Embed Size (px)
Citation preview

DNA sequencing methods
Dr. L.Yatawara
Dept.of MLS
FAHS,
University of Peradeniya Dr.L.Yatawara 1

DNA sequencing: methods
I. Brief history of sequencing
II. Sanger dideoxy method for sequencing
III. Sequencing large pieces of DNA
Dr.L.Yatawara 2

Why sequence DNA?
• All genes available for an organism to use -- a
very important tool for biologists
• Not just sequence of genes, but also positioning
of genes and sequences of regulatory regions
• New recombinant DNA constructs must be
sequenced to verify construction or positions of
mutations
• Etc.
Dr.L.Yatawara 3
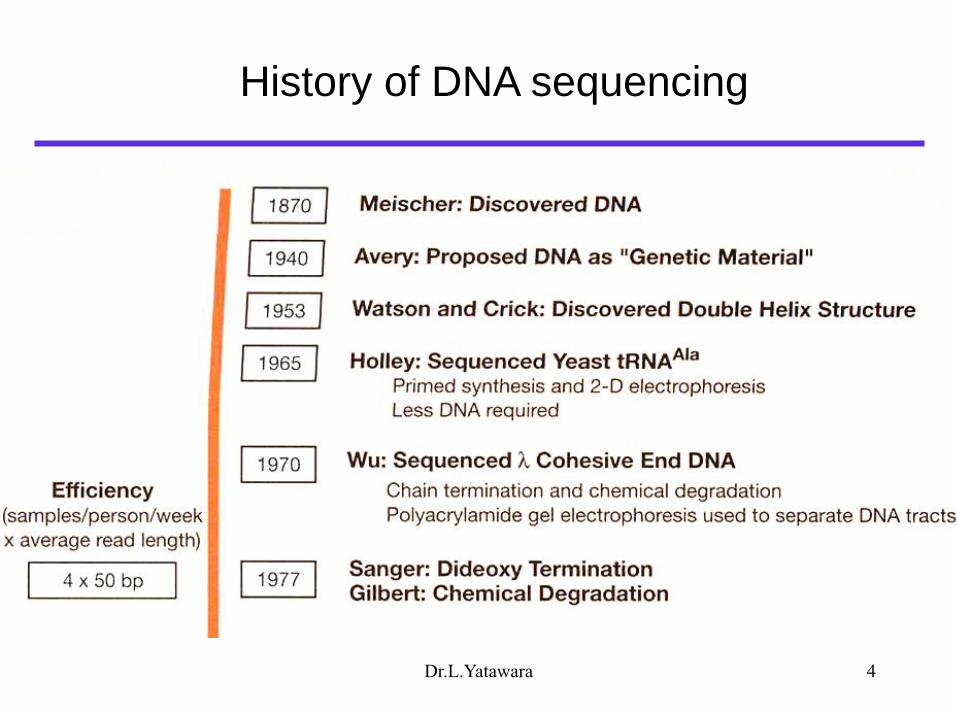
History of DNA sequencing
Dr.L.Yatawara 4

MC chapter 12
History of DNA sequencing
Dr.L.Yatawara 5

Methods of sequencing
A. Sanger dideoxy (primer extension/chain-termination)
method: most popular protocol for sequencing, very
adaptable, scalable to large sequencing projects
B. Maxam-Gilbert chemical cleavage method: DNA is
labelled and then chemically cleaved in a sequence-
dependent manner. This method is not easily scaled and
is rather tedious
C. Pyrosequencing: measuring chain extension by
pyrophosphate monitoring
Dr.L.Yatawara 6

for dideoxy sequencing you need:
1) Single stranded DNA template
2) A primer for DNA synthesis
3) DNA polymerase
4) Deoxynucleoside triphosphates and
dideoxynucleotide triphosphates
Dr.L.Yatawara 7

Primers for DNA sequencing
• Oligonucleotide primers can be synthesized by phosphoramidite chemistry--usually designed manually and then purchased
• Sequence of the oligo must be complimentary to DNA flanking sequenced region
• Oligos are usually 15-30 nucleotides in length
Dr.L.Yatawara 8

DNA templates for sequencing:
• Single stranded DNA isolated from
recombinant M13 bacteriophage containing
DNA of interest
• Double-stranded DNA that has been
denatured
• Non-denatured double stranded DNA (cycle
sequencing)
Dr.L.Yatawara 9

One way for obtaining single-stranded DNA from a double
stranded source--magnets
Dr.L.Yatawara 10

Reagents for sequencing:
DNA polymerases
• Should be highly processive, and incorporate ddNTPs efficiently
• Should lack exonuclease activity
• Thermostability required for “cycle sequencing”
Dr.L.Yatawara 11

