Embed Size (px)
Citation preview

MOLECULAR AND CELLULAR BIOLOGY,0270-7306/99/$04.0010
Jan. 1999, p. 353–363 Vol. 19, No. 1
Copyright © 1999, American Society for Microbiology. All Rights Reserved.
Cyclin D- and E-Dependent Kinases and the p57KIP2 Inhibitor:Cooperative Interactions In Vivo
ENRIQUE GOMEZ LAHOZ,1† NANETTE J. LIEGEOIS,1 PUMIN ZHANG,2 JEFFREY A. ENGELMAN,1
JAMES HORNER,1 ADAM SILVERMAN,1 RONALD BURDE,1 MARTINE F. ROUSSEL,3
CHARLES J. SHERR,3,4 STEPHEN J. ELLEDGE,2,5 AND RONALD A. DEPINHO1*
Department of Microbiology and Immunology and Department of Medicine, Albert Einstein College of Medicine,Bronx, New York 10461;1 Howard Hughes Medical Institute4 and Department of Tumor Cell Biology,3 St. Jude
Children’s Research Hospital, Memphis, Tennessee 38105; and Verna & Marrs McLean Department ofBiochemistry2 and Howard Hughes Medical Institute and Department of Molecular and Human
Genetics,5 Baylor College of Medicine, Houston, Texas 77030
Received 9 December 1997/Returned for modification 12 January 1998/Accepted 28 September 1998
This study examines in vivo the role and functional interrelationships of components regulating exit from theG1 resting phase into the DNA synthetic (S) phase of the cell cycle. Our approach made use of several keyexperimental attributes of the developing mouse lens, namely its strong dependence on pRb in maintenance ofthe postmitotic state, the down-regulation of cyclins D and E and up-regulation of the p57KIP2 inhibitor in thepostmitotic lens fiber cell compartment, and the ability to target transgene expression to this compartment.These attributes provide an ideal in vivo context in which to examine the consequences of forced cyclinexpression and/or of loss of p57KIP2 inhibitor function in a cellular compartment that permits an accuratequantitation of cellular proliferation and apoptosis rates in situ. Here, we demonstrate that, despite substan-tial overlap in cyclin transgene expression levels, D-type and E cyclins exhibited clear functional differences inpromoting entry into S phase. In general, forced expression of the D-type cyclins was more efficient than cyclinE in driving lens fiber cells into S phase. In the case of cyclins D1 and D2, ectopic proliferation required theirenhanced nuclear localization through CDK4 coexpression. High nuclear levels of cyclin E and CDK2, whilenot sufficient to promote efficient exit from G1, did act synergistically with ectopic cyclin D/CDK4. Thefunctional differences between D-type and E cyclins was most evident in the p57KIP2-deficient lens whereincyclin D overexpression induced a rate of proliferation equivalent to that of the pRb null lens, while overex-pression of cyclin E did not increase the rate of proliferation over that induced by the loss of p57KIP2 function.These in vivo analyses provide strong biological support for the prevailing view that the antecedent actions ofcyclin D/CDK4 act cooperatively with cyclin E/CDK2 and antagonistically with p57KIP2 to regulate the G1/Stransition in a cell type highly dependent upon pRb.
Progression into the DNA synthetic (S) phase of the mam-malian cell cycle requires inactivation of the retinoblastomaprotein (pRb) via its phosphorylation by cyclin-dependent ki-nases. This phosphorylation cancels pRb-mediated repressionof the transactivation of genes whose activities are necessaryfor S-phase entry (52, 61). During the G1 phase, pRb phos-phorylation is initially triggered by the cyclin D-dependentkinases CDK4 and CDK6 and then followed by cyclin E-de-pendent CDK2 (23). The cyclin D- and E-dependent kinaseshave a propensity to phosphorylate distinct serine and threo-nine residues of pRb (10), and under normal conditions whereboth kinases are sequentially expressed at physiologic levels,pRb phosphorylation by cyclin E-CDK2 may depend upon theprevious action of cyclin D-dependent kinases (10, 23). Inhi-bition of cyclin D-dependent kinases in cells containing a func-tional pRb protein prevents pRb phosphorylation and leads toG1 phase arrest (4, 46), whereas cells lacking pRb function arerefractory to such signals and continue to enter S phase (22, 26,29, 33). In contrast, inhibition of cyclin E-dependent kinaseactivity in pRb-negative cells prevents S-phase entry (41), im-
plying that cyclin E-CDK2 targets also non-pRb substrateswhose phosphorylation is essential for G1 exit. Overexpressionof either cyclin D1 or E leads to a decrease in the duration ofG1 phase in rodent fibroblasts (40, 46) with additive effectswhen ectopic expression of both is enforced (49), but only D1induction leads to rapid and immediate pRb hyperphosphory-lation (48). Because the induction and assembly of the cyclinD-dependent kinases are controlled by extracellular mitogenicand integrin-dependent matrix signals (3), the ability of theseenzymes to modulate pRb function ultimately helps to placethe cell’s commitment to enter S phase under non-cell-auton-omous controls.
The stimulatory actions of the G1 cyclins are countered bythose of the CDK inhibitors (CKIs). There are two classes ofCKIs, the INK4 proteins (INK4a to -d), which act specificallyon cyclin D-dependent kinases, and the CIP/KIP family(p21CIP1, p27KIP1, and p57KIP2), which functions more broadlyto inhibit cyclin E-, A-, and B-dependent kinases as well (13,54). The levels of p27KIP1 in quiescent (G0) T cells and fibro-blasts are relatively high and greatly exceed that of the G1cyclins, but once these cells are stimulated to reenter the cycleand progress into late G1 phase, much of the p27KIP1 is de-graded (25, 39, 43). Nonetheless, residual levels of p27KIP1 andp21CIP1 in continuously proliferating cells are believed to setan inhibitory threshold which active cyclin-CDK complexes areforced to overcome (54).
The three D-type cyclins, D1, D2, and D3, share many struc-
*Corresponding author. Mailing address: Dana-Farber Cancer In-stitute, Harvard Medical School, 44 Binney St. M463, Boston, MA02115. Phone: (617) 632-6085. Fax: (617) 632-6069. E-mail:[email protected].
† Present address: Department of Pathology, Memorial Sloan-Ket-tering Cancer Center, New York, NY 10021.
353
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Nov
embe
r 20
21 b
y 39
.110
.32.
138.

tural features and biochemical properties but exhibit distinctpatterns of expression with respect to cell type and develop-mental stage (52). Skeletal myoblasts induced to differentiateunder low mitogen conditions exhibit a marked decrease incyclin D1 and a reciprocal rise in cyclin D3 expression, with areversal of this pattern occurring upon exposure to the anti-differentiation agents bFGF and TGF-b (47). Such observa-tions suggest different modes of regulation for cyclins D3 andD1 but do not resolve whether the two play distinct roles inmuscle differentiation. A comparative analysis of each D-typecyclin in a granulocyte differentiation system also suggestedfunctional differences, in that cells overexpressing cyclins D2and D3 but not D1 were unable to differentiate in granulocytecolony-stimulating factor (24). These observations could relateto the fact that cyclins D2 and D3, unlike D1, can interact withCDK2 (in addition to CDK4/6) to form active complexes (14).Although their differential regulation and unique biologicaleffects in these cell culture-based systems imply that there maybe distinct roles for each of the D-type cyclins in cellulargrowth and differentiation, such biological distinctions are notsupported by the finding that, in D1- or D2-deficient mice, thevast majority of tissues which express more than one D-typecyclin appear to grow and develop normally, suggesting func-tional redundancy (15, 55, 56).
