acids in similar range
NH
F• Aniline required with unsubstituted piperazine series• Nitrogen required but N H is not in
Piperazine ring SAR• Nitrogen required, but N-H is not in unsubstituted piperazines• Variety of aromatics help potency (para and meta preferred)• Heteroaromatics and cycloaliphatics not active in unsubstituted piperazine series, but
• 8-substitution helps with potency and mouse exposure (dimethyl > valine, alanine > hydrogen) • 5-dimethyl and 6-dimethyl moderately helps potency, no real effect on PK• Cyclic lactams (6 oxo) derivatives active with
25 Chatterjee – Lecture at TSRI Oct 29 2014
active with 8-dimethyl groups• Can modestly effect hERG activity
• Cyclic lactams (6-oxo) derivatives active with 8,8-dimethyl substitution and reduces metabolism

hERG Optimization Targeting the amine functionality
P. falciparum 3D7 EC50 = 50 nM
Modulate amine basicity:
GNF824
Solubility (pH 6 8) = 34 mg/L
hERG (Dofetilide binding) IC50 = 12.47 M
pKa = 3.3Glycine amine pKa = 7.5
Solubility (pH 6.8) = 34 mg/L
Replacement of amine with alcohol:
hERG (Dofetilide binding)
P falciparum 3D7 EC50 = 300 nM OptimizeP. falciparum
potency
GNF711
hERG (Dofetilide binding) IC50 > 30 M
Solubility (pH 6.8) = 110 mg/L P. falciparum 3D7 EC50= 20 nMGNF557
26 Chatterjee – Lecture at TSRI Oct 29 2014
Unable to progress due to poor metabolic stability and Log D
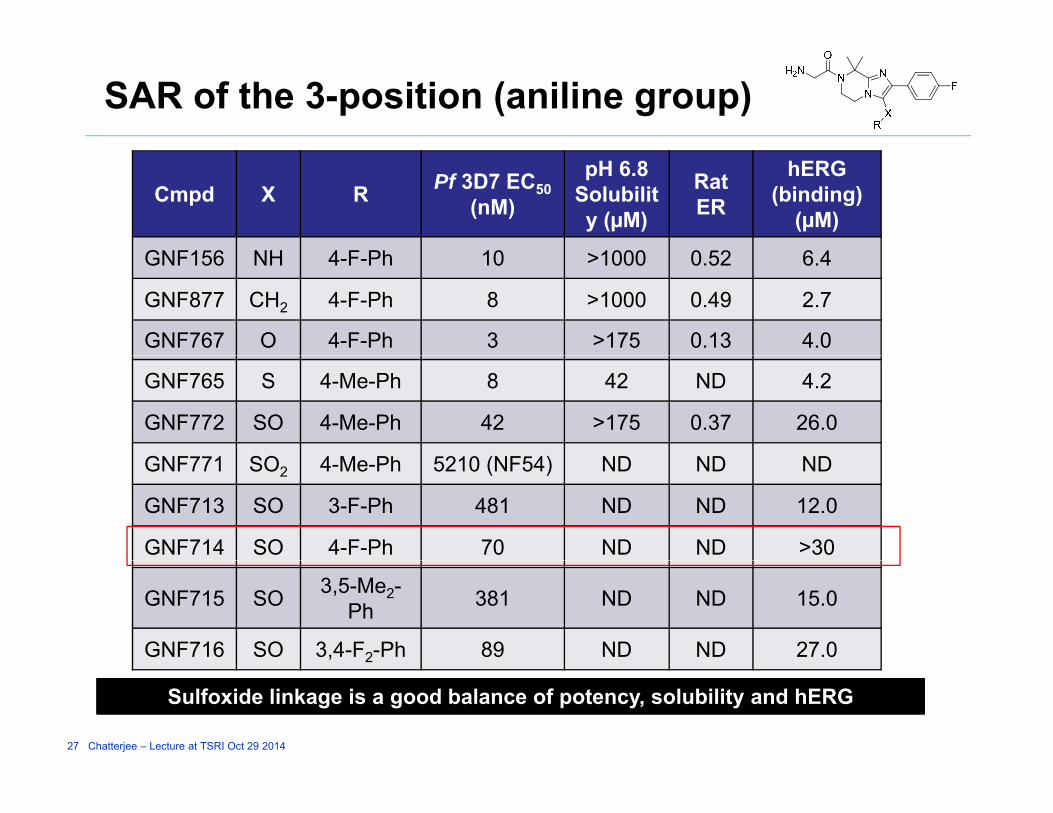
SAR of the 3-position (aniline group)
Cmpd X R Pf 3D7 EC50 (nM)
pH 6.8 Solubility (µM)
Rat ER
hERG (binding)
(µM)
GNF156 NH 4 F Ph 10 >1000 0 52 6 4GNF156 NH 4-F-Ph 10 >1000 0.52 6.4
GNF877 CH2 4-F-Ph 8 >1000 0.49 2.7
GNF767 O 4-F-Ph 3 >175 0.13 4.0
GNF765 S 4-Me-Ph 8 42 ND 4.2
GNF772 SO 4-Me-Ph 42 >175 0.37 26.0
GNF771 SO 4 M Ph 5210 (NF54) ND ND NDGNF771 SO2 4-Me-Ph 5210 (NF54) ND ND ND
GNF713 SO 3-F-Ph 481 ND ND 12.0
GNF714 SO 4-F-Ph 70 ND ND >30
GNF715 SO 3,5-Me2-Ph 381 ND ND 15.0
GNF716 SO 3,4-F2-Ph 89 ND ND 27.0
27 Chatterjee – Lecture at TSRI Oct 29 2014
Sulfoxide linkage is a good balance of potency, solubility and hERG

