Embed Size (px)
Citation preview

Determination of Potential Parameters for Amino Acid Zwitterions
Oh Young Kwon, Su Yeon Kim, and Kyoung Tai No*,†
Department of Chemistry, Soong Sil UniVersity, Sang Do 5 Dong 1-1, Dong Jak Gu, Seoul 156-743, Korea
Young Kee KangDepartment of Chemistry, Chungbuk National UniVersity, Cheongju, Chungbuk 361-763, Korea
Mu Shik Jhon†
Department of Chemistry, Korea AdVanced Institute of Science and Technology,373-1 Kusung-dong Yusung-gu, Taejon 305-701, Korea
Harold A. Scheraga*Baker Laboratory of Chemistry, Cornell UniVersity, Ithaca, New York 14853-1301
ReceiVed: April 24, 1996; In Final Form: August 16, 1996X
An empirical potential energy function for description of the interactions between amino acid zwitterions hasbeen developed. Potential energy surfaces of molecular ion pairs, methylammonium ion-acetate ion, glycinezwitterion-methyl ammonium ion, and glycine zwitterion-acetate ion, were obtained with 6-31G**ab initiomolecular orbital calculations. The point charges of the ions were obtained with the modified partialequalization of orbital electronegativity (M-PEOE) method, the coefficients of the attractive part of thenonbonded potential were calculated from charge-dependent effective atomic polarizabilities, and the coefficientsof the repulsive part of the nonbonded potential were obtained from the equilibrium conditions of moleculesin molecular crystals and by reproducing the lattice energies of amino acid molecular crystals. The hydrogen-bond model proposed by No et al. was introduced, and the potential parameters of the model were determinedin order that the potential energy function could reproduce both theab initio potential energy surfaces andthe experimental structures of some amino acid molecular crystals. The minimum-energy crystal structuresobtained through crystal packing computations with the empirical potential energy function agreed well withX-ray crystal structures.
I. Introduction
Amino acids in aqueous solution are ionized and can act asacids or bases. Several charged species exist at equilibriumdepending on the pH of the solution.R-Amino acids havingone amino group and one carboxylic acid group crystallize fromneutral aqueous solutions as fully ionized species known aszwitterions or dipolar ions, and each amino acid has both apositive and a negative charge. Since the N-H+‚‚‚-O-Chydrogen bonds are very strong in amino acid crystals, theystabilize the zwitterion form and play a major role in determiningthe structures of amino acid crystals. Therefore, this type ofhydrogen bond is characteristic of amino acid crystals. Inves-tigation of the interactions-COO-‚‚‚+H3N-, -COO-‚‚‚H2O,and-NH3
+‚‚‚OH2 is very important for illumination of thebehavior of peptides in solution. Therefore, reliable potentialenergy functions for these hydrogen-bonded pairs are necessaryfor theoretical investigations of the behavior of peptides insolution or in crystals.Such zwitterions, for example,γ-aminobutyric acid (GABA),
act as essential neurotransmitters in the central nervous system.1,2
Therefore, investigation of the behavior of zwitterions inaqueous solution and of the interaction of zwitterions with theirreceptors is very important. The ammonium ion is an essentialfunctional group in almost all neurotransmitters and in many
other important biological molecules.3 Therefore, the preciseexpression of the interaction between-NH3
+ and-CO2- is
very important for the investigation of receptor-ligand interac-tions that occur in biological systems.4
Pair potential energy functions describing the interactionsbetween water and biomolecules have been investigated byseveral workers.5-14 Analytical potential energy functions havebeen determined for both water-neutral amino acid5a-c,7d,8a,c,10band water-amino acid zwitterion5d-g,6,7d-g,8b,c,10a,c,11-14 pairs.There are numerous studies of the interactions between
-NH3+ and water and between-CO2
- and water by molecularorbital calculations4,5a,d-g,6,7d-g,8b,9,11,12and by computer sim-ulations.5c,8b,10,13,14 Analytical potential energy functions forion-molecule pairs have also been proposed.5d-g,6,7d-f,8b How-ever, only a few molecular orbital calculations4,6,11bhave beencarried out for the interaction between-CO2
- and-NH3+, and
the potential energy surface for this interaction6,7b is not yetdescribed with a proper analytical function. Experimentally,information about the structures and binding energies betweenmolecules that contain these ionic groups can be obtained fromX-ray structures and heats of sublimation of crystals of aminoacids. Since the hydrogen bond in a zwitterion pair is verystrong, the information inherent in crystal structures is confinedto a limited number of configurations, i.e., to only energeticallystable configurations. For this reason, it is necessary to carryout molecular orbital calculations to obtain information aboutthe whole potential energy surface for the interaction betweenionic pairs.
† Member of the Center for Molecular Science, Korea.* Authors to whom correspondence should be addressed.X Abstract published inAdVance ACS Abstracts,October 15, 1996.
17670 J. Phys. Chem.1996,100,17670-17677
S0022-3654(96)01180-X CCC: $12.00 © 1996 American Chemical Society
+ +
+ +

Since the contribution of the electrostatic interaction betweenthe two dipoles,µN-H and µO-C, to the N-H+‚‚‚-O-Cinteraction energy is dominant, a physically realistic representa-tion of the electrostatic interaction is very important fordescribing the angular dependence and the energy of thehydrogen bond. Momany et al.7c calculated the minimum-energy packing structures and the lattice energies of crystals ofvarious amino acids with their empirical potential energyfunction, ECEPP. In the calculations, all potential parametersof the atoms in amino acid zwitterions, except the net atomiccharges, were taken from the potential energy function that hadbeen developed for neutral peptides.7b Although they concludedthat “the potential energy functions are applicable to confor-mational studies of polypeptides and proteins”, the resultingbinding energies that correspond to the lattice energies weretoo small. This is due mainly to low estimates of theelectrostatic interactions between amino acids. Voogd et al.15
and No et al.16 calculated the contribution of the electrostaticenergy to the lattice energy of crystals of amino acids. Suchcontributions, estimated from heat of sublimation data,17 werepredominant (over 60%) compared with the other interactionterms.The purpose of this work is to develop a set of potential
energy functions for the-NH3+‚‚‚-O2C- pair. For simplicity,
the point charges are placed on the centers of atoms, and thepoint charges are calculated by the modified partial equalizationof orbital electronegativity (M-PEOE) method.18 The hydrogen-bond model, which was proposed by No et al.,19 is utilizedbecause it reproduces the angular dependence of hydrogen bondsformed between amides and between carboxylic acids. More-over, the merit of the hydrogen-bond model is that it does notcontain an angle variable in its functional form. The nonbondedpotential parameters are determined by using the equilibriumconditions of molecular crystals. Finally, to test the reliabilityof the newly developed set of potential energy functions, crystalpacking studies of some amino acid zwitterions are carried outand compared with experimental data.