Forced expression of D-type and E cyclins in several celltypes in transgenic mice has established an oncogenic orgrowth-promoting role for these G1 cyclins (5, 6, 27, 50, 60).Nonetheless, a direct functional comparison of each D-typecyclin and cyclin E in a well-defined pRb-dependent system invivo has yet to be conducted. We have relied upon the devel-oping mouse lens to study cyclin function, as this organ systemis (i) extremely simple and comprised of a single cell type, (ii)organized anatomically into an anterior layer of proliferatingepithelial cells and a posterior compartment of postmitoticdifferentiated lens fiber cells, (iii) a stable developmental chro-nometer enabling a traceable account of experimentally in-duced alterations in growth, differentiation, and apoptosis, and(iv) dispensable, thus tolerating the genetic manipulation ofpathways essential for viability. The cell cycle components op-erating in the lens have also been well defined by previousstudies. pRb plays an exclusive role in controlling the mitoticarrest of lens fiber cells (36), and loss of pRb alone is associ-ated with a dramatic increase of proliferation, accompanied byimpaired expression of late-stage differentiation markers inthese cells. In addition, the deregulation of lens cell prolifer-ation by the loss of pRb activates a p53-dependent apoptoticprogram that leads to the efficient elimination of abnormallygrowing cells (36). Furthermore, lenses rendered null for bothpRb-related proteins, p107 and p130, exhibit normal patternsof growth (unpublished observations).
G1 cyclins and CKIs exhibit contrasting patterns of expres-sion in the developing lens. In situ hybridization studies per-formed on embryonic lenses have revealed that endogenouscyclin D1 and D2 mRNAs are present in both the proliferativeepithelial cells and in the postmitotic equatorial lens fiber cells,whereas cyclin E transcripts are not readily detected in normallenses but are up-regulated in pRb-deficient lenses (17).Among the known CKIs, p57KIP2 appears to play an importantrole as evidenced by the observations that its transcripts andprotein levels are high in postmitotic lens fiber cells and itselimination by gene targeting results in inappropriate prolifer-ation and apoptosis of lens fiber cells (62). The null p57KIP2
phenotype is reminiscent of, but less dramatic than, the dereg-ulated growth and apoptosis observed in the pRb-deficient lens(36). While these findings suggest a functional link betweenp57KIP2 and pRb, the more attenuated consequences of
p57KIP2 deficiency point to additional regulators of pRb inac-tivation and S-phase entry in lens fiber cells in vivo. In thisstudy, we examined the consequences of forced expression ofeach G1 cyclin and its partner CDK in the mouse lens and haveattempted to ascertain whether these G1 cyclin-CDK activitiesare modulated by p57KIP2.
MATERIALS AND METHODS
Production of transgenic mice. To generate the various transgenic constructs,full-length cDNAs of the mouse cyclins D1 to D3, cyclin E, CDK2, and CDK4genes were inserted into the CPV-1 expression cassette (a kind gift from PaulOverbeek) between the aA-crystallin promoter and the simian virus 40 splice andpolyadenylation sequences (9, 21). By using standard transgenic mouse method-ology, gel-purified transgenic inserts were microinjected into the pronucleus offertilized eggs derived from B6/CBA F1 intercrosses. B6/CBA F1 mice (JacksonLaboratories) were also used to propagate the transgenic lines. Tail-derivedgenomic DNAs were assayed for the presence of the transgene by standardSouthern and/or DNA slot blot procedures (51) using specific probes directed tothe open reading frames of each transgene.
Protein isolation and Western immunoblot analyses. Lenses were homoge-nized in ice-cold 0.1 M Tris (pH 7.4), the water-soluble lens proteins wereseparated from water-insoluble lens membrane proteins by centrifugation at 4°Cfor 20 min (57), and the protein concentration was determined by the Bradfordassay (Bio-Rad). The membrane pellets were resuspended in a mixture of ice-cold 0.1 M Tris (pH 8.0), 7 M urea, and 5 mM EDTA (8). Proteins wereseparated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (12.5% polyacrylamide) and electroblotted to nitrocellulose. Blots wereblocked for 1 h at room temperature in phosphate-buffered saline (PBS) con-taining 5% milk and 0.1% Tween 20. The primary antibody incubations werecarried out for 1 h with affinity-purified polyclonal antisera against either cyclinD1 or E (Santa Cruz Biotechnology, Inc.) or MIP26 or a-crystallins (gifts fromJoe Horwitz and Sam Zigler) or with a rat monoclonal antibody against cyclinsD2 and D3 described previously (59), diluted in blocking solution, followed byeither anti-rabbit or anti-rat immunoglobulin G horseradish peroxidase-conju-gated antibody as the secondary antibody. Peroxidase activity was detected withenhanced chemiluminescence (Amersham).
Quantification of transgenic cyclin proteins. To generate recombinant cyclinproteins, cDNAs which included the coding region of cyclins D1, D3, or Esubcloned into the pGEX vector (Pharmacia Biotech, Inc.) were transfected intoEscherichia coli BL 21 DE3 cells (Novagene). After 4 h of IPTG (isopropyl-b-D-thiogalactopyranoside) induction, overnight cultures were sonicated, and thefusion proteins were purified by using glutathione-Sepharose 4B RediPack col-umns (Pharmacia Biotech, Inc.). The concentration of recombinant glutathioneS-transferase (GST)-cyclin fusion proteins was determined by quantification ofthe intensity of the bands by Coomassie-stained SDS-PAGE using a Gel Doc1000 apparatus (Bio-Rad) and employing recombinant cyclin D1 (gift from N.Pavletich) as the concentration standard. Fifty micrograms of total lens proteins,homogenized as described above, and a range of different amounts of recombi-nant cyclins were separated by SDS–12% PAGE and electroblotted to nitrocel-lulose. Blots were treated as described above. The intensity of the bands gener-ated by chemiluminescence reaction developed by using ECL 1 Plus (AmershamLife Science) exposed in a film was determined with a Gel Doc 1000 apparatus(Bio-Rad), and the formula that fit the standard curve best was determined byusing the CA-Cricket Graph III program. This formula was applied to estimatethe concentrations of the different transgenic cyclin proteins.
Histological analysis and indirect IF. Samples were fixed in 10% bufferedformalin and processed through paraffin embedding using standard procedures.Sections measuring 3 mm in thickness were cut parallel to the optic nerve, affixedto poly-L-lysine-coated slides, dewaxed in xylene, rehydrated through an ethanolseries, and stained with hematoxylin and eosin. For indirect immunofluorescence(IF), paraffin-embedded sections were rehydrated, rinsed in PBS, and blocked in3% bovine serum albumin in PBS at room temperature for 20 min. Affinity-purified polyclonal antisera against either cyclins D1, E, CDK4, or CDK2 (SantaCruz Biotechnology, Inc.); p57KIP2 (62); MIP26; a-, b-, or g-crystallins (giftsfrom Sam Zigler and Joe Horwitz); or a rat monoclonal antibody against cyclinD2 or D3 (59) were diluted in 3% BSA–PBS, incubated on sections overnight at4°C, and washed in PBS. The secondary antibody, fluorescein isothiocyanate-linked anti-rabbit or anti-rat immunoglobulin G antibody, was diluted, incubatedfor 1 h at room temperature, and washed in PBS. The amount of primary andsecondary antibody used was titrated in order to yield quantitative information.
BrdU incorporation and TUNEL assays. Pregnant mice were injected intra-peritoneally with bromodeoxyuridine (BrdU) dissolved in PBS at a dose of 100mg/g of body weight (35). After 2 h, the embryos were processed to detect in situBrdU incorporation into newly synthesized DNA as described previously (37).For detection of apoptosis in situ, the terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay was performed asdescribed previously (20) with minor modifications (37). To obtain a quantitativeestimate of the rates of S-phase entry and apoptosis in the lens fiber region,BrdU- or TUNEL-positive nuclei were scored in a minimum of 20 different lenssections derived from two independent embryos. Nontransgenic lens fiber cells
354 LAHOZ ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Nov
embe
r 20
21 b
y 39
.110
.32.
138.

are always postmitotic and do not exhibit apoptotic features. The statisticalanalysis of the data was carried out using the Microsoft SPSS program.