Profile of Preclinical candidate: GNF156
• In vitro Activity Profile of Imidazolopiperazines– P. falciparum W2 potency: EC50 = 0.006 μM– Compound exhibits < 5-fold shift across 15 strains– Selectivity vs. Huh7 cells: >10,000 fold– Complete transmission blocking at 500 nM
• In vivo ProfileG d l PK/bi il bilit d T > 4 h
GNF156
– Good oral PK/bioavailability and exposure; T1/2 > 4 h– In vivo efficacy in P. berghei mouse model: ED99 = 2.2 mg/kg– Causal prophylaxis data: Fully protective at 20 mg/kg
• ADME/Safety/Physicochemical ProfileADME/Safety/Physicochemical Profile– ADMET: solubility: >1000 µM @ pH1 and @ pH 6.8; no GSH trapping; no Cyp
induction or phototoxicity flags– Clean PCP receptor panel, Ames, MNT and good metabolic stability– Formulation suitable for tox studies identified (35x exposure multiples achieved in
rats and 16x in dogs) – hERG activity at 6.6 µM in binding assay; 13 µM in automated patch clamp assay
• Unique activity for blood/liver-stage infections and transmission blocking
28 Chatterjee – Lecture at TSRI Oct 29 2014
Unique activity for blood/liver stage infections and transmission blocking