Single stranded DNA 5‟ 3‟
5‟ 3‟
Sanger dideoxy sequencing--basic method
a) Anneal the primer
Dr.L.Yatawara 12
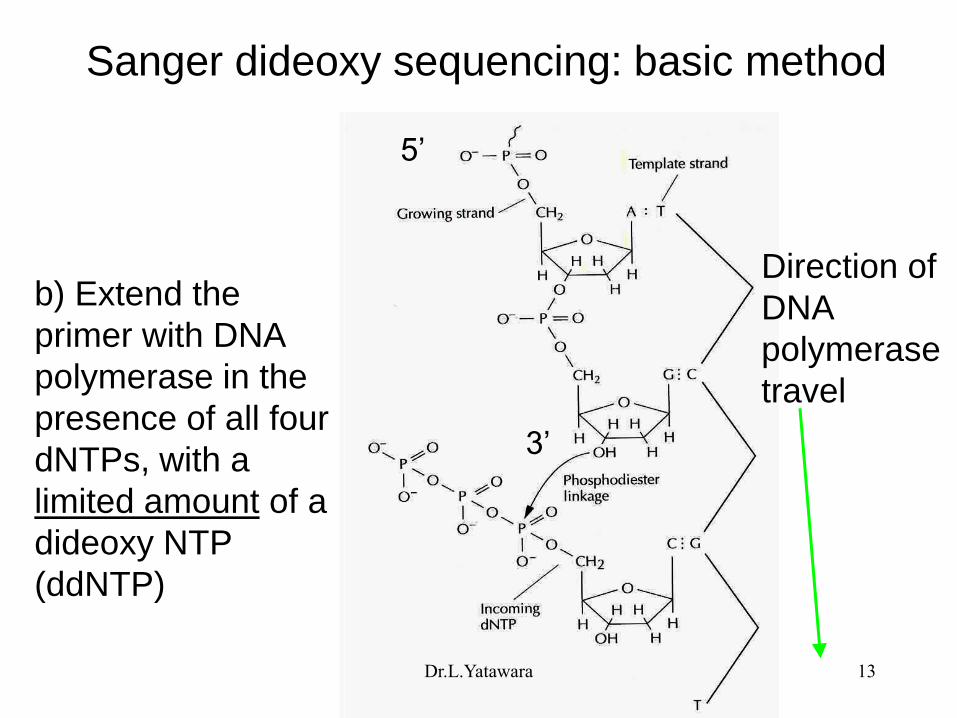
Sanger dideoxy sequencing: basic method
b) Extend the
primer with DNA
polymerase in the
presence of all four
dNTPs, with a
limited amount of a
dideoxy NTP
(ddNTP)
5‟
3‟
Direction of
DNA
polymerase
travel
Dr.L.Yatawara 13

Sanger dideoxy sequencing: basic method
5‟ 3‟
5‟ 3‟
T T T T
ddA
ddA
ddA
ddA
ddATP in the
reaction: anywhere
there‟s a T in the
template strand,
occasionally a ddA
will be added to the
growing strand
Dr.L.Yatawara 14

How to visualize DNA fragments?
• Radioactivity
– Radiolabeled primers (kinase with 32P)
– Radiolabelled dNTPs (gamma 35S or 32P)
• Fluorescence
– ddNTPs chemically synthesized to contain fluors
– Each ddNTP fluoresces at a different wavelength allowing identification
Dr.L.Yatawara 15

Analysis of sequencing products:
Polyacrylamide gel electrophoresis--good
resolution of fragments differing by a single
dNTP
– Slab gels: as previously described
– Capillary gels: require only a tiny amount of
sample to be loaded, run much faster than
slab gels, best for high throughput
sequencing
Dr.L.Yatawara 16

DNA sequencing gels: old school
Analyze sequencing
products by gel
electrophoresis,
autoradiography
Different ddNTP used in
separate reactions
Radioactively labelled primer or
dNTP in sequencing reaction
Dr.L.Yatawara 17

Dr.L.Yatawara 18

cycle sequencing: denaturation
occurs during temperature cycles
94°C:DNA denatures
45°C: primer anneals
60-72°C: thermostable DNA
pol extends primer
Repeat 25-35 times
Advantages: don‟t need a lot
of template DNA
Disadvantages: DNA pol
may incorporate ddNTPs
poorly Dr.L.Yatawara 19

An automated sequencer
The output Dr.L.Yatawara 20

Current trends in sequencing:
It is rare for labs to do their own sequencing:
--costly, perishable reagents
--time consuming
--success rate varies
Instead most labs send out for sequencing:
--You prepare the DNA (usually plasmid, M13, or PCR product),
supply the primer, company or university sequencing center
does the rest
--The sequence is recorded by an automated sequencer as an
“electropherogram”
Dr.L.Yatawara 21

• Next generation sequencing- whole genome
sequencing
• GWAS
• SNP studies
Dr.L.Yatawara 22

~160 kbp
~1 kbp
Assemble sequences by
matching overlaps
BAC sequence
BAC overlaps give genome sequence
BREAK UP THE GENOME,
PUT IT BACK TOGETHER
Dr.L.Yatawara 23

