Embed Size (px)
Citation preview

UNIVERSIDADE FEDERAL DE SANTA CATARINA CENTRO DE CIÊNCIAS FÍSICAS E MATEMATICAS
DEPARTAMENTO DE QUÍMICA
Determinação de parâmetros de qualidade do biodiese l utilizando espectrofotometria UV/Vis
DANIEL ALFONSO SPUDEIT
Florianópolis novembro/2009

Daniel Alfonso Spudeit
Determinação de parâmetros de qualidade do biodiese l utilizando espectrofotometria UV/Vis
Relatório apresentado ao Departamento de Química
da Universidade Federal de Santa Catarina,
como requisito parcial da disciplina de
Estágio Supervisionado II (QMC 5512)
Orientador: Gustavo Amadeu Micke
Florianópolis
02/2009
Daniel Alfonso Spudeit

Determinação de parâmetros de qualidade do biodiese l utilizando espectrofotometria UV/Vis
_______________________________________ Profa. Dra. Inês Maria Costa Brighente
Coordenadora de Estágios do Curso de Química-Bacharelado
Banca Examinadora:
__________________________________________ Prof. Gustavo Amadeu Micke
Orientador
____________________________________
Prof. Ivan Gonçalves de Souza
__________________________________________ Prof. Luiz Augusto dos Santos Madureira
Florianópolis novembro/2009

Dedico o cumprimento de mais esta etapa da minha vida aos meus pais Vilmar e
Maurizaura que sempre estiveram ao meu lado nos dias bons e ruins, que souberam
perdoar meus defeitos e reconhecer minhas virtudes.

Agradecimentos:
• Aos meus Pais Vilmar e Maurizaura e aos meus irmãos Samuel e
Willian por me amarem e me apoiarem em tudo.
• A minha família que me forneceu todo apoio para que
permanecesse estudando mesmo quando achei que seria
impossível.
• Aos amigos de longa data e aos que eu conquistei durante estes
anos por todo o companheirismo, carinho, puxões de orelha e
incentivo : Alex (latino), Deonildo, Graziela salvador, Geovanni, Jô,
Leandro (leko), Robert (cenoura), Rafaella (tutu), Thiago Costa, e
a todos do lab 306.
• Aos amigos do LABEC: Ana,Luciano, Melina, Michele, Marcel,
Rafael por me ajudarem e me aturarem.
• A minha namorada, Thiele, por todo amor, carinho e muita
paciência.
• Ao meu orientador, Professor Gustavo Micke, pela confiança,
paciência e dedicação.

I
SUMÁRIO
RESUMO................................................................................................... III
1 INTRODUÇÃO ...................................... ..................................................1
1.1 Espectrofotometria UV/Vis...................... .......................................... 2
1.1.1 Instrumentação ............................................................................ 3
2 REVISÃO DA LITERATURA............................ .......................................5
3 OBJETIVOS........................................ .................................................. 11
3.1 Objetivo Geral................................. ...................................................11
3.2 Objetivos Específicos.......................... .............................................11
4 MATERIAIS E MÉTODOS.............................. .......................................12
4.1 Equipamentos................................... .................................................12
4.2 Reagentes e Soluções........................... ............................................12
4.3 Preparo das amostras........................... .............................................12
4.3.1 Extração da glicerina ...................................................................12
4.3.2 Oxidação da glicerina e análise por espectroscopia UV/Vis ....13
5 RESULTADOS E DISCUSSÃO........................... .....................................14
5.1 Determinação da glicerina...................... .............................................14
5.2 Estudo da reação entre iodato e iodeto......... ....................................14
5.2.1 Estudo do efeito pH na reação ......................................................14
5.2.2 Estudo da concentração do iodeto ...............................................16

II
5.3 Estudo da formação do complexo entre IO 4- / Mo .............................17
5.3.1 Especiação do molibdato ...............................................................17
5.3.2 Estudo da influência do pH na formação do complexo ..............18
5.3.3 Determinação da estequiometria do complexo ...........................19
5.4 Aplicação do método na determinação de glicerin a em biodiesel...... 20
5.4.1 Figuras de mérito da metodologia................... ..................................20
5.4.2 Efeito de matriz ....................................................................................21
6 CONCLUSÕES..............................................................................................24
7 REFERÊNCIAS BIBLIOGRÁFICAS....................... ......................................25

III
RESUMO
Ao longo do seu desenvolvimento humanidade criou uma dependência dos
combustíveis derivados do petróleo como a gasolina e o diesel. Porém nos
últimos anos assuntos como aquecimento global e esgotamento das fontes de
petróleo fizeram com que a sociedade buscasse novas alternativas energéticas
que fossem de fonte renovável e de queima limpa e entre essas novas
alternativas está o biodiesel que é proveniente de fontes vegetais e animais tais
como soja, dendê gordura bovina e etc. O biodiesel é produzido através de
uma reação de transesterificação do óleo com um álcool, metanol ou etanol,
dando como principal subproduto a glicerina. Quando presente em grandes
quantidades no biodiesel a glicerina pode causar danos ao motor como
entupimento de bombas de injeção e acumulo no fundo do tanque de
armazenamento e quando submetida a altas temperaturas sofre decomposição
e libera acroleína, substância nociva ao meio ambiente. A glicerina na presença
de periodato sofre uma clivagem oxidativa gerando como produtos formaldeído,
ácido fórmico, iodato e água. Neste trabalho será apresentada uma
metodologia para determinação de glicerina baseada na reação desta com o
periodato, clivagem oxidativa. Por espectroscopia UV/VIS será medida a
reação do iodato com o iodeto para gerar o triiodeto que será analisado no
comprimento de onda de 350 nm. Para quantificação da amostras foi feita uma
curva de calibração com uma faixa linear de 1,51x10-6 a 9,05x10-6 mol L-1 e
com R2 de 0,9988, com limites de quantificação e de detecção de 1,71 e 0,51
mg L-1 respectivamente. Os dados os obtidos foram comparados com os dados
obtidos por eletroforese capilar.
Palavras-chave: Biodiesel,glicerina livre, espectrometria UV/VIS e eletroforese
capilar.