GNF156: Current Synthesis Route
10 step route, but minimal purifications
29 Chatterjee – Lecture at TSRI Oct 29 2014
27% overall yield Low cost of goods for starting materials
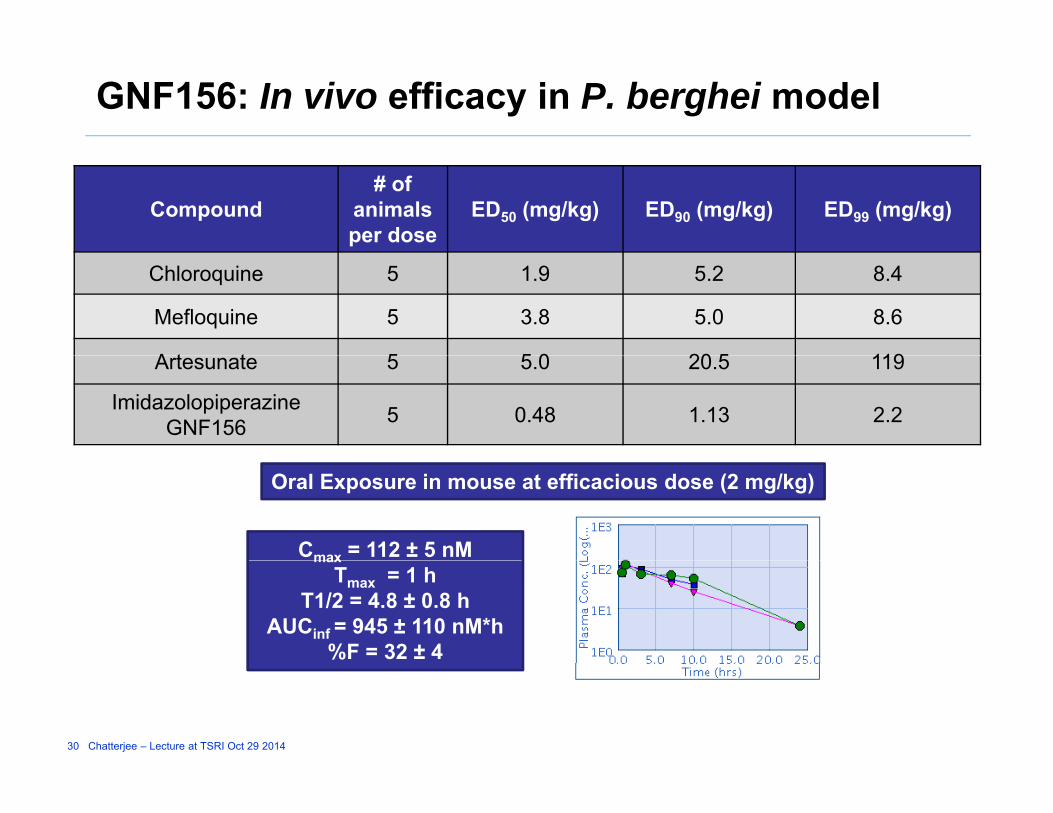
GNF156: In vivo efficacy in P. berghei model
Compound# of
animals per dose
ED50 (mg/kg) ED90 (mg/kg) ED99 (mg/kg)
Chloroquine 5 1.9 5.2 8.4
Mefloquine 5 3.8 5.0 8.6
A t t 5 5 0 20 5 119Artesunate 5 5.0 20.5 119
ImidazolopiperazineGNF156 5 0.48 1.13 2.2
Oral Exposure in mouse at efficacious dose (2 mg/kg)
Cmax = 112 ± 5 nMmaxTmax = 1 h
T1/2 = 4.8 ± 0.8 hAUCinf = 945 ± 110 nM*h
%F = 32 ± 4
30 Chatterjee – Lecture at TSRI Oct 29 2014
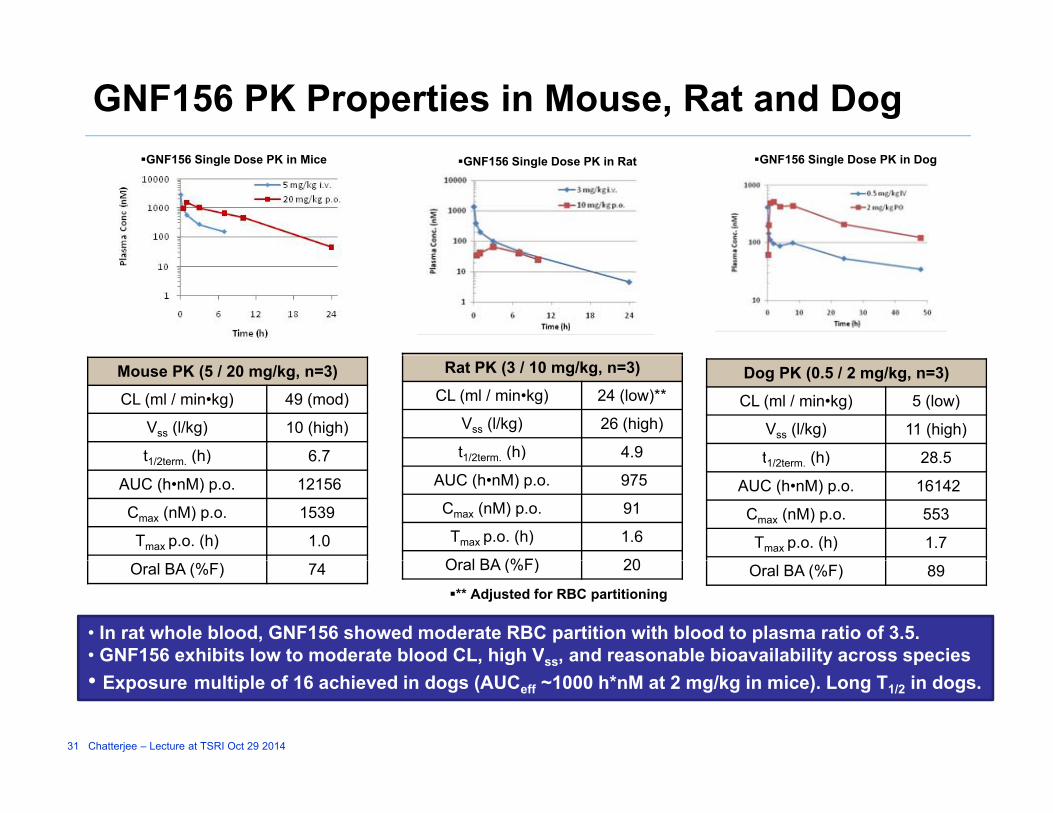
GNF156 PK Properties in Mouse, Rat and DogGNF156 Single Dose PK in Mice GNF156 Single Dose PK in Rat GNF156 Single Dose PK in Dog
Mouse PK (5 / 20 mg/kg, n=3)
CL (ml / min•kg) 49 (mod)
Vss (l/kg) 10 (high)
t1/2t (h) 6 7
Rat PK (3 / 10 mg/kg, n=3)
CL (ml / min•kg) 24 (low)**
Vss (l/kg) 26 (high)
t1/2term (h) 4.9
Dog PK (0.5 / 2 mg/kg, n=3)
CL (ml / min•kg) 5 (low)
Vss (l/kg) 11 (high)
t (h) 28 5t1/2term. (h) 6.7
AUC (h•nM) p.o. 12156
Cmax (nM) p.o. 1539
Tmax p.o. (h) 1.0
1/2term. ( )
AUC (h•nM) p.o. 975
Cmax (nM) p.o. 91
Tmax p.o. (h) 1.6
O l BA (%F) 20
t1/2term. (h) 28.5
AUC (h•nM) p.o. 16142
Cmax (nM) p.o. 553
Tmax p.o. (h) 1.7 Oral BA (%F) 74 Oral BA (%F) 20 Oral BA (%F) 89
• In rat whole blood, GNF156 showed moderate RBC partition with blood to plasma ratio of 3.5.• GNF156 exhibits low to moderate blood CL, high Vss, and reasonable bioavailability across species
** Adjusted for RBC partitioning
31 Chatterjee – Lecture at TSRI Oct 29 2014
, g ss, y p• Exposure multiple of 16 achieved in dogs (AUCeff ~1000 h*nM at 2 mg/kg in mice). Long T1/2 in dogs.

