Embed Size (px)
Citation preview

Data Integrity -The new pillar in GMP?
SAAPI Conference
Pretoria, 13 April 2016
Manuel Zahn

"Data"
Chromatograms;
Lab journals;
Analytical test results;
Batch records;
Acceptance criteria;
Archives;
CTD;
Anything, that is in conflict with "The Business".
© 3R Pharma Consulting GmbH – Slide # 2
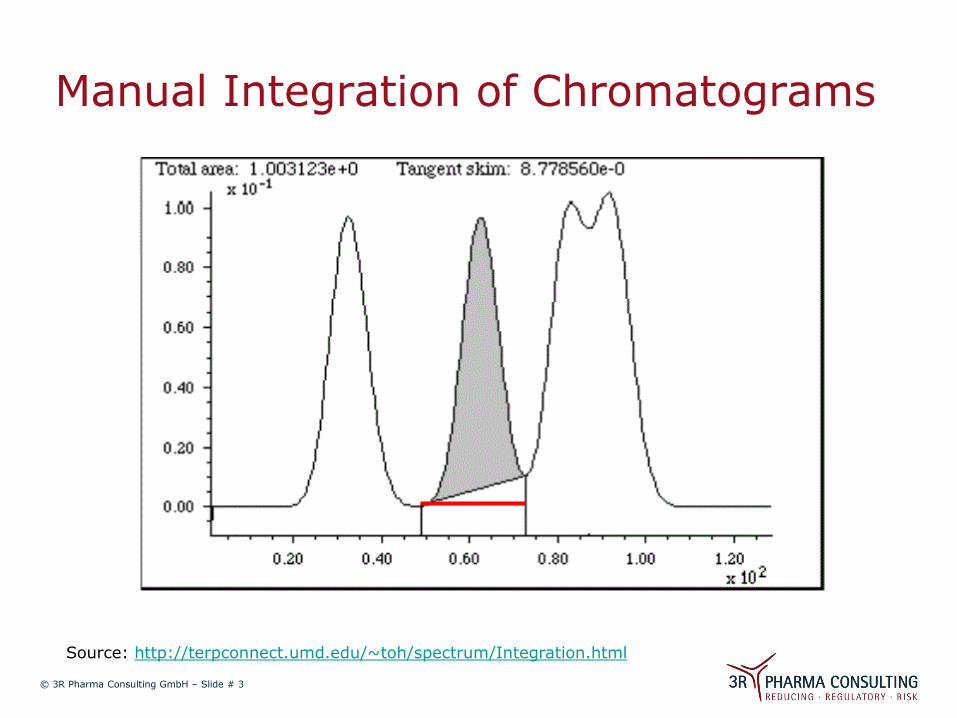
Manual Integration of Chromatograms
Source: http://terpconnect.umd.edu/~toh/spectrum/Integration.html
© 3R Pharma Consulting GmbH – Slide # 3

Manual Integration of Chromatograms
What GMP requires:
Define reasons for manual correction and the method to be used in an SOP;
Second person to check and approve manual integration;
Original and corrected chromatograms must be added to the documentation as proof.
© 3R Pharma Consulting GmbH – Slide # 4

FDA Warning Letter to Novacyl China
© 3R Pharma Consulting GmbH – Slide # 5

FDA Warning Letter to Novacyl China
The inspection documented that your firm made changes to integration parameters for the impurities test without appropriate documentation or justification.
Your firm relied upon hand written notes on a chromatogram discovered in a drawer at the laboratory as the documentation for this change.
Your firm implemented this change without an audit trail that would have captured the date of the change and who made the change.
© 3R Pharma Consulting GmbH – Slide # 6

Executed Batch Records
What GMP requires:
Plausibility controls and data integrity: The operator's input can be checked at different production phases against other data or specifications.
Automatic calculations or derivations (control loops) are possible in real time.
Entries are made by authorized persons and are checked as thoroughly as possible for plausibility and integrity during the input.
© 3R Pharma Consulting GmbH – Slide # 7

