Embed Size (px)
Citation preview

RHEUMATIC FEVER - aetiology & immunopathogenesis
Dr Sandeep MohananDepartment of Cardiology, Calicut Medical College

OVERVIEW
- HISTORY- EPIDEMIOLOGY- AETIOLOGY- IMMUNOPATHOGENESIS- PATHOLOGY

INTRODUCTIONRheumatic fever is a non-suppurative complication of
Streptococcal infection.
Most common cause of acquired cardiac disease in children & young adults
Annually ~4,70,000 cases of rheumatic fever worldwide( Moss& Adams)

HISTORYKnown to exist as early as the 17th centuryChorea was related to arthritis by Sydenham in 1600s.
Charles Wells (1812) – Association of rheumatism with carditis and subcutaneous nodules
Jean –Baptiste Bouillaud (1836) – 1st publication on rheumatic fever (‘Father of rheumatic heart disease’)

Jean-Baptiste Bouillaud

Association of ARF with pharyngitis was noted in 1880
Walter B Cheadle (1889)– 1st classic description of rheumatic fever.
Ludwig Achoff(1904) - 1st description of pathology of rheumatic carditis.

Ludwig Aschoff
- Aschoff-Tawara node
- Aschoff-Rokitansky sinuses

Alvin F Coburn (1931) – 1st proved causal relationship between Beta hemolytic Strep and ARF
Alvin F. Coburn.THE SIGNIFICANCE OF PROLONGED STREPTOCOCCAL ANTIBODY DEVELOPMENT IN RHEUMATIC FEVER. J Clin Invest. 1939 January; 18(1): 141–145.

Rebecca Lancefield (1933) – Serogrouping of Streptococci
T Duckett Jones (1944) – 1st diagnostic criteria for RFHe presented his paper on the diagnosis of rheumatic fever
in Chicago (AMA meeting)

EPIDEMIOLOGY OF RF Incidence in developed countries ~ 0.5-3/100,000 Incidence in developing countries – as high as ~ 100-200/100,000Overall mean incidence of RF worldwide : 5-50/100,000(Tibazarwa et al. Incidence of acute rheumatic f ever in the world: a systemic review of population-based studies.
Heart2008; 94:1534-40)
- India – 25-50% of global burden of RHD (WHO statistics 2002) - India – 0.2-0.75/1000/year in school children and around 1/3rd develop RHD.(Grover et al. Burden of rheumatic and congenital heart disease. Indian Heart J2002; 54: 104-7)
Recent ICMR registry study, prevalence of RF – 0.0007 to 0.2/1000 in urban population.

RF worldwide incidence

RF worldwide incidence recent trends
However recent data from India can not be conclusively relied upon to suggest a definite declining trend, due to the fallacies in epidemiological studies.
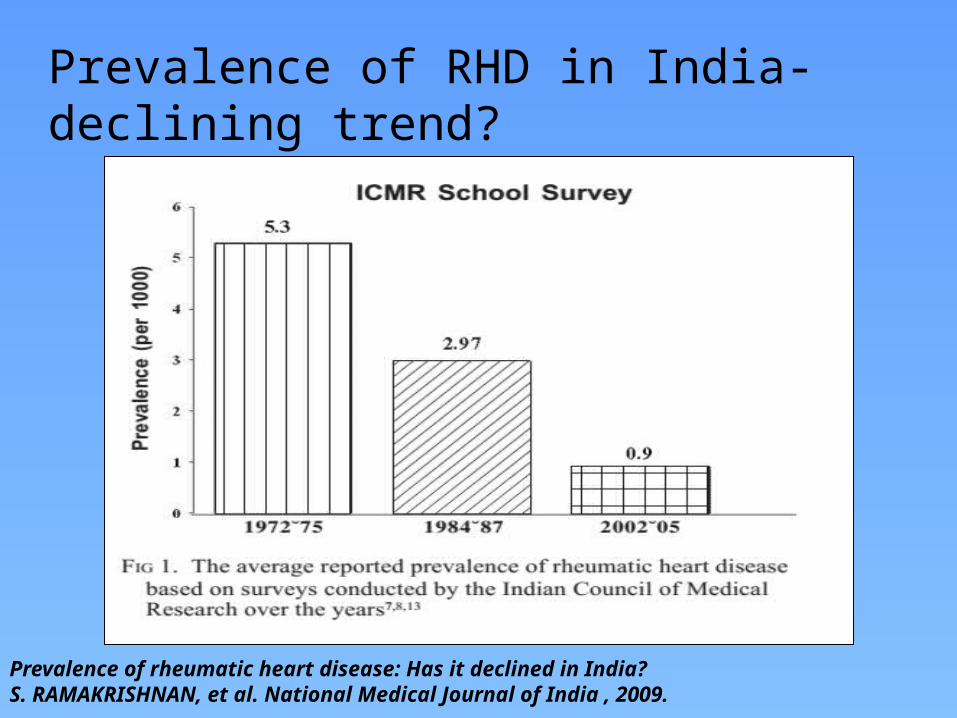
Prevalence of RHD in India-declining trend?
Prevalence of rheumatic heart disease: Has it declined in India?S. RAMAKRISHNAN, et al. National Medical Journal of India , 2009.