GNF156: Human efficacious dose projectionsSpecies Units Mice Rat Dog Human
Body weight kg 0.025 0.25 8.7 70
Dose mg/kg 5 3 0.5 1
Vss L/kg 10.2 25.7 10.9 10.2 to 25.7
CL mL/min/kg 49.2 84.4 4.8 0.9 to 4.5
T1/2 h 2 4 9 28 5 14 5 to 71T1/2 h 2 4.9 28.5 14.5 to 71
F % 66 47 89 67
Efficacy : ED99 in mice for KAF156 is 2.2 mg/kg.y 99 g g
Bioavailability : F was estimated to be 67% in humans
Clearance : 0.9 to 4.5 ml/min/kg using various methods.
Human efficacious dose (to show similar efficacy to the mouse 99% parasitemia reduction) Human efficacious dose (to show similar efficacy to the mouse 99% parasitemia reduction)was predicted to be 2.3 to 48mg based on the following assumption
Efficacy is similar in P.b infected mice and P.f infected human
PK is similar in normal mice and P b infected mice
32 Chatterjee – Lecture at TSRI Oct 29 2014
PK is similar in normal mice and P.b infected mice

Imidazolopiperazines Block Transmission
Stage V gametocyte transmissionmosquito feeding experimental procedure:
Novel activity for a potent blood-stage antimalarial
mosquito feeding experimental procedure:1. The compounds are added to a bloodmeal
with NF54 gametocytes2. Six days after the fed mosquitoes are
dissected and checked for oocysts3 Compounds can have an effect on3. Compounds can have an effect on
mosquitoes, therefore the mortality of the mosquitoes is checked every day
% infected mosquitoes # Oocysts/ mosquito
Control 95 49 • GNF776 (lead cmpd) inhibits oocyst development dose-dependently
Conclusions and path forward:
GNF776(0.7 M)
60 1.5
GNF776(7 M)
0 0
• Optimized GNF156 blocks transmission completely at 500 nM
• This novel activity is critical for potential eradication goals
33 Chatterjee – Lecture at TSRI Oct 29 2014
Data furnished by Sauerwein lab

GNF156 preclinical safety summary
Genotoxicity
Mini Ames mutagenicity assay: negative
No significant tox at up to 30-45 (rat) or 135-250 (dog) fold over ED99 exposure
Mini Ames mutagenicity assay: negative
Phototoxicity
3T3 NRU assay: negative (PIF = 1.4)
2-week rat toxicity at 10, 30, and 100 mg/kg/day
100 mg/kg/day: salivation, respiratory distress, lymphocyte counts and WBC mild thyroid follicular hypertrophy/hyperplasia and possibly testicularWBC, mild thyroid follicular hypertrophy/hyperplasia, and possibly testicular degeneration (1/5 rats but not in controls)
≥30 mg/kg/day: platelets (17-20%), in 3/5 rats at 30 and all rats at 100 mg/kg/day, mild-mod creatinine (up to 2-fold); mild (15%) globulin
All doses: CK (-16- 56%) and glucose (-6 - 15%) in several rats per group
Dog rising dose with telemetry at 2, 10, 30, 100, 250 mg/kg
34 Chatterjee – Lecture at TSRI Oct 29 2014
Dog rising dose with telemetry at 2, 10, 30, 100, 250 mg/kg
250 mg/kg: Transient heart rate increase without ECG changes