FDA Warning Letter to Dr. Reddy's Lab
© 3R Pharma Consulting GmbH – Slide # 8

FDA Warning Letter to Dr. Reddy's Lab
Your employees did not complete batch production and control records immediately after activities were performed.
Data were later entered into GMP documents after operations had already ended as though they had been entered at the time of the operation.
Our investigator observed copies of issued, partially-used and unused batch records, analytical raw data, analytical results, training records, and cleaning validation protocols in the waste area.
© 3R Pharma Consulting GmbH – Slide # 9

Laboratory Data
What GMP requires:
In order to demonstrate test results are documented at the time of execution, the QC laboratory can use laboratory notebooks (e.g., bound notebook, pre-numbered);
An electronic and validated data collection system can also be used to record the raw data at the time it is produced.
© 3R Pharma Consulting GmbH – Slide # 10

FDA Warning Letter to Dr. Reddy's Lab
The investigator found that batch samples were routinely re-tested following failing or atypical results until acceptable results were obtained.
You conducted purity testing by HPLC. This sample failed the purity specification limit, but you did not document, report or investigate the failure.
Your QC data package, which you used to support batch disposition decisions, showed passing results on the same day but does not include the initial failing results.
© 3R Pharma Consulting GmbH – Slide # 11

FDA Warning Letter to Novacyl Thailand
© 3R Pharma Consulting GmbH – Slide # 12

FDA Warning Letter to Novacyl Thailand
Your analyst selectively invalidated data during related substance testing.
For example, you did not retain data from all six injections used for the initial system suitability. Your analyst discarded one of the six injections performed with no justification.
© 3R Pharma Consulting GmbH – Slide # 13

FDA Warning Letter to Novacyl China
Our inspection documented multiple instances, where the analysts did not record raw material lot numbers during sample preparation, making it impossible to link the raw materials used to the appropriate test worksheet.
© 3R Pharma Consulting GmbH – Slide # 14

OOS
What GMP requires:
Any out-of-specification result obtained should be investigated and documented according to a procedure;
This procedure should require analysis of the data, assessment of whether a significant problem exists, allocation of the tasks for corrective actions, and conclusions.
© 3R Pharma Consulting GmbH – Slide # 15

FDA Warning Letter to Dr. Reddy's Lab
Our investigator documented that five batches of intermediate failed the optical purity test by HPLC;
You acknowledged that, since 2012, 11 batches had failed the optical purity test, and that you had been unable to determine a root cause for such failures;
Our investigator documented 13 instances of OOS results for a single impurity found in your intermediate.
You indicated that, since 2012, 65 batches of this intermediate failed to meet the single impurity specification.
© 3R Pharma Consulting GmbH – Slide # 16

Access Control
What GMP requires:
Physical and/or logical controls should be in place to restrict access to computerised system to authorised persons.
Suitable methods of preventing unauthorised entry to the system may include the use of keys, pass cards, personal codes with passwords, biometrics, restricted access to computer equipment and data storage areas.
EudraLex Volume 4 Annex 11 Section 12.1
© 3R Pharma Consulting GmbH – Slide # 17

FDA Warning Letter to Dr. Reddy's Lab
Your HPLC systems are configured so that no passwords are required to log in. Anyone who accesses the system can use software administrator privileges, which means that there is no electronic or procedural control to prevent manipulation of data.
Your HPLC system had no access controls to prevent alteration or deletion of data.
Your HPLC software lacked an audit trail feature to document all activities related to the chromatographic analysis.
© 3R Pharma Consulting GmbH – Slide # 18

FDA Warning Letter to Novacyl Thailand
Your firm did not have proper controls in place to prevent the unauthorized manipulation of your laboratory’s raw electronic data;
Your HPLC computer software lacked active audit trail functions to record changes;
Your laboratory systems did not have access controls to prevent deletion or alteration of raw data;
During the inspection, your analysts demonstrated that they were given inappropriate user permissions to delete HPLC data files;
The GC computer software lacked password protection allowing uncontrolled full access to all employees.
© 3R Pharma Consulting GmbH – Slide # 19