RESULTS
Formation of the ocular lens results from a series of devel-opmental events whose well-defined nature greatly enhancesthe use of this organ as a model system for cell cycle analysis invivo. Lens development begins on embryonic day 9.5 (E9.5)with the creation of the lens placode followed by its invagina-tion and eventual formation of a hollow lens vesicle by E11.5.Next, lens vesicle cells positioned on the posterior wall with-draw from the cell cycle and actively differentiate into elongat-ing primary lens fiber cells. By E14.5, the elongation process iscomplete, and the resultant structure is composed of an ante-rior layer of proliferating epithelial cells and a posterior com-partment of postmitotic differentiated lens fibers. The contin-ued growth of the lens ensues from the recruitment ofproliferating epithelial cells to the lateral equatorial regionwhere their growth is arrested and they differentiate into sec-ondary lens fiber cells. For purposes of this study, it is impor-tant to note that growth is restricted to the anterior epitheliallayer and that apoptosis, while occasionally observed in thisanterior region, does not take place in the lens fiber cell com-partment (45). Previous studies have shown that endogenouscyclin D1 and D2 mRNAs are expressed in both proliferativeepithelial cells and in differentiating equatorial lens fiber cells(17). On the protein level, endogenous cyclin D2 protein ex-pression is below the level of detection, while cyclin D1 immu-noreactivity is readily detected in the sensory retina, the pro-liferative anterior epithelial layer of the lens, and to a lesserdegree, in differentiating lens fiber cells of the equatorial re-gion. Most importantly, cyclin D1 is not detected in the central-most postmitotic, fully differentiated lens fiber region (see be-low). Cyclin E, although detectable by reverse transcription-PCR, is not detected via RNA in situ hybridization or indirectIF analyses (data not shown).
Production of cyclin transgenic mice and analysis of lensfiber cell transgene expression. Several independent trans-genic lines that ectopically express one of the three D-typecyclins or cyclin E in the postmitotic differentiated lens fibercell compartment were generated with the aid of the aA-crystallin promoter. This promoter has proven effective in di-recting the expression of many different transgenes to the lensfiber cell region exclusively, commencing on E12.5 and con-
tinuing thereafter (42). The level and regional pattern of ex-pression of each transgene in fully formed E16.5 lenses or inlens-derived extracts were examined and compared with non-transgenic, age-matched controls. The transgenic lines selectedfor further study were found to express detectable levels of thetransgene-encoded proteins by Western blot analysis (Fig. 1),and these levels were higher than those in their nontransgeniclittermate controls (data not shown). To determine the amountof ectopic cyclins present in the lenses of transgenic mice, theintensity of Western blot-positive bands of several lines perconstruct was compared to that of bands generated by knownamounts of recombinant cyclins (Fig. 1). This analysis allowedus to estimate that aA-cyclin D1 line 9 and aA-cyclin D1 line 23had amounts of cyclin D1 of 76 and 46 ng per 50 mg of totalprotein extract, respectively; aA-cyclin D3 line 25 and aA-cyclin D3 line 20 had 100 and 400 ng of cyclin D3 per 50 mg oftotal protein extract, respectively, while aA-cyclin E line 36 andaA-cyclin E line 40 had 27 and 44 ng of cyclin E per 50 mg oftotal protein extract, respectively. To compare the phenotypesof cyclin D1 and cyclin E transgenic mice, the studies belowwere performed using aA-cyclin D1 line 23 and aA-cyclin Eline 40, which had very similar levels of transgenic proteins (46ng and 45 ng per 50 mg of total protein extract, respectively).Since cyclins are also expressed endogenously in the anteriorepithelial layer (see above), IF was used to verify that abun-dant levels of each transgene-encoded product were properlydirected to the lens fiber cell compartment. The results ofrepresentative IF studies of the various cyclin transgene-ex-pressing lenses are presented in Fig. 2 (and see below). Thesestudies showed that cyclin D3 transgene-encoded protein wasevenly distributed throughout the nucleus and cytoplasm oflens fiber cells (Fig. 2E), whereas transgene-encoded cyclinsD1, D2, and E exhibited more complex subcellular distributionpatterns. Anti-D1 signals were detected in nuclei of the equa-torial region with less abundant nuclear staining in the centrallens fiber cells (Fig. 2B and C). In contrast, the interveningregion showed predominantly cytoplasmic staining with only afew nuclei staining positive (Fig. 2). An identical staining pat-tern for the transgene-encoded cyclin D2 protein was observed(data not shown). Cyclin E was detected in nuclei within theequatorial region and in the cytoplasm of the remainder of thelens fiber cell compartment (Fig. 2I). Since all of the cyclintransgene constructs were driven by identical promoter and
FIG. 1. Western blot analysis of recombinant GST-cyclin fusion proteins and total lens lysates derived from aA-cyclin D1 and D3 and aA-cyclin E transgenic miceas assayed with anti-cyclin D1 (lanes 1 to 7), anti-cyclin D3 (lanes 8 to 13) and anti-cyclin E (lanes 14 to 20) antibodies. The bands indicated with arrowheads correspondeither to the previously reported sizes for cyclin D1 (35 kDa), cyclin D3 (33 kDa), or cyclin E (50 kDa) proteins or to the GST-cyclin fusion proteins. Lanes 1 to 4,decreasing amounts of GST-cyclin D1 fusion protein used as standard curve (200 ng [lane 1], 100 ng [lane 2], 50 ng [lane 3], and 25 ng [lane 4]). Lanes 5 to 7, transgeniccyclin D1 protein from lenses of aA-cyclin D1 line 23 (lane 5), a transgenic mouse hemizygous for aA-cyclin D1 line 23 and aA-cdk4 line 14 transgenes (lane 6), andaA-cyclin D1 line 9 (lane 7). Lanes 8 to 13, decreasing amounts of GST-cyclin D3 fusion protein used as standard curve (100 ng [lane 8], 50 ng [lane 9], 25 ng [lane10], and 10 ng [lane 11]). Lanes 12 and 13, transgenic cyclin D3 protein from lenses of aA-cyclin D3 line 25 (lane 12) and aA-cyclin D3 line 20 (lane 13). Lanes 14 to17, decreasing amounts of GST-cyclin E fusion protein used as standard curve (100 ng [lane 14], 50 ng [lane 15], 25 ng [lane 16], and 10 ng [lane 17]). Lanes 18 to 20,transgenic cyclin E protein from lenses of aA-cyclin E line 36 (lane 18), aA-cyclin E line 40 (lane 19), and a transgenic mouse hemizygous for aA-cyclin E line 40 andaA-cdk2 line 31 transgenes (lane 20).
VOL. 19, 1999 CONTROL OF G1 EXIT IN VIVO 355
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Nov
embe
r 20
21 b
y 39
.110
.32.
138.

enhancer elements, these complex and distinct expression pat-terns likely reflect differences in posttranscriptional regulation.
Analysis of forced expression of D-type cyclins and cyclin Ein the lens and its effect on cell morphology, S-phase entry,and apoptosis. Unlike nontransgenic lenses in which the lensfiber cells are arrayed along a well-defined axis, E16.5 lensesfrom all three aA-D3 transgenic lines exhibited a severelydisorganized pattern. These morphological abnormalities (ap-parent even in the IF studies [compare Fig. 2E with the moreorganized lens shown in panel G]) were consistent with alteredpatterns of proliferation and apoptosis (see below). In con-trast, the other aA-cyclin transgenic lenses (D1, D2, and E)presented with lens fiber cell compartments that were morpho-logically indistinguishable from nontransgenic controls (datanot shown). These findings prompted a thorough analysis ofpatterns of growth, apoptosis, and differentiation markers ineach of the transgenic lines.