Visceral LeishmaniasisL i h i
Extracellularpromastigotesin insect gut
• Disease caused by kinetoplastids from Leishmania genus (L. donovani, L. infantum / chagasi)
• Endemic to 82 countries with > 90% cases found in 3 regions:- India / Bangladesh / Nepal
Leishmania spp.
Sandfly (Phl b t
in insect gut- Sudan / Ethiopia / Kenya - Brazil
• Prevalence- 500,000 new cases per year
(Phlebotomus, Lutzomyia)- Mortality conservatively estimated as 100,000 deaths per year
• Routes of infection – through bite of Leishmania-infected sandfly• Visceral Leishmaniasis natural history
- Most infections have sub-clinical characterR it t d b i t i t
- Incubation period typically 2-8 months - clinical features : fever >80%, wasting > 70%, splenomegaly > 90%
(almost universal), hepatomegaly > 50%, anemia > 60%- untreated disease almost always fatal, cause of death usually
Regurgitated by insect into bite wound
Propagates as amastigote y , y
concurrent illness (pneumonia, tuberculosis, dysentery)• Current therapies
- Pentavalent antimonials, amphotericin B, paromomycin, miltefosine- Drawbacks include cost (liposomal amphotericin B), toxicity
gin vacuole of macrophages & dendritic cells
35 Chatterjee – Lecture at TSRI Oct 29 2014
( p p ), y(antimonials, amphotericin B, miltefosine), drug resistance (antimonials) Parasites transmitted back to
insect during blood meal
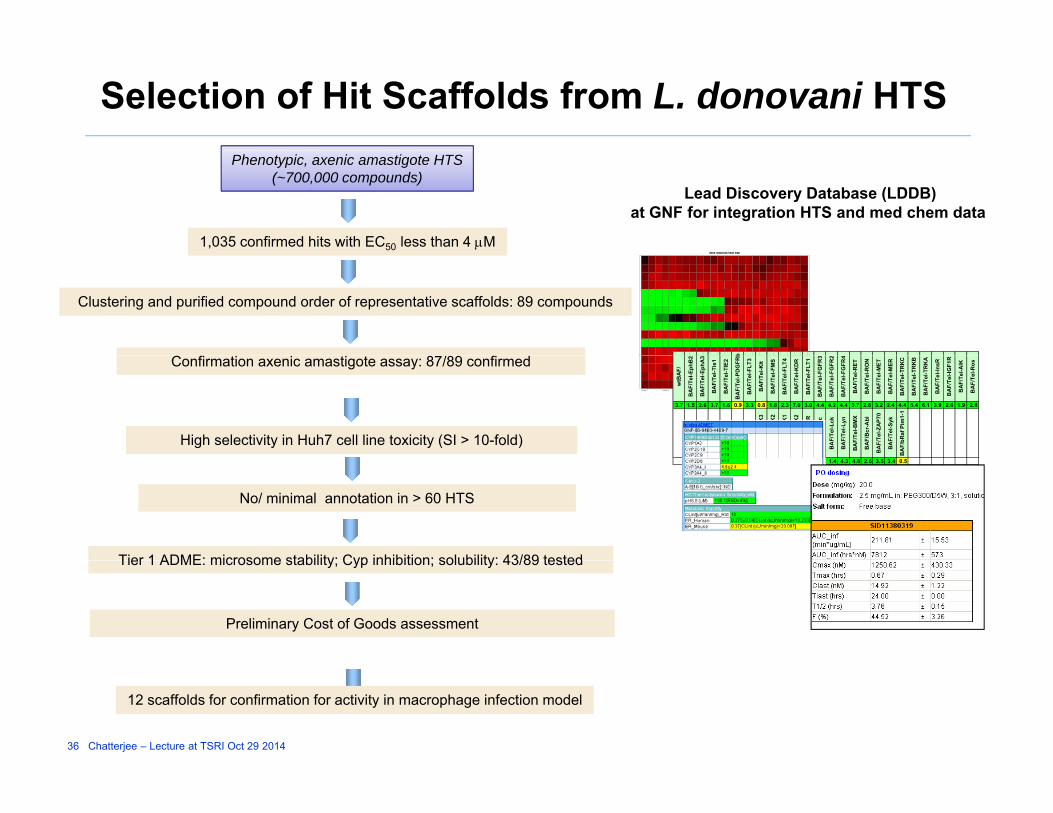
Selection of Hit Scaffolds from L. donovani HTS
dose response heat map
1,035 confirmed hits with EC50 less than 4 M
Phenotypic, axenic amastigote HTS(~700,000 compounds)
Lead Discovery Database (LDDB)at GNF for integration HTS and med chem data
dose response heat map
b
Clustering and purified compound order of representative scaffolds: 89 compounds
Column 1 Column 4 Column 7 Column 10 Column 13 Column 16 Column 19 Column 22
wtB
AF/
BA
F/Te
l-Eph
B2
BA
F/Te
l-Eph
A3
BA
F/Te
l-Tie
1
BA
F/Te
l-TIE
2
BA
F/Te
l-PD
GFR
b
BA
F/Te
l-FLT
3
BA
F/Te
l-Kit
BA
F/Te
l-FM
S
BA
F/Te
l-FLT
4
BA
F/Te
l-KD
R
BA
F/Te
l-FLT
1
BA
F/Te
l-FG
FR3
BA
F/Te
l-FG
FR2
BA
F/Te
l-FG
FR4
BA
F/Te
l-RET
BA
F/Te
l-RO
N
BA
F/Te
l-MET
BA
F/Te
l-MER
BA
F/Te
l-TR
KC
BA
F/Te
l-TR
KB
BA
F/Te
l-TR
KA
BA
F/Te
l-Ins
R
BA
F/Te
l-IG
F1R
BA
F/Te
l-AlK
BA
F/Te
l-Ros
3.7 1.5 2.6 3.7 1.6 0.9 3.3 0.8 1.8 2.3 7.0 3.0 4.4 4.2 4.4 3.7 2.8 3.2 2.4 4.4 5.4 6.1 3.9 2.6 1.9 2.8
BA
F/Te
l-JA
K3
BA
F/Te
l-JA
K2
BA
F/Te
l-JA
K1
BA
F/Te
l-TYK
2
BA
F/Te
l-FG
R
BA
F/Te
l-Src
BA
F/Te
l-Lck
BA
F/Te
l-Lyn
BA
F/Te
l-BM
X
BA
F/B
cr-A
bl
BA
F/Te
l-ZA
P70
BA
F/Te
l-Syk
BA
F/bR
af P
im1-
1
Confirmation axenic amastigote assay: 87/89 confirmed
High selectivity in Huh7 cell line toxicity (SI > 10-fold)3.5 3.7 3.7 2.5 2.1 1.9 1.4 4.3 4.8 2.6 3.5 3.4 0.5
No/ minimal annotation in > 60 HTS
Tier 1 ADME: microsome stability; Cyp inhibition; solubility: 43/89 tested
Preliminary Cost of Goods assessment
Tier 1 ADME: microsome stability; Cyp inhibition; solubility: 43/89 tested
36 Chatterjee – Lecture at TSRI Oct 29 2014
12 scaffolds for confirmation for activity in macrophage infection model