II. Computations
a. Model Compounds and Calculation of PotentialEnergy Surface. The methylammonium cation, acetate anion,and glycine zwitterion were used as rigid-geometry modelcompounds (Figure 1). The potential energy surfaces (PES)for the intermolecular interactions of the ion pairs of the modelcompounds, methylammonium ion-acetate ion (Figure 2a),glycine zwitterion-methyl ammonium ion (Figure 2b), andglycine zwitterion-acetate ion (Figure 2c), were calculated withthe 6-31G**ab initio MO method at the Hartree-Fock level.The minimum-energy conformations of the model compoundsand their ion pairs were also optimized with 6-31G**ab initioMO calculations. In the calculation of the PES of the molecularion pairs, the geometry and conformation of each monomer werefixed at their minimum-energy conformations. All theab initioMO calculations were carried out with the Gaussian92 pro-gram.20
To describe the relative orientation of the two molecules thatparticipate in the hydrogen bond in the binary complex, sixdegrees of freedom are necessary if the two molecules areassumed rigid. As shown in Figure 2a-c, the geometricalparameters are the following: the N5‚‚‚O5 distance (rNO), theC1am-N5‚‚‚O5 bond angle (θCNO), the H5‚‚‚O5-C5 bond angle
(θHOC), the C1am-H1‚‚‚N5‚‚‚O5 dihedral angle (φCHNO), the
H5-N5‚‚‚O5‚‚‚C5 dihedral angle (φHNOC), and the N5‚‚‚O5-C5-C1ac dihedral angle (φNOCC). The C1 atoms, C1
am and C1ac, in
Figure 2a-c are bonded to the ammonium ion and acetate ion,
respectively. Among these six geometrical parameters, threeare varied for the calculation of potential energy surface of themethylammonium ion-acetate ion and glycine zwitterion-acetate ion complexes. Four geometrical parameters are variedfor the glycine zwitterion-methylammonium ion complex. Theother geometrical parameters are fixed at the values of theequilibrium geometries (Figure 1). For each complex, about120 configurations were sampled, and theab initio stabilizationenergy (Vst), which corresponds to the dimerization energy at 0K, was calculated at each sampled point. The values ofVstobtained from theab initio calculations correspond to thedifference between the energy of the ionic complex and the sumof two monomer energies,Vst ) Vcomplex - (Vmonomer1 +Vmonomer2). In Table 1, the sampling regions and intervals ofthe variables are summarized for each pair.b. Empirical Potential Energy Function. The stabilization
Figure 1. Optimized geometry, bond lengths (in angstroms), and bondangles (in degrees), the M-PEOE net atomic charges,δ’s (in esu), andthe dispersion coefficients,Cii ’s (in kcal Å6/mol), of the modelcompounds treated here, namely, (a, top) methyl ammonium ion, (b,middle) acetate ion, and (c, bottom) glycine zwitterion.
Potential Parameters for Amino Acid Zwitterions J. Phys. Chem., Vol. 100, No. 44, 199617671
+ +
+ +

energy due to intermolecular interaction was described by thefollowing empirical effective pair potential energy function.
whereqi and qj are the charges on atomsi and j, rij is theirseparation distance,VHB is a hydrogen-bond potential definedbelow, and the coefficientsAij andCij are defined below. Theatom types, classified according to the chemical environments
of the atoms in the model compounds, are listed in Table 2. Inthe third and fourth columns are given the atom types used inthe M-PEOE18 and the charge-dependent effective atomicpolarizability (CDEAP) calculation method,21 respectively. Sincethe atom types of the CDEAP method are classified mainlyaccording to the valence states of the atoms in molecules andthose of the M-PEOE method are classified according to thechemical environments of the atoms in molecules, the twomethods use different classifications of the atom types.It was assumed that the net atomic charges are located on
the atomic centers, and they were calculated with the M-PEOE18
method because there are no experimental gas phase electricalmoments for the model compounds introduced in this work.M-PEOE is an empirical point charge calculation method inwhich the empirical parameters are adjusted in order that thecalculated point charges can reproduce experimental electricalmultipole moments andab initio electrostatic potentials. Themonomer charges (in both the monomeric units and thecomplex) were fixed throughout the calculations. The relativecontribution of the electrostatic interaction energy betweenamino acid zwitterions to the lattice energy of amino acidmolecular crystal was investigated by No et al.16awith severalpoint charge sets including the M-PEOE charges. The differ-ence between the electrostatic interaction energy and the latticeenergy at 0 K corresponds to the sum of the hydrogen-bond,nonbonded, polarization, etc., energies. It is very important tobalance (by repeated optimization trials) the electrostatic interac-tion energy component with the other energy components, fora correct prediction of the physical properties of molecules, i.e.,conformation, interaction energy,etc. From this point of view,it was concluded that the point charges taken as the M-PEOEcharges are physically realistic. The point charges are sum-marized in Table 3.Lennard-Jones 6-12 type functions were used for the
nonbonded interactions. The attractive coefficients,Cii ’s, werecalculated with the Slater-Kirkwood formula23
TABLE 1: Sampled Configurations of the Model Binary Complexes for Calculating theab Initio Potential Energy Surfaces
model complex rNO, ∆ra (Å) θCNO,b ∆θa (deg) φCHNO,c ∆φa (deg) φHNOC,d ∆φa(deg)
methylammonium-acetate ion 1.5-4.5, 0.2 0-170, 10 e -87 to-177, 30-102 to-162, 30
glycine zwitterion-methylammonium ion 1.5-4.5, 0.2 0-170, 10 99-159, 30 69 to 159, 30114-144, 30 84 to 174, 30
glycine zwitterion-acetate ion 1.5-4.5, 0.2 0-170, 10 e -48 to-78, 30-138 to-153, 15
a ∆ represents the grid intervals forr, θ, andφ, in the PES calculations.b θCNO is the bond angle formed by the C1am, N5, and O5 atoms.c φCHNOis the dihedral angle formed by the C1am, H1, N5, and O5 atoms.d φHNOC is the dihedral angle formed by the H5, N5, O5, and C5 atoms.e φCHNO wasnot changed; it was fixed at the value of the equilibrium conformation (see Figure 2).