For analysis of differentiation status, the temporal and spa-tial expression patterns of the family of lens crystallins and themembrane intrinsic protein 26 (MIP26) was examined byWestern blot analysis and IF methods. These specific markerswere selected since normal lens fiber cell differentiation ishighly dependent upon the proper expression of these pro-
teins. In this regard, a-crystallins are considered markers ofearly-stage differentiation, while g-crystallins and MIP26 areclassical late-stage markers (8, 32, 45). Despite the D3-inducedmorphological defects, analysis of the ratio of early- to late-stage differentiation markers as measured by Western blotting(Fig. 3) or of their pattern of distribution by IF (data notshown) failed to uncover significant differences in the levelsand regional distribution of these markers between age-matched wild-type and transgenic lenses.
Since overexpression of G1 cyclins has been correlated withinappropriate S-phase entry, we next examined whether en-forced expression of a D-type cyclin or cyclin E is sufficient topromote S-phase entry in normally postmitotic lens fiber cells.Lenses from aA-D3 mice showed ectopic nuclear-associatedBrdU staining in the lens fiber cell compartment (Fig. 2F andTable 1), indicating that forced expression of cyclin D3 alonewas sufficient to drive some lens fiber cells into S phase. Thisexperimental result was obtained in three separate studies per-formed with three independent transgenic lines. In contrast, anormal pattern of cell proliferation (restricted to the anteriorepithelial layer) was observed in all aA-D1 (Fig. 2D), aA-D2(not shown), and aA-E (Fig. 2J) transgenic lenses.
To assess whether lens fiber cell expression of the G1 cyclins
FIG. 2. In situ immunofluorescence analysis of transgene-derived protein expression in the embryonic (E16.5) lenses of the various aA-cyclin and aA-cdk transgenicmice and their pattern of cellular proliferation as assayed by an indirect immunoperoxidase method for the detection of BrdU incorporation into newly synthesizedDNA in situ. (A) Nontransgenic lens stained with anti-cyclin D1 antibody. Cyclin D1 is the only endogenous cyclin detected with the available antisera. (B) aA-cyclinD1 line 23 stained with anti-cyclin D1 antibody. (C) Higher magnification of the region framed in the upper left corner of panel B. A white arrow points to a positivestained nucleus, and a black arrow indicates a negative stained nucleus. (D) BrdU incorporation in aA-cyclin D1 line 23. Arrows point to positive nuclear-associatedstaining indicating physiological S-phase entry in the anterior epithelial layer. (E) aA-cyclin D3 line 25 stained with anti-cyclin D3 antibody. (F) BrdU incorporationpattern in aA-cyclin D3 line 25. Single arrows point to normal S-phase activity in the anterior epithelial layer, while double-headed arrows point to a subset of ectopiccycling in the lens fiber compartment. (G) aA-cdk4 line 14 stained with anti-CDK4 antibody. (H) BrdU incorporation pattern in aA-cdk4 line 14. (I) aA-cyclin E line40 stained with anti-cyclin E antibody. (J) BrdU incorporation pattern in aA-cyclin E line 40. (K) aA-cdk2 line 31 stained with anti-CDK2 antibody. (L) BrdUincorporation pattern in aA-cdk2 line 31. Abbreviations: e, anterior epithelial layer; f, lens fiber cells; r, retina.
356 LAHOZ ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Nov
embe
r 20
21 b
y 39
.110
.32.
138.

also leads to an apoptotic response, E16.5 lenses from eachtransgenic line were analyzed by TUNEL. Apoptotic nucleiwere not detected in the lens fiber region of nontransgeniclenses or in aA-D1, aA-D2, and aA-E transgenic lenses (Fig.
3A to D). In contrast, several apoptotic nuclei were observedin aA-D3 lens fiber cells (Fig. 3E and Table 1). D3-inducedproliferation and apoptosis, albeit modest, are reminiscent ofthose obtained in previous studies using the pRb-deficient lensthat showed that inappropriate lens fiber cell proliferationactivates an apoptotic response (reference 36 and see Fig. 7I).
Forced expression of CDK4 or CDK2 does not promoteS-phase entry. Since the G1 cyclins form active holenzymes byassembling with CDK subunits, it was important to addresswhether abundant levels of these catalytic units alone, as op-posed to the regulatory cyclin subunits, could promote S-phaseentry. To this end, high-levels of CDK4 or CDK2 were directedto the lens fiber cell compartment with the aid of the aA-crystallin promoter. IF analyses revealed that transgenic CDK4was evenly distributed in the cytoplasm and nuclei of all lensfiber cells (Fig. 2G). In contrast, transgene-encoded CDK2localized primarily to the cytoplasm with only a subset of equa-torial and posterior cell nuclei staining positive (Fig. 2K). Inthe nontransgenic lens, endogenous CDK4 exhibited a diffusepattern of subcellular distribution similar to that of the trans-gene-encoded protein, albeit at much lower levels, whereasendogenous CDK2 was localized to the cytoplasm of the lensfiber cells only (data not shown). Despite a clear increase in thelevel of transgene-encoded CDK4 and CDK2 above endoge-nous levels, these transgenic lenses failed to show BrdU incor-poration in the lens fiber cells, indicating that a high level ofCDK4 or CDK2 expression is not sufficient to promote S-phaseentry (Fig. 2H and L). Unexpectedly, despite a lack of ectopicproliferation, CDK4 transgenic lines exhibited lens fiber cellapoptosis (Fig. 3F), indicating that inappropriate proliferationis not a requirement for activation of an apoptotic response.This response appears to be specific to CDK4, since apoptosiswas not detected in the aA-CDK2 transgenic lines even undercircumstances where CDK2 was directed to the nucleus (seebelow).
CDK-induced nuclear translocation of cyclins D1, D2, and Epromotes S-phase entry with D1/CDK4 and D2/CDK4 but notE/CDK2. The failure of transgenic cyclins D1, D2, and E toinduce S-phase entry could relate, in part, to low nuclear cyclinlevels that are insufficient to counter suppression by CDKinhibitors or to low partner CDK levels (see below). To ad-dress the latter, each cyclin was coexpressed along with itspartner CDK through the production of double transgenics bycrossing mice carrying single transgenes. Western blot analysisof lenses from transgenic mice hemizygous for aA-cyclin D1line 23 and aA-cdk4 line 14 transgenes or aA-cyclin E line 40and aA-cdk2 line 31 transgenes showed no significant differ-
FIG. 3. Analysis of apoptosis and stage-specific differentiation markers in theembryonic lenses of G1 cyclin transgenic and control mice. Lettered panels,results of TUNEL assay of E16.5 eye sections from nontransgenic mice (A) andfrom aA-cyclin D1, D2, E, or D3 and aA-cdk4 transgenic lines (B to F, respec-tively). e, anterior epithelial layer; f, lens fiber cells; r, retina. Arrows indicateTUNEL-positive nuclear staining consistent with apoptotic internucleosomalcleavage. Bottom panels, Western blot analyses of early (a-crystallin)- and late(MIP26)-stage differentiation markers. Each lane contains soluble lysate fraction(a-crystallin) or membrane fraction derived from a lens equivalent which permitsan internal comparison between a-crystallin and MIP26 levels as describedpreviously. The upper blot was probed with a rabbit polyclonal anti-a-crystallinantibody and the lower blot was probed with anti-MIP26 antibody. Lanes: 1,nontransgenic lens; 2, aA-cyclin D1 line 23; 3, aA-cyclin D2 line 2; 4, 5, and 6,aA-cyclin D3 lines 20, 25, and 42, respectively; 7 and 8, aA-cyclin E lines 40 and36, respectively.