Sequencing large pieces of DNA:
the “shotgun” method
• Break DNA into small pieces (typically sizes of around 1000 base pairs is preferable)
• Clone pieces of DNA into M13
• Sequence enough M13 clones to ensure complete coverage (eg. sequencing a 3 million base pair genome would require 5x to 10x 3 million base pairs to have a reliable representation of the genome)
• Assemble genome through overlap analysis using computer algorithms, also “polish” sequences using mapping information from individual clones, characterized genes, and genetic markers
• This process is assisted by robotics Dr.L.Yatawara 24

Sequencing strategy
A whole chromosome shotgun sequencing
strategy was used to determine the genome
sequence of P. falciparum clone 3D7. This approach
was taken because a whole genome shotgun
strategy was not feasible or cost-effective with the
technology that was available at the beginning of the
project. Also, high-quality large insert libraries of (A -
T)-rich P. falciparum DNA have never been
constructed in Escherichia coli, which ruled out a
clone-by-clone sequencing strategy. The
chromosomes were separated on pulsed field gels,
and chromosomal DNA was extracted…
Dr.L.Yatawara 25

The shotgun sequences were assembled into
contiguous DNA sequences (contigs), in some cases with
low coverage shotgun sequences of yeast artificial
chromosome (YAC) clones to assist in the ordering of
contigs for closure. Sequence tagged sites (STSs)10,
microsatellite markers11,12 and HAPPY mapping7 were
also used to place and orient contigs during the gap
closure process. The high (A /T) content of the genome
made gap closure extremely difficult7–9.
Chromosomes 1–5, 9 and 12 were closed,
whereas chromosomes 6–8, 10, 11, 13 and 14 contained
3–37 gaps (most less than 2.5 kb) per chromosome at the
beginning of genome annotation. Efforts to close the
remaining gaps are continuing.
Dr.L.Yatawara 26

Methods: Sequencing, gap closure and annotation
The techniques used at each of the three participating
centres for sequencing, closure and annotation are described in
the accompanying Letters.
To ensure that each centres‟ annotation procedures produced
roughly equivalent results, the Wellcome Trust Sanger Institute
(„Sanger‟) and the Institute for Genomic Research („TIGR‟)
annotated the same100-kb segment of chromosome 14.
Thus 88% of the exons predicted by the two centres in the 100-
kb fragment were identical or overlapped.
Dr.L.Yatawara 27

Previous sequencing techniques: one DNA molecule at a time
Needed: many DNA molecules at a time -- arrays
One of these: “pyrosequencing”
Cut a genome to DNA fragments 300 - 500 bases long
Immobilize single strands on a very small plastic bead (one
piece of DNA per bead)
Amplify the DNA on each bead to cover each bead to boost the
signal
Separate each bead on a plate with up to 1.6 million wells
Dr.L.Yatawara 28

Sequence by DNA polymerase -dependent chain extension,
one base at a time in the presence of a reporter (luciferase)
Luciferase is an enzyme that will emit a photon of light in
response to the pyrophosphate (PPi) released upon nucleotide
addition by DNA polymerase
Flashes of light and their intensity are recorded
Dr.L.Yatawara 29

Height of peak indicates the number of
dNTPs added
This sequence: TTTGGGGTTGCAGTT Dr.L.Yatawara 30

Introduction to bioinformatics 1) Making biological sense of DNA
sequences
2) Online databases: a brief survey
3) Database in depth: NCBI
4) What is BLAST?
5) Using BLAST for sequence analysis
6) “Biology workbench”, etc.
www.ncbi.nlm.nih.gov
www.tigr.org
http://workbench.sdsc.edu Dr.L.Yatawara 31

There‟s plenty of DNA to make sense of
http://www.genomesonline.org/
(2006) Dr.L.Yatawara 32

Making sense of genome sequences:
1) Genes
a) Protein-coding
• Where are the open reading frames?
• What are the ORFs most similar to? (What is
the function/structure/evolution history?)
b) RNA
2) Non-genes
a) Regulation: promoters and factor-binding sites
b) Transactions: replication, repair, and
segregation, DNA packaging (nucleosomes)
Dr.L.Yatawara 33

Sequence output
Computer calls
GNNTNNTGTGNCGGATACAATTCCCCTCTAGAAATAATTTTGTTTAACTTTAAGAAGGAGATATACATATGCACCACCAC
CACCACCACCCCATGGGTATGAATAAGCAAAAGGTTTGTCCTGCTTGTGAATCTGCGGAACTTATTTATGATCCAGAAAG
GGGGGAAATAGTCTGTGCCAAGTGCGGTTATGTAATAGAAGAGAACATAATTGATATGGGTCCTAAGTGGCGTGCTTTTG
ATGCTTCTCAAAGGGAACGCAGGTCTAGAACTGGTGCACCAGAAAGTATTCTTCTTCATGACAAGGGGCTTTCAACTGCA
ATTGGAATTGACAGATCGCTTTCCGGATTAATGAGAGAGAAGATGTACCGTTTGAGGAAGTGGCANTCCANATTANGAGT
TAGTGATGCAGCANANAGGAACCTAGCTTTTGCCCTAAGTGAGTTGGATAGAATTNCTGCTCAGTTAAAACTTCCNNGAC
ATGTAGAGGAAGAAGCTGCAANGCTGNACANAGANGCAGNGNGANAGGGACTTATTNGANGCAGATCTATTGAGAGCGTT
ATGGCGGCANGTGTTTACCCTGCTTGTAGGTTATTAAAAGNTCCCGGGACTCTGGATGAGATTGCTGATATTGCTAGAGC
Raw data
Dr.L.Yatawara 34