1
1. Introdução
O primeiro motor movido a diesel foi criado em 1893 por Rudolph Diesel, em
Augsburg, na Alemanha, e o combustível utilizado era de óleo de amendoim. Diesel
acreditava que o combustível feito de biomassa, era uma alternativa viável aos
motores a vapor.
Diesel, entre 1911 e 1912, afirmava: “O motor a diesel pode ser alimentado por
óleos vegetais, e ajudará no desenvolvimento agrícola dos países que vierem a
utilizá-lo. O uso de óleos vegetais como combustível pode parecer insignificante hoje
em dia. Mas com o tempo irão se tornar tão importante quanto o petróleo e o carvão
são atualmente”.
Ao longo do seu desenvolvimento a humanidade criou uma dependência do
petróleo utilizando-o como fonte de energia. Porém fatores como poluição, preço e
disponibilidade deste combustível fizeram com que a humanidade buscasse fontes
de energia que fossem renováveis e menos nocivas ao ambiente, entre essas fontes
está o biodiesel.
Biodiesel é o nome de um combustível alternativo de queima limpa composto de
mono-alquilésteres de ácidos graxos de cadeia longa, derivado de óleos vegetais,
tais como girassol, mamona, soja, babaçu e demais oleaginosas, ou de gorduras
animais. Como propriedades físico-químicas os impedem de serem utilizados
diretamente como combustíveis, o óleo passa por uma transformação química
gerada por reações de esterificação, transesterificação ou craqueamento. Entre
estas a transesterificação é a mais utilizada.
Na produção do biodiesel a transesterificação compreende a reação entre um
triglicerídeo e um álcool, gerando um éster e um subproduto, a glicerina. O processo
global de transesterificação de óleos vegetais e gorduras é uma seqüência de três
reações reversíveis e consecutivas em que os monoglicerídeos e os diglicerídeos
são os intermediários, nesta reação são necessários três moles de álcool para cada
mol de triglicerídeo e o glicerol é formado como principal subproduto.

2
De acordo com a lei brasileira 11, 097, o biodiesel deve ser introduzido na matriz
energética, na proporção de 2% nos primeiros três anos após a publicação desta
podendo sofrer um aumento gradativo conforme disponibilidade de produção., tendo
como objetivo até o ano de 2012 que o percentual de biodiesel adicionado seja de
5%.
Com a introdução do biocombustível surgiu a necessidade da criação de normas
que garantissem a qualidade do combustível, pois algumas substâncias quando
presentes em grandes quantidades podem causar sérios danos ao motor e ao meio
ambiente, como por exemplo, a glicerina que pode ocasionar o entupimento de
bombas e injetores, corrosão dos tanques de armazenamento e quando submetidas
a altas temperaturas pode liberar acroleína , substância nociva. A ANP, Agência
Nacional do Petróleo, Gás Natural e Bicombustíveis, estabeleceu como 0,02% e
0,25% as quantidades máximas de glicerina livre e total, respectivamente.
Na presença de periodato, um oxidante forte, a glicerina sofre uma reação de
clivagem oxidativa formando como produto ácido fórmico, formaldeído, iodato e
água. Este estudo tem como objetivo o desenvolvimento de uma metodologia para
determinação de glicerina em amostras de biodiesel por espectrofotometria UV/VIS
e eletroforese capilar, baseando-se na clivagem oxidativa da glicerina.
1.1. Espectrofotometria no UV-Vis
Espectroscopia é o estudo da interação entre matéria e radiação
eletromagnética. A luz ultravioleta e a luz visível fornecem energia causando as
transições eletrônicas, promoção de um elétron para um orbital de maior energia.
Dependendo da energia necessária para a transição eletrônica, a molécula pode
absorver na região do ultravioleta ou no visível [1]. A luz ultravioleta é a radiação
eletromagnética com comprimento de onda entre 180 e 400 nm; a luz visível
apresenta o comprimento de onda na região entre 400 e 780 nm.
A técnica é fundamentada na lei de Lambert-Beer, que propõe que em um
determinado comprimento de onda, a absorvância depende da concentração de
espécies absorventes no meio em que a luz atravessa (Eq. 1).
A=εbc (1)

3
Onde A é a absorvância, ε é a absortividade molar, b é o caminho ótico e C é
a concentração da espécie absorvente.
Por apresentar boa robustez, custo relativamente baixo e grande número de
aplicações, a espectrofotometria na região do ultravioleta e do visível vem sendo
uma das técnicas analíticas mais empregadas [2].
1.1.1 Instrumentação
Os espectrofotômetros podem ser de feixe simples ou de feixe duplo onde um
feixe de luz passa por um divisor de feixe o qual alternadamente direciona este para
a amostra ou para a cela de referência várias vezes por segundo. Neste trabalho
será utilizado o equipamento de feixe simples.
O espectrofotômetro de feixe simples (Fig. 1) primeiramente registra o
espectro da referência e em seguida o da amostra. Este consiste basicamente de
uma fonte luz (A), onde normalmente usa-se lâmpada de deutério e tungstênio,
sendo a primeira para a região do UV e a segunda para a região do visível.
Outra parte de um espectrofotômetro é o monocromador (B). São destinados
a selecionar faixas do espectro de emissão de luz [3]. Os monocromadores podem
ser de prismas, como o da Figura 1, de grade ou de filtro. Após passar pelos
monocromadores o feixe de luz incide na amostra que está em uma célula. As
células normalmente são de quartzo ou vidro, porém a de vidro só pode ser utilizada
para análises na região do visível, pois absorve na região do UV.
Por último, o detector (D). Os espectrofotômetros podem empregar um ou
vários detectores. Os que empregam apenas um detector são chamados
monocanais ou temporais e monitoram de forma sequencial as intensidades de
radiação do espectro eletromagnético. Aqueles que empregam vários detectores são
chamados multicanais ou espaciais, monitorando simultaneamente vários
comprimentos de onda. Esses equipamentos também podem ser dispersivos,
empregando, via de regra, uma rede de difração, e não-dispersivos, empregando,
por exemplo, filtros ou diodos emissores de luz [4].

