Embed Size (px)
Citation preview

Dr. Sander B. Nabuurs
Computational Drug Discovery groupCenter for Molecular and Biomolecular InformaticsRadboud University Medical Centre

The road to new drugs.
How to find new hits?
High Throughput Screening (HTS)
Virtual Screening (VS)
Integration HTS and VS
Molecular docking.
Considering protein flexibility.
Structure-based drug design in practice: Influenza case study.

WHAT DO WE WANT?
The goal of a drug is tomodulate the function ofits target receptor whichresult in a pharmacologicaleffect in the human body.
HOW DO WE GET THERE?
Developing a new drug: is extremely difficult.
takes a lot of time (> 10 years).
is very expensive (~ 1 billion $).
target compound pharmacologicaleffect

Marketing &Sales
RegistrationClinical
DevelopmentPre-Clinical
DevelopmentLead
OptimizationLead
DiscoveryTarget
Discovery
RESEARCH DEVELOPMENT
O
NH
S
F
CN

Lead Discovery
Hit / Lead Candidate
Lead Optimization
Development Candidate
Computer Aided Drug Design
Target Discovery
TargetIdentification
TargetValidation
AssayDevelopment
O
NH
S
F
CN
Bioinformatics
In Vitro
In Vivo

Computer-Aided Drug Design (CADD) refers to theapplication of informatics methods within rationaldrug design, to discover, design and optimizebiologically active compounds.

Ligands unknown
Target protein structure unknown
high-throughputscreening
Target protein structure known
TWO SCENARIOS
Target geneTarget DiscoveryLigands unknown


HTS is the most importantsource of new hits.
Pharmaceutical companieshave screening libraries upto a few million compounds.
Chemical space of drug-likemolecules is > 1080
.
Building a good screeningcollection is crucial!
Active compounds
Compound collection

In-housecollection
Availableto buy
Everything Diverseselection
Focusedselection
Screening set?

DIVERSE SELECTION FOCUSED SELECTION
Diverse selection:identify
dissimilarcompounds
Focused selection:Identifysimilar
compounds

DIVERSE SELECTION
Chemical Descriptor 2
Ch
em
ical
Des
crip
tor
1
HOW CAN WE MEASURE SIMILARITY?

In the selection ofscreening compoundsthe ‘Rule of Five’ isoften used.
It summarizes typicalproperties of knowndrugs.
These rules are oftenused as a first filteringstep.
In 1997 Chris Lipinski observedfor many drugs:
molecular weight < 500
lipophilicity (LogP) < 5
H-bond donors < 5
H-bond acceptors < 10
rotatable bonds < 10

DIVERSE SELECTION FOCUSED SELECTION
Chemical Descriptor 2
Ch
em
ical
Des
crip
tor
1
Chemical Descriptor 2
Ch
em
ical
Des
crip
tor
1

Sampling around known active sub-structures or structuralfragments can improve the quality of the library.
dopamine derivative

The use of chemical (ormolecular) descriptors isbased on the similarproperty principle.
Molecules with similarstructures and similarproperties should alsoexhibit similar activity.
Chemical Descriptor 2
Ch
emic
al D
esc
rip
tor
1

Fingerprints consistof various descriptorsencoded into bitstrings.
These descriptorscan be fragments orthe presence orabsence of otherproperties.
dopamine derivative

HOW CAN WE MEASURE SIMILARITY?

xA = 8xAB = 5
xB = 6
ABBA
ABAB
xxx
xS
Tanimoto coefficient
56.09
5
568
5
ABS
Note: this is just one of many different similarity measures!