Figure 2. Minimum-energy conformations of model binary com-plexes: (a, top) CH3-NH3
+‚‚‚-O2C-CH3, (b, middle) CH3-NH3
+‚‚‚glycine zwitterion, (c, bottom) CH3-CO2-‚‚‚glycine zwitterion.
Vst ) ∑i>j
qiqj
rij+ VHB + ∑
i>j ( Aijrij12
-Cij
rij6) (1)
TABLE 2: Classification of Atom Types and TheirDescriptions for Zwitterions
atom typea
(this work) description
designationfor M-PEOEmethodb
designationfor CDEAPmethodc
H1 aliphatic H H1H5 bonded to N+ H(N+) H1
C1 aliphatic C(sp3) C4
C5 in carboxylate ion (CO2-) C(O-) C3
N5 in ammonium ion (sp3) N+ N4
O5 in carboxylate ion (CO2-) O- O1
a The atom types H1 and C1 were defined in our previous paper, ref22. b The atomic species classified for the M-PEOE method aresummarized in Table 3 of the Appendix of ref 18b.cReference 21.
Cii ) 43[ ep
me1/2] Ri
2
(Ri/Ni)1/2
(2)
17672 J. Phys. Chem., Vol. 100, No. 44, 1996 Kwon et al.
+ +
+ +

where e,p, andme have their usual meanings.Ni andRi arethe number of effective electrons and effective atomic polar-izability, respectively.
Standard combination rules were used.
The effective atomic polarizabilities,Ri’s, were calculated withthe CDEAP method.21 The polarizability of theith atomicspecies in a molecule,Ri*, is described as a function of the netatomic charges,
where R*i,o, ai, and dqi are the inherent effective atomicpolarizability, charge coefficient, and net atomic charge of theith atomic species in a molecule. TheCii ’s for the atomicspecies appearing in this work are listed in the last column ofTable 3.The repulsive coefficients,Aii ’s, were adjusted to satisfy the
equilibrium conditions of the molecules in molecular crystalsand to reproduce the lattice energies of the amino acid molecularcrystals. In the calculations, the molecules in the crystals wereassumed to be rigid bodies. The details of the constraint methodand the fitting procedures are described in our previous paper.22
Since zwitterions in amino acid crystals change to a neutral formupon sublimation through intramolecular proton transfer, thelattice energy cannot be estimated17 simply from the experi-mental enthalpy of sublimation. For the estimation of the latticeenergies of some amino acid crystals, Voogd et al.15 and No etal.16 carried out extensiveab initio calculations of proton transferenergies of several amino acids. The lattice energies of severalamino acids were taken from the work of No et al.16aand wereused as constraints for determining the repulsive coefficients,A ii ’s.For the description of the hydrogen bonds formed between
-CO2- and-NH3
+, the hydrogen-bond model proposed by Noet al.19 was introduced. The hydrogen-bond model describestheab initioPES of hydrogen-bonded molecular pairs, amide-
amide, amide-carboxylic acid, and carboxylic acid-carboxylicacid very well. With that model, the angular dependence ofthe hydrogen-bond energy could be described without introduc-ing an explicit angular-dependent potential energy term. Figure3 shows the geometrical parameters that are generally adoptedfor describing the hydrogen-bond system, N-H+‚‚‚-O-C. IfrNH andrOC are fixed, then four degrees of freedom,Viz., rHO,θNHO, θHOC, andφNHOC, are necessary for describing the relativeorientations in the hydrogen-bonded system. Then, the hydrogen-bond potential energy can be described as a function of the fourgeometrical parameters.
If the coordinate system described in Figure 4 is introducedinstead of the coordinate system of Figure 3, the hydrogen-bond potential can also be described with the four distances,rHO, rNO, rHC, andrNC.
For simplicity, in order to use effective two-body potentials,VHB was approximated as a sum of two-body potential functions.
For h, a 1-6-12 type function was used.
where the subscriptij represents one of the four atomic pairs,HO, NO, HC, and NC, respectively. Since the 1-4 interactionpotential,hNC(rNC), does not contribute appreciably toVHB (asobserved during the energy calculations),hNC was not includedin the optimization of the parameters ofVHB. Therefore, theoptimum values of theB andD of the 1-2 pair, (H‚‚‚O), andof the 1-3 pairs (N‚‚‚O and H‚‚‚C), were determined toreproduce theab initio PES of the hydrogen-bonded molecularpairs. SinceB andD for the 1-4 pair were not included in theoptimization,ANC andCNC of eq 3 were used forBNC andDNC,respectively, of eq 10.
TABLE 3: Net Atomic Point Charges Calculated with theM-PEOE Method and the Attractive Nonbonded PotentialParameters,Cii, Calculated with the CDEAP Method andthe Slater-Kirkwood Formula
moleculeatomtypea
point charge(esu)
Cii
(kcal Å6/mol)
methyl ammonium ion H1 0.1161 40.68C1 0.0854 427.43H5 0.3410 30.67N5 -0.4566 577.16
acetate ion H1 -0.0055 40.68C1 -0.0984 427.43C5 0.6808 223.01O5 -0.7816 450.12
glycine zwitterion H1 0.0123 40.68C1 0.1945 427.43H5 0.3368 30.67N5 -0.5314 577.16C5 0.8685 223.01O5 -0.7845 450.12
a From column 1 of Table 2.
VNB ) ∑i>j ( Aijrij12 -
Cij
rij6 ) (3)
εij ) 0.25Cij2/Aij σij ) (Aij/Cij)
1/6 (4)
εij ) (εiiεjj)1/2 σij ) (σii + σjj)/2 (5)
R*i ) R*i,0 - aidqi (6)
Figure 3. Coordinate system,rNH (in angstroms),θNHO (in degrees),θHOC (in degrees), andφNHOC (in degrees), which is usually used todescribe a hydrogen-bonded system.
