Embed Size (px)
Citation preview

376A Volume 54, Number 11, 2000
focal pointBY LAURA A. VANDERBERG
LOS ALAMOS NATIONAL LABORATORY
LOS ALAMOS, NEW MEXICO 87545
Detection of BiologicalAgents: Looking for
Bugs in All theWrong Places
INTRODUCTION
T he threat of new and potentpathogens has become a greatconcern over the last several
years. Recent advances in biotech-nology, the low cost and ease of pro-ducing potent pathogens, and theirrelative invisibility have increasedthe likelihood for biowarfare. Al-most any pathogenic organism canbe used as a biological warfare (BW)agent. Table I shows some of themore common pathogenic microor-ganisms that may be employed asagents of biowarfare. Currently, atleast seven countries are thought tohave active biothreat agent (BA)production and research programs.1
On a weight-for-weight basis, BAsare more toxic than chemical warfare(CW) agents and can potentially pro-vide broader coverage than CWagents per pound of payload. Smallquantities of biological material (forexample, 1 kg of anthrax) may harmhundreds of thousands of people, de-pending on the delivery method andweather conditions.2
The cost to produce BAs is mini-
mal and does not require any specialequipment. In fact, the same equip-ment that is used to produce biotech-nological products (e.g., fermenta-tion systems, centrifuges, chroma-tography column puri� cation equip-ment, autoclaves, cell concentrators)can be used to produce BAs! Thisdual-use issue is daunting in terms ofbeing able to detect proliferation ofbiological weapons research. Oncedisseminated, BAs can reproduce inthe host to cause infection and befurther disseminated.
History. One of the � rst recordeduses of biological warfare was in1346, when bodies of Tartar soldierswho had died of the plague were cat-apulted over the walls of Kaffa, a be-sieged city. During the French andIndian War (1754–1767) smallpoxwas used against Native Americans.A captain in the British forces gaveblankets and a handkerchief from thesmallpox hospital to the NativeAmericans and recorded an entry inhis diary that read, ‘‘I hope it willhave the desired effect.’’3 Both ofthese attempts to wage biologicalwarfare happened at about the same
time as outbreaks (epidemics) of thesame diseases, respectively. Thus,the plague outbreak in Kaffa and thesmallpox epidemic among NativeAmericans could conceivably havebeen the result of natural outbreaksand not due solely to these inci-dents.4
World War I enemies took advan-tage of microbiological advancesand employed biological warfare atwill. The Germans supposedly in-fected livestock that were to be ex-ported to Allied forces with B. an-thracis and B. mallei, and the U.S.attempted to contaminate livestockfeed.5,6 Despite the international dip-lomatic effort to limit proliferationof weapons of mass destruction(both chemical and biological) fol-lowing the war, a number of coun-tries began research efforts to devel-op biological weapons. These in-cluded Poland, the Netherlands, Bel-gium, France, the Soviet Union, andItaly.7
The Japanese used biologicalweapons against the Manchuriansduring 1932–1945. Examples oftheir use included contaminating wa-
APPLIED SPECTROSCOPY 377A
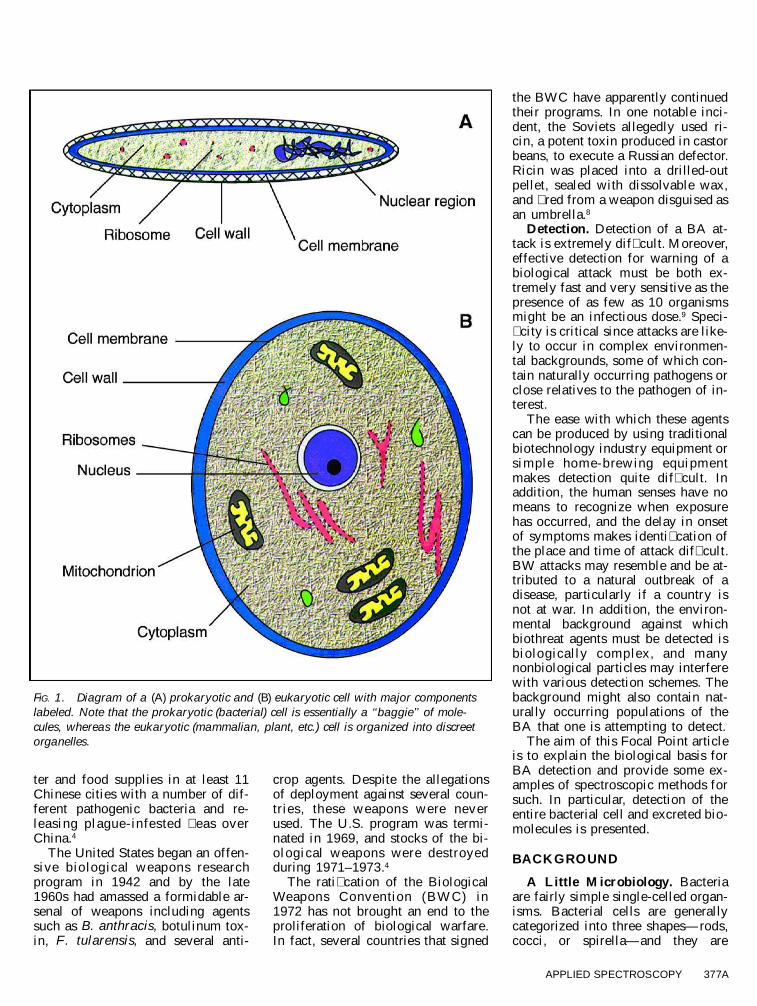
FIG. 1. Diagram of a (A) prokaryotic and (B) eukaryotic cell with major componentslabeled. Note that the prokaryotic (bacterial) cell is essentially a ‘‘baggie’’ of mole-cules, whereas the eukaryotic (mammalian, plant, etc.) cell is organized into discreetorganelles.
ter and food supplies in at least 11Chinese cities with a number of dif-ferent pathogenic bacteria and re-leasing plague-infested � eas overChina.4
The United States began an offen-sive bio logical weapons researchprogram in 1942 and by the late1960s had amassed a formidable ar-senal of weapons including agentssuch as B. anthracis, botulinum tox-in, F. tularensis, and several anti-
crop agents. Despite the allegationsof deployment against several coun-tries , these weapons were neverused. The U.S. program was termi-nated in 1969, and stocks of the bi-ological weapons were destroyedduring 1971–1973.4
The rati� cation of the BiologicalWeapons Convention (BW C) in1972 has not brought an end to theproliferation of biological warfare.In fact, several countries that signed
the BWC have apparently continuedtheir programs. In one notable inci-dent, the Soviets allegedly used ri-cin, a potent toxin produced in castorbeans, to execute a Russian defector.Ricin was placed into a drilled-outpellet, sealed with dissolvable wax,and � red from a weapon disguised asan umbrella.8
Detection. Detection of a BA at-tack is extremely dif� cult. Moreover,effective detection for warning of abiological attack must be both ex-tremely fast and very sensitive as thepresence of as few as 10 organismsmight be an infectious dose.9 Speci-� city is critical since attacks are like-ly to occur in complex environmen-tal backgrounds, some of which con-tain naturally occurring pathogens orclose relatives to the pathogen of in-terest.
