Embed Size (px)
Citation preview

Review article
n engl j med 363;17 nejm.org october 21, 20101638
franklin h. epstein lecture
Franklin H. Epstein, M.D., served the New England Journal of Medicine for more than 20 years. A keen clinician, accomplished researcher, and outstanding teacher, Dr. Epstein was Chair and Professor of Medicine at Beth Israel Deaconess Medical Center, Boston, where the Franklin H. Epstein, M.D., Memorial
Lectureship in Mechanisms of Disease has been established in his memory.
Cardiac Development and Implications for Heart Disease
Jonathan A. Epstein, M.D.
From the Department of Cell and Devel-opmental Biology and the Cardiovascular Institute, University of Pennsylvania School of Medicine, Philadelphia. Address reprint requests to Dr. Epstein at the Department of Cell and Developmental Biology, Univer-sity of Pennsylvania School of Medicine, 421 Curie Blvd., 1154 BRB II, Philadelphia, PA 19104, or at [email protected] .edu.
N Engl J Med 2010;363:1638-47.Copyright © 2010 Massachusetts Medical Society.
During the past decade, our understanding of the development of the embryonic heart has been improved by a number of discoveries. These new findings will require changes in standard teachings of how the four-
chambered heart forms, and they have implications for the management of con-genital and acquired heart disease. In the coming years, additional advances in our knowledge of cardiac development are likely to further influence the classification and treatment of congenital heart disease, inform clinicians on the best uses of re-generative treatment (e.g., stem-cell therapy), and revise our understanding of some cardiovascular disorders in adults. This review gives examples of recent findings in the field of cardiac development, with an emphasis on those likely to have the greatest effect on clinical practice.
Ov erv ie w
Classic teaching holds that cardiovascular development proceeds from the early spec-ification of bilateral clusters of progenitor cells that coalesce to form a cardiac crescent and a midline linear heart tube. This tube, which consists of an inner cell layer of endothelium surrounded by myocardial precursor cells,1 undergoes a series of looping or bending events followed by ballooning or expansion of regions des-tined to become cardiac chambers. Subsequently, a series of septation events results in a four-chambered heart with parallel systemic and pulmonary circulations.
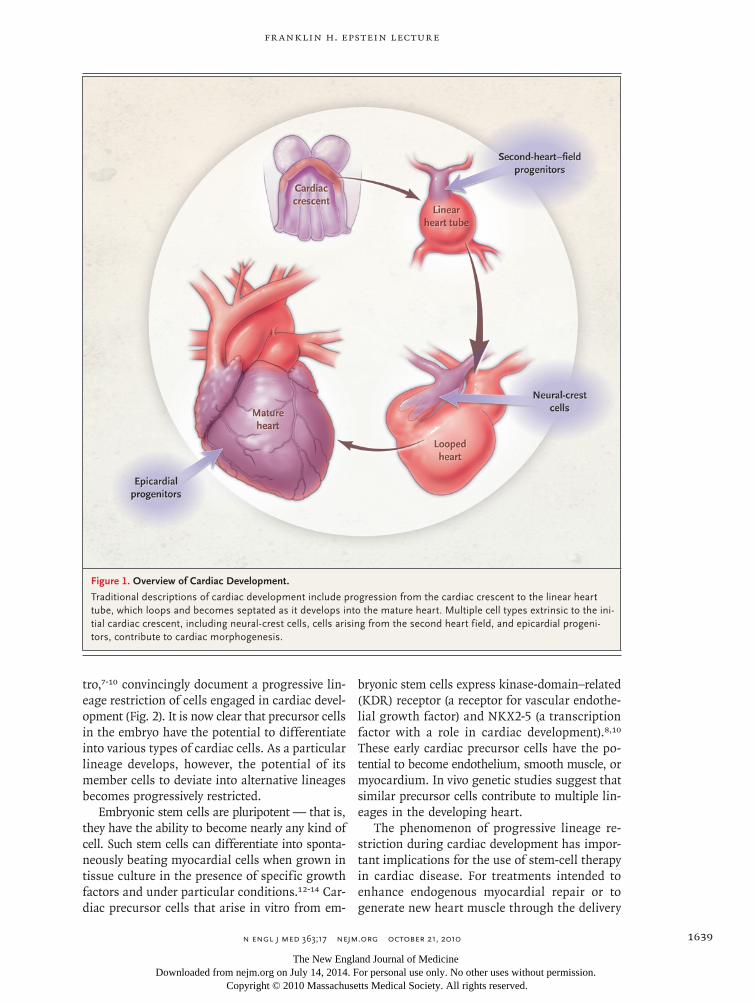
Additional cell types that lie outside the heart tube are important in the devel-opment of the heart and influence its morphogenesis (Fig. 1). For example, neural-crest cells, which form components of the peripheral nervous system and the cra-niofacial regions, migrate to the heart, where they are essential for septation of the cardiac outflow tract.2 The link between neural-crest cells3 and septation helps to explain the association of craniofacial defects with some forms of congenital heart disease.
Line age R es tr ic tion
The mature heart consists of many different cell types, including myocardial cells, endothelial cells, smooth-muscle cells, fibroblasts, and specialized conducting cells. Until recently, the origins and lineages of these various cell types were unclear. Recently, however, the technique of gene targeting has allowed rigorous “fate map-ping” (a method used to determine the cellular derivatives of a cell or population of cells) and lineage analysis in mammalian embryos and adults.4-7 The results of these studies, and of clonal analyses of embryonic stem-cell differentiation in vi-
The New England Journal of Medicine Downloaded from nejm.org on July 14, 2014. For personal use only. No other uses without permission.
Copyright © 2010 Massachusetts Medical Society. All rights reserved.
Fr anklin H. Epstein Lecture
n engl j med 363;17 nejm.org october 21, 2010 1639
tro,7-10 convincingly document a progressive lin-eage restriction of cells engaged in cardiac devel-opment (Fig. 2). It is now clear that precursor cells in the embryo have the potential to differentiate into various types of cardiac cells. As a particular lineage develops, however, the potential of its member cells to deviate into alternative lineages becomes progressively restricted.