Figure 4. Coordinate system,rHO, rNO, rCH, and rNC, which areintroduced in this work for describing the hydrogen bond. Distancesare in angstroms.
V′HB ) f (rHO,θNHO,θHOC,φNHOC) (7)
V′HB(rHO,θNHO,θHOC,φNHOC) = VHB(rHO,rNO,rHC,rNC) (8)
VHB = hHO(rHO) + hNO(rNO) + hHC(rHC) + hNC(rNC) (9)
hij(rij) )qiqjrij
-Bij
rij6
+Dij
rij12
(10)
Potential Parameters for Amino Acid Zwitterions J. Phys. Chem., Vol. 100, No. 44, 199617673
+ +
+ +

For VHO, the 1-2 pair in the hydrogen bond, Hagler et al.24
introduced both 1-6-9 and 1-6-12 type functions, andMcGuire et al.7a used a 1-10-12 type function.c. Procedure for Optimization of Parameters. In this
work, the potential parameters inVNB, i.e., theAii ’s, and inVHO,i.e., theBk’s andDk’s, were determined iteratively with twooptimization procedures because these two parameter setsinfluence each other. The stabilization energy,Vst, at aconfigurationRl, can be written as follows.
For the optimization of the potential parameters, the two setsof parameters{Aii} and {Bk,Dk} were determined iteratively.In the first iterative step,{Aii} was fixed at{Aii
(0)}, and{Bk,Dk}which satisfies the following conditions were obtained as{Bk
(1),Dk(1)}.
where the superscript (0), (1), and (n) represent initial, first,and nth iterative steps.Rm represents one of theBk
(n)’s andDk(n)’s, andVab(Rl) is the ab initio stabilization energy atRl.
Next, in the second iterative step,{Bk(1),Dk
(1)} were fixed toobtain{Aii} as a{Aii
(2)}, which satisfies the following equilib-rium conditions of the crystal.
where Fto,ka and τo,ka represent the magnitude of the nettranslational force and net torque of thekath molecule in thecrystal at its equilibrium position. The superscripts o andkarepresent the equilibrium position and thekath molecule,respectively, in the asymmetric unit of the unit cell of the crystal.ELo,cal andEL
expt represent the calculated and experimental latticeenergy of the crystal at 0 K.Wt, Wτ, andWL are weightingfactors. Details of this procedure are described in our previouspaper.22 In the third iterative step,{Bk
(3),Dk(3)} which satisfy eq
13, were determined at fixed{Aii(2)}. This iterative procedure
continued until the equilibrium conditions of the crystal weresatisfied within a certain error bound, and theab initioPES aredescribed well with the optimized parameters. The latticeenergies and the information about the amino acid crystals whichwere used as constraints are summarized in Table 4.d. Test of the Reliability of the Potential Energy Function
through Crystal Packing Studies. Crystal packing computa-tions were carried out with the crystal packing computerprogram Lattice Minimization (LMIN).26 This program mini-mizes the energy of a crystal subject to no constraint other thanthe existence of a fully variable lattice. The molecules in the
unit cell are treated as independent rigid bodies; energyminimization is carried out with the translational and rotational(Euler angle) coordinates of all molecules in the unit cell, andthe six lattice parameters, as independent variables. Symmetryrelations between the molecules in the unit cell are computedbefore and after energy minimization; comparison of thesecomputed symmetries shows whether the initial space groupsymmetry was retained after energy minimization.In the determination of the parameters of the potential (section
IIa), the molecular geometry and conformation were optimized.However, for the crystal packing studies, the molecular geom-etries of the molecules in the crystals were taken from theexperimental crystal structures. The initial positions andattitudes of the molecules in the unit cell were calculated byapplying the transformations of the appropriate space group tothe coordinates of the asymmetric unit, as described in ref 26.The parametersA andB, which determine the cutoff distanceand feather for nonbonded interactions,26 were set to 4.0 and4.5, respectively.
III. Results and Discussion
The optimized hydrogen-bonding potential parameters arelisted in Table 5. The depth,εk, and the equilibrium distance,r°k, of the potential well ofhHO, the 1-2 pair of theVHB, wereobtained as 4.211 kcal/mol and 1.850 Å, respectively. Theprobability distribution of the O‚‚‚H bond distance, usuallycalled thehydrogen-bond distance, in amino acid and peptidecrystals was investigated by several workers.27,28 The resultingmaximum probability was between 1.9 and 2.0 Å forr°max. Thevalue ofr°k ) 1.850 Å is a little shorter thanr°max because bothO and H atoms are located inside of the repulsive region of the1-3 interaction potential functions. The maximum distributionrange ofrNO in the crystals was found to lie between 2.8 and2.9 Å. However,r°NO of hNO was obtained as 3.902 Å. Thismeans that the N+ atom is located inside the repulsive core ofthe oxygen atom, O-, and vice versa. The same situation arisesfor the C and H atomic pair. This is characteristic of thehydrogen-bond model introduced in this work to describe theangular dependence of the hydrogen-bonded system.19 Thehydrogen bond is formed inside the repulsive cores of the 1-3potential functions,hNO andhHC. The equilibrium distance ofthe nonbonded potential functions for the N‚‚‚O and H‚‚‚Catomic pairs are 3.145 and 3.120 Å, respectively. The repulsionbetween the 1-3 atomic pairs in the hydrogen-bonded systemplays an important role in the description of the angulardependence of the hydrogen-bonded potential energy surface.The role of the 1-3 interactions in describing the angulardependence of the hydrogen-bond energy was discussed in ourprevious paper.19
The optimized nonbonded potential parameters are sum-marized in Table 6. TheCii ’s were obtained from the effective
VHB ) ∑k
HO,NO,HC,NC
hk(rk) (11)
Vst ) Vst(Rl,{Aii},{Bk,Dk}) )VNB(Rl,{Aii}) + VHB(Rl,{Bk,Dk}) + Vel(Rl) (12)
∂
∂Rm(∑l |Vab(Rl) - Vst(Rl,{Aii
(n)},{Bk(n),Dk
(n)})|
|Vab(Rl)| ) ) 0
for all Rm (13)
FM({Aii(n)},{Bk
(n),Dk(n)}) ) ∑
ka
Nm
[Wt|Fto,ka({Aii(n)},{Bk(n),Dk(n)})| +
Wτ|τo,ka({Aii(n)},{Bk(n),Dk(n)})|] + WL|ELo,cal- EL
expt| (14)
(∂FM∂Aii(n))
jj*ii
) 0 for all ii (15)
TABLE 4: Experimental Crystal Information for SomeAmino Acid Crystals and Their Lattice Energies Estimatedfrom the Experimental Enthalpies of Sublimation19
crystal
space group andnumber of moleculesin asymmetric unit
experimentallattice energy(kcal/mol) ref
R-glycine zwitterion P21/n, Z) 4 68.0 1725a
L-alanine zwitterion P212121, Z) 4, 23 K 65.5 1725b
â-alanine zwitterion Pbca, Z) 8 25cDL-valine zwitterion P21/c, Z) 4 70.2 19
25dTda L-cysteine zwitterion P212121, Z) 4 25e
a Td means tetragonal.