Efficacy Comparison - Hits vs. Known Drugs
• 8 of 12 scaffolds show activity < 10 M on
Compound ID Axenic Amastigote IC50 (M)
Macrophage infection IC50 (M)
GNF6372 1.41 > 50y
intracellular amastigotes• 2 of these 8 scaffolds have
no activity on axenic
GNF6513 2.56 > 50
GNF7674 2.24 > 50
GNF3336 0.63 > 50
A h i i B 1 42 0 13y
amastigotes – fail to reconfirm
• 4 of 12 scaffolds no activity
Amphotericin B 1.42 0.13
Miltefosine 4.12
yon intracellular amastigotes at 50 M
• Several interesting scaffolds G GNF6513g
(on right) were not optimized for chemistry despite amastigote activity
GNF6372 GNF6513
37 Chatterjee – Lecture at TSRI Oct 29 2014
GNF7674 GNF3336

Leishmania ProjectCurrent Hit-to-Lead Status
• 8 scaffolds have been identified with following properties– Good SAR from screen (active and inactive members within a
ff ld)scaffold)– All commercially available compounds, not annotated with any anti-
infective activity– Preliminary in vitro ADME assessed, including:
• Liver microsome stability (mouse and human)• CyP450 3A4 reversible inhibition (for DDIs)• Solubility assessment in pH 6.8 and in simulated GI fluid buffer
(Fassif)– ~1000 compounds made on these series since Feb 20111000 compounds made on these series since Feb 2011
• Goal for project is to find 1-2 lead scaffold with in vivo potency similar to miltefosine
38 Chatterjee – Lecture at TSRI Oct 29 2014

Triazolothiazoles hit GNF8023
Axenic HTS hit with moderate macrophage activityUnable to measure oral exposure
GNF8023
Unable to measure oral exposure mouse plasma instabilityInstability arises from cleavage of the methyl esterS i h t SAR
GNF5410
PotencyMacrophage Infection: 5.5 M
Solubility
Series has steep SAR:Acid compound (GNF5410) is inactive on the parasite, along with small amides (total of 15 Solubility
HT-Eq Sol: 119 M
Microsomal StabilityER Mouse: 0.97
(analogs made)Other amines other than piperidine also inactive including azetidines Cyp Inhibition
CYP3A4: 4 M
CytotoxicityMouse Macrophage: >50 M
including azetidines, pyrrolidines and piperazines
Chemistry on hold for this series
39 Chatterjee – Lecture at TSRI Oct 29 2014

Mouse Model of Acute Visceral LeishmaniasisAcute VL Model ValidationL donovani propagation in BALB/c Mouse
n = 5 animals per group2 miltefosine doses used (10 mg/kg and 30 mg/kg)p.o. dosing x 5 days (7 through 11 days)
Acute VL Model Validation
6ange
L. donovani propagation in BALB/c Mousetail-vein injection of amastigotesparasitemia quantification in liver by histology and qPCR
1.0E+1e
Efficacy of miltefosine in BALB/c VL
liver parasitemia quantified on day 14 (qPCR)
2
4
asite
mia
–fo
ld c
ha
1.0E-1
1.0E+0
mia
-fo
ld c
hang
e
0
2
0 7 14
Live
r par
a
Days Post-infection
1.0E-4
1.0E-3
1.0E-2
Live
r pa
rasi
te
Days Post infection
Vehicle 10 mg/kg 30 mg/kg
Treatment
Results are in agreement with previously publisheddata: reported ED50 for miltefosine against L. don.Giemsa stain of liver imprints from infected BALB/c mouse
40 Chatterjee – Lecture at TSRI Oct 29 2014
p 50 gin mice: ∼ 6 mg / kg / day × 5 days)
Giemsa stain of liver imprints from infected BALB/c mouseon day 14 post-infection. Infected macrophages containingL. donovani amastigotes are indicated with arrows