1IN HIV Protease inhibitorTanimoto Similarity 0.47Tanimoto Similarity 0.47
VAC HIV Protease inhibitor
XN1 HIV Protease inhibitorTanimoto Similarity 0.63
MK1 HIV Protease inhibitorTanimoto Similarity 0.63
BEB HIV Protease inhibitorTanimoto Similarity 0.60
BEH HIV Protease inhibitorTanimoto Similarity 0.60
PZQ NOT HIV Prot. inhibitor Tanimoto Similarity 0.49
TI3 NOT HIV. inhibitor TS Tanimoto Similarity 0.48
A high Tanimoto Similarity can beuseful for prioritization.
However, no guarantees!

flutamide retro-flutamide
progesterone receptor 4 nM 6 nM
glucocorticoid receptor 25 nM 38 nM
androgen receptor 0.5 nM 55 nM
flutamide retro-flutamide
NH
O OHCF3
BrO2N
NH
OHCF3
BrO2N
O

Despite being the majorsource of new hits, HTS hasits drawbacks:
It’s expensive. In practice only accessible to
industry. Logistical errors. e.g. ‘frequent hitters’
Measurement errors. e.g. suboptimal readout
Strategic errors. e.g. assay variability
Active compounds
Compound collection

Ligand(s) unknown
Target protein structure unknown
high-throughputscreening
Target protein structure known
virtual screening
TWO SCENARIOS
Target geneTarget DiscoveryLigand(s) unknown

In Virtual Screening (VS)compounds are selectedusing computer programsto predict receptor binding.
VS is much cheaper and isable to process much morecompounds in less time.
Experimental validation ishowever always required! Active compounds
Compound database

A few success stories from virtual screening

STRUCTURE-BASED VS
Predict the orientation(and affinity) of a smallmolecule binding to aprotein target.
Requires the availability ofa 3D target structure!
Structure-based virtual screening
Active compounds
• Compounds to purchase.• Compounds from in-house library.• Virtual compounds.
Compound database

Docking
program
Target
protein
Compounddatabase
Docking
program
Target-Compoundcomplexes
Activecompounds

Despite its advantages VS alsohas its drawbacks:
Experimental validation isalways required.
Protein structure errors. e.g. ‘induced fit’
Sampling errors. e.g. faulty poses due to solvent
Scoring errors. e.g. false positives / negatives Active compounds
Compound database

Screening library
Focused library Hits
High ThroughputScreening
Virtual Screening
Hypothesis generation
Focused and sequential screening

VS hits
Hits
Analysis
Virtual Screening
Parallel and independent screening
HTS hits
High ThroughputScreening
Screening library

The road to new drugs.
How to find new hits?
High Throughput Screening (HTS)
Virtual Screening (VS)
Integration HTS and VS
Molecular docking .
Considering protein flexibility.
Structure-based drug design in practice: Influenza case study.

The docking problem involvesmany degrees freedom:
Translational.
Rotational.
Configurational
(Ligand + Receptor!)
Since the early eightiesseveral docking algorithmshave been devised.
These can be characterizedby the number of degreesfreedom that they ignore.
Target
protein
Compound
Docking
program
Target-Compoundcomplex

Fully flexible docking
Induced fit docking
Flexible ligand docking
Rigid body docking
Receptor
flexibility
Ligand
flexibility
Ligand
translations
Ligand
rotations

A number of flexible ligand dockingprograms:
Dock[Kuntz et al, J Mol Biol, 161:269-288, 1982]
Autodock[Morris et al, J Comput Chem, 19:1639-1662, 1994]
FlexX[Rarey et al, J Mol Biol, 261:470-489, 1996]
Gold[Jones et al, J Mol Biol, 267:727-748, 1997]
Glide[Friesner et al, J Med Chem, 47:1739-1749, 2004]