TABLE 1. Average number of positive nuclei per lens 6 standarddeviation in E16.5 transgenic embryos
Transgene No. of BrdU-positivenuclei
No. of TUNEL-positivenuclei
NTG 0 0D1 0 0D2 0 0D3 10.4 6 1.6 5.7 6 0.9E 0 0CDK4 0 3.0 6 0.5CDK2 0 0D1/CDK4 5.2 6 0.7 4.8 6 0.7E/CDK2 1.6 6 0.6 0E/CDK4 6.1 6 1.7 3.0 6 0.7D1/E/CDK2 3.4 6 0.7 0E/CDK4/CDK2 12.0 6 1.6 2.5 6 0.6D1/E/CDK4/CDK2 23.0 6 3.2 6.3 6 1.5
VOL. 19, 1999 CONTROL OF G1 EXIT IN VIVO 357
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Nov
embe
r 20
21 b
y 39
.110
.32.
138.
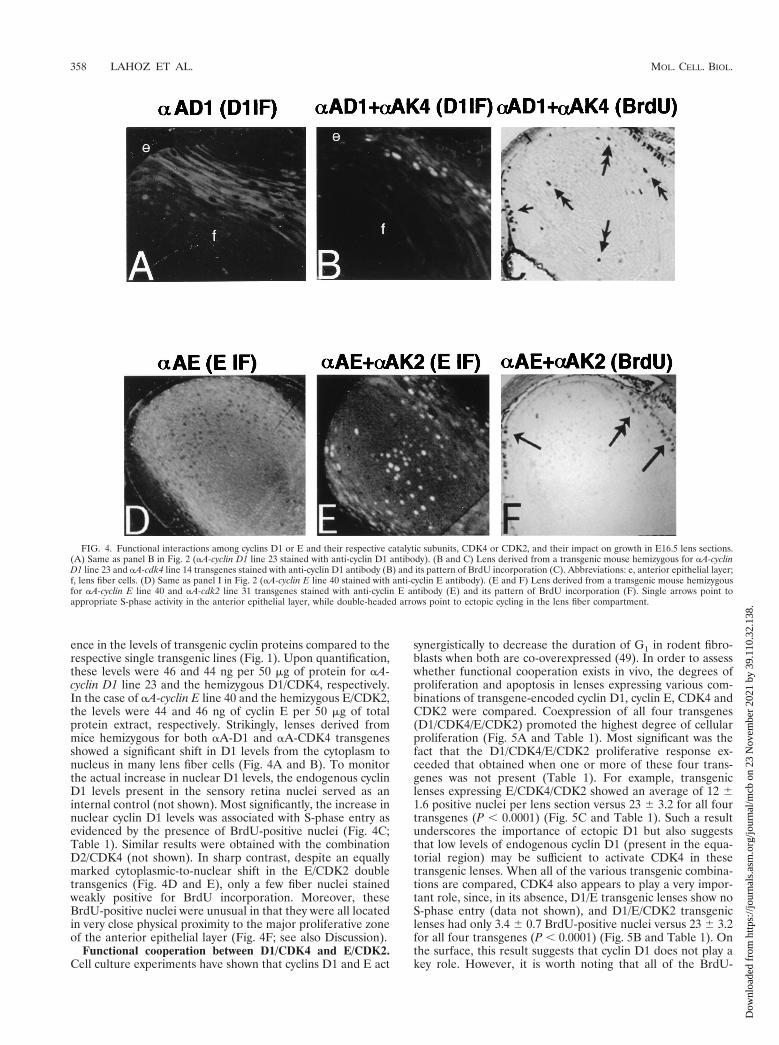
ence in the levels of transgenic cyclin proteins compared to therespective single transgenic lines (Fig. 1). Upon quantification,these levels were 46 and 44 ng per 50 mg of protein for aA-cyclin D1 line 23 and the hemizygous D1/CDK4, respectively.In the case of aA-cyclin E line 40 and the hemizygous E/CDK2,the levels were 44 and 46 ng of cyclin E per 50 mg of totalprotein extract, respectively. Strikingly, lenses derived frommice hemizygous for both aA-D1 and aA-CDK4 transgenesshowed a significant shift in D1 levels from the cytoplasm tonucleus in many lens fiber cells (Fig. 4A and B). To monitorthe actual increase in nuclear D1 levels, the endogenous cyclinD1 levels present in the sensory retina nuclei served as aninternal control (not shown). Most significantly, the increase innuclear cyclin D1 levels was associated with S-phase entry asevidenced by the presence of BrdU-positive nuclei (Fig. 4C;Table 1). Similar results were obtained with the combinationD2/CDK4 (not shown). In sharp contrast, despite an equallymarked cytoplasmic-to-nuclear shift in the E/CDK2 doubletransgenics (Fig. 4D and E), only a few fiber nuclei stainedweakly positive for BrdU incorporation. Moreover, theseBrdU-positive nuclei were unusual in that they were all locatedin very close physical proximity to the major proliferative zoneof the anterior epithelial layer (Fig. 4F; see also Discussion).
Functional cooperation between D1/CDK4 and E/CDK2.Cell culture experiments have shown that cyclins D1 and E act
synergistically to decrease the duration of G1 in rodent fibro-blasts when both are co-overexpressed (49). In order to assesswhether functional cooperation exists in vivo, the degrees ofproliferation and apoptosis in lenses expressing various com-binations of transgene-encoded cyclin D1, cyclin E, CDK4 andCDK2 were compared. Coexpression of all four transgenes(D1/CDK4/E/CDK2) promoted the highest degree of cellularproliferation (Fig. 5A and Table 1). Most significant was thefact that the D1/CDK4/E/CDK2 proliferative response ex-ceeded that obtained when one or more of these four trans-genes was not present (Table 1). For example, transgeniclenses expressing E/CDK4/CDK2 showed an average of 12 61.6 positive nuclei per lens section versus 23 6 3.2 for all fourtransgenes (P , 0.0001) (Fig. 5C and Table 1). Such a resultunderscores the importance of ectopic D1 but also suggeststhat low levels of endogenous cyclin D1 (present in the equa-torial region) may be sufficient to activate CDK4 in thesetransgenic lenses. When all of the various transgenic combina-tions are compared, CDK4 also appears to play a very impor-tant role, since, in its absence, D1/E transgenic lenses show noS-phase entry (data not shown), and D1/E/CDK2 transgeniclenses had only 3.4 6 0.7 BrdU-positive nuclei versus 23 6 3.2for all four transgenes (P , 0.0001) (Fig. 5B and Table 1). Onthe surface, this result suggests that cyclin D1 does not play akey role. However, it is worth noting that all of the BrdU-
FIG. 4. Functional interactions among cyclins D1 or E and their respective catalytic subunits, CDK4 or CDK2, and their impact on growth in E16.5 lens sections.(A) Same as panel B in Fig. 2 (aA-cyclin D1 line 23 stained with anti-cyclin D1 antibody). (B and C) Lens derived from a transgenic mouse hemizygous for aA-cyclinD1 line 23 and aA-cdk4 line 14 transgenes stained with anti-cyclin D1 antibody (B) and its pattern of BrdU incorporation (C). Abbreviations: e, anterior epithelial layer;f, lens fiber cells. (D) Same as panel I in Fig. 2 (aA-cyclin E line 40 stained with anti-cyclin E antibody). (E and F) Lens derived from a transgenic mouse hemizygousfor aA-cyclin E line 40 and aA-cdk2 line 31 transgenes stained with anti-cyclin E antibody (E) and its pattern of BrdU incorporation (F). Single arrows point toappropriate S-phase activity in the anterior epithelial layer, while double-headed arrows point to ectopic cycling in the lens fiber compartment.
358 LAHOZ ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Nov
embe
r 20
21 b
y 39
.110
.32.
138.