The downtrend of rheumatic fever incidence has however led to decline in the global interest for further research on its genetic and immuno-pathogenic basis.
Thus even now after >70 years of its first etio-pathological explanation, the actual immune basis for the development of rheumatic carditis is still an unsolved mystery.

AETIOLOGYAlvin F Coburn in 1931 1st proved the association
between Streptococcus pyogenes and RF
Other agents that have been explored and linked to RF are HSV-1, Coxsackie B & Measles virus. However they all remained unproven hypothesis.

THE ‘GAS’Streptococcus pyogenesGram positive cocci1µm in diameterChains or pairsUsually capsulatedNon motileNon spore formingFacultative anaerobesCatalase negative (Staphylococci are catalase positive)

Lancefield’s serological Classification
Streptococci classified into many groups from A-K & H-VOne or more species per groupClassification based on C- carbohydrate antigen of cell wall
Groupable streptococci A, B and D (more frequent) C, G and F (Less frequent)
Non-groupable streptococci S. pneumoniae (pneumonia) S. viridans species
e.g. S. mutans
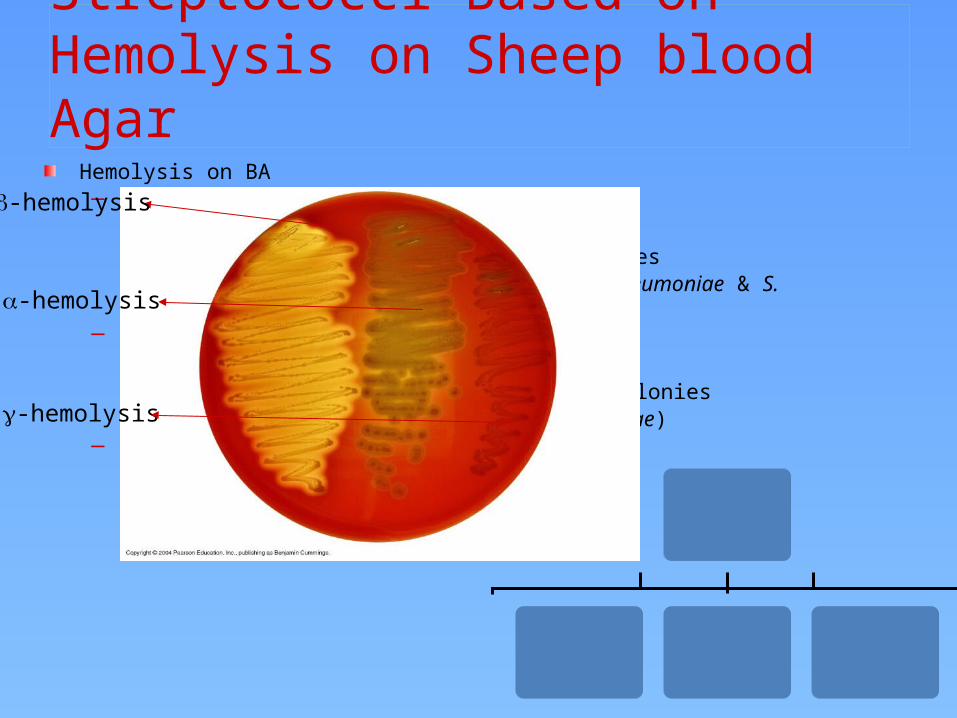
Classification of Streptococci Based on Hemolysis on Sheep blood Agar
Hemolysis on BA– -hemolysis
Partial hemolysisGreen discoloration around the coloniese.g. non-groupable streptococci (S. pneumoniae & S. viridans)
– -hemolysisComplete hemolysisClear zone of hemolysis around the coloniese.g. Group A & B (S. pyogenes & S. agalactiae)
– -hemolysisNo lysise.g. Group D (Enterococcus spp)
-hemolysis
-hemolysis
-hemolysis