FDA Warning Letter to Novacyl China
During the inspection, our investigator found a chromatogram in the trash, which reported an additional chromatographic peak when compared to the standard;
During the inspection, your firm stated that the analyst discarded the chromatogram because it was present in the blank injection;
However, the analyst was unable to retrieve the blank chromatogram from the system because it was overwritten by a subsequent injection.
© 3R Pharma Consulting GmbH – Slide # 20

Archiving
What GMP requires:
Integrity of the records must be ensuredthroughout the retention period by secure controls;
The accessibility, readability and integrity of the data are of primary importance, particularly after modifications have been made in hardware and software.
© 3R Pharma Consulting GmbH – Slide # 21
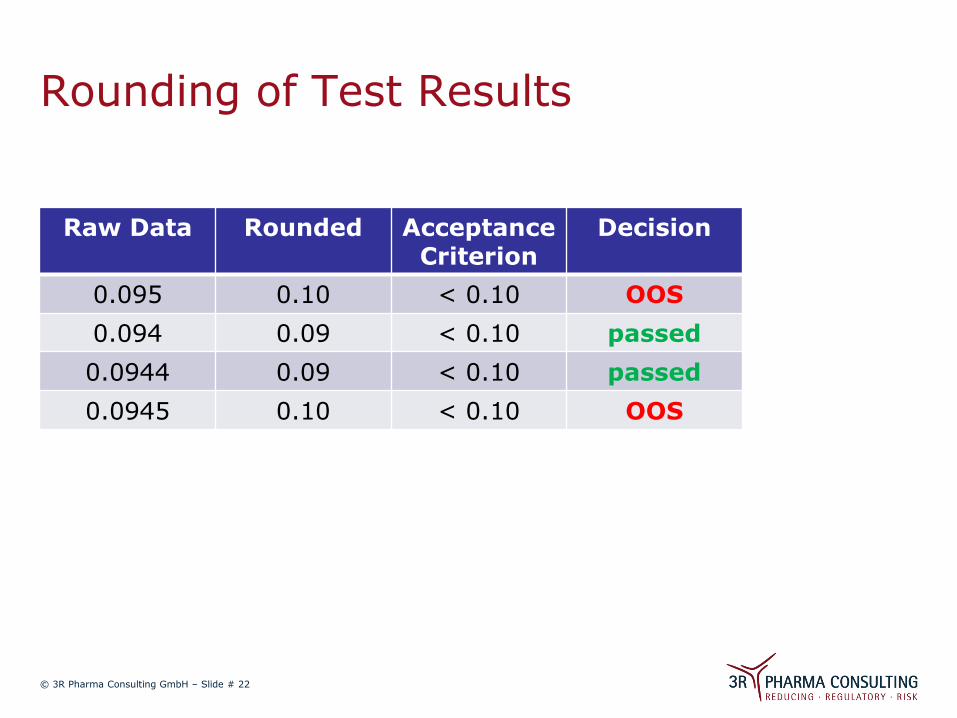
Rounding of Test Results
Raw Data Rounded Acceptance Criterion
Decision
0.095 0.10 < 0.10 OOS
0.094 0.09 < 0.10 passed
0.0944 0.09 < 0.10 passed
0.0945 0.10 < 0.10 OOS
© 3R Pharma Consulting GmbH – Slide # 22

CTD
Source of starting material
Batch results
Stability data
Results of process validation
© 3R Pharma Consulting GmbH – Slide # 23

Principle of Data Integrity
Define the universe of data that will be collected, the procedures to collect it, and the means to verify its accuracy and validity;
Describe the procedure to audit data;
Describe the process for correcting errors;
Keep data so that they are not accidentally lost;
Only authorized individuals can make data entries;
Data entries must not be deleted;
Changes must be made in the form of amendments;
The data base must be made as tamperproof as possible.
© 3R Pharma Consulting GmbH – Slide # 24

Thank You
3R Pharma Consulting GmbHDr. Manuel ZahnWildbader Str. 375335 Dobel Germanywww.3rpc.com
© 3R Pharma Consulting GmbH – Slide # 25





