4
Figura 1. Representação esquemática de um espectrofotômetro de feixe simples e com monocromador de prisma.
A
B C
D

5
2. Revisão da Literatura
A quantidade de glicerina livre e total presente em uma amostra de biodiesel
B100, bicombustível puro, podem ser determinados por diversas técnicas analíticas.
A ANP recomenda dois métodos. O método ASTM (do inglês, “American Society for
Testing and Materials”) D 6584, que utiliza cromatografia gasosa com detector FID
(do inglês, “Flame Ionization Detection”), porém este método não se aplica ao
combustível proveniente da mamona. Para a análise de biodiesel proveniente desta
oleaginosa recomenda-se usar um método baseado na iodometria [5]. Na literatura
são apresentados diferentes métodos para determinação de glicerina na sua forma
livre e ligada em amostras de biodiesel, sempre buscando um método rápido e de
baixo custo que apresente resultados satisfatórios no que se refere à precisão e
exatidão.
O primeiro método descrito na literatura para determinação da quantidade de
glicerina total em biodiesel baseia-se em um processo enzimático. O processo
enzimático envolve extração em fase sólida, reação de saponificação, seguida pela
análise enzimática da amostra. Este método, que foi desenvolvido para o biodiesel
de colza, não faz distinção entre mono, di, e triglicerídeos, além de ser de grande
complexidade e baixa reprodutibilidade em seus resultados. [6]
Bondioli e col. descrevem o método de cromatografia a gás com detector de
ionização por chama (CG-FID) para a determinação de glicerol livre em biodiesel, o
qual não é necessário derivatização da amostra. O método é válido para óleo de
colza, óleo de girassol e biodiesel de soja e é adequado para quantidades de
glicerina livre na amostra maiores que 0,02% [7] .
Mitellbach e col. desenvolveram uma metodologia para determinação de
glicerina livre em biodiesel e simultaneamente a determinação de metanol através
da cromatografia a gás. O método consistiu na dissolução das amostras em
dimetilformamida (DMF), subseqüente silanização com bis-trimetilsilil
trifluoroacetamida (BSTFA) e posterior separação utilizando uma coluna DB-5 (60 m
x 0,25 mm) com detecção por FID ou MS (do inglês” mass spectrometry”).[8]

6
Lozano e col. apresentaram o primeiro método para determinação de glicerina
em biodiesel utilizando HPLC (do inglês “high performance liquid chromatography”)
[9]. O método foi desenvolvido para a determinação do teor de glicerol livre em
ésteres derivados de óleo vegetal. Após a extração do glicerol foi possível sua
determinação em pequenas amostras de biodiesel por HPLC combinado com um
detector por pulso amperométrico, (HPLC-PAD). O método demonstrou simplicidade,
rapidez e exatidão. Em outro estudo, Sala e Bondioli [10] avaliaram duas
metodologias distintas para determinação de glicerina. Um dos métodos empregou
uma titulação com periodato e o outro se baseou na quantificação por HPLC. A partir
dos resultados, os autores concluíram que o método com periodato mostrou
exatidão e precisão satisfatórias, que foram melhoradas com o uso de uma titulação
potenciométrica. O segundo método apresentou maior rapidez e mais informações
sobre o analito. Além disso, uma metodologia para determinação de glicerina por
HPLC-RID (do inglês “refractive index detector”) foi descrita por Hájek e col. [11]. O
método mostrou-se mais rápido que os métodos que utilizam GC e apresentou
desempenho analítico similar.
Foglia e col. [12] descrevem uma comparação entre HTGC (do inglês, “high
temperature gas chromatography”) e HPLC para determinação de glicerina ligada,
em biodiesel de soja e colza. HTGC-MS e HPLC-ELSD, (do inglês “evaporative light
scattering detector”), mostraram resultados semelhantes. Entretanto, considerando-
se o lado operacional, o método que utilizou HPLC demonstrou maior simplicidade
quando comparado ao HTGC, pelo fato de as amostras não necessitarem de
derivatização prévia. Com isso, a metodologia utilizando HPLC apresentou um
menor tempo de análise e pôde ser diretamente empregada em diversas fontes de
biodiesel.
Catharino e col. desenvolveram uma metodologia utilizando MS, a qual
empregou infusão direta e ionização por ESI (do inglês, “electrospray”) permitindo a
determinação de resíduos de glicerol, mono, di, e triglicerídeos, além da
determinação de álcool e o monitoramento da degradação e adulteração de
amostras de biodiesel. Essa técnica demonstrou ser eficiente na determinação e

7
rápida para a quantificação de parâmetros de qualidade estabelecidos para o
biodiesel.[13]
Um método espectrofotométrico baseado na quantificação da 3,5-diacetil-1,4-
dihidrolutidina no comprimento de onda de 410 nm foi desenvolvido para
determinação de glicerol [14]. Esse composto é obtido através da reação de
Hantzsch do glicerol, a qual consiste em duas reações sucessivas: (i) oxidação da
glicerina livre pelo periodato, formando formaldeído e (ii) a reação deste composto
com acetilacetona na presença de acetato de amônio, resultando no produto de
interesse. Segundo o autor, a metodologia demonstrou simplicidade, rapidez e
reduzido custo. Louzeiro e col. avaliaram estatisticamente o método desenvolvido
por Bondioli e col. [15]. De acordo com os autores a precisão do método ficou
comprometida com as variações de temperatura do laboratório e do banho
termostatizado, porém mostrou-se eficiente para a determinação de glicerina livre, e
os limites de detecção alcançados estavam muito abaixo do teor estabelecido por
lei.
Em trabalho recente Lorenço e Stradiotto [16], apresentaram uma nova
metodologia para determinação de glicerina livre em biodiesel. O método é baseado
na eletrooxidação da glicerina em eletrodos de platina através da técnica de
potenciometria cíclica. Um método rápido para extração da glicerina do biodiesel
com água seguido da eliminação de interferentes orgânicos foi desenvolvido pelos
autores. Com isso, esse novo método pôde ser aplicado em diversas amostras de
biodiesel. De acordo com os autores, o método mostrou-se rápido e com ótima
reprodutibilidade para a determinação de glicerol em várias amostras de biodiesel,
sem a necessidade de recalibração.
Naviglio e col. descreveram em artigo uma metodologia para determinação de
glicerina livre em óleos [17]. O glicerol livre foi oxidado seletivamente a ácido fórmico
e a adição do periodato ocorreu no mesmo meio em que ocorreu a transesterificação
sem qualquer interferência. O ácido fórmico gerado foi titulado potenciometricamente
através de reação ácido-base. Com o método proposto foi possível a determinação
específica e exata de glicerol livre em triglicerídeos e biodiesel.