Molecular docking typically consists of two separate stages:
1. Exploration of conformational and configurational space.
2. Evaluation of the strength of the receptor-ligand interaction.
Targetprotein
Dockingprogram
Compound
Target-Compoundcomplex
Sampling
Scoring
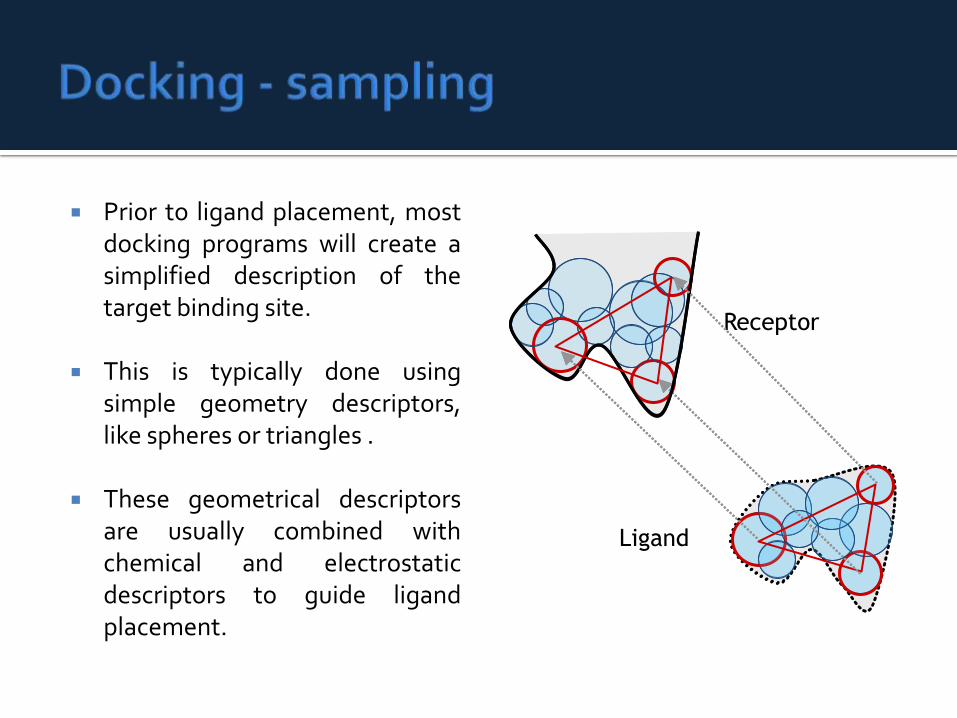
Prior to ligand placement, mostdocking programs will create asimplified description of thetarget binding site.
This is typically done usingsimple geometry descriptors,like spheres or triangles .
These geometrical descriptorsare usually combined withchemical and electrostaticdescriptors to guide ligandplacement.
Receptor
Ligand

Example 1
Example 2

Docking programs generate alarge number of differentdocking poses.
In general one can distinguishtwo different scenarios:
1. Many different poses of thesame ligand need to be rankedfor accuracy.
2. Different poses of differentligands need to be rankedbased on their receptoraffinity.
The ideal scoring function workswell in both cases...
1 …52 3 4
1 …52 3 4

• First principles scoring functionsgenerally use a Molecular Mechanicsforce field.
• Such force fields typically containintra-molecular terms:
– Bond lengths
– Bond angles
– Dihedral terms
• And inter-molecular terms:
– Van der Waals contacts (non-polar)
– Electrostatic interactions (polar)Ebind = Eintra + Enonpolar + Epolar

• Empirical scoring functions have beendeveloped to score ligands very rapidly.
ΔGbind= ΔG0 +
ΔGpolar · Σ f(Complex) +
ΔGnon-polar · Σ f(Complex) +
ΔGrot · Nrotatable-bonds
• ΔG0, ΔGpolar, ΔGnon-polar, and ΔGrot areempirically parameterized weights.
• f(Complex) is a penalty function aimedat penalizing any unfavorableinteraction geometries.