• Disease caused by kinetoplastid Trypanosoma cruzi
Chagas diseaseT i• Disease caused by kinetoplastid Trypanosoma cruzi
• Endemic to 18 countries in Central and South America• Prevalence
- ~ 200,000 new cases per yearM t lit ti t d 50 000 d th
extracellularepimastigotesin insect gut
Trypanosoma cruzi
- Mortality estimated as 50,000 deaths per year• Routes of infection:
- 80% of infections attributed to bite of infected insect vector- Blood transfusion Kissing bug
(T i t i )
in insect gut
- Congenital transmission (4% of babies born to infected mothers)- Orally via contaminated food
• Chagas disease natural history- Acute phase: 7-9 days; often passed unnoticed; in 90% cases
(Triatominae)
T f d b i t facute disease resolves spontaneously- Indeterminate phase: ~ 70% patients remain asymptomatic, risk of
sudden death due to cardiac abnormalities- Chronic phase: develops 10-30 years after infection in ~ 30%
ti t l di f ti h t f il i L ti
Transferred by insect feces deposited near bite wound
propagatesintracellularlyin cytoplasm
patients; leading cause of congestive heart failure in Latin America; costly treatment
• Current therapies- Benznidazole, nifurtimox – nitroheterocycles with side effects
(dermatitis diarrhea nausea vomiting etc )
as amastigote
invades manydifferent celltypes
41 Chatterjee – Lecture at TSRI Oct 29 2014
(dermatitis, diarrhea, nausea, vomiting, etc.)- Active on acute form of disease (80% cure rate), marginally on
chronic form (20% cure rate) Parasites are transmitted back to insect during blood meal

Cross-Activity of HAT / VL Hits against T. cruzi
L. donovaniaxenic amastigote
T. bruceibloodstream form
Active Cmpds
L. don. selective 25.8 %
410 37793
T.brucei selective 23.7 %
L. don.+T.brucei 5.8 %
L.don.+T.cruzi 20.2%410 37793
214
322 175
T.brucei+T.cruzi 10.1 %
Pan-kinetoplastids 13.5 %
T t l T i ti 44 7%322 175 Total T. cruzi active 44.7%
L. don. HTS represents a more abundant source of T. cruzi-active
d th T b i HTS
T i
compounds than T. brucei HTS
Unusually low overlap between L. donovani and T. brucei hits outside of pan-kinetoplastids
42 Chatterjee – Lecture at TSRI Oct 29 2014
T. cruzi8 day infection assay
p p

Identification of T. cruzi Amastigote Inhibitors
T. cruzi amastigote proliferation( % ( %) @ )1
T. cruzi8 day infection assay
Subset of 574 L. don. / T. brucei – active inhibitors tested f ti it t ti t(>75% (50%) inhibition @ 10 M)1(IC50 < 4 M) for activity at amastigoteproliferation stage1
148 compounds found active (>75% i hibiti ) @ 10 M
409 132 16
(>75% inhibition) @ 10 Mconcentration
~90% of amastigote stage-ifi i hibit(332) (209) (63) specific inhibitors were
previously identified as T. cruzi-active in 8 day assay
Significant portion of T cr iSignificant portion of T. cruzi –active hits is likely to act at another stage of parasite life cycle
1data obtained and provided by Juan Engel Sandler Center
43 Chatterjee – Lecture at TSRI Oct 29 2014
1data obtained and provided by Juan Engel, Sandler Center

T. cruzi Hit-to-lead Chemistry
• Hit-to-lead chemistry started Oct 2010• All compounds from academic
HN
N
N
GNF2609Unable to seperate parasite
ti it f h C
GNF5063Reasonable oral exposureP l ti it h t ll
• All compounds from academic collaboration compound collection at GNF
• 8 total scaffolds tested in footpad efficacy model, developed by Rick Tarleton and co-
k
ClO
activity from human CypGood oral PK50% reduction in parasitemia in footpad model
Poor selectivity on host cellUnable to remove chelating 2-pyridine
workers• Several scaffolds show good in vitro
potency and oral exposure, however no efficacy in footpad model
GNF8308Poor selectivity on 3T3 host cellActive on human Cyps (< 2 µM)G d l PK b t f t d ffi
44 Chatterjee – Lecture at TSRI Oct 29 2014
Good oral PK but no footpad efficacy