8
Pinzi e col. propuseram uma abordagem automatizada com base na
determinação de glicerol livre e ligado à produção de biodiesel. O método foi
baseado na extração líquido-líquido de glicerina a partir do biodiesel para uma fase
aquosa etanólica, onde foi oxidada a formaldeído e numa reação subseqüente
reagiu com acetilacetona. O produto da reação foi fotometricamente medido em 410
nm. Esse experimento consistiu em um coletor por injeção em fluxo para extração
líquido-líquido sem separação de fases e mudanças interativas da direção de fluxo.
O método proposto foi mais rápido e com custos mais reduzidos quando comparado
aos métodos baseados em GC e HPLC e mostrou limites de detecção e
quantificação de glicerina livre e total bem abaixo dos limites estabelecidos pela lei
européia. [18]
Um método para análise simultânea dos produtos da reação de
transesterificação de monoglicerideos, diglicerideos, triglicerídeos, glicerol e metil
éster foi desenvolvido utilizando cromatografia de permeação em gel (GPC)
acoplada a um detector de índice de refração [19]. O tetrahidrofurano foi utilizado
como fase móvel. A preparação da amostra envolveu apenas diluição e
neutralização. O método apresentou boa reprodutibilidade. Um método similar foi
desenvolvido para avaliar a influência de diferentes fatores que pudessem afetar a
transesterificação do óleo de colza com etanol anidro e etóxido de sódio como
catalisador [20]. Por esse método foi realizada a quantificação de etil éster, mono, di,
triglicerídeos e glicerol.
Gonçalves Filho e Micke [21] apresentaram um método para determinação de
glicerina livre em biodiesel utilizando a eletroforese capilar (EC) como ferramenta
analítica. Antes da análise a reação entre o glicerol e o periodato foi realizada, em
menos de dois minutos, formando como produto de reação o iodato. A relação
iodato/periodato foi monitorada por EC. Amostras de biodiesel comerciais
produzidos a partir da gordura de frango, óleo de soja e mamona foram analisadas
pela metodologia proposta. Todas as amostras apresentaram valores inferiores aos
determinados pela ANP. De acordo com os autores, essa metodologia para extração
e análise de glicerol em biodiesel mostrou-se rápida, simples e confiável.

9
A reação de oxidação pelo periodato tem demonstrado grande importância
no desenvolvimento de métodos de micro-análises para um grande número de
espécies orgânicas e inorgânicas, podendo ser empregada em uma grande
variedade de equipamentos cromatográficos, fotométricos, eletroquímicos com os
mais diferentes instrumentos de detecção encontrados [22].
A reação de oxidação do iodeto a triiodeto pelo iodato, conhecida como
reação de Dushman, é muito importante para a química analítica e teve um papel
importante no desenvolvimento de reações como, por exemplo, a reação de
oscilação de Bray-liebhasky. Inúmeros trabalhos publicados buscam determinar uma
lei da velocidade da reação e propõem um mecanismo para esta. O primeiro
trabalho publicado sobre a reação entre iodato-iodeto foi em 1904 por Dushman [23].
Em seu trabalho, Dushman estabelece a primeira lei de velocidade para a reação
como:
v= [IO3-][H+]2{k [I-]2] + k’[I-][I3
-]} (2)
onde k= 1.3x109 M-4.s-1 e k’= 9.2x108 M-4.s-1.O autor estabelece as ordens totais de
reação como quatro, cinco e seis em concentrações de iodeto menores do que 10-8
mol L-1.
Em seu trabalho Barton e Wright, investigaram a reação de Dushman por
método anperométrico direto através da quantificação do triiodeto e o efeito catalítico
dos íons fosfatos e carboxilatos. As análises foram realizadas a temperatura
ambiente e a força iônica mantida constante com o uso de eletrólito [24].
Schimitz em seu trabalho apresenta um novo estudo sobre a cinética da
reação de Dushman e propõe um mecanismo para a catálise desta reação. Segundo
o autor, em meio não tamponado a lei de velocidade para o iodeto é de segunda
ordem em concentrações médias e altas deste, porém em concentrações muito
baixas torna-se de primeira ordem. Em meio tamponado a lei de velocidade para o
iodeto torna-se menor devido ao efeito catalisador do tampão [25].
Nakashima e col. apresentaram em estudo uma forma para determinação
simultânea de periodato e iodato utilizando eletroforese capilar com detector UV-vis,

10
através da reação de complexação entre periodato e molibdato em meio ácido. De
acordo com os autores a formação do complexo ocorre rapidamente e de maneira
seletiva, ou seja, não há formação do complexo entre o molibdato e o iodato. As
amostras foram analisadas em 220 nm. [26]

11
3. Objetivos
3.1. Objetivos Gerais
Desenvolvimento de metodologia analítica para determinação de glicerina
livre em amostras de biodiesel utilizando espectrofotometria UV/VIS. Avaliação
comparativa da metodologia desenvolvida neste estudo com a obtida por
eletroforese capilar.
3.2. Objetivos Específicos
- Validação da metodologia analítica proposta quanto aos parâmetros de exatidão,
efeito de matriz e linearidade.
- Comparação entre a nova metodologia com a metodologia publicada
recentemente utilizando a EC apresentada por Gonçalves Filho e Micke [21].