Diagrammatic structure of the group A beta hemolytic streptococcus
Capsule
Cell wall
Protein antigens
Group carbohydrate
Peptidoglycan
Cyto.membrane
Cytoplasm
…………………………………………………...

Pathogenic and Virulence FactorsStructural components
M protein Lipoteichoic acid F protein for cell adhesion Hyaluronic acid capsule-- which acts
to camouflage the bacteria
Enzymes (facilitate the spread of bacteria in tissues) Streptokinases Deoxynucleases C5a peptidase
Pyrogenic toxins-- stimulate macrophages and helper T cells to release cytokines
Streptolysins Streptolysin O lyse red blood cells,
white blood cells, and platelets Streptolysin S

M protein- Major surface antigen (T,R)- Rebecca Lancefield in 1962- ‘Emm’ gene- Resistance to phagocytosis
- Has epitopes causing cross-reactivity with myocardium, synovia, skin & brain.
> 130 M serotypes

Alpha helical coiled-coil fibrillar protein– significant homology in structure & a.a sequences to tropomyosin, myosin, keratin, vimentin & laminin.
It has a hypervariable NH2- terminal and a conserved C-terminal.
The NH2- terminal is responsible for the formation of opsono-phagocytic Abs after around 2 weeks of infection.
The body is divided into A, B and C repeats based on the peptide sequence periodicity.
The B repeat region is the immunodominant region and elicit an exaggerated immune response(B cell)– however this is not opsonic.

• The C repeat region is considered to have conserved T cell epitopes that also elicit tissue specific immune response ( basis for RF-vaccine research)•Based on the conserved C repeat regions Class I & Class II GAS strains are named.
•It is the Class I M-type of which belongs the strains 1,3,5,6,14,18,19 and 24 ---that have been associated with RF•The Class II strains have non-reactive M-types.
•The important cross-reactive epitopes are distributed between the B & C repeats of the M-protein.

Cross reactivity of M protein is related to structural as well as sequential homology – 30-40%
Antiphagocytic properties are cause by specific inhibitory effects on alternative complement pathway (Factor H affinity mediated inhibition)
M protein also promote streptococcal adherence
M proteins exhibit antigenic variation through intragenic recombination– causing varying lengths of the a.a sequence
-- This hampers the formation of broad non-type specific immunity to counter against reinfection from different strains of GAS.
-- this variation in strains have also hampered the development of a vaccine based on M protein.

Probable Streptococcal vaccines

Streptococcal pharyngitis~15-20% of pharyngitis in children 5- 15 years.Most common is Group A – ~60% especially in temperate countriesGroup C and G also form a good majority in tropical countries (not
related RF)
Carrier state does not mean infection and is not clinically relevant ( maybe epidemiologically relevant)
Infection itself maybe asymptomatic in up to 30-50% cases of RF.
Infection is defined as a rising trend in antibody titresSecondary infection is primarily determined by socioeconomic and
environmental factors.

Streptococcal pharyngitis (contd)IP- 2-4 daysSudden onsetSevere odynophagiaRhinitis, Laryngitis and bronchitis are not
featuresFever, headache, vomiting and abdominal
painCharacteristic physical findings
Penicillin treatment may not alter the duration of illness.