The ease with which these agentscan be produced by using traditionalbiotechnology industry equipment orsimple home-brew ing equipmentmakes detection quite dif� cult. Inaddition, the human senses have nomeans to recognize when exposurehas occurred, and the delay in onsetof symptoms makes identi� cation ofthe place and time of attack dif� cult.BW attacks may resemble and be at-tributed to a natural outbreak of adisease, particularly if a country isnot at war. In addition, the environ-mental background against whichbiothreat agents must be detected isbiologically complex, and manynonbiological particles may interferewith various detection schemes. Thebackground might also contain nat-urally occurring populations of theBA that one is attempting to detect.
The aim of this Focal Point articleis to explain the biological basis forBA detection and provide some ex-amples of spectroscopic methods forsuch. In particular, detection of theentire bacterial cell and excreted bio-molecules is presented.
BACKGROUND
A Little Microbiology. Bacteriaare fairly simple single-celled organ-isms. Bacterial cells are generallycategorized into three shapes—rods,cocci, or spirella—and they are
378A Volume 54, Number 11, 2000
focal point
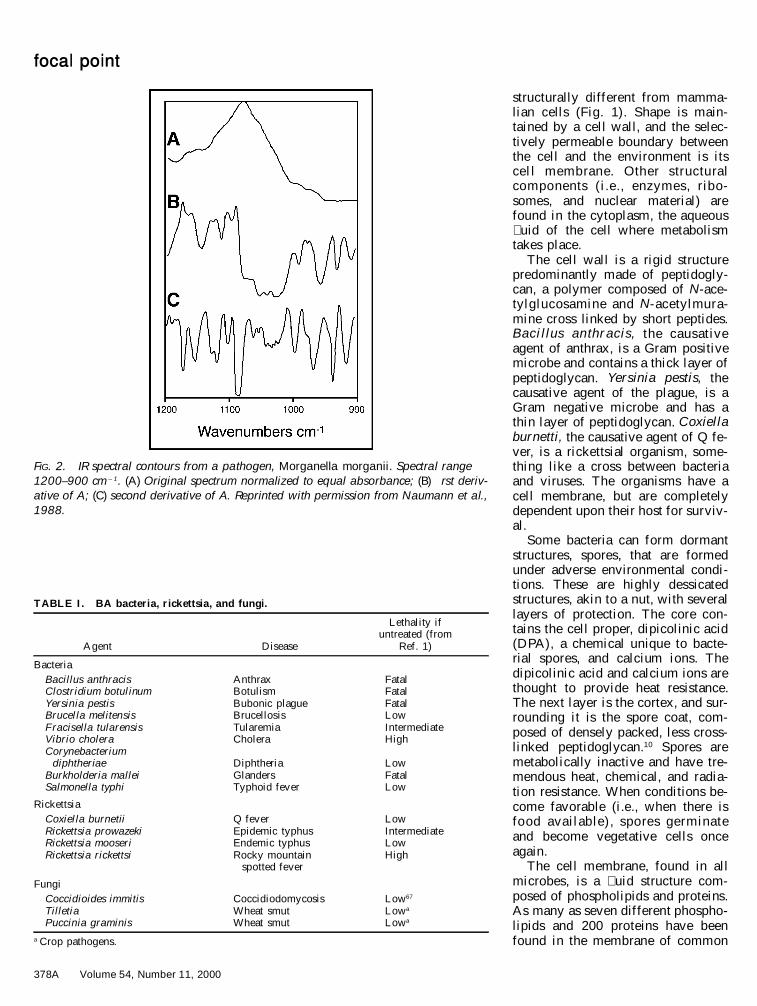
FIG. 2. IR spectral contours from a pathogen, Morganella morganii. Spectral range1200–900 cm21. (A) Original spectrum normalized to equal absorbance; (B) �rst deriv-ative of A; (C) second derivative of A. Reprinted with permission from Naumann et al.,1988.
TABLE I. BA bacteria, rickettsia, and fungi.
Agent Disease
Lethality ifuntreated (from
Ref. 1)
BacteriaBacillus anthracisClostridium botulinumYersinia pestisBrucella melitensisFracisella tularensisVibrio choleraCorynebacterium
diphtheriaeBurkholderia malleiSalmonella typhi
AnthraxBotulismBubonic plagueBrucellosisTularemiaCholera
DiphtheriaGlandersTyphoid fever
FatalFatalFatalLowIntermediateHigh
LowFatalLow
RickettsiaCoxiella burnetiiRickettsia prowazekiRickettsia mooseriRickettsia rickettsi
Q feverEpidemic typhusEndemic typhusRocky mountain
spotted fever
LowIntermediateLowHigh
FungiCoccidioides immitisTilletiaPuccinia graminis
CoccidiodomycosisWheat smutWheat smut
Low67
Lowa
Lowa
a Crop pathogens.
structurally different from mamma-lian cells (Fig. 1). Shape is main-tained by a cell wall, and the selec-tively permeable boundary betweenthe cell and the environment is itscell membrane. Other structuralcomponents (i.e ., enzymes, ribo-somes, and nuclear material) arefound in the cytoplasm, the aqueous� uid of the cell where metabolismtakes place.
The cell wall is a rigid structurepredominantly made of peptidogly-can, a polymer composed of N-ace-tylglucosamine and N-acetylmura-mine cross linked by short peptides.Bacillus anthracis, the causativeagent of anthrax, is a Gram positivemicrobe and contains a thick layer ofpeptidoglycan. Yersinia pestis, thecausative agent of the plague, is aGram negative microbe and has athin layer of peptidoglycan. Coxiellaburnetti, the causative agent of Q fe-ver, is a rickettsial organism, some-thing like a cross between bacteriaand viruses. The organisms have acell membrane, but are completelydependent upon their host for surviv-al.