12
4. Materiais e Métodos
4.1. Equipamentos
As análises foram realizadas em um equipamento de espectrofotometria UV/Vis
modelo 800 xi (FEMTO, São Paulo, Brasil) de feixe simples. Foram utilizadas
cubetas de quartzo com o caminho ótico igual a 1 cm.
4.2. Reagentes e soluções
Todos os reagentes utilizados são de grau analítico. Glicerina, iodato de potássio,
ácido periódico, iodeto de potássio, clorofórmio foram adquiridos da Synth (São
Paulo, Brasil). Molibdato de sódio foi adquirido da Merck (São Paulo, Brasil). Água
desionizada (Mili-Q, Milipore, Bedford, MA, E.U.A.) foi utilizada para o preparo das
soluções. Soluções padrão estoque de periodato de sódio, glicerina, iodeto de
potássio e molibdato de sódio foram preparadas com água desionizada e
armazenadas sob refrigeração (4 ºC). As soluções padrão de trabalho foram
preparadas diariamente a partir da diluição das soluções estoque com água.
4.3. Preparo das amostras.
4.3.1. Extração da glicerina .
Apoiando-se no fato de que a glicerina é mais solúvel em água do que no
biodiesel e que esse apresenta inúmeras propriedades que dificultariam as análises,
fez-se uma lavagem do biodiesel com água para que a glicerina livre presente fosse
transferida para a fase aquosa.
As amostras foram preparadas pesando-se 40 mg de biodiesel para cada
1500 µL de água. As amostras foram colocadas sob agitação durante alguns
minutos para garantir uma extração mais eficiente. Após a agitação adicionou-se
250 µL de clorofórmio, apenas para que ocorresse uma inversão de fases e com
isso fosse facilitada a remoção da fase aquosa.

13
4.3.2. Oxidação da glicerina e análise por espectroscopia UV-Vis
Em um eppendorf de 2 mL adicionou-se 500 µL da fase aquosa, 500 µL de
água e 1000 µL de periodato (IO4-), em excesso, para a oxidação da glicerina.
Para análise no espectrofotômetro as amostras foram preparadas da
seguinte forma: primeiro adicionou-se 300 µL de molibdato, 1100 µL de água e 500
µL da amostra oxidada, aguardou-se um minuto para que a reação entre molibdato e
o periodato, que estava em excesso em relação a glicerina, ocorresse. Em seguida
adicionou-se 600 µL de iodeto e a leitura em 352 nm foi realizada. As concentrações
dos reagentes estão resumidas na Tabela 1.
Tabela 1. Concentrações dos reagentes utilizados
Reagentes [ ]inicial mol L-1
Volume adicionado( μL)
Periodato 9,4x10-4
1000
Molibdato 0,1 300
Tampão ácido fórmico/formiato 0,5/0,25 500
Iodeto 0,5 600
H20 ****** 1100

14
5. Resultados e discussão 5.1. Determinação da glicerina.
Glicerina e periodato reagem rapidamente para gerar como produto
formaldeído, ácido fórmico, água e iodato (Equação 3). Iodato reage com o iodeto
para gerar o triiodeto, reação de Dushman, porém o periodato também reage com
iodeto para também formar triiodeto então para que este não seja um interferente,
buscou-se uma maneira de impedir esta reação, através da complexação do
periodato com o molibdato, já que em meio ácido os dois reagem rapidamente para
formar um complexo estável e o mesmo não ocorre entre o molibdato e o iodato.
Baseando-se nessas reações desenvolveu-se uma metodologia para determinação
da glicerina livre presente no biodiesel por espectrofotometria UV-Vis.
(3)
5.2. Estudo da reação entre iodato e iodeto.
Iodato e iodeto reagem rapidamente para formar o triiodeto, essa reação
pode ser influenciada por diversos fatores, porém estudaremos apenas o efeito do
pH e da concentração de iodeto.
5.2.1.Estudo do efeito do pH na reação
Como pode-se observar através da lei da velocidade de reação sugerida
por Dushman em 1904 [19], a reação depende da concentração hidrogeniônica
(Equação 4):
V=k[IO3-]m[I-]n[H+]p (4)
onde v é velocidade da reação, k é constante de velocidade e os expoentes m, n, p
são as ordens parciais dos reagentes.