17674 J. Phys. Chem., Vol. 100, No. 44, 1996 Kwon et al.
+ +
+ +
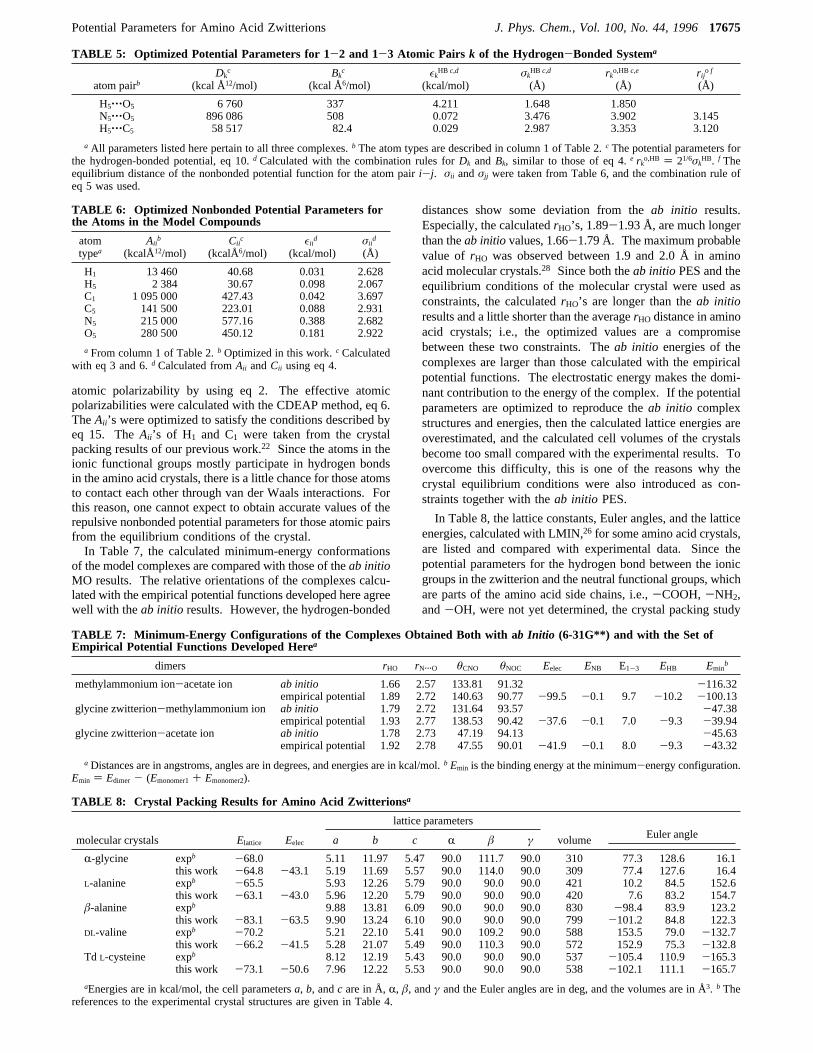
atomic polarizability by using eq 2. The effective atomicpolarizabilities were calculated with the CDEAP method, eq 6.TheAii ’s were optimized to satisfy the conditions described byeq 15. TheAii ’s of H1 and C1 were taken from the crystalpacking results of our previous work.22 Since the atoms in theionic functional groups mostly participate in hydrogen bondsin the amino acid crystals, there is a little chance for those atomsto contact each other through van der Waals interactions. Forthis reason, one cannot expect to obtain accurate values of therepulsive nonbonded potential parameters for those atomic pairsfrom the equilibrium conditions of the crystal.In Table 7, the calculated minimum-energy conformations
of the model complexes are compared with those of theab initioMO results. The relative orientations of the complexes calcu-lated with the empirical potential functions developed here agreewell with theab initio results. However, the hydrogen-bonded
distances show some deviation from theab initio results.Especially, the calculatedrHO’s, 1.89-1.93 Å, are much longerthan theab initio values, 1.66-1.79 Å. The maximum probablevalue of rHO was observed between 1.9 and 2.0 Å in aminoacid molecular crystals.28 Since both theab initioPES and theequilibrium conditions of the molecular crystal were used asconstraints, the calculatedrHO’s are longer than theab initioresults and a little shorter than the averagerHO distance in aminoacid crystals; i.e., the optimized values are a compromisebetween these two constraints. Theab initio energies of thecomplexes are larger than those calculated with the empiricalpotential functions. The electrostatic energy makes the domi-nant contribution to the energy of the complex. If the potentialparameters are optimized to reproduce theab initio complexstructures and energies, then the calculated lattice energies areoverestimated, and the calculated cell volumes of the crystalsbecome too small compared with the experimental results. Toovercome this difficulty, this is one of the reasons why thecrystal equilibrium conditions were also introduced as con-straints together with theab initio PES.