Some bacteria can form dormantstructures, spores, that are formedunder adverse environmental condi-tions. These are highly dessicatedstructures, akin to a nut, with severallayers of protection. The core con-tains the cell proper, dipicolinic acid(DPA), a chemical unique to bacte-rial spores, and calcium ions. Thedipicolinic acid and calcium ions arethought to provide heat resistance.The next layer is the cortex, and sur-rounding it is the spore coat, com-posed of densely packed, less cross-linked peptidoglycan.10 Spores aremetabolically inactive and have tre-mendous heat, chemical, and radia-tion resistance. When conditions be-come favorable (i.e., when there isfood availab le), spores germinateand become vegetative cells onceagain.
The cell membrane, found in allmicrobes, is a � uid structure com-posed of phospholipids and proteins.As many as seven different phospho-lipids and 200 proteins have beenfound in the membrane of common

APPLIED SPECTROSCOPY 379A
bacteria such as Escherichia coli.11
The interactions of these differentproteins and phospholipids may pro-vide a diagnostic � ngerprint for aparticular microbe.12
Many intracellular molecules inbiological systems (not just bacteria)are associated with energy-yieldingreactions. Some have speci� c elec-tronic excitation and emission spec-tra, providing a spectroscopic signa-ture. For example, the amino acidstryptophan, phenylalanine, tyrosine,and histidine, which are componentsof proteins, can be excited by radi-ation at 250–300 nm. Nicotinamideadenine dinucleo tide (phosphate)(NAD(P)H) is excited at 325 nm andhas an emission band from 420 to580 nm. These features can be usedto distinguish biological from non-biological particles, but not neces-sarily to distinguish between bacteriaand other biologically derived ma-terial.
Pathogenic bacteria have speci� cmechanisms by which they affecthost cells. Generally, these organ-isms must be equipped with a meansof invading the host and obtainingnutrients to permit their survival andreplication in vivo.13 Most pathogensproduce proteinaceous toxins thatcan be quite lethal. For example, just0.001 mg/kg of botulinum toxin (pro-duced by the pathogen Clostridiumbotulinum) constitutes an infectivedose.14 Toxins are typically consid-ered as CW agents, but since spec-troscopic detection may be quite use-ful in detecting these small mole-cules, they have been included inthis discussion.
DISCUSSION
Conventional Detection Meth-ods. The culturing of unknown mi-crobes on solid surfaces has been ac-cepted as standard practice for over100 years.15 This procedure involvesplacement of a sample on a growthsurface, allowing it to reproduce toform visible colonies—with furtherexamination of the morphological,biochemical, and physiological char-acteristics of one particular isolate.At this time, no single test can pro-vide a de� nitive identi� cation of any
particular organism. In some cases,direct microscopic examination of asample may provide top-level infor-mation (morphology, staining char-acteristics) about a potential patho-gen, but additional time-consumingcon� rmatory testing must be done.These types of techniques can re-quire days to complete testing andidenti� cation.
Rapid biochemical testing andidenti� cation may be accomplishedby using any one of a variety ofcommercially available k its (e.g.,API, Analytab Products, Plainview,NY; Biolog, Biolog, Inc., Hayward,CA). These kits provide a suite ofkey tests that can distinguish be-tween similar microbes, but they re-quire that an unknown microbe beisolated and grown in a pure cul-ture— again, a time-consumingfront-end process. Interpretation ofthe results from rapid biochemicaltesting can also be a challenge asculture density and other parametershave a signi� cant effect on testingoutcomes.
Immunological methods have longbeen employed for the identi� cationof pathogenic microbes. Almost allmicrobial species have at least oneunique antigen. Identi� cation andpuri� cation of that antigen enableone to generate antibodies to that an-tigen. These antibodies can then beused to detect the presence of theoriginal antigen through a number ofmethods. ELISA, or enzyme-linkedimmunosorbent assay, is perhaps themost well-known and frequentlyused immunological technique. Inthis technique, speci� c antibodiesare adsorbed onto the walls of a wellthat is part of a microtiter plate. Asuspension that contains, or is sus-pected to contain, the antigen of in-terest is added to the well. If it ispresent, the antigen will react withthe bound antibody. The well isrinsed to remove unbound materials.A second antibody (one that alsobinds to the antigen) with a conju-gated reporter enzyme is added. Thisantibody binds to the antigen, excessmaterial is rinsed away, and a sub-strate for the reporter enzyme is add-ed. The sandwiched antigen is de-
tected by assaying for reporter en-zyme activity. This activity is typi-cally a colorimetric reaction.11
Molecular biological techniquessuch as in situ hybridization andpolymerase chain reaction (PCR)have been developed and are ex-tremely sensitive. These DNA/RNA-based techniques allow for the rapididenti� cation of an organism whenextremely small amounts of geneticmaterial are present.16 In situ hybrid-ization uses speci� c sequences ofDNA that contain a � uorescent tag.When these sequences bind to theDNA of interest, they can be readilyseen under the microscope or by� ow cytometry.17 Extensive samplepreparation time is needed for in situhybridization, making it only a smallimprovement over culture-basedmethods. PCR is used to amplify mi-nute quantities of speci� c DNA se-quences and can be used to unequiv-ocally identify microorganisms with-out prior culturing; however, auto-mation may be dif� cult, and thepresence of contamination is detri-mental.18 As with in situ hybridiza-tion, extensive and time-consumingsample clean-up and preparation isrequired.
Toxin production can be detectedthrough a number of methods. Acommon method involves exposureof mammalian or plant cell cultureto a putative toxin and observationof cell necrosis or death.19,20 Immu-noassays such as ELISA and assaysusing PCR are also employed.21–23
Spectroscopic Methods for De-tection and Identi� cation. To ad-dress the need for rapid and sensitivepathogen detection, particularly thedetection of BA, there is a need forrapid, compact, � eld-portable, user-friendly equipment. Spectroscopicmethods have the potential to meetthese challenging criteria; indeed, anumber of biosensors have been de-veloped that address one or more ofthese requirements. The remainderof this Focal Point article will ex-amine some of these technologies indetail and indicate shortfalls and fur-ther developmental needs, improve-ments, and other requirements.