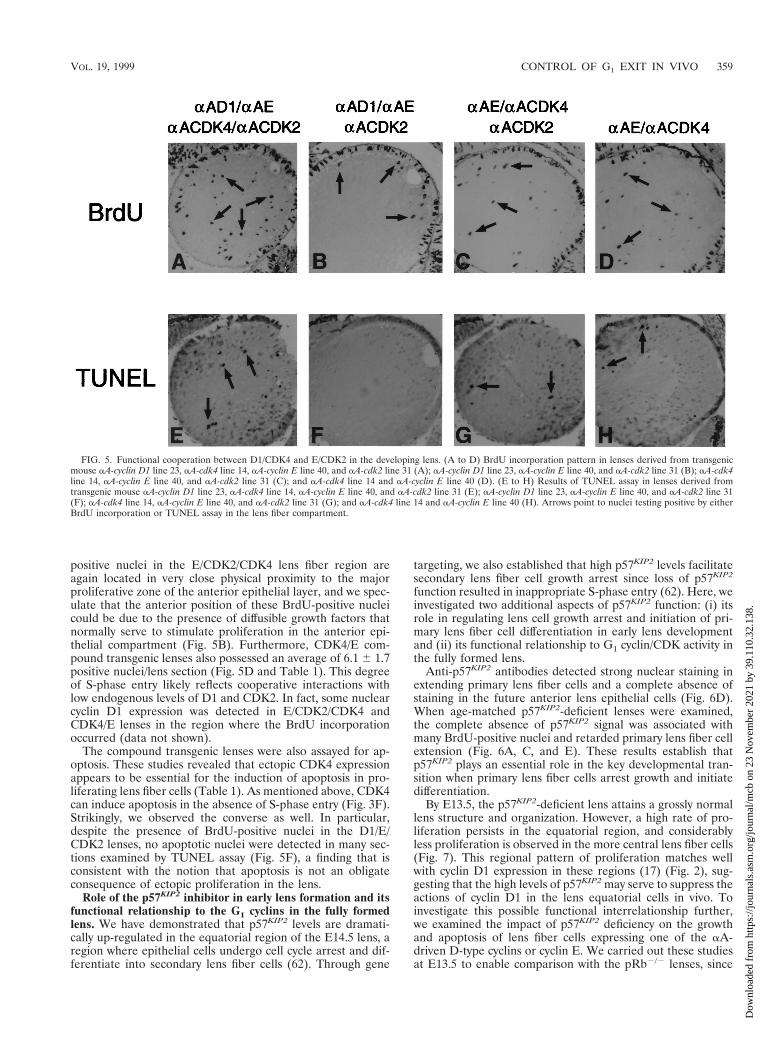
positive nuclei in the E/CDK2/CDK4 lens fiber region areagain located in very close physical proximity to the majorproliferative zone of the anterior epithelial layer, and we spec-ulate that the anterior position of these BrdU-positive nucleicould be due to the presence of diffusible growth factors thatnormally serve to stimulate proliferation in the anterior epi-thelial compartment (Fig. 5B). Furthermore, CDK4/E com-pound transgenic lenses also possessed an average of 6.1 6 1.7positive nuclei/lens section (Fig. 5D and Table 1). This degreeof S-phase entry likely reflects cooperative interactions withlow endogenous levels of D1 and CDK2. In fact, some nuclearcyclin D1 expression was detected in E/CDK2/CDK4 andCDK4/E lenses in the region where the BrdU incorporationoccurred (data not shown).
The compound transgenic lenses were also assayed for ap-optosis. These studies revealed that ectopic CDK4 expressionappears to be essential for the induction of apoptosis in pro-liferating lens fiber cells (Table 1). As mentioned above, CDK4can induce apoptosis in the absence of S-phase entry (Fig. 3F).Strikingly, we observed the converse as well. In particular,despite the presence of BrdU-positive nuclei in the D1/E/CDK2 lenses, no apoptotic nuclei were detected in many sec-tions examined by TUNEL assay (Fig. 5F), a finding that isconsistent with the notion that apoptosis is not an obligateconsequence of ectopic proliferation in the lens.
Role of the p57KIP2 inhibitor in early lens formation and itsfunctional relationship to the G1 cyclins in the fully formedlens. We have demonstrated that p57KIP2 levels are dramati-cally up-regulated in the equatorial region of the E14.5 lens, aregion where epithelial cells undergo cell cycle arrest and dif-ferentiate into secondary lens fiber cells (62). Through gene
targeting, we also established that high p57KIP2 levels facilitatesecondary lens fiber cell growth arrest since loss of p57KIP2
function resulted in inappropriate S-phase entry (62). Here, weinvestigated two additional aspects of p57KIP2 function: (i) itsrole in regulating lens cell growth arrest and initiation of pri-mary lens fiber cell differentiation in early lens developmentand (ii) its functional relationship to G1 cyclin/CDK activity inthe fully formed lens.
Anti-p57KIP2 antibodies detected strong nuclear staining inextending primary lens fiber cells and a complete absence ofstaining in the future anterior lens epithelial cells (Fig. 6D).When age-matched p57KIP2-deficient lenses were examined,the complete absence of p57KIP2 signal was associated withmany BrdU-positive nuclei and retarded primary lens fiber cellextension (Fig. 6A, C, and E). These results establish thatp57KIP2 plays an essential role in the key developmental tran-sition when primary lens fiber cells arrest growth and initiatedifferentiation.
By E13.5, the p57KIP2-deficient lens attains a grossly normallens structure and organization. However, a high rate of pro-liferation persists in the equatorial region, and considerablyless proliferation is observed in the more central lens fiber cells(Fig. 7). This regional pattern of proliferation matches wellwith cyclin D1 expression in these regions (17) (Fig. 2), sug-gesting that the high levels of p57KIP2 may serve to suppress theactions of cyclin D1 in the lens equatorial cells in vivo. Toinvestigate this possible functional interrelationship further,we examined the impact of p57KIP2 deficiency on the growthand apoptosis of lens fiber cells expressing one of the aA-driven D-type cyclins or cyclin E. We carried out these studiesat E13.5 to enable comparison with the pRb2/2 lenses, since
FIG. 5. Functional cooperation between D1/CDK4 and E/CDK2 in the developing lens. (A to D) BrdU incorporation pattern in lenses derived from transgenicmouse aA-cyclin D1 line 23, aA-cdk4 line 14, aA-cyclin E line 40, and aA-cdk2 line 31 (A); aA-cyclin D1 line 23, aA-cyclin E line 40, and aA-cdk2 line 31 (B); aA-cdk4line 14, aA-cyclin E line 40, and aA-cdk2 line 31 (C); and aA-cdk4 line 14 and aA-cyclin E line 40 (D). (E to H) Results of TUNEL assay in lenses derived fromtransgenic mouse aA-cyclin D1 line 23, aA-cdk4 line 14, aA-cyclin E line 40, and aA-cdk2 line 31 (E); aA-cyclin D1 line 23, aA-cyclin E line 40, and aA-cdk2 line 31(F); aA-cdk4 line 14, aA-cyclin E line 40, and aA-cdk2 line 31 (G); and aA-cdk4 line 14 and aA-cyclin E line 40 (H). Arrows point to nuclei testing positive by eitherBrdU incorporation or TUNEL assay in the lens fiber compartment.
VOL. 19, 1999 CONTROL OF G1 EXIT IN VIVO 359
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Nov
embe
r 20
21 b
y 39
.110
.32.
138.