atgttgtatttgtctgaagaaaataaatccgtatccactccttgcc
ctcctgataagattatctttgatgcagagaggggggagtacattt
gctctgaaactggagaagttttagaagataaaattatagatca
agggccagagtggagggccttcacgccagaggagaaaga
aaagagaagcagagttggagggcctttaaacaatactattca
cgataggggtttatccactcttatagactggaaagataaggatg
ctatgggaagaactttagaccctaagagaagacttgaggcatt
gagatggagaaagtggcaaattaga
What does this sequence do?
Could it encode a protein?
Dr.L.Yatawara 35

Looking for ORFs (Open Reading Frames)
using “DNA Strider”
Dr.L.Yatawara 36

ORF map 1) Where are the potential starts (ATG)
and stops (TAA, TAG, TGA)?
2) Which reading frame is correct?
= ATG
= stop
codon
Reading frame #1 appears to encode a protein Dr.L.Yatawara 37

Cautions in ORF identification
• Not all genes initiate with ATG, particularly in certain microbes (archaea)
• What is the shortest possible length of a real ORF? 50 amino acids? 25 amino acids? Cut-off is somewhat arbitrary.
• In eukaryotes, ORFs can be difficult to identify because of introns
• Are there other sequences surrounding the ORF that indicate it might be functional? – promoter sequences for RNA polymerase binding
– Shine-Dalgarno sequences for ribosome binding?
Dr.L.Yatawara 38

What is the function of
the sequenced gene? Classical methods:
-- mutate gene, characterize phenotype for clues to function
(genetics)
-- purify protein product, characterize in vitro (biochemistry)
Comparison to previously characterized genes:
-- genes sequences that have high sequence similarity
usually have similar functions
-- if your gene has been previously characterized
(using classical methods) by someone else, you want
to know right away! (avoid duplication of labor) Dr.L.Yatawara 39

NCBI NCBI home page --Go to www.ncbi.nlm.nih.gov for the following
pages
Pubmed: search tool for literature--search by author, subject, title
words, etc.
All databases: “a retrieval system for searching several linked
databases”
BLAST: Basic Local Alignment Sequence Tool
OMIM: Online Mendelian Inheritance in Man
Books: many online textbooks available
Tax Browser: A taxonomic organization of organisms and their
genomes
Structure: Clearinghouse for solved molecular structures Dr.L.Yatawara 40

What does BLAST do?
1) Searches chosen sequence database
and identifies sequences with similarity
to test sequence
2) Ranks similar sequences by degree of
homology (E value)
3) Illustrates alignment between test
sequence and similar sequences
Dr.L.Yatawara 41

Alignment of sequences:
The principle: two homologous sequences derived from the
same ancestral sequence will have at least some identical
(similar) amino acid residues
Fraction of identical amino acids is called “percent identity”
Similar amino acids: some amino acids have similar
physical/chemical properties, and more likely to substitute for
each other--these give specific similarity scores in
alignments
Gaps in similar/homologous sequences are rare, and are
given penalty scores
Dr.L.Yatawara 42