Measurements of the intrinsic � uo-
380A Volume 54, Number 11, 2000
focal point
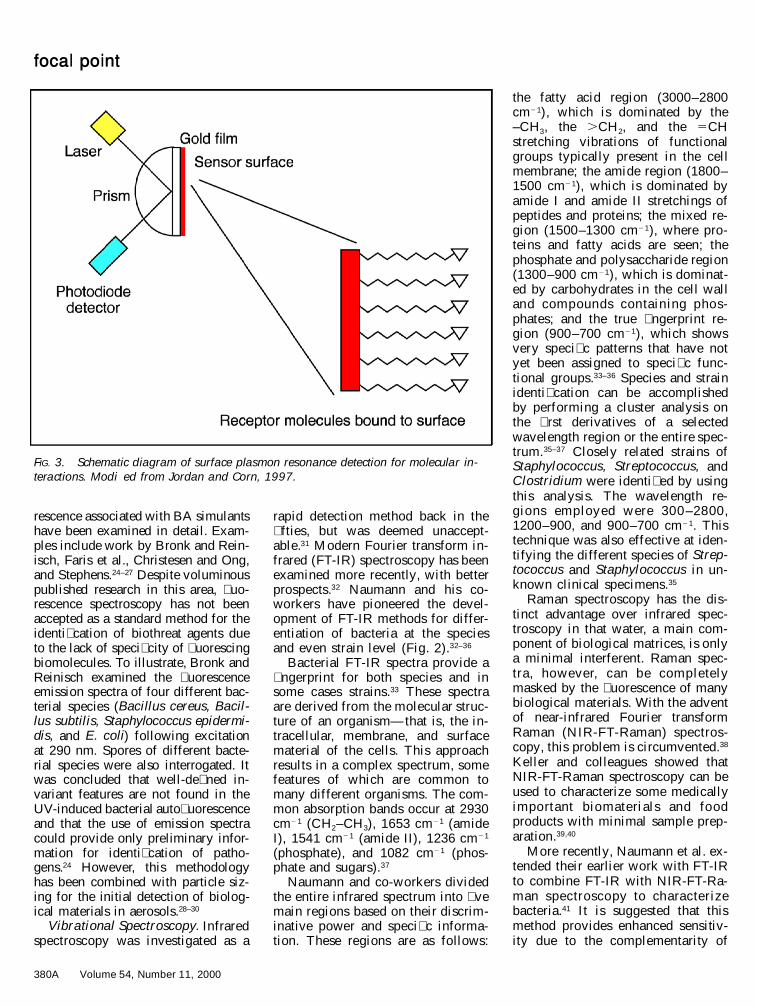
FIG. 3. Schematic diagram of surface plasmon resonance detection for molecular in-teractions. Modi�ed from Jordan and Corn, 1997.
rescence associated with BA simulantshave been examined in detail. Exam-ples include work by Bronk and Rein-isch, Faris et al., Christesen and Ong,and Stephens.24–27 Despite voluminouspublished research in this area, � uo-rescence spectroscopy has not beenaccepted as a standard method for theidenti� cation of biothreat agents dueto the lack of speci� city of � uorescingbiomolecules. To illustrate, Bronk andReinisch examined the � uorescenceemission spectra of four different bac-terial species (Bacillus cereus, Bacil-lus subtilis, Staphylococcus epidermi-dis, and E. coli) following excitationat 290 nm. Spores of different bacte-rial species were also interrogated. Itwas concluded that well-de� ned in-variant features are not found in theUV-induced bacterial auto� uorescenceand that the use of emission spectracould provide only preliminary infor-mation for identi� cation of patho-gens.24 However, this methodologyhas been combined with particle siz-ing for the initial detection of biolog-ical materials in aerosols.28–30
Vibrational Spectroscopy. Infraredspectroscopy was investigated as a
rapid detection method back in the� fties, but was deemed unaccept-able.31 Modern Fourier transform in-frared (FT-IR) spectroscopy has beenexamined more recently, with betterprospects.32 Naumann and his co-workers have pioneered the devel-opment of FT-IR methods for differ-entiation of bacteria at the speciesand even strain level (Fig. 2).32–36
Bacterial FT-IR spectra provide a� ngerprint for both species and insome cases strains.33 These spectraare derived from the molecular struc-ture of an organism—that is, the in-tracellular, membrane, and surfacematerial of the cells. This approachresults in a complex spectrum, somefeatures of which are common tomany different organisms. The com-mon absorption bands occur at 2930cm21 (CH2–CH3), 1653 cm21 (amideI), 1541 cm21 (amide II), 1236 cm21
(phosphate), and 1082 cm21 (phos-phate and sugars).37
Naumann and co-workers dividedthe entire infrared spectrum into � vemain regions based on their discrim-inative power and speci� c informa-tion. These regions are as follows:
the fatty acid region (3000–2800cm21), which is dominated by the–CH3, the .CH2, and the 5CHstretching vibrations of functionalgroups typically present in the cellmembrane; the amide region (1800–1500 cm21), which is dominated byamide I and amide II stretchings ofpeptides and proteins; the mixed re-gion (1500–1300 cm21), where pro-teins and fatty acids are seen; thephosphate and polysaccharide region(1300–900 cm21), which is dominat-ed by carbohydrates in the cell walland compounds contain ing phos-phates; and the true � ngerprint re-gion (900–700 cm21), which showsvery speci� c patterns that have notyet been assigned to speci� c func-tional groups.33–36 Species and strainidenti� cation can be accomplishedby performing a cluster analysis onthe � rst derivatives of a selectedwavelength region or the entire spec-trum.35–37 Closely related strains ofStaphylococcus, Streptococcus, andClostridium were identi� ed by usingthis analysis. The wavelength re-gions employed were 300 –2800,1200–900, and 900–700 cm21. Thistechnique was also effective at iden-tifying the different species of Strep-tococcus and Staphylococcus in un-known clinical specimens.35
Raman spectroscopy has the dis-tinct advantage over infrared spec-troscopy in that water, a main com-ponent of biological matrices, is onlya minimal interferent. Raman spec-tra, however, can be completelymasked by the � uorescence of manybiological materials. With the adventof near-infrared Fourier transformRaman (NIR-FT-Raman) spectros-copy, this problem is circumvented.38
Keller and colleagues showed thatNIR-FT-Raman spectroscopy can beused to characterize some medicallyimportant biomaterials and foodproducts with minimal sample prep-aration.39,40
More recently, Naumann et al. ex-tended their earlier work with FT-IRto combine FT-IR with NIR-FT-Ra-man spectroscopy to characterizebacteria.41 It is suggested that thismethod provides enhanced sensitiv-ity due to the complementarity of