Embryonic stem cells are pluripotent — that is, they have the ability to become nearly any kind of cell. Such stem cells can differentiate into sponta-neously beating myocardial cells when grown in tissue culture in the presence of specific growth factors and under particular conditions.12-14 Car-diac precursor cells that arise in vitro from em-
bryonic stem cells express kinase-domain–related (KDR) receptor (a receptor for vascular endothe-lial growth factor) and NKX2-5 (a transcription factor with a role in cardiac development).8,10 These early cardiac precursor cells have the po-tential to become endothelium, smooth muscle, or myocardium. In vivo genetic studies suggest that similar precursor cells contribute to multiple lin-eages in the developing heart.
The phenomenon of progressive lineage re-striction during cardiac development has impor-tant implications for the use of stem-cell therapy in cardiac disease. For treatments intended to enhance endogenous myocardial repair or to generate new heart muscle through the delivery
Figure 1. Overview of Cardiac Development.
Traditional descriptions of cardiac development include progression from the cardiac crescent to the linear heart tube, which loops and becomes septated as it develops into the mature heart. Multiple cell types extrinsic to the ini-tial cardiac crescent, including neural-crest cells, cells arising from the second heart field, and epicardial progeni-tors, contribute to cardiac morphogenesis.
The New England Journal of Medicine Downloaded from nejm.org on July 14, 2014. For personal use only. No other uses without permission.
Copyright © 2010 Massachusetts Medical Society. All rights reserved.

T h e n e w e ngl a nd j o u r na l o f m e dic i n e
n engl j med 363;17 nejm.org october 21, 20101640
of appropriate cardiac progenitor cells, or to grow bioprostheses of contractile myocardial patches,8 researchers and practitioners must consider which cells can best meet these goals.8,15,16 For example, the regenerated tissue that results from treatment with a progenitor cell that has the potential to produce only cardiac myocytes will be unlike nor-mal cardiac tissue, which has multiple cellular components. Progenitors with a restricted differ-entiation capacity may have to be replaced with multipotent progenitor cells if the goal is to re-generate multilineage tissue composed of endo-thelium, smooth muscle (regenerating vascula-ture), and contractile myocardium. At this time, the most appropriate type of progenitor cell to be used in stem-cell therapy for ischemic cardiomy-
opathy and other forms of heart failure is un-known, and the markers and gene-expression signatures that characterize various progenitors are only now being elucidated.
Progressive lineage restriction is also a feature of the differentiation of a multipotent hematopoi-etic stem cell into various blood-cell lineages.17 The delineation of each stage of lineage restriction in blood-cell precursors has allowed for identifi-cation of clinically useful growth factors, such as granulocyte colony-stimulating factor, granulo-cyte–macrophage colony-stimulating factor, and erythropoietin, each of which affects a different progenitor. As is the case with hematopoietic stem cells, the identification and characterization of specific cardiac progenitor cells and cardiac
Figure 2. Progressive Lineage Restriction.
In the hematopoietic system, stem cells become progressively restricted during differentiation. Similarly, cardiac progenitors, which may share a common precursor with hematopoietic stem cells, become progressively restricted in terms of the types of potential mature de-rivatives that can ultimately be produced. Adapted from Wu et al.11
The New England Journal of Medicine Downloaded from nejm.org on July 14, 2014. For personal use only. No other uses without permission.
Copyright © 2010 Massachusetts Medical Society. All rights reserved.

Fr anklin H. Epstein Lecture
n engl j med 363;17 nejm.org october 21, 2010 1641
growth factors may lead to useful treatments for myocardial infarction or heart failure.
The Second He a rt Field
Not all precursors of cardiac muscle reside in the cardiac crescent and the early linear heart tube. Many right ventricular myocytes and, to a variable degree, myocytes in the atria, left ventricle, and cardiac inflow and outflow tracts enter the devel-oping heart after its initial looping stages are com-plete.18-20 These additional cells arise from a sec-ond heart field that is medial and ventral to the primary cardiac crescent. (In the embryo, a field consists of a group of related cells within a de-fined boundary.) Cells in the second heart field migrate first to the pharyngeal regions, where they can be identified in mouse embryos in early gestation or midgestation according to the prod-ucts of specific marker genes, including the tran-scription factor islet 1.20 These second-heart–field cardiac precursors in the pharyngeal regions invade the developing heart and migrate along its inflow and outflow tracts. Second-heart–field progeni-tors that express islet 1 are multipotent cells that can give rise to smooth-muscle cells at the base of the aorta and pulmonary arteries, to endothe-lial cells, or to myocardium.7
The existence of a second heart field has im-portant implications for understanding congeni-tal heart diseases. For example, abnormalities in the distinct genetic pathways that mediate for-mation of myocytes in either the right or the left ventricle could explain congenital defects whose predominant effect is on the right or left ventri-cle. The results of induced genetic perturbations in only second-heart–field cells of embryonic mice suggest that abnormalities in this population can cause double-outlet right ventricle, right ventricu-lar hypoplasia, pulmonic stenosis, and tetralogy of Fallot.21,22 Moreover, genetic studies in humans have shown that haplotypes within the ISL1 lo-cus are strongly associated with these forms of congenital heart disease.23 Perhaps other right-sided disorders, such as hypoplastic right ventri-cle, Ebstein’s anomaly, and some forms of arrhyth-mogenic right ventricular dysplasia,24 are also the result of abnormalities in second-heart–field cells.