In practice moleculardocking is generally used toanswer two different typesof questions:
1. Which compounds in mycompound collection couldbe active on receptor A?
2. How does the complex lookthat is formed by receptorA and compound B?
Compound BReceptor A
+
Complex?
docking
Compoundcollection
Receptor A
+
Actives?
docking

The road to new drugs.
How to find new hits?
High Throughput Screening (HTS)
Virtual Screening (VS)
Integration HTS and VS
Molecular docking .
Considering protein flexibility.
Structure-based drug design in practice: Influenza case study.

Drug targets are flexiblebiomolecules and theirdynamics play an important rolein ligand binding.
Insight in receptor flexibility canbe valuable when interpretingstructure activity relationships(SAR) and optimizing leadcompounds.
Predicting ligand binding inflexible binding sites is howeverproblematic !
1 ligand1 receptor
conformation
10 ligands10 receptor
conformations

LOCK AND KEY INDUCED FIT
+Receptor A
X
Complex A-X
+Receptor A
Y
Complex A’-Y

Introduceflexibility
Target Induced fitcomplex
Optimizecomplexes
Generatecomplexes
Compound
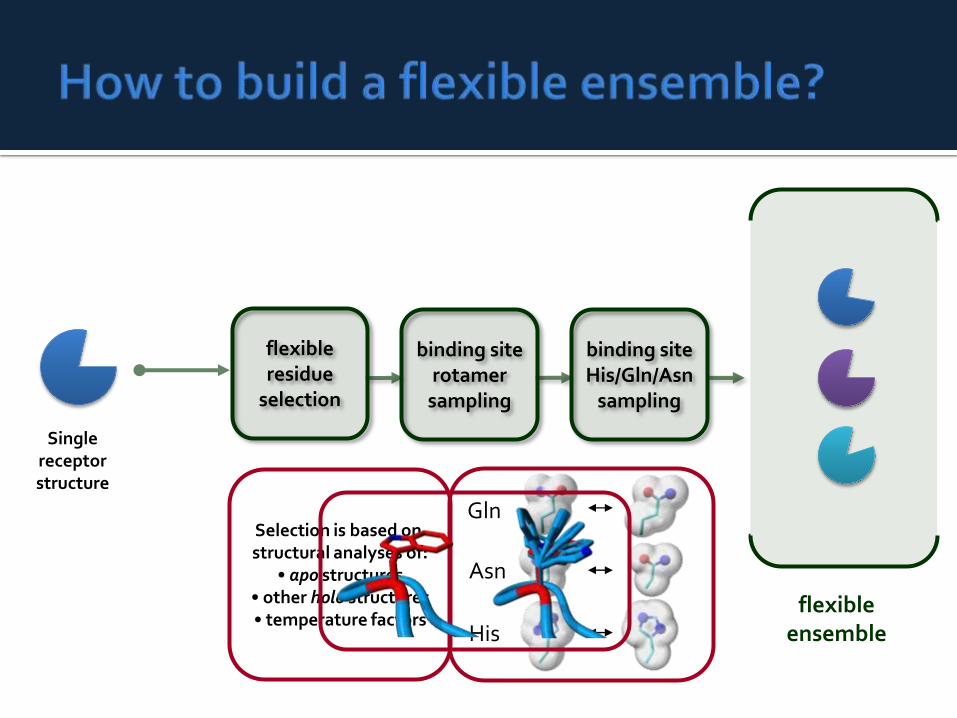
Gln
Asn
His
Singlereceptorstructure
flexibleensemble
flexibleresidue
selection
binding siteHis/Gln/Asn
sampling
binding siterotamer
sampling
Selection is based on structural analyses of:
• apo structures• other holo structures• temperature factors

The Fleksy approach docks intoan ensemble of receptorstructures.
The approach is based on aunited protein descriptiongenerated from an ensemble ofprotein structures.
In our case the ensemblecontains the generated set ofside chain rotamers andsampled Asn/Gln/His sidechains.

2 Asnstates 15 side chain
rotamers
8 Hisstates
crystal structureapo form
crystal structureholo form
identifyflexible
residues
sampleAsn/His
?