APPLIED SPECTROSCOPY 381A
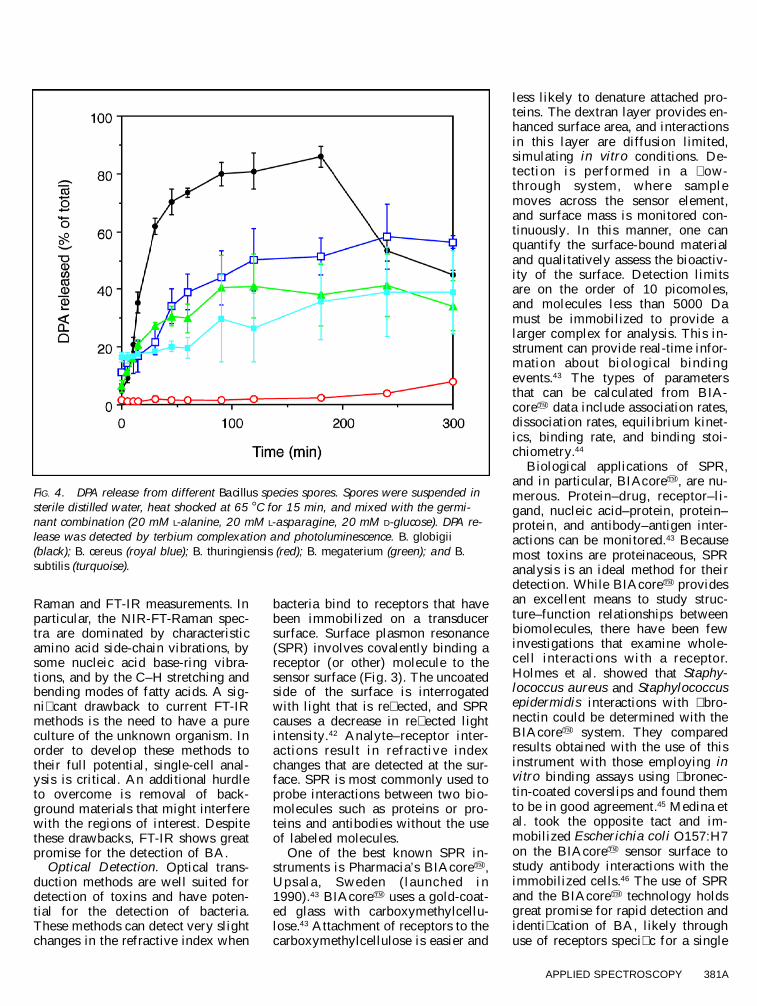
FIG. 4. DPA release from different Bacillus species spores. Spores were suspended insterile distilled water, heat shocked at 65 8 C for 15 min, and mixed with the germi-nant combination (20 mM L-alanine, 20 mM L-asparagine, 20 mM D-glucose). DPA re-lease was detected by terbium complexation and photoluminescence. B. globigii(black); B. cereus (royal blue); B. thuringiensis (red); B. megaterium (green); and B.subtilis (turquoise).
Raman and FT-IR measurements. Inparticular, the NIR-FT-Raman spec-tra are dominated by characteristicamino acid side-chain vibrations, bysome nucleic acid base-ring vibra-tions, and by the C–H stretching andbending modes of fatty acids. A sig-ni� cant drawback to current FT-IRmethods is the need to have a pureculture of the unknown organism. Inorder to develop these methods totheir full potential, single-cell anal-ysis is critical. An additional hurdleto overcome is removal of back-ground materials that might interferewith the regions of interest. Despitethese drawbacks, FT-IR shows greatpromise for the detection of BA.
Optical Detection. Optical trans-duction methods are well suited fordetection of toxins and have poten-tial for the detection of bacteria.These methods can detect very slightchanges in the refractive index when
bacteria bind to receptors that havebeen immobilized on a transducersurface. Surface plasmon resonance(SPR) involves covalently binding areceptor (or other) molecule to thesensor surface (Fig. 3). The uncoatedside of the surface is interrogatedwith light that is re� ected, and SPRcauses a decrease in re� ected lightintensity.42 Analyte–receptor inter-actions resu lt in refractive indexchanges that are detected at the sur-face. SPR is most commonly used toprobe interactions between two bio-molecules such as proteins or pro-teins and antibodies without the useof labeled molecules.
One of the best known SPR in-struments is Pharmacia’s BIAcorey,Upsala , Sweden (launched in1990).43 BIAcorey uses a gold-coat-ed glass with carboxymethylcellu-lose.43 Attachment of receptors to thecarboxymethylcellulose is easier and
less likely to denature attached pro-teins. The dextran layer provides en-hanced surface area, and interactionsin this layer are diffusion limited,simulating in vitro conditions. De-tection is performed in a � ow-through system, where samplemoves across the sensor element,and surface mass is monitored con-tinuously. In this manner, one canquantify the surface-bound materialand qualitatively assess the bioactiv-ity of the surface. Detection limitsare on the order of 10 picomoles,and molecules less than 5000 Damust be immobilized to provide alarger complex for analysis. This in-strument can provide real-time infor-mation about biological b indingevents.43 The types of parametersthat can be calculated from BIA-corey data include association rates,dissociation rates, equilibrium kinet-ics, binding rate, and binding stoi-chiometry.44
Biological applications of SPR,and in particular, BIAcorey, are nu-merous. Protein–drug, receptor–li-gand, nucleic acid–protein, protein–protein, and antibody–antigen inter-actions can be monitored.43 Becausemost toxins are proteinaceous, SPRanalysis is an ideal method for theirdetection. While BIAcorey providesan excellent means to study struc-ture–function relationships betweenbiomolecules, there have been fewinvestigations that examine whole-cell interactions with a receptor.Holmes et al. showed that Staphy-lococcus aureus and Staphylococcusepidermidis interactions with � bro-nectin could be determined with theBIAcorey system. They comparedresults obtained with the use of thisinstrument with those employing invitro binding assays using � bronec-tin-coated coverslips and found themto be in good agreement.45 Medina etal. took the opposite tact and im-mobilized Escherichia coli O157:H7on the BIAcorey sensor surface tostudy antibody interactions with theimmobilized cells.46 The use of SPRand the BIAcorey technology holdsgreat promise for rapid detection andidenti� cation of BA, likely throughuse of receptors speci� c for a single

382A Volume 54, Number 11, 2000
focal point
FIG. 5. Method by which reporter bacteriophages can be used to detect viable mi-crobes.