The usual classification of congenital heart diseases depends on the anatomical characteris-tics of the abnormality. These traditional classi-fication systems may have to be changed in light of emerging evidence that the same developmen-
tal abnormality, or similar developmental abnor-malities, can underlie anatomically distinct con-genital heart disorders. An example is the group of clinically dissimilar congenital heart defects (e.g., a double-outlet right ventricle and right ven-tricular hypoplasia) that are related through their association with abnormalities of second-heart–field progenitor cells.25 Aberrations of these pre-cursor cells may also cause anatomical abnor-malities of the left or right side of the heart (e.g., defects of atrial septation, ventricular septation, conus positioning, and great-vessel alignment) because the cells contribute to both the inflow and the outflow tracts of the heart.26 Moreover, recent studies of what have come to be known as second-heart–field cardiac defects suggest that in-flow and outflow abnormalities frequently coex-ist.26 Anatomical classification is undoubtedly clinically useful, but it is likely that classification systems based on developmental relationships and genetic causes will provide additional diagnostic and prognostic information. Consensus opinions that explicitly define developmentally based clas-sifications of congenital heart disorders will also promote enhanced communication among basic researchers and their clinical colleagues.
The Epic a r dium in C a r di ac De v el opmen t a nd R epa ir
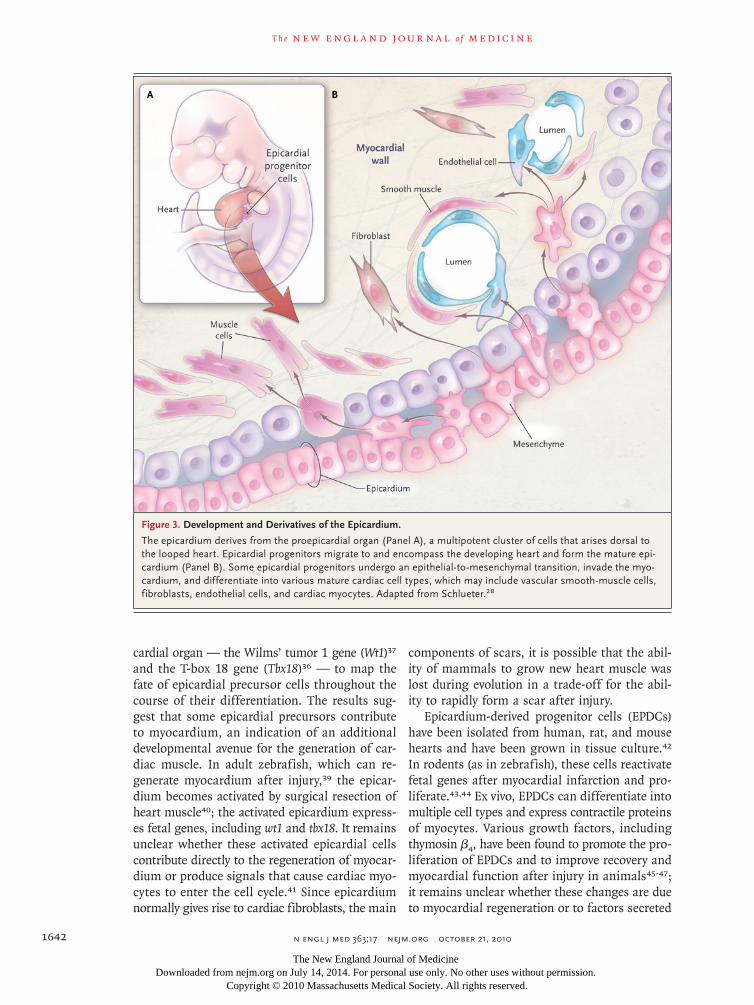
The epicardium, a layer of connective tissue lo-cated between the myocardium and the pericar-dium,27 arises from a transient embryonic struc-ture called the proepicardial organ (Fig. 3). Cells in this organ derive from the septum transver-sum, which separates the embryo’s thorax from its abdomen, and to the diaphragm and liver. Some proepicardial cells migrate to the developing heart and contribute to the formation of the epicardial layer. Descendants of proepicardial cells invade the myocardium, where they develop into fibro-blasts in the heart and smooth-muscle cells of the coronary arteries.29-32 Signals from the epi-cardium are required for proper maturation of the myocardium and normal development of the cor-onary arteries.33-35
Recent studies suggest that epicardial pro-genitor cells are multipotent, with the ability to differentiate into smooth muscle, fibroblasts, and perhaps also cardiac muscle and endothelium36,37
(although the idea that these cells form coronary endothelium has been challenged38). These stud-ies used genetic markers expressed by the proepi-
The New England Journal of Medicine Downloaded from nejm.org on July 14, 2014. For personal use only. No other uses without permission.
Copyright © 2010 Massachusetts Medical Society. All rights reserved.
T h e n e w e ngl a nd j o u r na l o f m e dic i n e
n engl j med 363;17 nejm.org october 21, 20101642
cardial organ — the Wilms’ tumor 1 gene (Wt1)37 and the T-box 18 gene (Tbx18)36 — to map the fate of epicardial precursor cells throughout the course of their differentiation. The results sug-gest that some epicardial precursors contribute to myocardium, an indication of an additional developmental avenue for the generation of car-diac muscle. In adult zebrafish, which can re-generate myocardium after injury,39 the epicar-dium becomes activated by surgical resection of heart muscle40; the activated epicardium express-es fetal genes, including wt1 and tbx18. It remains unclear whether these activated epicardial cells contribute directly to the regeneration of myocar-dium or produce signals that cause cardiac myo-cytes to enter the cell cycle.41 Since epicardium normally gives rise to cardiac fibroblasts, the main
components of scars, it is possible that the abil-ity of mammals to grow new heart muscle was lost during evolution in a trade-off for the abil-ity to rapidly form a scar after injury.
Epicardium-derived progenitor cells (EPDCs) have been isolated from human, rat, and mouse hearts and have been grown in tissue culture.42 In rodents (as in zebrafish), these cells reactivate fetal genes after myocardial infarction and pro-liferate.43,44 Ex vivo, EPDCs can differentiate into multiple cell types and express contractile proteins of myocytes. Various growth factors, including thymosin β4, have been found to promote the pro-liferation of EPDCs and to improve recovery and myocardial function after injury in animals45-47; it remains unclear whether these changes are due to myocardial regeneration or to factors secreted
Figure 3. Development and Derivatives of the Epicardium.