RF following GAS pharyngitisLatent period of 1 to 5 weeks ( 18 days)0.3-3%-Rheumatogenic M serotypes : 1,3,5,6, 14,18,19, 24, 27,29 ( 2, 4, 12, 22 and 28- unlikely to cause RF) Virulence of the particular strains Encapsulation & formation of mucoid colonies Anti-Streptococcal host-immune response
RF following skin infections with GAS have been reported in the aborginal tribes of Australia.
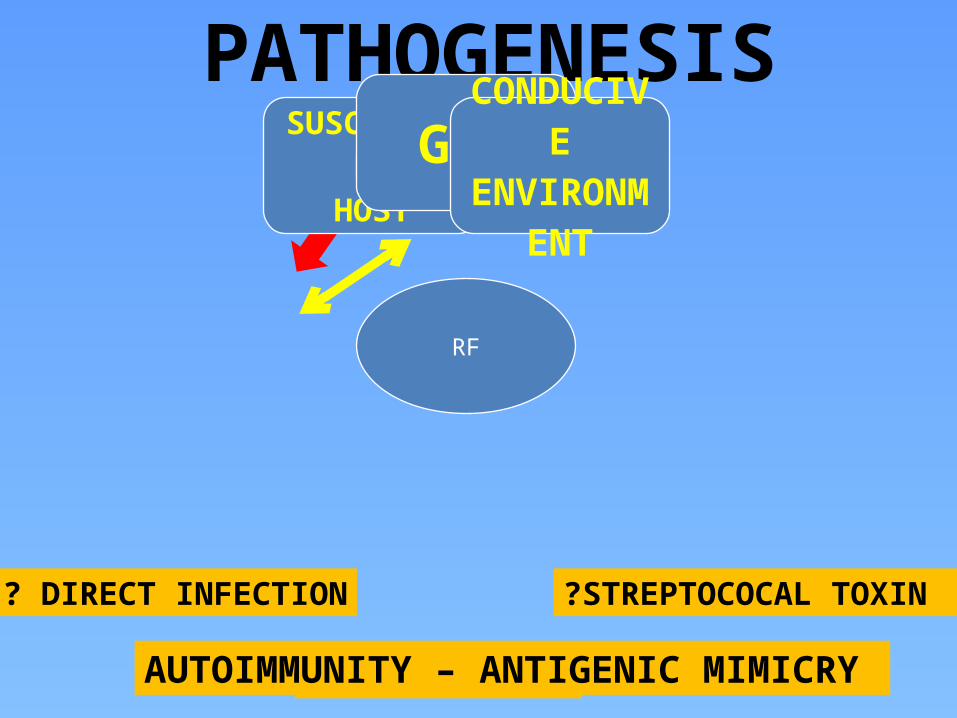
PATHOGENESIS
RF
SUSCEPTIBLEHOST
GASCONDUCIV
EENVIRONM
ENT
? DIRECT INFECTION ?STREPTOCOCAL TOXIN
? AUTOIMMUNITYAUTOIMMUNITY – ANTIGENIC MIMICRY


THE CONDUCIVE ‘ENVIRONMENT’OvercrowdingPovertyPoor nutritionPoor hygienePoor access to health careRapid spread
Lack of primary preventionLack of secondary prevention
INDIA

Changes in ‘environmental factors’ with time

GAS VIRULENCE
Virulent clones may change in a cyclical manner with time. These result in epidemics such as the one that occurred in USA in the early 1990s.

HOST RESPONSE - IMMUNITYThe exact immune reaction that occurs to a preceding GAS
infection is yet to be elucidated.However there have been several postulates of which
“MOLECULAR MIMICRY” is presently considered to be involved.
Stollerman et al (1960) 1st noted the relation between RF and certain rheumatogenic M-type strains of GAS infection.
Kaplan et al(1965) was the first to postulate molecular mimicry as the cause of RF by demonstrating Igs and complement bound to cardiac tissue (in the absence of bacteria) following streptococcal infection.

IMMUNOPATHOGENESIS
Mechanisms involved: ?Molecular Mimicry (? M-protein , ? Carbohydrate antigen) --
Direct relationship not proven yet ?Super-antigens ?Genetic susceptibility for development of ‘forbidden clones’
Causative antigen is yet to be conclusively identified!!( Humans are the only natural hosts for GAS and experiments
have failed to develop an appropriate animal model)


MOLECULAR MIMICRY“Sharing of epitopes between host tissue and bacterial
antigens”- Antibody (B cell) mediated:1) Recognition of aminoacid sequences2) Recognition of homologous non-identical a.a sequences3) Recognition of epitopes on different molecules
- Cell mediated (T cell) : 1) By antigen presentation to TCR 2) Epitope spreading (i.e T cells recognize epitopes in other
proteins with equal or more priority than the original bacterial epitope)

Ab mediated injury Antibodies and complement mediated injury were conclusively demonstrated in
cardiac tissues by 1970. Earlier studies pointed towards ?M protein and group A carbohydrate (N-acetyl
glucosamine) as the antigens. Priliminary studies using monoclonal Abs suggested ? myosin as the dominant
autoantigen.
Other autoantigens against which mimicry was identified were vimentin, tropomyosin, keratin and laminin.
(Laminin- An ECM protein that secreted by endothelial cells and lines the heart valves)
Gln-Lys-Ser-Lys-Gln was identified as an epitope on the M protein which cross reacted with Abs in the sera.
Ab mediated injury is presumed as the initiator of cardiac injury.