Homology of proteins
Homology: similarity of biological structure, physiology,
development, and evolution, based on genetic inheritance
Homologous proteins: statistically similar sequence, therefore
similar functions (often, but not always…)
Alignment of TFB and TFIIB sequences
Pho TFB1 1 -- ----- ----- ----- MTKQK VCPVC GST-- EFIYD PERGE IVCAR CGYPab TFB 1 -- ----- ----- ----- MTKQR VCPVC GST-- EFIYD PERGE IVCAR CGYPfu TFB1 1 -- ----- ----- ----- MNKQK VCPAC ESA-- ELIYD PERGE IVCAK CGYTko TFB1 1 -- ----- ----- ----- MSGKR VCPVC GST-- EFIYD PSRGE IVCKV CGYTko TFB2 1 -- ----- ----- MRG-- ISPKR VCPIC GST-- EFIYD PRRGE IVCAK CGYPfu TFB2 1 -- ----- MSSTE PGGGW LIYPV KCPYC KSR-- DLVYD RQHGE VFCKK CGSPho TFB2_ deduc edNTD isfro mBLAS T_ 1 -- ----- ----- YGG-- --SKI RCPVC GSS-- KIIYD PEHGE YYCAE CGHSso TFB1 1 -- ----- ----- MLYLS EENKS VSTPC PPD-- KIIFD AERGE YICSE TGESso TFB2 1 -- ----- ----- ----- ----M KCPYC KTDN- AITYD VEKGM YVCTN CASSce TFIIB 1 MM TRESI DKRAG RRGPN LNIVL TCPEC KVYPP KIVER FSEGD VVCAL CGLcon sensu s 1 m k vcpvC gst eliyd perGe ivCar cgy
Pho TFB1 32 VI EENII DMGPE WRAFD ASQR- -EKRS RTGAP ESILL HDKGL STDIG IDRPab TFB 32 VI EENIV DMGPE WRAFD ASQR- -EKRS RTGAP ESILL HDKGL STDIG IDRPfu TFB1 32 VI EENII DMGPE WRAFD ASQR- -ERRS RTGAP ESILL HDKGL STEIG IDRTko TFB1 32 VI EENVV DEGPE WRAFD PGQR- -EKRA RVGAP ESILL HDKGL STDIG IDRTko TFB2 35 VI EENVV DEGPE WRAFE PGQR- -EKRA RTGAP MTLMI HDKGL STDID WRDPfu TFB2 42 IL ATNLV DSEL- ----- ----- ---SR KTKTN DIPRY -TKRI G---- ---Pho TFB2_ deduc edNTD isfro mBLAS T_ 33 VI KS--F DTRV- ----- ----- ---RT FSSP- --PKF RSKGT S---- ---Sso TFB1 37 VL EDKII DQGPE WRAFT PEEK- -EKRS RVGGP LNNTI HDRGL STLID WKDSso TFB2 29 VI EDSAV DPGPD WRAYN AKDR- -NEKE RVGSP STPKV HDWGF HTIIG YGRSce TFIIB 51 VL SDKLV DTRSE WRTFS NDDHN GDDPS RVGEA SNPLL DGNNL STRIG KGEcon sensu s 51 vi eeniv D gpe wrafd qr ekrs rtgap esill hdkgl stdig r
Pho TFB1 80 -- ----S LTGLM REKMY RLRKW QSRLR VSDAA ERNLA FALSE LDRIT AQLPab TFB 80 -- ----S LTGLM REKMY RLRKW QSRLR VSDAA ERNLA FALSE LDRIT AQLPfu TFB1 80 -- ----S LSGLM REKMY RLRKW QSRLR VSDAA ERNLA FALSE LDRIT AQLTko TFB1 80 -- ----S LTGLM REKMY RLRKW QSRLR VSDAA ERNLA FALSE LDRLA SNLTko TFB2 83 KD IHGNQ ITGMY RNKLR RLRMW QRRMR INDAA ERNLA FALSE LDRMA AQLPfu TFB2 70 -- ----- --EFT REKIY RLRKW QKKI- ---SS ERNLV LAMSE LRRLS GMLPho TFB2_ deduc edNTD isfro mBLAS T_ 57 -- ----- --DMV REKIH RLKRL DS--- ---FG NKTEK LGVEE ISRIS SQLSso TFB1 85 KD AMGRT LDPKR RLEAL RWRKW QIRAR IQSSI DRNLA QAMNE LERIG NLLSso TFB2 77 -- ----- --AKD RLKTL KMQRM QNKIR VS-PK DKKLV TLLSI LNDES SKLSce TFIIB 1 01 -- ----- ---TT DMRFT KELNK AQGKN VMDKK DNEVQ AAFAK ITMLC DAAcon sensu s 1 01 s ltglm rekmy rlrkw qsrlr vsdaa ernla false ldrit aql