The epicardium derives from the proepicardial organ (Panel A), a multipotent cluster of cells that arises dorsal to the looped heart. Epicardial progenitors migrate to and encompass the developing heart and form the mature epi-cardium (Panel B). Some epicardial progenitors undergo an epithelial-to-mesenchymal transition, invade the myo-cardium, and differentiate into various mature cardiac cell types, which may include vascular smooth-muscle cells, fibroblasts, endothelial cells, and cardiac myocytes. Adapted from Schlueter.28
The New England Journal of Medicine Downloaded from nejm.org on July 14, 2014. For personal use only. No other uses without permission.
Copyright © 2010 Massachusetts Medical Society. All rights reserved.

Fr anklin H. Epstein Lecture
n engl j med 363;17 nejm.org october 21, 2010 1643
by EPDCs that have paracrine effects influencing myocyte survival or function. These studies sug-gest that interventions involving the manipulation of epicardial activation and EPDCs after injury — whether through the use of systemic therapy, treat-ments administered within the pericardial space, or the application of a drug-eluting patch to the epicardial surface of the damaged heart — may be worthwhile avenues for further investigation.
The C a r di ac Conduc tion S ys tem
The specialized cells of the cardiac conduction sys-tem arise from myocardial precursor cells. The mature cells have relatively poor contractility and express specialized ion channels and gap-junction proteins, including connexins, that mediate elec-trical coupling with neighboring cells.48,49 Early in development, at the time of chamber specifi-cation, the myocardium between the developing atria and ventricles has slow conduction charac-teristics and other properties reminiscent of the atrioventricular node. Similarly, the myocardium of the inflow tract acquires autonomous activity and develops pacemaker function. The sinus node develops from this tissue. The cells that give rise to the sinus node express the fetal TBX18 gene, whereas the cells that give rise to the atrioven-tricular node and the Purkinje system express the NKX2-5 transcription factor.48 The possibility that precursors of pacemaker cells of the sinus node are related to the myocardium surrounding the pulmonary veins could be important (and is cur-rently under investigation) because atrial fibrilla-tion commonly arises from an arrhythmia within the pulmonary veins. The myocardium of the pos-terior wall of the left atrium extends to and en-sheathes the proximal pulmonary vein, thereby providing electrical continuity, and atrial fibrilla-tion can be successfully treated through electrical isolation of the pulmonary veins.50 The develop-ment of pulmonary-vein myocardium requires the PITX2 transcription factor,51 and recent ge-nomewide association studies have identified haplotypes at 4q25, near PITX2, that are associated with atrial fibrillation.52,53 Thus, the response of pulmonary-vein myocardial cells to altered PITX2 function may underlie the susceptibility to atrial fibrillation. It is also possible that melanocyte-like cells in the heart,54 which are present in the atrioventricular ring, the atria, and the pulmo-nary veins in the developing embryo, play a role in atrial fibrillation. In animal models, genetic
abnormalities induced in these cells increase the susceptibility to atrial arrhythmia.
The region of slow-conducting myocardium that separates atria from ventricles in the embryo and provides atrioventricular delay initially oc-cupies the entire atrioventricular ring.48,49,55 As cardiac development proceeds, fibroblasts derived from the epicardium invade the atrioventricular sulcus and form the annulus fibrosis,56,57 which isolates the atria from the ventricles electrically. The atrioventricular canal myocardium regresses, and the property of slow conduction becomes restricted to the specialized cells of the atrioven-tricular node. In animal models, deficient devel-opment of the annulus fibrosis causes abnormal electrical connectivity between the atria and ven-tricles, pre-excitation, and characteristics of the Wolff–Parkinson–White syndrome.56,58 Ectopic myocardium that bridges the atrioventricular re-gion is, however, more common than breakdown of the annulus fibrosis in humans with this syn-drome. Consequently, some forms of the syn-drome may result when the normal regression of the atrioventricular canal myocardium fails to oc-cur during development.
Proper formation of the atrioventricular node depends on transcription factors that play reiter-ated roles during cardiac development. These fac-tors include NKX2-5, TBX5, and GATA4.48 In mice, Tbx5 and Gata4 regulate the expression of con-nexin 30.2, which is required for slow conduction in the atrioventricular node.59 Haploinsufficiency of Gata4 in mice causes a short PR interval.59 Mu-tation of NKX2-5, TBX5, or GATA4 has been associ-ated with an atrial septal defect in humans as well as in animal models.60-63 Hence, structural inter-ference of conducting fibers by the septal defect may not entirely explain the association between atrial septal defects and conduction abnormalities. Rather, a single underlying genetic defect may affect septal closure and specialized conduction cells independently.64 Indeed, patients with NKX2-5 mutations can have isolated conduction defects. These factors, and the cellular processes that these factors regulate, may continue to play important roles in conduction tissues through-out adult life. For example, progressive degenera-tion of the atrioventricular node and heart block develop when the Nkx2-5 gene has been inacti-vated in adult mice.65 It seems likely that an as-sociation between certain risk alleles of this gene or related genes will be found in — and may predict — heart block in elderly patients.
The New England Journal of Medicine Downloaded from nejm.org on July 14, 2014. For personal use only. No other uses without permission.
Copyright © 2010 Massachusetts Medical Society. All rights reserved.

T h e n e w e ngl a nd j o u r na l o f m e dic i n e
n engl j med 363;17 nejm.org october 21, 20101644
C a r diova scul a r M at ur ation
At the time of birth, the cardiovascular system un-dergoes a series of abrupt and critical changes. Blood must be diverted to the lungs for oxygen-ation, and the portal circulation must perfuse the liver when enteric feeding begins. Increased oxy-gen tension associated with a baby’s first breaths, coupled with withdrawal from exposure to mater-nal prostaglandins, stimulates closure of the duc-tus arteriosus, sending blood from the right ven-tricle to the lungs and establishing the parallel pulmonary and systemic circulations. The foramen ovale of the atrial septum closes. The ductus veno-sus constricts, sending portal blood to the liver.