15
Para demonstrar a influência do pH na reação, um estudo foi realizado por
espectrofotometria em um único comprimento de onda, 352 nm. Os pontos
analisados foram preparados variando-se a concentração de iodato, a fim de simular
sua formação pela reação de oxidação da glicerina (i), todos os pontos foram
preparados em diferentes pHs e por fim adicionou-se uma quantidade fixa de iodeto.
Após a adição do iodeto os pontos foram acompanhados em média por uma hora
em intervalos de dez minutos. Com os dados obtidos foi possível obter retas com
inclinações diferentes e plotando-se essas inclinações em função do tempo, (Figura
2), pode-se observar que a sensibilidade aumenta com a diminuição do pH do meio
e que em pHs maiores a reação torna-se muito lenta a ponto de sua utilização não
ser viável. Os dados obtidos confirmam o que Dushman sugeriu em seu artigo, que
a reação entre iodato e iodeto é dependente do pH, quanto menor este for mais
rapidamente a reação (Equação 5) ocorrerá.
(5)
0 50 100 150 200 250
0
2000
4000
6000
8000
10000
12000
14000
pH 4,45 pH 4,96 pH 5,24 pH 5,51
Incl
inaç
ao
Tempo, min.
Figura 2. Influencia do pH na reação entre iodato e iodeto.
Quando o triiodeto é formado, por não ser estável, este entra em equilíbrio
formando iodo e iodeto, Equação 6. Então para que ocorra um aumento da sua
estabilidade o iodeto deve ser adicionado em excesso, pois o excesso deste
IO3- + 8I- + 6H+ 3I3
- + 3H2O
16
0
0.5
1
1.5
2
2.5
0 0.01 0.02 0.03 0.04 0.05 0.06 0.07 0.08 0.09
[Iodeto]
Ab
sorv
ânci
a
deslocaria o equilíbrio da reação, Equação 5, quase que totalmente para a direita
favorecendo a formação do triiodeto.
I2 + I- I3
-
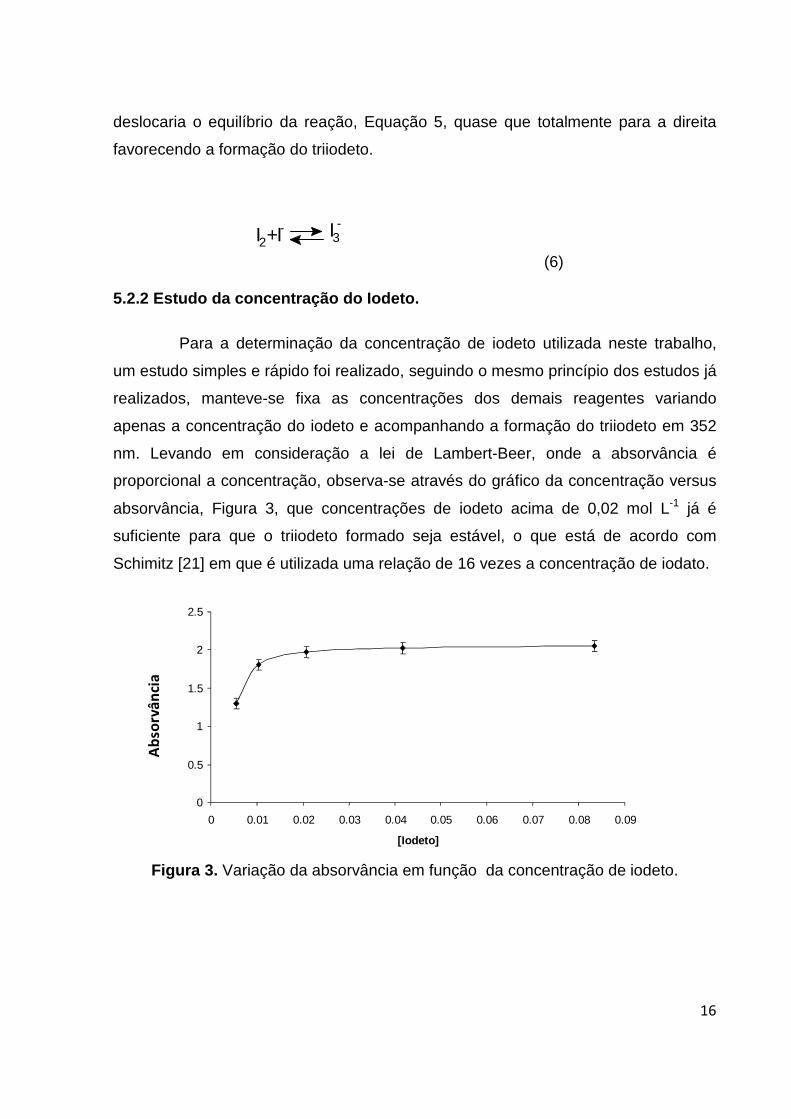
(6) 5.2.2 Estudo da concentração do Iodeto.
Para a determinação da concentração de iodeto utilizada neste trabalho,
um estudo simples e rápido foi realizado, seguindo o mesmo princípio dos estudos já
realizados, manteve-se fixa as concentrações dos demais reagentes variando
apenas a concentração do iodeto e acompanhando a formação do triiodeto em 352
nm. Levando em consideração a lei de Lambert-Beer, onde a absorvância é
proporcional a concentração, observa-se através do gráfico da concentração versus
absorvância, Figura 3, que concentrações de iodeto acima de 0,02 mol L-1 já é
suficiente para que o triiodeto formado seja estável, o que está de acordo com
Schimitz [21] em que é utilizada uma relação de 16 vezes a concentração de iodato.
Figura 3. Variação da absorvância em função da concentração de iodeto.

17
5.3. Estudo da formação do complexo entre IO 4-/ Mo.
Para uma melhor compreensão da reação de complexação entre o
periodato e o molibdato foram estudados algumas características desta reação, tais
como a especiação do molibdato no pH de trabalho, o pH ótimo de formação e
estequiometria da reação .
5.3.1. Especiação do molibdato
Inúmeros estudos foram realizados com o intuito de compreender a
complexa química do molibdato como, por exemplo, quando uma solução básica de
molibdato é acidificada, os íons de molibdato se condensam nas mais diferentes
formas para formar diferentes espécies do isopolimolibdato. O processo de
condensação pode ser considerado uma rápida agregação das espécies
protonadas. Prasad e col. [27] sugerem que o processo de condensação para os
poliânions, pode ser representado pela Equação 7 a seguir :
aH + bMoO4
2- = (MoO4)b-a/2(MoO3)a/2 + (a/2)H2O (7)
Baseando-se no trabalho de Prasad e col. [27] realizou-se uma titulação
potenciométrica do molibdato com ácido clorídrico a fim de se determinar a espécie
predominante em pH menor que 4. Através da análise do gráfico apresentado na
Figura 4 , onde se tem o pH em função do volume de HCl adicionado. A partir deste
estudo observou-se um ponto de equivalência em 3 mL. Com isso foi possível
encontrar o número de mols de H+ necessários para neutralizar todo o molibdato.
Como o número de mols utilizados do molibdato de sódio é conhecido, foi possível
baseando-se na Equação 7, sugerir que o isopolimolibdato predominante em pH
abaixo de 4 é um octomêro de fórmula molecular [Mo8O26 ]4-. Para este caso os
valores de a e b são de 12 e 8, respectivamente.