Pho TFB1 1 24 KL PKHVE EEAAR LYREA VRKGL IRGRS IESVI AACVY AACRL LKVPR TLDPab TFB 1 24 KL PKHVE EEAAR LYREA VRKGL IRGRS IESVI AACVY AACRL LKVPR TLDPfu TFB1 1 24 KL PRHVE EEAAR LYREA VRKGL IRGRS IESVM AACVY AACRL LKVPR TLDTko TFB1 1 24 SL PKHVE EEAAR LYREA VRKGL IRGRS IEAVI AACVY AACRL LKVPR TLDTko TFB2 1 33 RL PRHLK EVAAS LYRKA VMKKL IRGRS IEGMV SAALY AACRM EGIPR TLDPfu TFB2 1 07 KL PKYVE EEAAY LYREA AKRGL TRRIP IETTV AACIY ATCRL FKVPR TLNPho TFB2_ deduc edNTD isfro mBLAS T_ 92 CL PKHVE REAVR IYRKL IKSGV TKGRS IESVA AACIY ISCRL YKVPR TLDSso TFB1 1 35 NL PKSVK DEAAL IYRKA VEKGL VRGRS IESVV AAAIY AACRR MKLAR TLDSso TFB2 1 17 EL PEHVK ETASL IIRKM VETGL TKRID QYTLI VAALY YSCQV NNIPR HLQSce TFIIB 1 41 EL PKIVK DCAKE AYKLC HDEKT LKGKS MESIM AASIL IGCRR AEVAR TFKcon sensu s 1 51 kL Pkhve eeAar lyrea vrkgl irgrs iesvi aAcvy aaCrl lkvpR tld
Pho TFB1 1 74 EI SDIAR VEKKE IGRSY RFIAR NLN-- ----- ---LT PKKLF VKPTD YVNPab TFB 1 74 EI SDIAR VEKKE IGRSY RFIAR NLN-- ----- ---LT PKKLF VKPTD YVNPfu TFB1 1 74 EI ADIAR VDKKE IGRSY RFIAR NLN-- ----- ---LT PKKLF VKPTD YVNTko TFB1 1 74 EI ADVSR VDKKE IGRSF RFIAR HLN-- ----- ---LT PKKLF VKPTD YVNTko TFB2 1 83 EI ASVSK VSKKE IGRSY RFMAR GLG-- ----- ---LN LRP-- TSPIE YVDPfu TFB2 1 57 EI ASYSK TEKKE IMKAF RVIVR NLN-- ----- ---LT PKMLL ARPTD YVDPho TFB2_ deduc edNTD isfro mBLAS T_ 1 42 EI AKVAK EDKKV IARVY RLVVK KLG-- ----- ---LS SKDML IRPEY YIDSso TFB1 1 85 EI AQYTK ANRKE VARCY RLLLR ELD-- ----- ---VS VPVS- -DPKD YVTSso TFB2 1 67 EF KVRYS ISSSE FWSAL KRVQY VANS- ----- ---IP GFRPK IKPAE YIPSce TFIIB 1 91 EI QSLIH VKTKE FGKTL NIMKN ILRGK SEDGF LKIDT DNMSG AQNLT YIPcon sensu s 2 01 Ei a i r vekke igrsy rfiar ln lt pkkl vkptd Yv
Pho TFB1 1 -- ----- ----- ----- MTKQK VCPVC GST-- EFIYD PERGE IVCAR CGYPab TFB 1 -- ----- ----- ----- MTKQR VCPVC GST-- EFIYD PERGE IVCAR CGYPfu TFB1 1 -- ----- ----- ----- MNKQK VCPAC ESA-- ELIYD PERGE IVCAK CGYTko TFB1 1 -- ----- ----- ----- MSGKR VCPVC GST-- EFIYD PSRGE IVCKV CGYTko TFB2 1 -- ----- ----- MRG-- ISPKR VCPIC GST-- EFIYD PRRGE IVCAK CGYPfu TFB2 1 -- ----- MSSTE PGGGW LIYPV KCPYC KSR-- DLVYD RQHGE VFCKK CGSPho TFB2_ deduc edNTD isfro mBLAS T_ 1 -- ----- ----- YGG-- --SKI RCPVC GSS-- KIIYD PEHGE YYCAE CGHSso TFB1 1 -- ----- ----- MLYLS EENKS VSTPC PPD-- KIIFD AERGE YICSE TGESso TFB2 1 -- ----- ----- ----- ----M KCPYC KTDN- AITYD VEKGM YVCTN CASSce TFIIB 1 MM TRESI DKRAG RRGPN LNIVL TCPEC KVYPP KIVER FSEGD VVCAL CGLcon sensu s 1 m k vcpvC gst eliyd perGe ivCar cgy
Pho TFB1 32 VI EENII DMGPE WRAFD ASQR- -EKRS RTGAP ESILL HDKGL STDIG IDRPab TFB 32 VI EENIV DMGPE WRAFD ASQR- -EKRS RTGAP ESILL HDKGL STDIG IDRPfu TFB1 32 VI EENII DMGPE WRAFD ASQR- -ERRS RTGAP ESILL HDKGL STEIG IDRTko TFB1 32 VI EENVV DEGPE WRAFD PGQR- -EKRA RVGAP ESILL HDKGL STDIG IDRTko TFB2 35 VI EENVV DEGPE WRAFE PGQR- -EKRA RTGAP MTLMI HDKGL STDID WRDPfu TFB2 42 IL ATNLV DSEL- ----- ----- ---SR KTKTN DIPRY -TKRI G---- ---Pho TFB2_ deduc edNTD isfro mBLAS T_ 33 VI KS--F DTRV- ----- ----- ---RT FSSP- --PKF RSKGT S---- ---Sso TFB1 37 VL EDKII DQGPE WRAFT PEEK- -EKRS RVGGP LNNTI HDRGL STLID WKDSso TFB2 29 VI EDSAV DPGPD WRAYN AKDR- -NEKE RVGSP STPKV HDWGF HTIIG YGRSce TFIIB 51 VL SDKLV DTRSE WRTFS NDDHN GDDPS RVGEA SNPLL DGNNL STRIG KGEcon sensu s 51 vi eeniv D gpe wrafd qr ekrs rtgap esill hdkgl stdig r