T cell mediated injury Involves T cell mediated ‘mimicry’ and epitope spreading.
Ab s also activate T cells which in turn initiate inflammatory response .Recent studies show that T cells specifically recognize an epitope on the M
protein - M5(81-96) epitope presented to the TCR by HLADR53.
Typically a TH1 response is initiated.
They produce the cytokines - IFN-gamma, IL-1 and TNF-alpha in valves, pericardium and myocardium.
IL4 ( an immune-regulator cytokine) is found in only small quantities in rheumatic valvulitis explaining the persistence and perpetuation of valvular inflammation..
Fae et al.How an autoimmune reaction triggered by molecular mimicry between streptococcal M protein and cardiac tissue proteins leads to heart lesions in rheumatic heart disease.J Autoimmun. 2005 Mar;24(2):101-9.

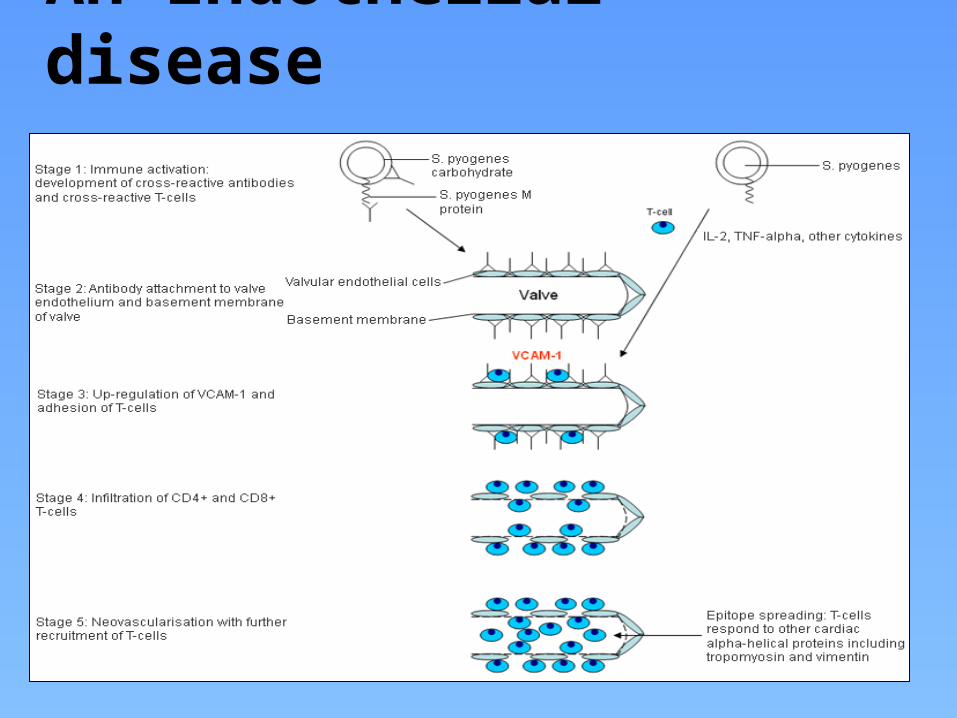
Immunopathogenesis of rheumatic valvulitisA 2 stage process:
Auto-Abs home in, damage and inflame the valve endothelium (? Laminin)
Complement mediated injury takes over
Upregulation of VCAM-1 occurs on endothelial surface
T cell mediated extensive injury and tissue infiltration via VCAM-1 (Epitope spreading – vimentin, myosin, ? Ags on VICs)
CD4+ TH1 mediated granulomatous inflammation
IFN gamma and TNF alpha mediated fibrosis, Low IL4 production
Neovascularization, persistence of inflammation & scarring