Less obvious but equally important changes occur in myocardial cells during the neonatal pe-riod. There is a shift in gene-expression profiles within the heart; many fetal isoforms of genes become down-regulated or replaced by their adult counterparts. Examples include genes encoding contractile components of the sarcomere, calcium-handling machinery, energy-utilization enzymes, and natriuretic factors.66-68 Re-expression of fe-tal genes occurs in nearly every form of heart failure in adults, and this phenomenon is thought to contribute to the progression of heart failure. Elucidation of the mechanisms that regulate the re-expression of fetal genes during disease states may reveal new targets for the treatment of heart failure. In addition, an improved understanding of the genetic programs governing myocyte mat-uration should inform the development of regen-erative treatments; the use of such treatments for cardiac disease has been hampered by a lim-ited ability to engineer fully mature adult cardiac myocytes from progenitor cells.
Our knowledge of the mechanisms that regu-late gene expression has increased considerably in recent years. Gene expression requires spe-cific transcription factors — proteins that acti-vate or inhibit transcription from genomic DNA to messenger RNA (mRNA) by binding to pro-moter or enhancer regions of genes. Changes in DNA packaging that are effected by chromatin and the enzymatic modification of histones, the principal protein component of chromatin, also influence gene expression through a mechanism termed epigenetic modification. This mechanism regulates gene expression by affecting the enzy-matic acetylation of histones (and causing other chemical modifications) and triggering the un-winding of chromatin (which exposes actively
transcribed loci); it also represses gene transcrip-tion by enzymatically deacetylating histones and condensing chromatin. Epigenetic control of chro-matin structure, a mechanism that mediates glob-al changes in gene-expression programs, is critical to cellular reprogramming (the directed altera-tion in which one cell type is changed to another — e.g., a fibroblast becomes a pluripotent stem cell) and to the determination of cell fate.
There are indications that the activities of spe-cific histone deacetylase enzymes are necessary for regulation of the expression of the fetal gene program in the heart during development and that these enzymes are involved in heart failure in adults. Genetic inactivation of histone deacety-lase 2 in mice, for example, decreases the expres-sion of fetal cardiac genes and inhibits reactiva-tion of the fetal gene program in situations involving cardiac stress in adults.69 Chemical in-hibitors of this enzyme, now being studied in phase 3 clinical trials for the treatment of cer-tain cancers, can prevent reactivation of the fetal gene program, cardiac hypertrophy, and heart failure in animal models (e.g., ClinicalTrials.gov numbers NCT00773747 and NCT01023308).70-75
These results suggest that epigenetic mechanisms regulate the transition of fetal cardiac gene pro-grams to adult programs and that these mecha-nisms could be targets for new treatments of heart failure.
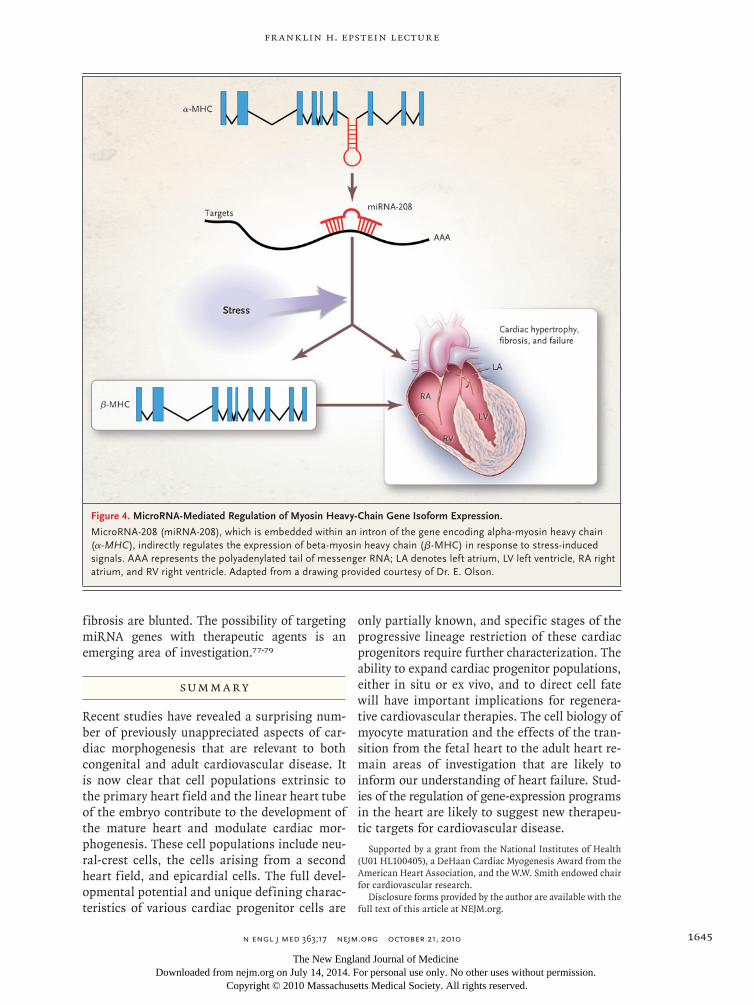
MicroRNA (miRNA) molecules are short, single strands of RNA that modulate gene expression by binding to complementary regions in mRNA transcripts. The miRNA genes in chromosomal DNA can be expressed as independent genes un-der the control of their own promoters or can be coexpressed with other genes in which they are embedded. In mice, the fetal heart expresses beta-myosin heavy chain (β-MHC), whereas the adult heart expresses alpha-myosin heavy chain (α-MHC). In adult mice, cardiac stressors such as pressure overload or chronic adrenergic stimulation in-duce re-expression of β-MHC and suppression of α-MHC.66 The basis of this reciprocal regulation is the embedding of a regulatory miRNA gene in the intron of the genes for each of the two MHC isoforms.76 The expression of α-MHC results in coexpression of miR-208 (a cardiac-specific miRNA gene), which, in the presence of stress-induced signals, indirectly activates transcription of β-MHC (Fig. 4). In the absence of miR-208, stress signals fail to activate β-MHC and other fetal genes, and both the hypertrophic response and associated
The New England Journal of Medicine Downloaded from nejm.org on July 14, 2014. For personal use only. No other uses without permission.