Chagas Mouse Footpad Model, Tarleton lab
• Efficacy model run in-house that employs a T. cruzi strain (CL Brener) expressing tandem dimeric tomato red fluorescent protein (tdTomato)• Mice are infected in the hind footpad with 2.5 x 105 T. cruzi tdTomato trypomastigotes and p yp gthe fluorescence signal intensity is measured as a surrogate of parasite load.
Vehicle
Day 6 post-infection Day 8 post-infection
Day 11 post-infection
D 6 t i f ti D 8 t i f ti D 11 t
Benznidazole
O
NHBnNN
O2N
45 Chatterjee – Lecture at TSRI Oct 29 2014
Day 6 post-infection Day 8 post-infection Day 11 post-infectionFluorescent tdTomato-expressing T. cruzi parasites imaged post-treatment

Summary / Lessons Learned
• Molecular weights greatly effect the cellular optimization– Forces the chemistry to be very atom economical (high ligand efficiencies)
• SAR can be challenging, but getting hits from HTS with good SAR is critical g g, g g g– Better to have SAR than absolute potencies off the deck– Potencies can be optimized relatively quickly compared to other parameters
• Weekly cytotoxicity, protein binding shift potency, and cross-resistance data to other scaffold lab-evolved resistance strains really allows for better optimization of other parameters
• MoA studies in parallel with optimizationSt t ith i l t t fil ( bl d t ti l i l th dd• Start with a simple target profile (e.g. blood-stage antimalarial, then address liver-stages, transmission, etc)
• Use extensive anti-target screening that has been developed over the last decade due to focus on target-based drug discoverydecade due to focus on target based drug discovery
• Academic collaborations have been very useful in using new assays for medicinal chemistry optimization
46 Chatterjee – Lecture at TSRI Oct 29 2014

Acknowledgements (Kinetoplastids)
47 Chatterjee – Lecture at TSRI Oct 29 2014

Acknowledgements (Malaria Research)Pharmacology/Analytical• Tove Tuntland• Perry Gordon• Jonathan Chang
Chemistry• Advait Nagle• Tao Wu • Tomoyo Sakata
R b t M
Swiss Tropical and Public Health Institute• Matthias Rottmann• Christoph Fischli
• Matthew Zimmerman• Liang Wang• Todd Groessl• Barbara Saechao• Bo Liu
• Robert Moreau• Jason Roland• Pranab Mishra• David Tully• Valentina Molteni
• Sonja Maerki
NITD• Bryan Yeung• Zou Bin
Biology• Kelli Kuhen • Carolyn Francek
Zh Ch
• Bo Liu• Chun Li• David Jones• Wendy Richmond• Kevin Johnson
Valentina Molteni• Anne Goh• Suresh B. Lakshminarayana• Veronique Dartois• Thomas Keller
Thi Di• Zhong Chen • Kerstin Henson• Rachel Borboa• James Gilligan• Tae-gyu Nam
• Tom Hollenbeck• Lucas Westling• Michael Kwok• Tiffany Chuan
• Thierry Diagana
External collaborators• Montip Gettayacamin (AFRIMS - Bangkok)• Robert Sauerwein (NCMLS - The
Tae gyu Nam • Neekesh Dharia• David Plouffe• Case McNamara• Stefan Meister
• John Isbell
Informatics• John Che
GNF Management• Jennifer Taylor
Ri h d Gl
Netherlands)• Ian Bathhurst (MMV)
48 Chatterjee – Lecture at TSRI Oct 29 2014
• Elizabeth Winzeler • Yingyao Zhou • Richard Glynne• Martin Seidel• Peter Schultz

Acknowledgements (Malaria - Development)
Core teamMargaret WeaverXingmei HanGi l FGiancarlo FranceseSreehari BabuRita RamosKaren Beltz
Clinical teamJens Praestgaard Ruobing Li
Bo HanWen ShiehMarkus Baenziger
Jens Praestgaard, Ruobing Li
Novartis Tropical MedicinesHeiner GrueningerAnne-Claire Marrast
External support and adviceMarcel Tanner, Swiss Tropical Institute Nick White, Oxford University
Paul AliuGilbert Lefevre
49 Chatterjee – Lecture at TSRI Oct 29 2014





















![[PPT]Introduction to Instrumental Analysis and …scs.illinois.edu/mgweb/Course_Notes/Chem243_Lien/course... · Web viewDynamic Range Precision vs. Accuracy in the common verbiage](https://img.dokumen.tips/doc/110x75/5b04373f7f8b9a89208d415b/pptintroduction-to-instrumental-analysis-and-scs-viewdynamic-range-precision.jpg)



![[people.csail.mit.edu]people.csail.mit.edu/jakobn/research/TalkPhDsem060403.pdfOutline of Part I: Proof Complexity and Resolution Introduction Propositional Proof Systems Proof Systems](https://img.dokumen.tips/doc/110x75/5b2555ef7f8b9a092d8b4c45/-of-part-i-proof-complexity-and-resolution-introduction-propositional-proof.jpg)