Rheumatic valvulitis
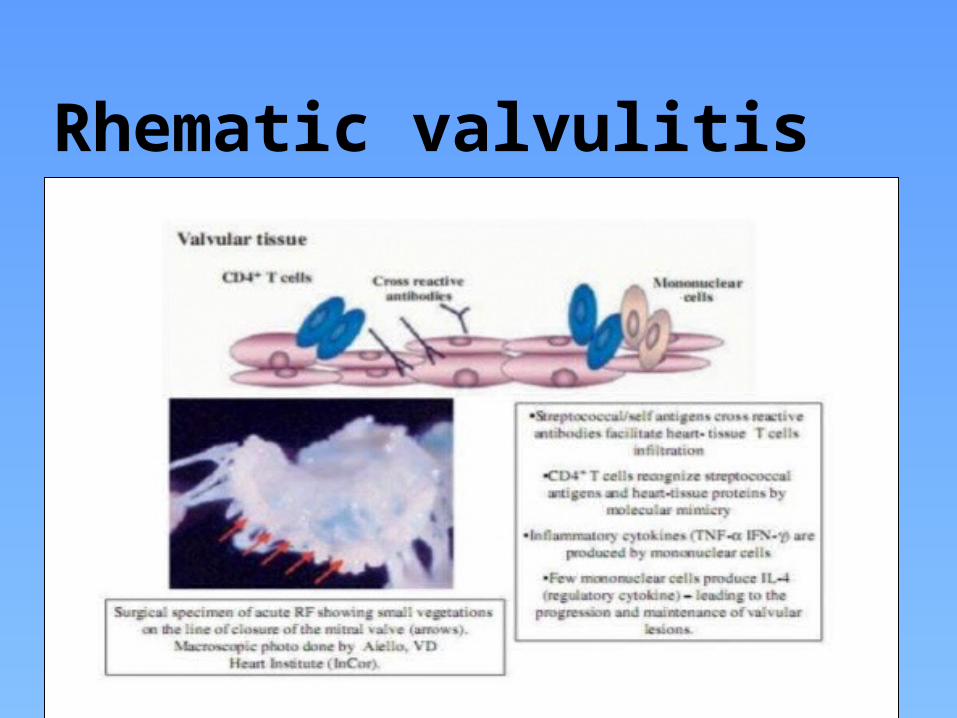
Rhematic valvulitis

Genetic susceptibility to RF- Familial tendency for RF had been investigated but no conclusive pattern of
inheritance could be elucidated.- HLA-II (Chr 6) is the gene most associated with RF and development of RHD- HLA-DR7 mostly associated with progressive valvular lesions in RHD- HLA-DR53, HLA-DQA, HLA-DR4, HLA-DR9(determines antigen presentation to T-cell receptors)
- TNF alpha alleles also associated
- Non HLA B cell antigen D8/17 is associated with increased susceptibility
- In a recent metanalysis that quantitated the genetic susceptibility of RF it was found that the concordance in monozygotic twins was 44% and in dizygotic twins 12% and the overall estimated heritability was 60%
(Engel et al. Genetic Susceptibility to Acute Rheumatic Fever: A Systematic Review and Meta-Analysis of Twin Studies. Plos-one, 2011.)

RF HLA class II susceptibility worldwide

Valve +/- Myocardium ?- Our present knowledge of the pathology reveals that RF primarily involves
mesothelial and endothelial tissues.-The cardiac valves are basically an extension of the A-V sulcus region formed by a
process called ‘undermining’, which excludes the myocardium.- The valve loses its muscle component and becomes a core of connective tissue
sandwiched between 2 layers of endothelium.
Even though “myosin” was previously thought as the main causative antigen – currently there is enough evidence on the fact that valves are targeted by priority... Rather than the myocardium.