Copyright © 2010 Massachusetts Medical Society. All rights reserved.
Fr anklin H. Epstein Lecture
n engl j med 363;17 nejm.org october 21, 2010 1645
fibrosis are blunted. The possibility of targeting miRNA genes with therapeutic agents is an emerging area of investigation.77-79
Summ a r y
Recent studies have revealed a surprising num-ber of previously unappreciated aspects of car-diac morphogenesis that are relevant to both congenital and adult cardiovascular disease. It is now clear that cell populations extrinsic to the primary heart field and the linear heart tube of the embryo contribute to the development of the mature heart and modulate cardiac mor-phogenesis. These cell populations include neu-ral-crest cells, the cells arising from a second heart field, and epicardial cells. The full devel-opmental potential and unique defining charac-teristics of various cardiac progenitor cells are
only partially known, and specific stages of the progressive lineage restriction of these cardiac progenitors require further characterization. The ability to expand cardiac progenitor populations, either in situ or ex vivo, and to direct cell fate will have important implications for regenera-tive cardiovascular therapies. The cell biology of myocyte maturation and the effects of the tran-sition from the fetal heart to the adult heart re-main areas of investigation that are likely to inform our understanding of heart failure. Stud-ies of the regulation of gene-expression programs in the heart are likely to suggest new therapeu-tic targets for cardiovascular disease.
Supported by a grant from the National Institutes of Health (U01 HL100405), a DeHaan Cardiac Myogenesis Award from the American Heart Association, and the W.W. Smith endowed chair for cardiovascular research.
Disclosure forms provided by the author are available with the full text of this article at NEJM.org.
Figure 4. MicroRNA-Mediated Regulation of Myosin Heavy-Chain Gene Isoform Expression.
MicroRNA-208 (miRNA-208), which is embedded within an intron of the gene encoding alpha-myosin heavy chain (α-MHC), indirectly regulates the expression of beta-myosin heavy chain (β-MHC) in response to stress-induced signals. AAA represents the polyadenylated tail of messenger RNA; LA denotes left atrium, LV left ventricle, RA right atrium, and RV right ventricle. Adapted from a drawing provided courtesy of Dr. E. Olson.
The New England Journal of Medicine Downloaded from nejm.org on July 14, 2014. For personal use only. No other uses without permission.
Copyright © 2010 Massachusetts Medical Society. All rights reserved.

T h e n e w e ngl a nd j o u r na l o f m e dic i n e
n engl j med 363;17 nejm.org october 21, 20101646
References
1. Harvey RP, Rosenthal N, eds. Heart development. San Diego, CA: Academic Press, 1999.2. Kirby ML, Gale TF, Stewart DE. Neu-ral crest cells contribute to normal aorti-copulmonary septation. Science 1983;220: 1059-61.3. Stoller JZ, Epstein JA. Cardiac neural crest. Semin Cell Dev Biol 2005;16:704-15.4. Cui C, Cheuvront TJ, Lansford RD, Moreno-Rodriguez RA, Schultheiss TM, Rongish BJ. Dynamic positional fate map of the primary heart-forming region. Dev Biol 2009;332:212-22.5. Hsieh PC, Segers VF, Davis ME, et al. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat Med 2007;13:970-4.6. Ma Q, Zhou B, Pu WT. Reassessment of Isl1 and Nkx2-5 cardiac fate maps us-ing a Gata4-based reporter of Cre activity. Dev Biol 2008;323:98-104.7. Moretti A, Caron L, Nakano A, et al. Multipotent embryonic isl1+ progenitor cells lead to cardiac, smooth muscle, and endothelial cell diversification. Cell 2006; 127:1151-65.8. Kattman SJ, Huber TL, Keller GM. Multipotent flk-1+ cardiovascular progen-itor cells give rise to the cardiomyocyte, endothelial, and vascular smooth muscle lineages. Dev Cell 2006;11:723-32.9. Wu SM, Fujiwara Y, Cibulsky SM, et al. Developmental origin of a bipotential myocardial and smooth muscle cell precur-sor in the mammalian heart. Cell 2006; 127:1137-50.10. Yang L, Soonpaa MH, Adler ED, et al. Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature 2008;453: 524-8.11. Wu SM, Fujiwara Y, Cibulsky SM, et al. Developmental origin of a bipotential myocardial and smooth muscle cell pre-cursor in the mammalian heart. Cell 2006;127:1137-50.12. Chen VC, Stull R, Joo D, Cheng X, Keller G. Notch signaling respecifies the hemangioblast to a cardiac fate. Nat Bio-technol 2008;26:1169-78.13. Laflamme MA, Chen KY, Naumova AV, et al. Cardiomyocytes derived from human embryonic stem cells in pro-sur-vival factors enhance function of infarcted rat hearts. Nat Biotechnol 2007;25:1015-24.14. Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic de-velopment. Cell 2008;132:661-80.15. Sui R, Liao X, Zhou X, Tan Q. The cur-rent status of engineering myocardial tis-sue. Stem Cell Rev 2010 March 3 (Epub ahead of print).16. Chien KR, Domian IJ, Parker KK. Car-diogenesis and the complex biology of