18
0 1 2 3 4 50
1
2
3
4
5
6
7
8
-log
[H+]
volume HCl / mL
pH dpH / dV
Figura 4. Titulação potenciométrica do MoO4
2- com HCl 0,1 mol L-1. 5.3.2. Estudo da influencia do pH na formação do c omplexo.
Para a escolha do pH no qual a reação ocorre de maneira mais eficiente,
realizou-se um estudo em que se manteve fixa as concentrações dos reagentes e,
variou-se apenas o pH. As leituras foram realizadas em 280 nm, região na qual o
complexo absorve sem interferência do molibdato ou periodato. Os resultados são
mostrados na Figura 5.
Figura 5: Estudo da formação do complexo em função do pH

19
Através dos dados obtidos, estabeleceu-se que a faixa ótima de pH para
formação do complexo encontra-se entre 2,8 e 3,5. Portanto, o pH de trabalho
escolhido foi 3,5.
5.3.3. Determinação da estequiometria do complexo.
Para a determinação da razão entre IO-4/Mo foi utilizado o método das
variações contínuas (Gráfico de Job), sendo que são utilizadas as mesmas
concentrações de periodato e molibdato variando-se apenas o volume destes, de tal
maneira que o volume final e o número de mols total mantenham-se constantes. As
amostras foram medidas no comprimento de onda adequado. Através da construção
de um gráfico da absorvância versus fração volumétrica de um dos reagentes, foi
possível obter a estequiometria do complexo, e através da comparação das
curvaturas das linhas experimentais com as linhas retas teóricas é possível a
obtenção da constante de formação.
As soluções de molibdato e periodato foram preparadas na concentração
de 0,001 mol L-1, os pontos da curvas foram preparados em tampão formiato/ácido
fórmico, pH 3,5, e medidas em 280 nm. Os resultados obtidos são mostrados na
Figura 6.
0,0 0,2 0,4 0,6 0,8 1,00,05
0,10
0,15
0,20
0,25
0,30
0,35
Abs
orvâ
ncia
Fraçao volumétrica
Figura 6 . Gráfico de Job para determinação da estequiometria e da constante de formação do complexo periodato/molibdato.

20
Através do gráfico das absorvâncias medidas versus a fração volumétrica
do molibdato, foi possível observar que o complexo formado possui uma alta
estabilidade, e que molibdato e periodato reagem na proporção de 6:1, concordando
então com a Equação 8:
(8)
5.4. Aplicação do método na determinação de gliceri na livre em biodiesel
As amostras foram monitoradas através da determinação da concentração
do triiodeto, este apresenta duas bandas na região do UV relativas a transferência
de carga, uma em 288 nm e outra em 352 nm como mostra o espectro apresentado
na Figura 7. O comprimento de onda de 352 nm foi utilizado para a determinação,
pois nenhum dos reagentes utilizados apresentou bandas próximas desta região,
evitando interferências.
240 260 280 300 320 340 360 380 400 420
0
200
400
600
800
Abs
orvâ
ncia
Comprimento de onda, nm.
Figura 7. Espectro de absorvância do complexo triiodeto (ε = 2533 L mol-1 cm-1 ) na concentração de 21 µmol L-1. Cubeta de quartzo com caminho óptico de 1 cm. 5.4.1. Figuras de mérito da metodologia
Para a quantificação das amostras foi realizada uma curva de calibração
com a concentração do periodato fixa e com concentrações crescentes de glicerina
IO4- + 6Mo [IMo6O24]
5-H+

21
obtendo-se uma resposta linear na faixa de 1,51 x 10-6 a 9,05 x 10-6 mol L-1 e com
um R2 de 0,9988. A Figura 8 apresenta a curva de calibração obtida com a
metodologia otimizada.
1 2 3 4 5 6 7 8 9 10
0,2
0,4
0,6
0,8
1,0
1,2
[ ] glicerina, 10 -6 mol.L -1
abso
rbân
cia
Figura 8: Curva de calibração para a metodologia proposta.
5.4.2. Efeito de matriz Para verificar o efeito da matriz da amostra, foram construídas uma curva de adição
de padrão e uma curva de calibração externa. A Figura 9 apresenta as curvas
obtidas.

22
1 2 3 4 5 6 7 8 9 10
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
curva glicerina adi. padrao
abso
rbân
cia
[ ] glicerina ,10 -6mol.L -1
Figura 9: curvas de adição de padrão e calibração externa para avaliação do efeito de matriz. É possível observar a partir dos resultados obtidos que a curva de adição de
padrão apresenta inclinação semelhante à curva de calibração externa, o que
demonstra que o método não apresenta efeito de matriz significativo.
Com o objetivo de demonstrar a validade do método desenvolvido alguns
fatores foram avaliados tais como linearidade, limite de detecção, limite de
quantificação, interferência de matriz e exatidão. A linearidade é demonstrada pelo
coeficiente de determinação (R2) de 0, 9988 (Tabela 2) que pela sua proximidade da
unidade demonstra que o modelo linear explica as variações de y. Outro valor que
demonstra a significância da regressão é o valor F de 4960,95 (Tabela 2) já que
quanto maior for o valor deste mais significante é a regressão.
Tabela 2. Figuras de mérito do método.
Linearidade –inclinação 0,127 Desvio padrão da inclinação 0,00181 Linearidade – intercepção 0,0136 Desvio padrão da Intercepção 0,00987 Linearidade- coeficiente de determinação 0,9988 F 4960 LOQ (mg L-1) 0,0713 LOD (mg L-1) 0,0213

23
O método apresentou um limite de detecção de 0,0213 mg L-1 e limite de
quantificação de 0,0713 mg L-1. Os limites de detecção e de quantificação foram
determinados através das equações 9 e 10 respectivamente.
LD = 3,33 s (9) LQ = 10 s (10) S S
Onde s é a estimativa do desvio padrão da reta de calibração e S é o coeficiente angular da reta.
Para avaliação da exatidão do método foram analisadas cinco amostras de
diferentes fontes de biodiesel e os dados obtidos comparados com os dados obtidos
por eletroforese capilar. Analisando os dados da Tabela 3 podemos concluir que o
método desenvolvido apresenta uma boa exatidão quando comparado com os
dados obtidos por eletroforese capilar .
Tabela 3: Dados da quantificação da amostra.
Amostra %UV-Vis % EC
Óleo de soja 0,0043 0
Biodiesel de soja 0,026 0,02
Biodiesel de soja B 0,014 0,017
FRITURA A 0,033 0,027
FRITURA B 0,015 0,016

24
6 Conclusões
Comparando-se os dados obtidos por espectrofotometria UV/Vis com os obtidos por
eletroforese capilar, podemos concluir que o método proposto é eficiente para
quantificação de glicerina livre em amostras de biodiesel. E analisando os valores
obtidos pode-se dizer que todas as amostras analisadas se encontram dentro dos
valores estipulados pela ANP.