BA multiplexed with other speci� creceptors.
Viability Detection. The spectro-scopic methods described above maybe employed to detect and identifypathogens and potential BAs. Ashortcoming of these techniques istheir inability to determine the via-bility of the pathogens they detect.While identity is important, beingable to determine whether or not thepathogens are alive is critical to as-sessing the danger of a situation. Vi-ability is an important characteristicof a BA that must be determinedquickly and properly. For example,recall the B’nai B’rth incident in
1997. Workers received a packagewith a petri dish containing red jelly-looking material. An attached noteclaimed the package contained an-thrax. People were kept in of� ces formore than eight hours, and streets indowntown Washington, D.C., wereclosed for a chemical hazard alert—ultimately for naught, s ince thepackage did not contain anthrax orany other pathogen.47 Or picture abattle� eld where a particle counter’salarm is set off by an incoming cloudof ‘‘biological material’’ and all sol-diers have to don uncomfortablemission oriented protective posture(MOPP) gear. If the de� nition of ter-
rorism is to cause panic or psycho-logical harm, a terrorist does notneed to use live BAs. Any nonviableBA would be detected as an attack,causing the desired effects withoutloss of life. A rapid means to deter-mine viability would quickly mini-mize a terrorist’s success.48
There are many ways to assessbacterial viability. Plating allows oneto assess viability, but requires ex-tended periods of time. Other meth-ods to determine viability includephysiological assays that do not de-tect viable bacterial spores, nor willthey identify the viable organisms,i.e ., d ifferentiate between back-ground and BA.49–52
One method for determining bac-terial spore viability involves detect-ing the release of dipicolinic acidduring germination. During germi-nation, a spore irreversibly commitsto becoming a vegetative cell within1–5 min of exposure to a suitablegerminant.53 At this point, divalentcations and DPA are released. Be-cause of its uniqueness and the speedwith which it is released, DPA rep-resents a tag for real-time viabilitydetection.
We have developed an assaybased on the commitment step of thegermination process and demonstrat-ed its rapid response and validity bytesting it with a variety of spore-forming microbes.54,55 Germinationof spores was optimized with the useof Bacillus globigii and tested onseveral other Bacillus spp. L-Alaninealone is often employed as a germi-nant; however, we found that a com-bination of L-alanine, L-asparagine,and D-glucose provided a more rapidgermination response.54,55
Once the DPA is released, it mustbe detected with high sensitivity.DPA readily complexes with lantha-nide metals such as terbium (Tb). Infact, DPA complexation to terbiumwas used as an assay method for ter-bium. The terbium cation (Tb31 ) re-acts w ith the dipicolinate anion(D PA 2 2 ) to fo rm the com plex[Tb(DPA)3]3 2 , which has � uores-cence intensity 10 000 times greaterthan terbium alone and has a distinc-

APPLIED SPECTROSCOPY 383A
tive photoluminescence emission at545 nm.56
The germinant combination wasused to germinate spores of a num-ber of different Bacillus spp. Follow-ing incubation, spores were removedand the liquid was mixed with anequal volume of terbium nitrate.DPA release was determined by � uo-rescence detection, and, with the ex-ception of B. thuringiensis, all or-ganisms tested readily released DPA(Fig. 4). Using a standardized 15min germinant exposure, Vanderberget al. showed that the detection limitsfor this assay are 45 ng DPA, or ap-proximately 105 viable spores/mL.54
Hindle and Hall showed that a 90min exposure to L-alanine followedby detection of DPA with terbiumresulted in a detection limit of 104
viable spores/mL.57 Both groupsconcluded that the assay has a num-ber of advantages including simplic-ity and ease of use; however, detec-tion limits must be improved as a le-thal inhaled dose of B. anthracis ison the order of 104 viable spores.1
Further development of this assayinto a biosensor-type system is war-ranted.
Recent biotechnological advanceshave allowed researchers to uselight-producing (bioluminescent) en-zymes as reporter molecules to as-sess the physiological state of an or-ganism. Use of these reporter mole-cules offers a number of advantagesover both traditional culturing andmolecular biological methods. Bio-luminescence reactions produce aphysical product that is not toxic,and their signal can be measured ac-curately with exquisite sensitivity.18
Luciferases catalyze reactions thatresult in the emission of photons andhave found wide use as reporters.The two most common luciferasesystems come from bacteria and � re-� ies. Bacterial luciferase catalyzesthe reaction below:
FMNH 2 1 RCHO 1 O2 ® FMN 1RCOOH 1 H2O 1 light (490 nm)
A functional electron transport chainin the microbe is required for pro-duction of � avin mononucleotide, re-duced (FMNH 2), and, as such, only
viable bacteria will produce light.57
Fire� y and click beetle luciferasecatalyze the reaction:
luciferin 1 ATP 1 O2 ®oxyluciferin 1 AMP 1 PP i 1 CO2
1 light (560 nm)
This reaction depends on the pres-ence of ATP, a universal energy-con-taining molecule. Since only viableorganisms contain ATP, the physio-logical state of an organism may beassessed.50
Light emission from these en-zymes can be measured by a varietyof methods. If suf� cient light isemitted, it may be visualized in adarkened room with the naked eye.For more sensitive detection, photo-graphic � lm may be exposed to sam-ples, scintillation counting or lumi-nometry can be used to quantifyemission, or photomultipliers can becoupled with � ber optics for remotesampling; the most sensitive detec-tion uses charge-coupled device(CCD) cameras for single photon(thus, single microbe) detection. De-spite the number of different meth-ods to detect light emission, there isno standardized methodology and nouniversally accepted unit of light ac-tivity with regard to biological func-tion, so useful comparisons are notpossible.59
Bioluminescent reporters for path-ogen detection have been success-fully developed. Ulitzur and Kuhn� rst developed reporter bacterio-phages, Lambda phage, to detect vi-able Escherichia coli.60 This conceptis illustrated in Fig. 5. The move-ment of genes into a bacteriumthrough use of a bacteriophage isknown as transduction and is a com-mon molecular biological technique.The luciferase gene is transduced viathe reporter bacteriophage into abacterial host genome. Following in-fection, transcription and translationof the luciferase protein occur onlyif the host microbe is alive and re-producing. The presence of the � uo-rescent protein is then detectedthrough luminometry. Subsequently,reporter bacteriophages have beenconstructed to detect Mycobacteriumtuberculosis, M. paratuberculosis,
M. avium, Salmonella typhinurium,and Listeria spp.61–67
Loessner et al. isolated a bacterio-phage from Listeria monocytogenes,phage A511, characterized it, andmodi� ed it to contain the gene forbacterial luciferase.65,66 The A511 re-porter phage detected as few as 10 2–103 viable Listeria cells/mL follow-ing infection and 2 h incubation.With a 16 h enrichment step (addi-tion of culture medium for Listeria)prior to testing with the reporterphage, as little as one bacterial cellper gram of arti� cially contaminatedsalad could be detected.66 In a fol-low-up of this work, Loessner et al.found that the A511 construct wasuseful in detecting Listeria spp. in avariety of foods, providing muchmore rapid screening for Listeriaspp. in foods than traditional cultur-ing techniques.67
These reporter phages have greatpromise to provide a tool for rapiddetection of viable bacterial patho-gens. A major shortfall of reporterphages is their relative lack of spec-i� city. They can be used to detectmicrobes within a particular genus;however, they are not species specif-ic. Because of the fact that othermembers of many biothreat agentgenera are naturally occurring micro-� ora, species speci� city is critical forreporter phage applicability.