regenerative cardiovascular medicine. Sci-ence 2008;322:1494-7.17. Morrison SJ, Uchida N, Weissman IL. The biology of hematopoietic stem cells. Annu Rev Cell Dev Biol 1995;11:35-71.18. Kelly RG, Brown NA, Buckingham ME. The arterial pole of the mouse heart forms from Fgf10-expressing cells in pha-ryngeal mesoderm. Dev Cell 2001;1:435-40.19. Waldo KL, Kumiski DH, Wallis KT, et al. Conotruncal myocardium arises from a secondary heart field. Development 2001;128:3179-88.20. Cai CL, Liang X, Shi Y, et al. Isl1 iden-tifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev Cell 2003;5:877-89.21. High FA, Jain R, Stoller JZ, et al. Mu-rine Jagged1/Notch signaling in the sec-ond heart field orchestrates Fgf8 expres-sion and tissue-tissue interactions during outflow tract development. J Clin Invest 2009;119:1986-96.22. Ward C, Stadt H, Hutson M, Kirby ML. Ablation of the secondary heart field leads to tetralogy of Fallot and pulmonary atresia. Dev Biol 2005;284:72-83.23. Stevens KN, Hakonarson H, Kim CE, et al. Common variation in ISL1 confers genetic susceptibility for human congeni-tal heart disease. PLoS One 2010;5(5)e10855.24. Lombardi R, Dong J, Rodriguez G, et al. Genetic fate mapping identifies second heart field progenitor cells as a source of adipocytes in arrhythmogenic right ven-tricular cardiomyopathy. Circ Res 2009; 104:1076-84.25. Dyer LA, Kirby ML. The role of sec-ondary heart field in cardiac develop-ment. Dev Biol 2009;336:137-44.26. Bajolle F, Zaffran S, Losay J, Ou P, Buckingham M, Bonnet D. Conotruncal defects associated with anomalous pulmo-nary venous connections. Arch Cardio-vasc Dis 2009;102:105-10.27. Männer J, Pérez-Pomares JM, Macías D, Muñoz-Chápuli R. The origin, forma-tion and developmental significance of the epicardium: a review. Cells Tissues Organs 2001;169:89-103.28. Schlueter J. The origin and therapeu-tic potential of the coronary blood vessel system. Presented at the European Society of Cardiology, Barcelona, August 29–Sep-tember 2, 2009. abstract.29. Guadix JA, Carmona R, Muñoz-Chá-puli R, Pérez-Pomares JM. In vivo and in vitro analysis of the vasculogenic poten-tial of avian proepicardial and epicardial cells. Dev Dyn 2006;235:1014-26.30. Vrancken Peeters MP, Gittenberger-de Groot AC, Mentink MM, Poelmann RE. Smooth muscle cells and fibroblasts of the coronary arteries derive from epitheli-al-mesenchymal transformation of the epi-
cardium. Anat Embryol (Berl) 1999;199: 367-78.31. Mikawa T, Gourdie RG. Pericardial mesoderm generates a population of cor-onary smooth muscle cells migrating into the heart along with ingrowth of the epi-cardial organ. Dev Biol 1996;174:221-32.32. Mikawa T, Fischman DA. Retroviral analysis of cardiac morphogenesis: discon-tinuous formation of coronary vessels. Proc Natl Acad Sci U S A 1992;89:9504-8.33. Stuckmann I, Evans S, Lassar AB. Erythropoietin and retinoic acid, secreted from the epicardium, are required for car-diac myocyte proliferation. Dev Biol 2003; 255:334-49.34. Chen TH, Chang TC, Kang JO, et al. Epicardial induction of fetal cardiomyocyte proliferation via a retinoic acid-inducible trophic factor. Dev Biol 2002;250:198-207.35. Pérez-Pomares JM, Phelps A, Sedmero-va M, et al. Experimental studies on the spatiotemporal expression of WT1 and RALDH2 in the embryonic avian heart: a model for the regulation of myocardial and valvuloseptal development by epicar-dially derived cells (EPDCs). Dev Biol 2002;247:307-26.36. Cai CL, Martin JC, Sun Y, et al. A myo-cardial lineage derives from Tbx18 epicar-dial cells. Nature 2008;454:104-8.37. Zhou B, Ma Q, Rajagopal S, et al. Epi-cardial progenitors contribute to the car-diomyocyte lineage in the developing heart. Nature 2008;454:109-13.38. Red-Horse K, Ueno H, Weissman IL, Krasnow MA. Coronary arteries form by developmental reprogramming of venous cells. Nature 2010;464:549-53.39. Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science 2002;298:2188-90.40. Lepilina A, Coon AN, Kikuchi K, et al. A dynamic epicardial injury response sup-ports progenitor cell activity during ze-brafish heart regeneration. Cell 2006;127: 607-19.41. Jopling C, Sleep E, Raya M, Martí M, Raya A, Belmonte JC. Zebrafish heart re-generation occurs by cardiomyocyte de-differentiation and proliferation. Nature 2010;464:606-9.42. Smart N, Riley PR. Derivation of epi-cardium-derived progenitor cells (EPDCs) from adult epicardium. In: Current proto-cols in stem cell biology. New York: John Wiley, 2009:Unit2C.2.43. Limana F, Bertolami C, Mangoni A, et al. Myocardial infarction induces embry-onic reprogramming of epicardial c-kit(+) cells: role of the pericardial f luid. J Mol Cell Cardiol 2010;48:609-18.44. Limana F, Zacheo A, Mocini D, et al. Identification of myocardial and vascular precursor cells in human and mouse epi-cardium. Circ Res 2007;101:1255-65.45. Smart N, Risebro CA, Melville AA, et al. Thymosin beta-4 is essential for coro-
The New England Journal of Medicine Downloaded from nejm.org on July 14, 2014. For personal use only. No other uses without permission.
Copyright © 2010 Massachusetts Medical Society. All rights reserved.