Pancarditis- a misnomer?Myocarditis presumed on the basis of finding interstitial inflammation and Aschoff
bodies.
No definite evidence of myocarditis!!- No consistent elevation of cardiac biomarkers- No evidence of loss of cardiac contractility- CHF does not occur without significant valvular lesions- Radionuclide studies (Tc pyrophosphate, antimyosin fab, indium labelled myosin etc ) failed to
demonstrate significant myocardial staining- Biopsy in acute RF failed to show cellular necrosis (Narula et al)--- inflammation was
subepicardial, subendocardial and perivascular- Surgical valve replacement during RF and AHF reverted features of heart failure.- Aschoff nodules do not contain myocardial cells.
=== Thus evidence points against the theory that molecular mimicry to myosin/tropomyosin is central to the pathogenesis!!!
An Endothelial disease

- Endothelial diseases tend to focal.
- They are usually transient due to the high regenerative capacity....however chronic and recurrent endothelial insults may result in subendothelial and connectuve tissue fibrosis.
- Explains the transient manifestations of Rheumatic fever and chronic involvement of valves
Conclusion:
Cross-reacting Ags – M protein +/- CarbohydrateSelf-Antigens - Laminin, VICs, Vimentin > >Myosin

SUPERANTIGENS A possible mechanism that may contribute to the
systemic inflammation.
They are glycoproteins found on bacterial cell wall that promote the binding of MHC Class II with TCRs
--- thus inciting T-cell activation and release of cytotoxins.
Streptococcal pyrogen exotoxin– a possible culprit May explain the systemic inflammatory changes

Recent alternative hypothesisOver the past 5 years an M protein N-terminal domain has been
noted to bind to the CB3 region of Collagen type IV.This seems to direct an antibody and immune response against
collagen and surrounding ground tissue.
These Abs do not bind to the M protein thus refuting the “Molecular Mimicry” theory
This is somewhat similar to the collagen related inflammation seen in Alports syndrome & Goodpastures syndrome.
Rajendra Tandon et al. Revisiting the pathogenesisof rheumatic fever and carditis. Nat Rev Cardio, Mar 2013.

PATHOLOGYPrimarily an ‘endothelial disease’
RF involves exudative & proliferative inflammation of collagen tissue or its ground substance with a marked tendency to involve the endothelium and sub endothelium-- blood vessels, endocardium, synovia & pericardium—
There is additional generalized vasculitis-like inflammation throughout the body.

CARDITIS Inflammation of sub-epicardial, sub-endocardial and perivascular
connective tissue. Characterized by ASCHOFF BODY (‘hallmark of rheumatic carditis’)
- They may be found in endocardium, myocardium & pericardium
Commissural fusion occurs due to repeated valve-angle inflammation
RHD is characterized by phases of inflammation as well as scared and hyalinized tissue.

Inflammation of valve endothelium extending to central core connective tissue and associated with neo-vascularization
Immunopathologically, antistreptococcal Abs are seen to bind to valvar interstitial cells(VICs) and sub-endothelial elastin fibrils
Humoral response and inflammation (exudative phase) is followed by a cellular response followed by neovascularisation and later fibrosis (proliferative phase)
Permanent valvular damage is probably due to scarring of the central thin core of connective tissue.
( unlike the other tissue endothelial invovement which heals quickly without scarring)
Present data suggest that valve interstitial cells(VICs) and vimentin may be the specific targets tat lead to RF carditis and RHD rather than myosin as previously presumed.
( Rajendra Tandon. Rheumatic fever pathogenesis. Approach in research needs change.Annals of Paed Cardiol 2012 )

The Aschoff body-Around 1-2mm -Lymphocytes, macrophages, B cells, Anitschkow cells and giant cells (Aschoff cells)– NO MYOCYTES.-Dominant T cells-CD4 :CD8 ~ 2:1-Giant cells are positive for vimentin
- Strictly perivascular-Minimal surrounding myocyte damage
ANITSCHKOW CELLS( “CATERPILLAR CELLS”) – Giant macrophages
ASCHOFF CELLS – Multinucleated cells due a coalition of Anitschkow cells

The Aschoff body itself goes through several stages of evolution
Fibrinoid/Exudative stage (2-3 weeks) Granuloma/Proliferative stage(1-6months) Perivascular scarring
More common in younger people.
The finding of Aschoff bodies in several phases of evolution in postmortem studies have suggested recurrent attacks of ‘rheumatic fever’ as the cause of RHD.

McCallum’s patch
McCallum’s patch: Gross finding of endocardial thickening in the posterior wall of LA due to inflammation as well as ‘jet’-trauma .

Rheumatic verrucae
-Due to progressive inflammation causing necrotic collagen to project outwards from the valve, On which platelet thrombi deposit. - Rheumatic verrucae occur on the atrial surface at sites of valve closure and on the chordae. -The valves become edematous thickened and vascularised.