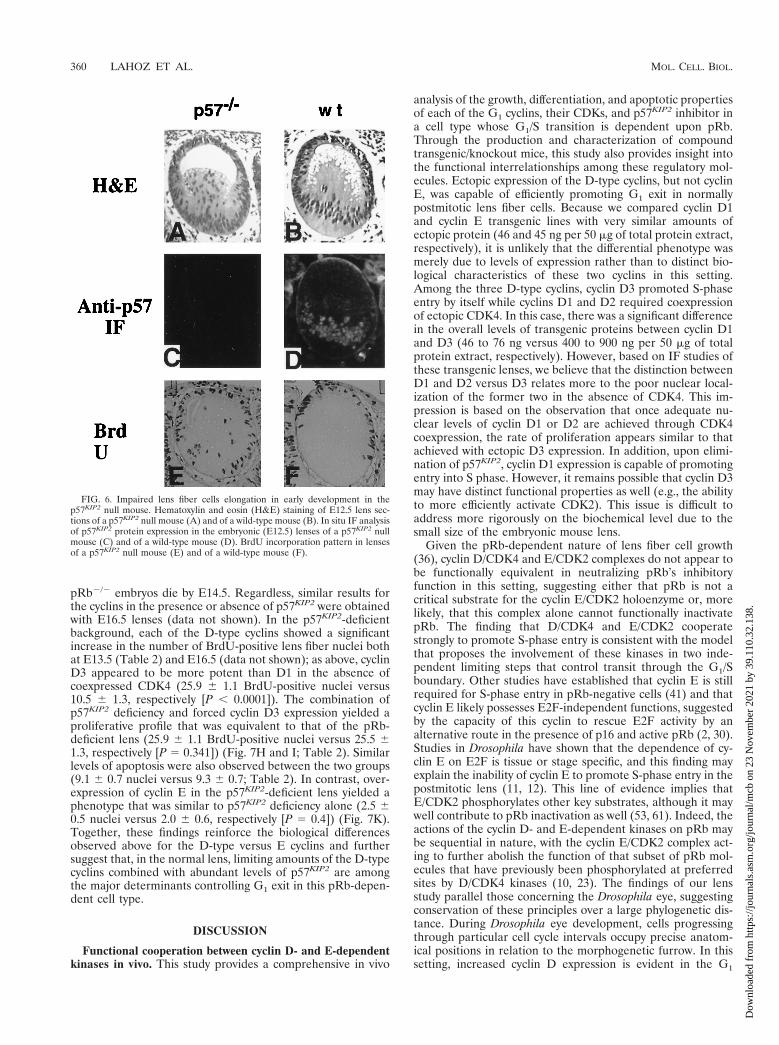
pRb2/2 embryos die by E14.5. Regardless, similar results forthe cyclins in the presence or absence of p57KIP2 were obtainedwith E16.5 lenses (data not shown). In the p57KIP2-deficientbackground, each of the D-type cyclins showed a significantincrease in the number of BrdU-positive lens fiber nuclei bothat E13.5 (Table 2) and E16.5 (data not shown); as above, cyclinD3 appeared to be more potent than D1 in the absence ofcoexpressed CDK4 (25.9 6 1.1 BrdU-positive nuclei versus10.5 6 1.3, respectively [P , 0.0001]). The combination ofp57KIP2 deficiency and forced cyclin D3 expression yielded aproliferative profile that was equivalent to that of the pRb-deficient lens (25.9 6 1.1 BrdU-positive nuclei versus 25.5 61.3, respectively [P 5 0.341]) (Fig. 7H and I; Table 2). Similarlevels of apoptosis were also observed between the two groups(9.1 6 0.7 nuclei versus 9.3 6 0.7; Table 2). In contrast, over-expression of cyclin E in the p57KIP2-deficient lens yielded aphenotype that was similar to p57KIP2 deficiency alone (2.5 60.5 nuclei versus 2.0 6 0.6, respectively [P 5 0.4]) (Fig. 7K).Together, these findings reinforce the biological differencesobserved above for the D-type versus E cyclins and furthersuggest that, in the normal lens, limiting amounts of the D-typecyclins combined with abundant levels of p57KIP2 are amongthe major determinants controlling G1 exit in this pRb-depen-dent cell type.
DISCUSSION
Functional cooperation between cyclin D- and E-dependentkinases in vivo. This study provides a comprehensive in vivo
analysis of the growth, differentiation, and apoptotic propertiesof each of the G1 cyclins, their CDKs, and p57KIP2 inhibitor ina cell type whose G1/S transition is dependent upon pRb.Through the production and characterization of compoundtransgenic/knockout mice, this study also provides insight intothe functional interrelationships among these regulatory mol-ecules. Ectopic expression of the D-type cyclins, but not cyclinE, was capable of efficiently promoting G1 exit in normallypostmitotic lens fiber cells. Because we compared cyclin D1and cyclin E transgenic lines with very similar amounts ofectopic protein (46 and 45 ng per 50 mg of total protein extract,respectively), it is unlikely that the differential phenotype wasmerely due to levels of expression rather than to distinct bio-logical characteristics of these two cyclins in this setting.Among the three D-type cyclins, cyclin D3 promoted S-phaseentry by itself while cyclins D1 and D2 required coexpressionof ectopic CDK4. In this case, there was a significant differencein the overall levels of transgenic proteins between cyclin D1and D3 (46 to 76 ng versus 400 to 900 ng per 50 mg of totalprotein extract, respectively). However, based on IF studies ofthese transgenic lenses, we believe that the distinction betweenD1 and D2 versus D3 relates more to the poor nuclear local-ization of the former two in the absence of CDK4. This im-pression is based on the observation that once adequate nu-clear levels of cyclin D1 or D2 are achieved through CDK4coexpression, the rate of proliferation appears similar to thatachieved with ectopic D3 expression. In addition, upon elimi-nation of p57KIP2, cyclin D1 expression is capable of promotingentry into S phase. However, it remains possible that cyclin D3may have distinct functional properties as well (e.g., the abilityto more efficiently activate CDK2). This issue is difficult toaddress more rigorously on the biochemical level due to thesmall size of the embryonic mouse lens.
Given the pRb-dependent nature of lens fiber cell growth(36), cyclin D/CDK4 and E/CDK2 complexes do not appear tobe functionally equivalent in neutralizing pRb’s inhibitoryfunction in this setting, suggesting either that pRb is not acritical substrate for the cyclin E/CDK2 holoenzyme or, morelikely, that this complex alone cannot functionally inactivatepRb. The finding that D/CDK4 and E/CDK2 cooperatestrongly to promote S-phase entry is consistent with the modelthat proposes the involvement of these kinases in two inde-pendent limiting steps that control transit through the G1/Sboundary. Other studies have established that cyclin E is stillrequired for S-phase entry in pRb-negative cells (41) and thatcyclin E likely possesses E2F-independent functions, suggestedby the capacity of this cyclin to rescue E2F activity by analternative route in the presence of p16 and active pRb (2, 30).Studies in Drosophila have shown that the dependence of cy-clin E on E2F is tissue or stage specific, and this finding mayexplain the inability of cyclin E to promote S-phase entry in thepostmitotic lens (11, 12). This line of evidence implies thatE/CDK2 phosphorylates other key substrates, although it maywell contribute to pRb inactivation as well (53, 61). Indeed, theactions of the cyclin D- and E-dependent kinases on pRb maybe sequential in nature, with the cyclin E/CDK2 complex act-ing to further abolish the function of that subset of pRb mol-ecules that have previously been phosphorylated at preferredsites by D/CDK4 kinases (10, 23). The findings of our lensstudy parallel those concerning the Drosophila eye, suggestingconservation of these principles over a large phylogenetic dis-tance. During Drosophila eye development, cells progressingthrough particular cell cycle intervals occupy precise anatom-ical positions in relation to the morphogenetic furrow. In thissetting, increased cyclin D expression is evident in the G1
FIG. 6. Impaired lens fiber cells elongation in early development in thep57KIP2 null mouse. Hematoxylin and eosin (H&E) staining of E12.5 lens sec-tions of a p57KIP2 null mouse (A) and of a wild-type mouse (B). In situ IF analysisof p57KIP2 protein expression in the embryonic (E12.5) lenses of a p57KIP2 nullmouse (C) and of a wild-type mouse (D). BrdU incorporation pattern in lensesof a p57KIP2 null mouse (E) and of a wild-type mouse (F).
360 LAHOZ ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Nov
embe
r 20
21 b
y 39
.110
.32.
138.

compartment, while cyclin E takes place as cells enter theS-phase zone (16).