In Table 8, the lattice constants, Euler angles, and the latticeenergies, calculated with LMIN,26 for some amino acid crystals,are listed and compared with experimental data. Since thepotential parameters for the hydrogen bond between the ionicgroups in the zwitterion and the neutral functional groups, whichare parts of the amino acid side chains, i.e.,-COOH,-NH2,and-OH, were not yet determined, the crystal packing study
TABLE 5: Optimized Potential Parameters for 1-2 and 1-3 Atomic Pairs k of the Hydrogen-Bonded Systema
atom pairbDk
c
(kcal Å12/mol)Bkc
(kcal Å6/mol)εkHB c,d
(kcal/mol)σk
HB c,d
(Å)rko,HB c,e
(Å)rijo f
(Å)
H5‚‚‚O5 6 760 337 4.211 1.648 1.850N5‚‚‚O5 896 086 508 0.072 3.476 3.902 3.145H5‚‚‚C5 58 517 82.4 0.029 2.987 3.353 3.120
a All parameters listed here pertain to all three complexes.b The atom types are described in column 1 of Table 2.c The potential parameters forthe hydrogen-bonded potential, eq 10.dCalculated with the combination rules forDk andBk, similar to those of eq 4.e rko,HB ) 21/6σk
HB. f Theequilibrium distance of the nonbonded potential function for the atom pairi-j. σii andσjj were taken from Table 6, and the combination rule ofeq 5 was used.
TABLE 6: Optimized Nonbonded Potential Parameters forthe Atoms in the Model Compounds
atomtypea
Aiib
(kcalÅ12/mol)Cii
c
(kcalÅ6/mol)εiid
(kcal/mol)σii
d
(Å)
H1 13 460 40.68 0.031 2.628H5 2 384 30.67 0.098 2.067C1 1 095 000 427.43 0.042 3.697C5 141 500 223.01 0.088 2.931N5 215 000 577.16 0.388 2.682O5 280 500 450.12 0.181 2.922
a From column 1 of Table 2.bOptimized in this work.cCalculatedwith eq 3 and 6.dCalculated fromAii andCii using eq 4.
TABLE 7: Minimum-Energy Configurations of the Complexes Obtained Both with ab Initio (6-31G**) and with the Set ofEmpirical Potential Functions Developed Herea
dimers rHO rN‚‚‚O θCNO θNOC Eelec ENB E1-3 EHB Eminb
methylammonium ion-acetate ion ab initio 1.66 2.57 133.81 91.32 -116.32empirical potential 1.89 2.72 140.63 90.77-99.5 -0.1 9.7 -10.2 -100.13
glycine zwitterion-methylammonium ion ab initio 1.79 2.72 131.64 93.57 -47.38empirical potential 1.93 2.77 138.53 90.42-37.6 -0.1 7.0 -9.3 -39.94
glycine zwitterion-acetate ion ab initio 1.78 2.73 47.19 94.13 -45.63empirical potential 1.92 2.78 47.55 90.01-41.9 -0.1 8.0 -9.3 -43.32
aDistances are in angstroms, angles are in degrees, and energies are in kcal/mol.b Emin is the binding energy at the minimum-energy configuration.Emin ) Edimer - (Emonomer1+ Emonomer2).
TABLE 8: Crystal Packing Results for Amino Acid Zwitterions a
lattice parametersEuler anglemolecular crystals Elattice Eelec a b c R â γ volume
R-glycine expb -68.0 5.11 11.97 5.47 90.0 111.7 90.0 310 77.3 128.6 16.1this work -64.8 -43.1 5.19 11.69 5.57 90.0 114.0 90.0 309 77.4 127.6 16.4
L-alanine expb -65.5 5.93 12.26 5.79 90.0 90.0 90.0 421 10.2 84.5 152.6this work -63.1 -43.0 5.96 12.20 5.79 90.0 90.0 90.0 420 7.6 83.2 154.7
â-alanine expb 9.88 13.81 6.09 90.0 90.0 90.0 830 -98.4 83.9 123.2this work -83.1 -63.5 9.90 13.24 6.10 90.0 90.0 90.0 799 -101.2 84.8 122.3
DL-valine expb -70.2 5.21 22.10 5.41 90.0 109.2 90.0 588 153.5 79.0-132.7this work -66.2 -41.5 5.28 21.07 5.49 90.0 110.3 90.0 572 152.9 75.3-132.8
Td L-cysteine expb 8.12 12.19 5.43 90.0 90.0 90.0 537 -105.4 110.9 -165.3this work -73.1 -50.6 7.96 12.22 5.53 90.0 90.0 90.0 538 -102.1 111.1 -165.7
aEnergies are in kcal/mol, the cell parametersa, b, andc are in Å,R, â, andγ and the Euler angles are in deg, and the volumes are in Å3. b Thereferences to the experimental crystal structures are given in Table 4.
Potential Parameters for Amino Acid Zwitterions J. Phys. Chem., Vol. 100, No. 44, 199617675
+ +
+ +

was limited to a few amino acid crystals in which onlyN-H+‚‚‚-O-C hydrogen bonds are formed.
R-Glycine. The cell constants ofR-glycine agree well withthe experimental values and the relative orientations of themolecules in the crystal are also well reproduced. The error inthe cell volume is about 0.3%. The lattice energy is estimateda little less well, 4.7%, compared with the estimated experi-mental lattice energy.
L-Alanine. Since the errors in the cell lengths are less than0.5%, and the lattice angles are the same as the experimentalvalues, the cell volume agrees well with the experimental value.The alanines in the crystal are a little rotated, about 1-3 °, fromtheir experimental positions. The lattice energies are alsoreproduced well. The contributions of the electrostatic energyto the lattice energy ofR-glycine andL-alanine crystals arealmost the same,∼43 kcal/mol.
â-Alanine. Since the cell parameterbwas estimated less wellcompared with experiment, with an error of about 4%, the cellvolume is also estimated less well by about 4%. The other cellparameters also agree well with the experimental values. Thecalculated lattice energy is much larger, 20 kcal/mol, than thatof L-alanine. This extraordinary stabilization is due mainly tothe contribution from the electrostatic interactions.