RMSD
0.6 Å

Ligand(s) unknownLigand(s) known!
Ligand(s) unknown
Target protein structure unknown
high-throughputscreening
Target protein structure known
virtual screening by docking
TWO SCENARIOS
Target geneTarget Discovery

SBDD
Structure based drugdesign relies on structuralknowledge of the targetprotein to design andoptimize lead compounds.
This knowledge is obtainedfrom either experimentalstructures or compu-tational predictions.
A requirement is the availability of receptor structures: NMR spectroscopy
X-ray crystallography
In practice protein X-ray crystallography is the major source of structural information.
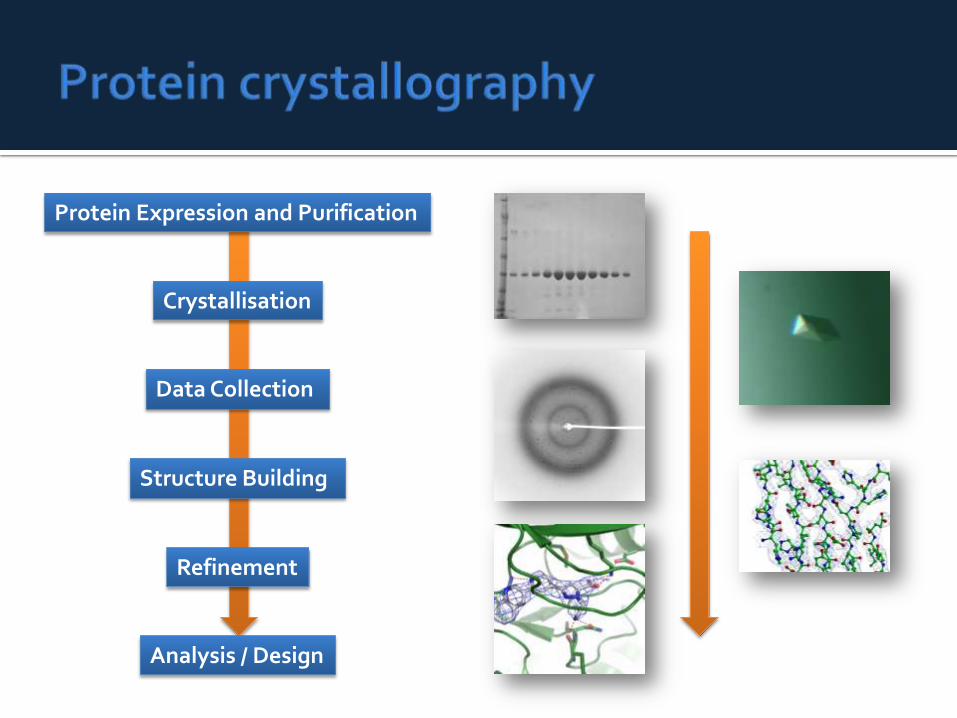
Protein Expression and Purification
Data Collection
Analysis / Design
Structure Building
Refinement
Crystallisation

A receptor structure canoften explain:
Binding
Specificity
Inhibition
Flexibility
Reaction mechanism
And it allows predictions tobe made!

1918 Influenza Epidemic Influenza Virus

NEURAMIDASE POCKET SIALIC ACID

RELENZA SIALIC ACID

RELENZA TAMIFLU
5.000.000 doses in NL

Trouw – 3 maart 2009

RELENZA TAMIFLU
WT Ki = 1.0H274Y Ki= 1.9
WT Ki = 1.0H274Y Ki =265
SIALIC ACID
H274YH274Y

UMC St. Radboud
Jacob de Vlieg
Gijs Schaftenaar
Schering-Plough
Scott Lusher
Markus Wagener
Ross McGuire
Hans Raaijmakers
Software
BiosolveIT
Accelrys
Molecular Networks
Funding
Dutch Organization for Scientific Research (NWO)





