Pho TFB1 80 -- ----S LTGLM REKMY RLRKW QSRLR VSDAA ERNLA FALSE LDRIT AQLPab TFB 80 -- ----S LTGLM REKMY RLRKW QSRLR VSDAA ERNLA FALSE LDRIT AQLPfu TFB1 80 -- ----S LSGLM REKMY RLRKW QSRLR VSDAA ERNLA FALSE LDRIT AQLTko TFB1 80 -- ----S LTGLM REKMY RLRKW QSRLR VSDAA ERNLA FALSE LDRLA SNLTko TFB2 83 KD IHGNQ ITGMY RNKLR RLRMW QRRMR INDAA ERNLA FALSE LDRMA AQLPfu TFB2 70 -- ----- --EFT REKIY RLRKW QKKI- ---SS ERNLV LAMSE LRRLS GMLPho TFB2_ deduc edNTD isfro mBLAS T_ 57 -- ----- --DMV REKIH RLKRL DS--- ---FG NKTEK LGVEE ISRIS SQLSso TFB1 85 KD AMGRT LDPKR RLEAL RWRKW QIRAR IQSSI DRNLA QAMNE LERIG NLLSso TFB2 77 -- ----- --AKD RLKTL KMQRM QNKIR VS-PK DKKLV TLLSI LNDES SKLSce TFIIB 1 01 -- ----- ---TT DMRFT KELNK AQGKN VMDKK DNEVQ AAFAK ITMLC DAAcon sensu s 1 01 s ltglm rekmy rlrkw qsrlr vsdaa ernla false ldrit aql
Pho TFB1 1 24 KL PKHVE EEAAR LYREA VRKGL IRGRS IESVI AACVY AACRL LKVPR TLDPab TFB 1 24 KL PKHVE EEAAR LYREA VRKGL IRGRS IESVI AACVY AACRL LKVPR TLDPfu TFB1 1 24 KL PRHVE EEAAR LYREA VRKGL IRGRS IESVM AACVY AACRL LKVPR TLDTko TFB1 1 24 SL PKHVE EEAAR LYREA VRKGL IRGRS IEAVI AACVY AACRL LKVPR TLDTko TFB2 1 33 RL PRHLK EVAAS LYRKA VMKKL IRGRS IEGMV SAALY AACRM EGIPR TLDPfu TFB2 1 07 KL PKYVE EEAAY LYREA AKRGL TRRIP IETTV AACIY ATCRL FKVPR TLNPho TFB2_ deduc edNTD isfro mBLAS T_ 92 CL PKHVE REAVR IYRKL IKSGV TKGRS IESVA AACIY ISCRL YKVPR TLDSso TFB1 1 35 NL PKSVK DEAAL IYRKA VEKGL VRGRS IESVV AAAIY AACRR MKLAR TLDSso TFB2 1 17 EL PEHVK ETASL IIRKM VETGL TKRID QYTLI VAALY YSCQV NNIPR HLQSce TFIIB 1 41 EL PKIVK DCAKE AYKLC HDEKT LKGKS MESIM AASIL IGCRR AEVAR TFKcon sensu s 1 51 kL Pkhve eeAar lyrea vrkgl irgrs iesvi aAcvy aaCrl lkvpR tld
Pho TFB1 1 74 EI SDIAR VEKKE IGRSY RFIAR NLN-- ----- ---LT PKKLF VKPTD YVNPab TFB 1 74 EI SDIAR VEKKE IGRSY RFIAR NLN-- ----- ---LT PKKLF VKPTD YVNPfu TFB1 1 74 EI ADIAR VDKKE IGRSY RFIAR NLN-- ----- ---LT PKKLF VKPTD YVNTko TFB1 1 74 EI ADVSR VDKKE IGRSF RFIAR HLN-- ----- ---LT PKKLF VKPTD YVNTko TFB2 1 83 EI ASVSK VSKKE IGRSY RFMAR GLG-- ----- ---LN LRP-- TSPIE YVDPfu TFB2 1 57 EI ASYSK TEKKE IMKAF RVIVR NLN-- ----- ---LT PKMLL ARPTD YVDPho TFB2_ deduc edNTD isfro mBLAS T_ 1 42 EI AKVAK EDKKV IARVY RLVVK KLG-- ----- ---LS SKDML IRPEY YIDSso TFB1 1 85 EI AQYTK ANRKE VARCY RLLLR ELD-- ----- ---VS VPVS- -DPKD YVTSso TFB2 1 67 EF KVRYS ISSSE FWSAL KRVQY VANS- ----- ---IP GFRPK IKPAE YIPSce TFIIB 1 91 EI QSLIH VKTKE FGKTL NIMKN ILRGK SEDGF LKIDT DNMSG AQNLT YIPcon sensu s 2 01 Ei a i r vekke igrsy rfiar ln lt pkkl vkptd Yv
Dr.L.Yatawara 43