DL-Valine. The cell parametersa andc are overestimated,about 0.15%, andb is estimated less well, about 4.5%. Thecell anglesR, â, andγ agree well with the experimental values.The cell volume is about 3% less than the experimental cellvolume. The lattice energy is a little greater than the experi-mental value obtained with a Born-Haber cycle.21 Thecontribution of the electrostatic interaction is a little smallerthan in theR-glycine andL-alanine crystals.Td L-Cysteine. The cell parameters are relatively well
reproduced, and the cell volume is also reproduced well. Theelectrostatic energy contributes more than inR-glycine,L-ala-nine, andDL-valine.In general, the energy-minimized lattice constants, Euler
angles, and lattice energies agree reasonably well with theexperimental values. The calculated lattice energies are esti-mated less well in all the crystals. The sums of the contributionof the nonelectrostatic terms,hOH, hNO, hHC, hNC, and VNB, tothe lattice energy were found to lie between 20 and 25 kcal/mol.The set of empirical potential functions developed in this work
reproduce the crystal structures well even though the potentialfunctions from theab initiominimum energy complex structuresdo not reproduce the dimer conformations very well, as shownin Table 7. The crystal structure itself does not contain anyinformation about regions of the potential surface other thannear the minimum-energy corresponding to the observed crystalstructure. In particular, it provides no information about theangular dependence of the hydrogen bond. Gas-phase potentialsfrom ab initio calculations, on the other hand, do provide thisinformation about the angular dependence and can reproducethe structure of the complex, but not the crystal structure.Therefore, we have derived our empirical potential functionswith a double constraint,Viz., to match the crystal structureandreproduce theab initio structure of the complex. While ourempirical potential does not reproduce complex structures verywell, it does reproduce crystal structures and lattice energiesvery well. We regard the empirical potential function as a goodone for polypeptides because the environment of a polypeptidein solution is much more like that in a crystal than the vacuumenvironment of theab initio calculations.If the empirical potential parameters are adjusted to reproduce
the ab initio minimum-energy complex structures well, the
energy-minimized cell volumes of the crystals are reduced toabout 80% of the experimental volumes. The cell volumescannot be increased simply by increasing the repulsive coreparameters of the nonbonded potential. If theσ of the Lennard-Jones potential function is increased, then the molecules in thecrystal rotate and become more compact with respect to theirsurroundings, the cell volume decreases, and the cell constantschange in an unpredictable manner.Since the crystals do not contain much information about the
angular dependence of the optimized hydrogen bond, duringthe optimization procedure, the angular dependence of theempirical potential energy function was mainly adjusted to theab initio potential energy surface, and the bond distance wasmainly adjusted to the information that comes from the crystals.
IV. Conclusions
A potential energy function for amino acid zwitterions hasbeen developed. The electrostatic interaction energy wascalculated with the point charges obtained from the M-PEOEmethod.18b The attractive coefficient of ther-6 term in thenonbonded potential was calculated with the Slater-Kirkwoodformula.23 The atomic polarizabilities appearing in the Slater-Kirkwood formula were calculated with the CDEAP method.21
The hydrogen-bond potential function for the zwitterions wasalso developed, based on our proposed hydrogen-bond model.19
The repulsive coefficient of ther-12 term in the nonbondedpotential was obtained through crystal packing calculations onamino acid crystals. Both the crystal equilibrium conditionsand the lattice energies were used as constraints for determiningthe repulsive parameters of the nonbonded potential. The setof potential energy functions does not reproduce the structureof ab initio complexes of the model compounds well. However,it does reproduce the crystal structures of some amino acidzwitterions. Since the purpose of this work is to developpotential energy functions for describing the interactionsbetween amino acid zwitterions and the role of the amino acidsin protein folding, we place stress on describing the crystalstructures rather than the structures of the complexes in thehydrogen-bonding models.
Acknowledgment. This work was supported by the Ministryof Science and Technology, Korea (N81540), by the KoreaScience and Engineering Foundation (Korea-U.S. CooperativeScience Program), and by research grants from the NationalScience Foundation (MCB95-13167 and INT93-06345). Thecomputations were carried out by using the Cornell NationalSupercomputer Facility, a resource of the Center for Theoryand Simulation in Science and Engineering at Cornell Univer-sity, which is funded in part by the National Science Foundation,New York State, the IBM Corporation, and members of itsCorporate Research Institute, and also by using the computersof the Chemistry Department of Soong Sil University.
References and Notes
(1) Turner, A. J.; Whittle, S. R.Biochem. J.1983, 209, 29.(2) Robert, E. In Bowery, N. G. Ed.Actions and Interactions of GABA
and Benzodiazepines; Raven Press: New York, 1984; p 1.(3) Wolff, M., Ed. The Basis of Medicinal Chemistry. InBurger’s
Medicinal Chemistry, 4th ed.; Wiley: New York, 1979.(4) Remko, M.J. Mol. Struct. (THEOCHEM)1989, 201, 287.(5) (a) Clementi, E.; Cavallone, F.; Scordamaglia, R.J. Am. Chem.
Soc. 1977, 99, 5531. (b) Scordamaglia, R.; Cavallone, F.; Clementi, E.J.Am. Chem. Soc. 1977, 99, 5545. (c) Bolis, G.; Clementi, E.J. Am. Chem.Soc. 1977, 99, 5550. (d) Carazzo, L.; Corongiu, G.; Petrongolo, C.; Clementi,E.J. Chem. Phys. 1978, 68, 787. (e) Corongiu, G.; Clementi, E.Gazz. Chim.Ital. 1978, 108, 273. (f) Clementi, E.; Corongiu, G.; Ranghino, G.J. Chem.Phys. 1981, 74, 578. (g) Ranghino, G.; Clementi, E.; Romano, S.Biopolymers1983, 22, 1449.
17676 J. Phys. Chem., Vol. 100, No. 44, 1996 Kwon et al.
+ +
+ +

(6) Ni, X.; Shi, X.; Ling, L. Int. J. Quantum Chem.1988, 34, 527.(7) (a) McGuire, R. F.; Momany, F. A.; Scheraga, H. A.J. Phys. Chem.
1972, 76, 375. (b) Momany, F. A.; Carruthers, L. M.; McGuire, R. F.;Scheraga, H. A.J. Phys. Chem. 1974, 78, 1595. (c) Momany, F. A.;Carruthers, L. M.; Scheraga, H. A.J. Phys. Chem. 1974, 78, 1621. (d)Nemenoff, R. A.; Snir, J.; Scheraga, H. A.J. Phys. Chem. 1978, 82, 2504.(e) Kim, S.; Jhon, M. S.; Scheraga, H. A.J. Phys. Chem. 1988, 92, 7216.(f) Wee, S. S.; Kim, S.; Jhon, M. S.; Scheraga, H. A.J. Phys. Chem. 1990,94, 1656. (g) Chipot, C.; Angyan, J. G.; Maigret, B.; Scheraga, H. A.J.Phys. Chem.1993, 97, 9797.