Fr anklin H. Epstein Lecture
n engl j med 363;17 nejm.org october 21, 2010 1647
nary vessel development and promotes neovascularization via adult epicardium. Ann N Y Acad Sci 2007;1112:171-88.46. Bock-Marquette I, Shrivastava S, Pipes GC, et al. Thymosin beta4 mediated PKC activation is essential to initiate the em-bryonic coronary developmental program and epicardial progenitor cell activation in adult mice in vivo. J Mol Cell Cardiol 2009;46:728-38.47. Smart N, Risebro CA, Melville AA, et al. Thymosin beta4 induces adult epicar-dial progenitor mobilization and neovas-cularization. Nature 2007;445:177-82.48. Christoffels VM, Smits GJ, Kispert A, Moorman AF. Development of the pace-maker tissues of the heart. Circ Res 2010;106:240-54.49. Moorman AF, Christoffels VM. Devel-opment of the cardiac conduction system: a matter of chamber development. Novar-tis Found Symp 2003;250:25-34.50. Peters NS, Schilling RJ, Kanagarat-nam P, Markides V. Atrial fibrillation: strategies to control, combat, and cure. Lancet 2002;359:593-603.51. Mommersteeg MT, Brown NA, Prall OW, et al. Pitx2c and Nkx2-5 are required for the formation and identity of the pul-monary myocardium. Circ Res 2007;101: 902-9.52. Damani SB, Topol EJ. Molecular ge-netics of atrial fibrillation. Genome Med 2009;1:54.53. Gudbjartsson DF, Arnar DO, Hel-gadottir A, et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature 2007;448:353-7.54. Levin MD, Lu MM, Petrenko NB, et al. Melanocyte-like cells in the heart and pulmonary veins contribute to atrial ar-rhythmia triggers. J Clin Invest 2009; 119:3420-36.55. Rutenberg JB, Fischer A, Jia H, Gessler M, Zhong TP, Mercola M. Developmental patterning of the cardiac atrioventricular canal by Notch and Hairy-related tran-scription factors. Development 2006;133: 4381-90.56. Kolditz DP, Wijffels MC, Blom NA, et al. Epicardium-derived cells in develop-ment of annulus fibrosis and persistence of accessory pathways. Circulation 2008; 117:1508-17.
57. Zhou B, von Gise A, Ma Q, Hu YW, Pu WT. Genetic fate mapping demonstrates contribution of epicardium-derived cells to the annulus fibrosis of the mammalian heart. Dev Biol 2010;338:251-61.58. Arad M, Moskowitz IP, Patel VV, et al. Transgenic mice overexpressing mutant PRKAG2 define the cause of Wolff-Par-kinson-White syndrome in glycogen stor-age cardiomyopathy. Circulation 2003;107: 2850-6.59. Munshi NV, McAnally J, Bezprozvan-naya S, et al. Cx30.2 enhancer analysis identifies Gata4 as a novel regulator of atrioventricular delay. Development 2009; 136:2665-74.60. Maslen CL. Molecular genetics of atrioventricular septal defects. Curr Opin Cardiol 2004;19:205-10.61. Basson CT, Cowley GS, Solomon SD, et al. The clinical and genetic spectrum of the Holt–Oram syndrome (heart–hand syn-drome). N Engl J Med 1994;330:885-91. [Erratum, N Engl J Med 1994;330:1627.]62. Schott JJ, Benson DW, Basson CT, et al. Congenital heart disease caused by muta-tions in the transcription factor NKX2-5. Science 1998;281:108-11.63. Garg V, Kathiriya IS, Barnes R, et al. GATA4 mutations cause human congeni-tal heart defects and reveal an interaction with TBX5. Nature 2003;424:443-7.64. Benson DW, Silberbach GM, Kava-naugh-McHugh A, et al. Mutations in the cardiac transcription factor NKX2.5 af-fect diverse cardiac developmental path-ways. J Clin Invest 1999;104:1567-73.65. Pashmforoush M, Lu JT, Chen H, et al. Nkx2-5 pathways and congenital heart disease: loss of ventricular myocyte lineage specification leads to progressive cardio-myopathy and complete heart block. Cell 2004;117:373-86.66. Feldman AM, Weinberg EO, Ray PE, Lorell BH. Selective changes in cardiac gene expression during compensated hy-pertrophy and the transition to cardiac decompensation in rats with chronic aor-tic banding. Circ Res 1993;73:184-92.67. Parker TG, Packer SE, Schneider MD. Peptide growth factors can provoke “fe-tal” contractile protein gene expression in rat cardiac myocytes. J Clin Invest 1990; 85:507-14.
68. Taegtmeyer H, Sen S, Vela D. Return to the fetal gene program: a suggested metabolic link to gene expression in the heart. Ann N Y Acad Sci 2010;1188:191-8.69. Trivedi CM, Luo Y, Yin Z, et al. Hdac2 regulates the cardiac hypertrophic re-sponse by modulating Gsk3 beta activity. Nat Med 2007;13:324-31.70. Antos CL, McKinsey TA, Dreitz M, et al. Dose-dependent blockade to cardio-myocyte hypertrophy by histone deacety-lase inhibitors. J Biol Chem 2003;278: 28930-7.71. Bush EW, McKinsey TA. Targeting histone deacetylases for heart failure. Ex-pert Opin Ther Targets 2009;13:767-84.72. Gallo P, Latronico MV, Grimaldi S, et al. Inhibition of class I histone deacety-lase with an apicidin derivative prevents cardiac hypertrophy and failure. Cardio-vasc Res 2008;80:416-24.73. Kong Y, Tannous P, Lu G, et al. Sup-pression of class I and II histone deacety-lases blunts pressure-overload cardiac hy-pertrophy. Circulation 2006;113:2579-88.74. Kee HJ, Sohn IS, Nam KI, et al. Inhibi-tion of histone deacetylation blocks car-diac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation 2006;113:51-9.75. Kook H, Lepore JJ, Gitler AD, et al. Cardiac hypertrophy and histone deacety-lase-dependent transcriptional repression mediated by the atypical homeodomain protein Hop. J Clin Invest 2003;112:863-71.76. van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 2007;316:575-9.77. Brown BD, Naldini L. Exploiting and antagonizing microRNA regulation for therapeutic and experimental applications. Nat Rev Genet 2009;10:578-85.78. Latronico MV, Condorelli G. MicroR-NAs and cardiac pathology. Nat Rev Car-diol 2009;6:419-29.79. van Rooij E, Marshall WS, Olson EN. Toward microRNA-based therapeutics for heart disease: the sense in antisense. Circ Res 2008;103:919-28.Copyright © 2010 Massachusetts Medical Society.
The New England Journal of Medicine Downloaded from nejm.org on July 14, 2014. For personal use only. No other uses without permission.
Copyright © 2010 Massachusetts Medical Society. All rights reserved.





