25
7. Referências Bibliográficas
1. BRUICE, Paula Y. Organic chemistry. 4th ed. California: Prentice Hall,. 1228 p, c2004. 2. LOBINSKI, R.; MARCZENKO, Z.; Recent Advances in Ultraviolet-Visible Spectrophotometry.Recent Advances in Ultraviolet-Visible Spectrophotometry Crit. Rev. Anal. Chem . 23, 55,1992. 3. Fonte de luz, detectores e monocromadores. Disponível em: <http://www.ifi.unicamp.br/~accosta/roteiros/9/nota%2009.htm>. Acesso em 06 Jun.2009 4. RAIMUNDO, I.M. PASQUINI, C. Espectrofotometria Multicanal e Arranjos de Fotodiodos. Química Nova, v. 20, p.83-88,1997. 5. Agência Nacional de Petróleo, Resolução ANP nº 42 , (24 de novembro de 2004), retificada DOU (19 de abril de 2005) – ANEXO B. 6. BAILER, J., HUEBER, K., Determination of saponifiable glycerol in biodiesel. Fraenius J. Anal Chem ., Heidelberg, v. 340, n. 3, p. 186. mar 1991. 7. BONDIOLI, P., MARIANI C., LANZANI A., and FEDELI E., Vegetable Oil Derivatives as Diesel Fuel Substitutes. Analytical Aspects. Note 2: Determination of Free Glycerol, Riv. Ital. Sostanze Grasse 69:7–9 ,1992. 8. MITTELBACH, M., ROTH G., and BERGMANN A., Simultaneous Gas Chromatographic Determination of Methanol and Free Glycerol in Biodiesel, ——Ibid.d. 42:431–434 ,1996. 9. LOZANO, P., CHIRAT N., GRAILLE J., and PIOCH D., Measurement of Free Glycerol in Biofuels, Fresenius J. Anal. Chem . 354:319–322, 1996. 10. SALA,M., BONDIOLI,P. Caratterizzazione analitica del glicerolo. Considerazioni sperimentali Riv. Ital. Sostanze Grasse 75, 305,1998. 11. HÁJEK, M, SKOPAL, F., MACHEK J. Determination of free glycerol in biodiesel. Eur. J.Lipid. Sci. Technol .108,666–9, 2006. 12. FOGLIA TA, JONES KC, PHILIPS JG, MITTELBACH M. Comparison of chromatographic methods for the determination of bound glycerol in biodiesel. Chromatographia 60, 305-311,2004.

26
13. CATHARINO R.R. e col. Biodiesel Typification and Quality Control by Direct Infusion Electrospray Ionization Mass Spectrometry Fingerprinting. Energy Fuels. 21, 3698, 2007. 14. BONDIOLI, P. & BELLA, L. D. An alternative spectrophotometric method for the determination of free glycerol in biodiesel. Eur. J. Sc. Technol., v. 107, p. 153-157, 2005. 15. LOUZEIRO, H. C. ; SILVA,F.C MOUZINHO, A. M. C. ; NASCIMENTO, A. A. ; SOUZA, A. G. ; CONCEIÇÃO, M. M. Determinação do Teor de Glicerina Livre em Biodiesel por Espectrofotometria do UV-Visível. In: 1º Congresso da rede Brasileira de Tecnologia de Biodiesel, 2006. Artigos técnico-científicos , v. 1. p. 286-290, 2006. 16. LOURENÇO,L.M. STRADIOTTO,N.R. Determination of free glycerol in biodiesel at a platinum oxide surface using potential cycling technique. Talanta 79, 92–96,2009. 17. NAVIGLIO, D. ROMANO,R. PIZZOLONGO, F. SANTINNI, A. DE VITO, A. et al. Rapid determination of esterified glycerol and glycerides in triglyceride fats and oils by means of periodate method after transesterification. Food Chemistry. v102, p399–405, 2007. 18 . PINZI, S. CAPOTE, F.P. JIMÉNEZ, R.J. DORADO, M.P. CASTRO, M.D.L. Flow injection analysis-based methodology for automatic on-line monitoring and quality control for biodiesel production. Bioresource Technology . v100, p421–427, 2009. 19. DARNOKO D, CHERYAN M, PERKINS EG. Analysis of vegetable oil transesterification products by gel permiationchromatography. J Liq Chrom Rel Technol ;v23(15), p2327-35, 2000. 20. FILLIERES, R. MLAYAH B.B, Delmas M. Ethanolysis of rapeseed oil: quantitation of ethyl esters, mono-, di-, and triglycerides and glycerol by high performance size-exclusion chromatography. J Am Oil Chem Soc ; v72(4), p 427-32. 1995 21. GONÇALVES FILHO, L.C., MICKE, G.A., J. Chromatogr. A 1154 (2007) 477. 22. VLESSIDIS, A.G., EVMIRIDS, N.P., Periodate oxidation and its contribution to instrumental methods of micro-analysis—A review, Analytica Chimica Acta 652 (2009) 85–127. 23. DUSHMAN, S.,The rate of the reaction between iodic and hidriodic acids. J. Phys. Chem., 1904, 8, 453. 24. BARTON, A. F. M., e WRIGHT, G. A., Kinetics of the lodate-Iodide Reaction : Catalysis by Carboxylate and Phosphate Ions, J. Chem. Soc . (A),2096-2103, 1968. 25. SCHIMITZ,G., Kinetics and mechanism of the iodate/iodide reaction and other

27
related reactions, Phys. Chem. Chem. Phys ., 1999, 1, 1909-1914. 26. NAKASHIMA, Y., SHEN, H., KUSYAMA, K., Simultaneous Determination of Periodate and Iodate by Capillary Electrophoresis, Analytical Science , v15, p 725-728.1999. 27. PRASAD, S. LEITE, V.D., SANTANA, R.A.C. e BRITO, J.B.Eletrometric Ivenstigations on formation of lanthanum molybdates as a function of pH.J.Braz.Chem.Soc., Vol.15 No. 2,246-252,2004.





