- Fibrinous pericarditis (“Bread & butter” ) is present microscopically in 100% patients -- no residual damage.
- Inflammation and fibrinoid necrosis of coronary arteries was also noted in a/c RF, similar to pathology of PAN.
- Rheumatic aortitis predominantly involved the aortic adventitia – no clinical significance
- Similar vascular inflammation has been noted systemically.

CNS - Chorea Disseminated meningoencephalitis affecting basal
ganglia, caudate nucleus, putamen, internal capsule and cerebellum
Obliterative endarteritis of cerebral and meningeal small vessels
Perivascular inflammation and petechial haemorrhage
Grossly normal brain tissue No AN formation

ARTHRITIS Endothelial inflammation of the synovia Fibrinoid granuloma, edema and diffuse inflammation. Lasts for around 2-3 weeks No permanent damage
Jaccoud’s arthropathy - due to periarthritis and fibrous thickening causing restriction of movement.

SUBCUTANEOUS NODULES Few mm to up to 2cm arising in crops , firm , painless and
freely movable under the skin. Over extensor surface of joints, skull, knuckles and spine
Central zone of necrosis surrounded by surrounded by histiocytes and fibroblasts along with perivascular inflammation
Induration occurs principally due to perivascular oozing of plasma and cells into connective tissue.

ERYTHEMA MARGINATUM
Annular evanescent eruption with well defined erythematous serpigenous borders and central clearing.
Trunk, Inner arms and thighs Never in face Painless, usually non-itchy Transient Histologically– Dermal inflammation
with minimal keratinocyte necrosis

Others... Renal microvascular inflammation
Pulmonary alveolar and microcapillary inflammation
Generalised serositis
?Rheumatic pneumonia

Immunopathological basis .........still a mystery
What is the causative factor -- ? M-protein, ? Carbohydrate If not myosin then why only the heart is chronically involved? If all valves have a common embryonic origin why is there
predisposition to the mitral valve? Gender bias in RHD.....but not in RF? Why no myocarditis? Why the variation in severity /clinical presentation in an individual
even with the same GAS strain?
--- ample space for research......implications for a “vaccine”

References Jagat Narula. Rheumatic Fever Moss and Adams.Paediatric Heart Disease. Braunwald’s Textbook of Heart Diseases E L Kaplan. Pathogenesis of acute rheumatic fever and rheumatic heart
disease: evasive after half a century of Heart2005;91:3–4.
Pulin Gupta et al. Rheumatic Fever- A Reppraisal, JIACM2012. Rajendra Tandon. Rheumatic fever pathogenesis: Approach in research
needs change. Annals of Paediatric cardiology, 2012. WHO technical report series. Rheumatic fever and rheumatic heart
disease.2004. The Pathology of Rheumatic Fever by Florence McKeown. Rheumatic Fever and RHD: The last 50 years. IJMR, April 2013.

THANK YOU
Sky is the limit………………..

QUIZ1) Father of Rheumatic heart diseasea) Charles Wellsb) Jean Baptiste Bouillaudc) Duckett Jonesd) Ludwig Aschoff

2) Which of the following GAS strains does not cause RF ?a) 2b) 3c) 5d) 6

3) Lancefield serotyping of Streptococci into various groups is based on:
a) M-proteinb) Carbohydrate Agc) Streptolysind) DNA structure

4) Least likely self antigen in the immunopathogenessis of Rheumatic fever:
1) Laminin2) Vimentin3) Antigens on valve interstitial cells4) Myosin

5)Most probably immunopathogenesis for RFA) Direct infectionB) AutoimmunityC) ToxinsD) Superantigens

6) The T cell response in RF includes all except:A) Cytokine secretionB) Epitope spreadingC) Th2 responseD ) B cell recruitment

7) Which genetic determinant has been associated with progressive valve disease in RF:
A) HLA DR4B) HLA DR7C) B cell D8/17D) HLA DR3

8) Mc Callums patch are seen on:A) Atrial side of MVB) LAC) Ventricular side of MVD) LV

9) Multinucleated cells seen in Rheumatic carditis are called:
A) Aschoff cellsB) Anitschkow cellsC) Owl eye cellsD) Mc Callums cells

10) Erythema marginatum may be seen at all places except:
A) TrunkB) PalmsC) FaceD) Thighs





