High sequence similarity correlates with functional similarity
40-20% identity: fold can be predicted by similarity but precise
function cannot be predicted (the 40% rule)
enzymes
Non-enzymes
Dr.L.Yatawara 44

Programs available for BLAST searches
Protein sequence (this is the best option)
blastp--compares an amino acid query sequence against a protein
sequence database
tblastn--compares a protein query sequence against a nucleotide
sequence database translated in all reading frames
DNA sequence
blastn--compares a nucleotide query sequence against a nucleotide
sequence database
blastx--compares a nucleotide query sequence translated in all reading
frames against a protein sequence database
tblastx--compares the six-frame translations of a nucleotide query
sequence against the six-frame translations of a nucleotide sequence
database. Dr.L.Yatawara 45

BLAST considers all possible combinations of
matches
mismatches
gaps
in any given alignment
Gives the “best” (highest scoring) alignment of sequences
Three scores
1) percent identity
2) similarity score
3) E-value--probability that two sequences will have
the similarity they have by chance (lower number, higher
probability of evolutionary homology, higher probability of
similar function) Dr.L.Yatawara 46

What is the E-value?
The E value represents the chance that the similarity is
random and therefore insignificant. Essentially, the E value
describes the random background noise that exists for
matches between sequences. For example, an E value of 1
assigned to a hit can be interpreted as meaning that in a
database of the current size one might expect to see 1
match with a similar score simply by chance.
You can change the Expect value threshold on most main
BLAST search pages. When the Expect value is increased
from the default value of 10, a larger list with more low-
scoring hits can be reported.
Dr.L.Yatawara 47

E values (continued)
From the BLAST tutorial:
Although hits with E values much higher than 0.1 are
unlikely to reflect true sequence relatives, it is useful
to examine hits with lower significance (E values
between 0.1 and 10) for short regions of similarity. In
the absence of longer similarities, these short
regions may allow the tentative assignment of
biochemical activities to the ORF in question. The
significance of any such regions must be assessed
on a case by case basis.
Dr.L.Yatawara 48

Relationship between E-value and function
Single domain proteins
Multi-domain proteins
E value greater than 10-10, similar structure but possibly
different functions Dr.L.Yatawara 49

Computer calls
GNNTNNTGTGNCGGATACAATTCCCCTCTAGAAATAATTTTGTTTAACTTTAAGAAGGAGATATACATATGCACCACCAC
CACCACCACCCCATGGGTATGAATAAGCAAAAGGTTTGTCCTGCTTGTGAATCTGCGGAACTTATTTATGATCCAGAAAG
GGGGGAAATAGTCTGTGCCAAGTGCGGTTATGTAATAGAAGAGAACATAATTGATATGGGTCCTAAGTGGCGTGCTTTTG
ATGCTTCTCAAAGGGAACGCAGGTCTAGAACTGGTGCACCAGAAAGTATTCTTCTTCATGACAAGGGGCTTTCAACTGCA
ATTGGAATTGACAGATCGCTTTCCGGATTAATGAGAGAGAAGATGTACCGTTTGAGGAAGTGGCANTCCANATTANGAGT
TAGTGATGCAGCANANAGGAACCTAGCTTTTGCCCTAAGTGAGTTGGATAGAATTNCTGCTCAGTTAAAACTTCCNNGAC
ATGTAGAGGAAGAAGCTGCAANGCTGNACANAGANGCAGNGNGANAGGGACTTATTNGANGCAGATCTATTGAGAGCGTT
ATGGCGGCANGTGTTTACCCTGCTTGTAGGTTATTAAAAGNTCCCGGGACTCTGGATGAGATTGCTGATATTGCTAGAGC
Raw data
What does this sequence do? Cue up BLAST…..
Dr.L.Yatawara 50

MKCPYCKSRDLVYDRQHGEVFCKKCGSILATNLVDSEL
SRKTKTNDIPRYTKRIGEFTREKIYRLRKWQKKISSERN
LVLAMSELRRLSGMLKLPKYVEEEAAYLYREAAKRGLT
RRIPIETTVAACIYATCRLFKVPRTLNEIASYSKTEKKEIM
KAFRVIVRNLNLTPKMLLARPTDYVDKFADELELSERVR
RRTVDILRRANEEGITSGKNPLSLVAAALYIASLLEGERR
SQKEIARVTGVSEMTVRNRYKELA
Find the open reading frame(s)
Translate it:
Dr.L.Yatawara 51

BLAST against (go to genomes page):
-- Microbial genomes
-- environmental sequences (genomes)
Results:
1) Distribution of hits: query sequence and positions in
sequence that gave alignments
2) Sequences producing significant alignments
1) Accession number (this takes you to the sequence that
yielded the hit: gene or contig)
2) Name of sequence (sometimes identifies the gene)
3) Similarity score
4) E-value
3) Alignments arranged by E value, with links to gene reports Dr.L.Yatawara 52

2) Large percentages of
coding proteins cannot be
assigned function based
on homology
1) Homology? the function is
only inferred (NOT known) Two problems with BLAST
Dr.L.Yatawara 53

For a current list of databases and bioinformatics
tools see: Nucleic Acids Research annual
bioinformatics issue (comes out every January).
List of all the databases described, by category:
http://www.oxfordjournals.org/nar/database/cap/
Guide to NCBI: see Webct
Dr.L.Yatawara 54

Bioinformatics:
making sense of biological sequence
• New DNA sequences are analyzed for ORFs (Open Reading Frames: protein)
• Any DNA or protein sequence can then be compared to all other sequences in databases, and similar sequences identified
• There is much more -- a great diversity of programs and databases are available
Dr.L.Yatawara 55

DNA
RNA
protein
genome
“transcriptome”
“proteome”
(we have this)
(we want these)
Dr.L.Yatawara 56