CONCLUSION
The threat of biological warfare isnot new to humankind, but recentbiotechnological advances havemade the threat even more real. The‘‘poor man’s atomic bomb’’ is vir-tually invisible and can potentiallywreak havoc on a nation’s army, acity’s population, or staple crops.Spectroscopic methods which havetraditionally been used in studyingbiomolecular interactions may proveto be the best means for detectingBA. In the future, development ofspectroscopic tools for this applica-tion will focus on detection in acomplex background, decipheringsignatures from as little as one BAand moving away from the need forpreculturing. Combining identi� ca-tion and viability detection in a sin-

384A Volume 54, Number 11, 2000
focal point
gle system will allow us to detect theoccurrence of a BA attack and de-termine the true danger associatedwith that attack. In conclusion, spec-troscopy holds promise for protec-tion in a technologically advancingworld of biological danger.
ACKNOWLEDGMENTS
Many thanks go to those whose work andideas are summarized in this Focal Point ar-ticle. Special thanks to J. R. Stephens, whosethoughts helped to shape this paper. Technicalassistance from G. L. Wagner is greatly ap-preciated.
1. J. Ali, L. Rodrigues, and M. Moodie,Jane’s US Chemical-Biological DefenseGuidebook (Jane’s Information Group,Alexandria, Virginia, 1997).
2. R. Danzig and P. B. Berkowsky, JAMA278, 431 (1997).
3. C. H. Sipe, The Indian Wars of Pennsyl-vania (Telegraph Press, Harrisburg, Penn-sylvania, 1929).
4. G. W. Christopher, T. J. Cieslak, J. A.Pavlin, and E. M. Eitzen, J. Amer. Med.Assoc. 278, 412 (1997).
5. A. G. Robertson and L. J. Robertson, Mil.Med. 160, 369 (1995).
6. J. Witcover, Sabotage at Black Tom: Im-perial Germany’s Secret War in America,1914–1917 (Algonquin Books of ChapelHill, Chapel Hill, North Carolina, 1989).
7. S. H. Harris, Factories of Death (Rou-tledge, New York, 1994).
8. N. C. Livingstone and D. J. D. Douglass,CBW: The Poor Man’s Atomic Bomb (In-stitute for Foreign Policy Analysis, Cam-bridge, Massachusetts, 1984).
9. Canadian Laboratory Centre for DiseaseControl, Of� ce of Biosafety, Materials Safe-ty Data Sheet for Francisella tularensis;http://www.hc-sc.ca/hpb/lcdc/biosafty/msds/index.html.
10. T. D. Brock and M. T. Madigan, Biologyof Microorganisms (Prentice Hall, Engle-wood Cliffs, New Jersey, 1991), 6th ed.
11. J. J. Perry and J. T. Staley, Microbiology:Dynamics and Diversity (Saunders Col-lege Publishing, Forth Worth, Texas,1997).
12. M. Jackson and H. H. Mantsch, Spectro-chim. Acta Rev. 15, 53 (1993).
13. S. Falkow, ASM News 63, 359 (1997).14. D. R. Franz, P. B. Jahrling, A. M. Fried-
lander, D. J. McClain, D. L. Hoover, W.R. Bryne, J. A. Pavlin, C. W. Christopher,and E. M. Eitzen, JAMA 278, 399 (1997).
15. N. R. Krieg and P. Gerhardt, ‘‘Solid, Liq-uid/Solid and Semisolid Culture’’, inMethods for General and Molecular Bac-teriology, P. Gerhardt, R. G. E. Murray,W. A. Wood, and N. R. Krieg, Eds.(American Society for Microbiology,Washington, D.C., 1994), p. 216.
16. M. Johns, L. Harrington, R. W. Titball,and D. L. Leslie, Lett. Appl. Microbiol.18, 236 (1994).
17. R. A. Keller, W. P. Ambrose, P. M. Good-win, J. H. Jett, J. C. Martin, and M. Wu,Appl. Spectrosc. 50, 12A (1996).
18. P. Billard and M. S. DuBow, Clin. Bio-chem. 31, 1 (1998).
19. K. M. Lam, Vet. Microbiol. 35, 133(1993).
20. Z. Charania, R. Vanmaele, and G. D.Armstrong, J. Microbiol. Meth. 14, 171(1991).
21. C. Toma, S. Nakamura, S. Kamiya, N.Nakasone, and M. Iwanaga, Microbiol.Immunol. 43, 737 (1999).
22. K. Moller and P. Ahrens, Anaerobe 2, 103(1996).
23. B. Swaminathan and P. Feng, Annu. Rev.Microbiol. 48, 401 (1994).
24. B. V. Bronk and L. Reinisch, Spectros-copy 47, 436 (1993).
25. R. A. Faris, D. J. Copeland, A. Eckstrom,C. Williams, C. Witham, C. B. Carlisle, J.Leonelli, and B. Bronk, SRIN TechnicalNote, Project # 2913, Contract # DAAA15-91-D-0002, May, 1992.
26. S. Christesen and K. Ong, presented at theMASINT Biological Defense Science andTechnology Symposium, Patrick AirForce Base, Florida (1996).
27. J. R. Stephens, in Proceedings of the Con-ference on Chemical and Biological De-fense Research, Aberdeen ProvingGround, Maryland (1996).