(8) (a) Jorgensen, W. L.; Swenson, C. J.J. Am. Chem. Soc. 1985, 107,1489. (b) Jorgensen, W. L.; Gao, J.J. Phys. Chem. 1986, 90, 2174. (c)Jorgensen, W. L.; Tirado-Rives, J.J. Am. Chem. Soc. 1988, 110, 1657.
(9) Bonaccorsi, R.; Palla, P.; Tomasi, J.J. Am. Chem. Soc. 1984, 106,1945.
(10) (a) Alagona, G.; Ghio, C.; Kollman, P.J. Am. Chem. Soc. 1986,108, 185. (b) Singh, U. C.; Brown, F. K.; Bash, P. A.; Kollman, P. A.J.Am. Chem. Soc. 1987, 109, 1607. (c) Alagona, G.; Ghio, C.; Kollman, P.A. J. Mol. Struct. (THEOCHEM)1988, 166, 385.
(11) (a) Kokpol, S. U.; Doungdee, P. B.; Hannongbua, S. V.; Rode, B.M.; Limtrakul, J. P.J. Chem. Soc., Faraday Trans. 21988, 84, 1789. (b)Remko, M.; Liedl, K. R.; Rode, B. M.J. Mol. Struct. (THEOCHEM)1995,336,7.
(12) Rzepa, H. S.; Yi, M.-Y.J. Chem. Soc., Perkin Trans. 21991, 531.(13) Stamato, F. M. L. G.; Goodfellow, J. M.Int. J. Quantum Chem.:
Quantum Biol. Symp.1986, 13, 277.(14) Smith, P. E.; Dang, L. X.; Pettitt, B. M.J. Am. Chem. Soc. 1991,
113, 67.(15) Voogd, J.; Derissen, J. L.; van Duijneveldt, F. B.J. Am. Chem.
Soc. 1981, 103, 7701.(16) (a) No, K. T.; Cho, K. H.; Kwon, O. Y.; Jhon, M. S.; Scheraga, H.
A. J. Phys. Chem. 1994, 98, 10742. (b) Kwon, O. Y.; Kim, S. Y.; No, K.T. Bull. Korean Chem. Soc. 1995, 16, 410.
(17) Power, L. F.; Turner, K. E.; Moore, F. H.Acta Crystallogr. 1976,B32, 11.
(18) (a) No, K. T.; Grant, J. A.; Scheraga, H. A.J. Phys. Chem. 1990,94, 4732. (b) No, K. T.; Grant, J. A.; Jhon, M. S.; Scheraga, H. A.J. Phys.Chem. 1990, 94, 4740.
(19) No, K. T.; Kwon, O. Y.; Kim, S. Y.; Jhon, M. S.; Scheraga, H. A.J. Phys. Chem. 1995, 99, 3478.
(20) Gaussian program, Revision D.2: Frisch, M. J.; Trucks, G. W.;Head-Gordon, M.; Gill, P. M. W.; Wong, M. W.; Foresman, J. B.; Johnson,B. G.; Schlegel, H. B.; Robb, M. A.; Replogle, E. S.; Gomperts, R.; Andres,J. L.; Raghavachari, K.; Binkley, J. S.; Gonzalez, C.; Martin, R. L.;Fox, D.J.; Defrees, D. J.; Baker, J.; Stewart, J. J. P.; Pople, J. A. Gaussian, Inc.,Pittsburgh, PA, 1992.
(21) No, K. T.; Cho, K. H.; Jhon, M. S.; Scheraga, H. A.J. Am. Chem.Soc. 1993, 115, 2005.
(22) No, K. T.; Kwon, O. Y.; Kim, S. Y.; Cho, K. H.; Yoon, C. N.;Kang, Y. K.; Gibson, K. D.; Jhon, M. S.; Scheraga, H. A.J. Phys. Chem.1995, 99, 13019.
(23) Scott, R. A.; Scheraga, H. A.J. Chem. Phys.1966, 45, 2091.(24) (a) Hagler, A. T.; Lifson, S.J. Am. Chem. Soc. 1974, 96, 5327. (b)
Hagler, A. T.; Leiserowitz, L.; Tuval, M.J. Am. Chem. Soc. 1976, 98, 4600.(c) Bernstein, J.; Hagler, A. T.J. Am. Chem. Soc. 1978, 100, 673. (d) Lifson,S.; Hagler, A. T.; Dauber, P.J. Am. Chem. Soc. 1979, 101, 5111. (e) Hagler,A. T.; Lifson, S.; Dauber, P.J. Am. Chem. Soc. 1979, 101, 5122. (f) Hagler,A. T.; Dauber, P.; Lifson, S.J. Am. Chem. Soc. 1979, 101, 5131.
(25) (a) Jo¨nsson, P.-G.; Kvick, A° . Acta Crystallogr. 1972, B28, 1827.(b) Destro, R.; Marsh, R. E.; Bianchi, R.J. Phys. Chem. 1988, 92, 966. (c)Papavinasam, E.; Natarajan, S.; Shivaprakash, N. C.Int. J. Peptide ProteinRes.1986, 28, 525. (d) Mallikarjunan, M.; Rao, S. T.Acta Crystallogr.1969, B25, 296. (e) Kerr, K. A.; Ashmore, J. P.; Koetzle, T. F.ActaCrystallogr. 1975, B31, 2022.
(26) (a) Gibson, K. D.; Scheraga, H. A.J. Phys. Chem. 1995, 99, 3752.(b) Gibson, K. D.; Scheraga, H. A.J. Phys. Chem.1995, 99, 3765.
(27) (a) Vinogradov, S. N.Biopolymers1979, 18, 1559. (b) Vinogradov,S. N. Int. J. Peptide Protein Res. 1979, 14, 281.
(28) Gorbitz, C. H.Acta Crystallogr. 1989, B45, 390.
JP961180V
Potential Parameters for Amino Acid Zwitterions J. Phys. Chem., Vol. 100, No. 44, 199617677
+ +
+ +





