28. Y. S. Cheng, E. B. Barr, B. J. Fan, P. J.Hargis, D. J. Rader, T. J. O’Hern, J. R.Torczynski, G. C. Tisone, B. L. Prepper-nau, S. A. Young, and R. J. Radloff,Aerosol Sci. Technol. 30, 186 (1999).
29. M. Seaver, J. D. Eversole, J. J. Hardgrove,W. K. Cary, and D. C. Roselle, AerosolSci. Technol. 30, 174 (1999).
30. P. P. Hairston, J. Ho, and F. R. Quant, J.Aerosol Sci. 28, 471 (1997).
31. K. P. Norris, J. Hyg. 57, 326 (1959).32. D. Naumann, V. Fijala, H. Labischinski,
and P. Giesbrecht, J. Molec. Struct. 174,165 (1988).
33. H. C. van der Mei, D. Naumann, and H.J. Busscher, Infrared Phys. Technol. 37,561 (1996).
34. D. Naumann, D. Helm, and H. Labischin-ski, Nature 351, 81 (1991).
35. D. Helm, H. Labischinski, G. Schallehn,and D. Naumann, J. Gen. Microbiol. 137,69 (1991).
36. D. Helm, H. Labischinski, and D. Nau-mann, J. Microbiol. Meth. 14, 127 (1991).
37. H. C. van der Mei, J. Noordmans, and H.J. Busscher, Biochim. Biophys. Acta 991,395 (1989).
38. B. Schrader, A. Hoffman, and S. Keller,Spectrochim. Acta, Part A 47, 1135(1991).
39. S. Keller, T. Lochte, B. Dippel, and B.Schrader, Fresenius’ J. Anal. Chem. 346,863 (1993).
40. S. Keller, B. Schrader, A. Hoffman, W.Schrader, K. Metz, A. Rehlaender, J.Pahnke, M. Ruwe, and W. Budach, J. Ra-man Spectrosc. 25, 663 (1994).
41. D. Naumann, S. Keller, D. Helm, C.
Schultz, and B. Schrader, J. Molec. Struct.347, 399 (1995).
42. A. M. Brodsky, L. W. Burgess, and S. A.Smith, Appl. Spectrosc. 52, A332 (1998).
43. U. Jonsson, L. Fagerstam, B. Ivarsson, B.Johnsson, R. Karlsson, K. Lundh, S. Lo-fas, B. Persson, H. Roos, I. Ronnberg, S.Sjolander, E. Strenberg, R. Stahlberg, C.Urbaniczky, H. Ostlin, and M. Malqvist,BioTechniques 11, 620 (1991).
44. R. Granzow and R. Reed, Bio/Technology10, 390 (1992).
45. S. D. Holmes, K. May, V. Johansson, F.Markey, and I. A. Critchley, J. Microbiol.Meth. 28, 77 (1997).
46. M. B. Medina, L. Van Houten, P. H.Cook, and S. I. Tu, Biotechnol. Techn. 11,173 (1997).
47. M. Powell and A. Lengel, WashingtonPost, Friday April 27, 1997.
48. T. M. Carlsen and L. A. Vanderberg,UCRL-AR-131343 DR and LA-UR 98-3336 (1998).
49. G. A. McFeters, F. P. Yu, B. H. Pyle, andP. S. Stewart, J. Microbiol. Meth. 21, 1(1995).
50. Sigma Technical Bulletin BSCA-1 12-87(Sigma, St. Louis, Missouri, 1987).
51. D. Rossel and J. Tarradellas, Environ.Tox. Water Qual. 6, 17 (1991).
52. L. C. F. Stubber� eld and P. J. A. Shaw, J.Microbiol. Meth. 12, 151 (1990).
53. P. Setlow, in Sporulation and Germina-tion, Proceedings of the 8th InternationalSpore Conference, H. S. Levinson, A. L.Sonenshein, and D. J. Tipper, Eds. (Amer-ican Society for Microbiology, Washing-ton, D.C., 1981), pp. 13–28.
54. L. A. Vanderberg, G. L. Wagner, M. Abel,R. J. Obiso, and T. J. Herdendorf, unpub-lished research .
55. R. J. Obiso, Jr., T. J. Herdendorf, and L.A. Vanderberg, Abstr. Amer. Soc. Micro-biol. 98 Gen. Meet. Abstr. I-5 (AmericanSociety for Microbiology, Washington,D.C., 1998), p. 308.
56. T. D. Barela and A. D. Sherry, Anal.Biochem. 71, 351 (1976).
57. A. A. Hindle and E. A. H. Hall, Analyst124, 1599 (1999).
58. J. J. Ryan, J. T. Cardwell, G. W. Childers,T. F. Kellogg, and A. W. Wang, J. DairySci. 62(S1), 206 (1979).
59. R. S. Burlage and C.-T. Kuo, Annu. Rev.Microbiol. 48, 291 (1994).
60. S. Ulitzur and J. Kuhn, in Biolumines-cence and Chemiluminescence: New Per-spectives, J. Schlomerich, R. Andreeson,A. Kapp, M. Ernst, and W. G. Woods,Eds. (Wiley, Bristol, 1987), p. 463.
61. G. J. Sarkis, W. R. Jacobs, and G. F. Hat-full, Mol. Microbiol. 15, 1055 (1995).
62. W. R. Jacobs, R. G. Barletta, R. Udani, J.Chan, G. Kalkut, G. Sosne, T. Kieser, G.J. Sarkis, G. F. Hatfull, and H. Bloom,Science 260, 819 (1993).
63. E. M. Folley-Thomas, D. L. Whipple, L.E. Bermudez, and G. R. Barletta, Micro-biol. 141, 1173 (1995).
64. P. E. Turpin, K. A. Maycroft, J. Bedford,

APPLIED SPECTROSCOPY 385A
C. L. Rowlands, and E. M. H. Wellington,Lett. Appl. Microbiol. 16, 24 (1993).
65. M. J. Loessner and M. Busse, Appl. En-viron. Microbiol. 56, 1912 (1990).
66. M. J. Loessner, C. E. D. Rees, G. S. A.B. Stewart, and S. Scherer, Appl. Environ.Microbiol. 62, 1133 (1996).
67. M. J. Loessner, M. Rudolf, and S. Scherer,
Appl. Environ. Microbiol. 63, 2961(1997).
68. C. E. Jordan, R. M. Corn, Anal. Chem.69, 1449 (1997).





