Subcellular distribution of the G1 cyclins in relation to CDKcoexpression. An unanticipated finding in the lens studies wasthe dramatic shift in subcellular distribution for D1, D2, or Eupon coexpression of its partner CDK. This observed shiftindicates that alterations in the levels of the G1 CDK subunitscan influence the subcellular localization of their cyclin part-ners in vivo. The mechanisms underlying these effects remainunclear, but coexpression of cyclins and CDKs is not likely tobe sufficient for their nuclear localization as evidenced by thefinding that cyclin D1 was excluded from a subset of lens fibernuclei despite high levels of ectopic CDK4 expression in thesecells in the D1/CDK4 lenses. Previous reports have describedcell-cycle-dependent changes in the subcellular localization ofcyclin D1 in proliferating fibroblasts, with D1 progressivelyaccumulating in the nuclei during G1 phase and excluded dur-ing S phase (4, 28), even though high levels of enzyme activitypersist (31, 34). Cyclin E is also excluded from the nucleus inlate S phase unless it is overexpressed (41), and this subcellularcompartmentalization of the E/CDK2 complex appears to bean important determinant of its kinase activity (7). Hence, thenuclear targeting of cyclins D1 and D2 or cyclin E must involveregulatory mechanisms that extend beyond abundant CDK4 orCDK2 levels.
CDK4-induced apoptosis does not require S-phase progres-sion. The results presented here show that overexpression ofCDK4 can induce apoptosis in the absence of proliferation.Moreover, despite the presence of BrdU-positive nuclei in theD1/E/CDK2 lenses, no apoptotic nuclei were detected in manylenses examined. Together, these two results indicate that ec-topic CDK4 plays a critical proapoptotic role. The activation ofapoptosis under G0/G1 block is not without precedent and hasbeen reported following Wilms tumor suppressor gene (WT1)overexpression in myeloblastic leukemia M1 cells, calciumionophore treatment in TSU-pr1 androgen-independent pros-tatic cancer cells, and induction of differentiation in HL-60cells (18, 38, 58). To place into context the activation of apo-ptosis by ectopic CDK4, we need to consider the status ofendogenous CDKs in the embryonic lens; among these kinases,CDK5 might be highly relevant. First, CDK5 and its activatingsubunit p35 are highly expressed in lens fiber cells of rats atE16.5, and a shift to nuclear staining is observed immediatelyprior to nuclear degradation (19). Moreover, immunoprecipi-tates of CDK5 from lens fibers showed kinase activity in vitrowith histone H1 as a substrate, suggesting a role of p35/CDK5in differentiating lens fiber cells in addition to its reportedfunction in neurons (19). Second, both CDK4 and p35/CDK5have each been associated with apoptosis previously; a domi-nant negative of CDK4 promotes survival of nerve growth
FIG. 7. Functional interactions between D-type cyclins and p57KIP2. Hematoxylin and eosin (H&E) staining of E13.5 lens sections of a p57KIP2 null mouse (A), aaA-cyclin D3 line 25/p57KIP2 null mouse (D), and a pRb null mouse (G). BrdU incorporation pattern in lenses of a p57KIP2 null mouse (B), a aA-cyclin D3 line25/p57KIP2 null mouse (E), a pRb null mouse (H), a aA-cyclin D1 line 23/p57KIP2 null mouse (J), and a aA-cyclin E line 40/p57KIP2 null mouse (K). TUNEL (TUN.)assay in lenses of a p57KIP2 null mouse (C), a aA-cyclin D3 line 25/p57KIP2 null mouse (F), and a pRb null mouse (I). Arrows point to nuclei testing positive by eitherBrdU incorporation or TUNEL assay in the lens fiber compartment.
VOL. 19, 1999 CONTROL OF G1 EXIT IN VIVO 361
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Nov
embe
r 20
21 b
y 39
.110
.32.
138.

factor-deprived sympathetic neurons (44), and double labelingof p35/CDK5 and confocal microscopy detected this kinasecomplex in both adult and embryonic dying cells (1). In the lenssystem, one is tempted to speculate that the ectopic expressionof CDK4 titrates away endogenous cyclin D1 which normallybinds to and inhibits the p35/CDK5 complex, resulting in anindirect activation of CDK5 triggering apoptosis with no effectin proliferation.
Functional relationship between the G1 cyclins and p57KIP2
in vivo. The loss of pRb function results in a high rate ofectopic mitotic activity and apoptosis in the lens fiber compart-ment of the developing mouse (36). We have reported that lossof p57KIP2 also leads to lens cell fiber proliferation and apo-ptosis, albeit to a degree that is far less pronounced than thatobserved in the pRb null lens (62). The more attenuated phe-notype of the p57KIP2 null lens is very similar to that of trans-genic lenses overexpressing cyclin D described here. Nonethe-less, in the p57KIP2 null lens, a very high rate of proliferation(equivalent to the pRb null condition) is observed in the equa-torial region where endogenous cyclin D1 and CDK4 normallypersist (62). Here, D-type cyclin overexpression in a p57KIP2
null background resulted in a degree of abnormal proliferationand apoptosis that was equivalent to that brought about by theloss of pRb. In contrast, overexpression of cyclin E in thissetting had no effect on either proliferation or apoptosis. To-gether, these observations indicate that a multilevel regulatorycircuit exists to govern the pRb-regulated G1/S transition inlens fiber cells in vivo and that a balance between proliferationand cell cycle exit is achieved primarily through the opposingforces of activating D-type cyclin kinases and inhibitoryp57KIP2.
Our findings suggest that in the anterior epithelial layer ofthe lens, high-cyclin D-CDK4 and only sporadic p57KIP2 ex-pression account for the normally high rate of proliferation ofundifferentiated epithelial cells. As cells reach the equatorialregion of the lens fiber compartment, despite continued D1,D2, and CDK4 expression, the abundant nuclear levels ofp57KIP2 correlate with proliferative arrest and the onset of lensfiber differentiation. Throughout the latter process, CDK4 ex-pression continues but is accompanied by diminished cyclin D1and persistent p57KIP2 levels. Together, these results provide invivo support for the view that cyclins D1 and D2, CDK4, andp57KIP2 are the critical components regulating pRb-mediatedgrowth arrest in the developing lens.
ACKNOWLEDGMENTS
We thank Nicole Schreiber-Agus and Andrew Koff for critical read-ing of the manuscript. We also thank Sam Zigler and Joe Horwitz forantibodies against MIP26 and the crystallins; Paul Overbeek for theCPV-1 cassette; and the Analytical Imaging Facility at Albert EinsteinCollege of Medicine.
This work was supported by the following NIH grants:RO1HD28317, RO1EY09300, RO1EY11267, and the Cancer CenterCore grant T2P30CA13330; the DAMD Breast Cancer Grant (toS.J.E.); NIH training grant 2T32AG00194 (to E.G.L.); and NIH train-
ing grant T32GM07288 (to N.J.L.). R.A.D. is a recipient of the Irma T.Hirschl Award.
REFERENCES
1. Ahuja, H. S., Y. Zhu, and Z. Zakeri. 1997. Association of cyclin-dependentkinase 5 and its activator p35 with apoptotic cell death. Dev. Genet. 21:258–267.
2. Alevizopoulos, K., J. Vlach, S. Hennecke, and B. Amati. 1997. Cyclin E andc-Myc promote cell proliferation in the presence of p16INK4a and of hypo-phosphorylated retinoblastoma family proteins. EMBO J. 16:5322–5333.
3. Assoian, R. K. 1997. Control of the G1 phase cyclin-dependent kinases bymitogenic growth factors and the extracellular matrix. Cytokine GrowthFactor Rev. 8:165–170.
4. Baldin, V., J. Lukas, M. J. Marcote, M. Pagano, and G. Draetta. 1993. CyclinD1 is a nuclear protein required for cell cycle progression in G1. Genes Dev.7:812–821.
5. Bodrug, S. E., B. J. Warner, M. L. Bath, G. J. Lindeman, A. W. Harris, andJ. M. Adams. 1994. Cyclin D1 transgene impedes lymphocyte maturation andcollaborates in lymphomagenesis with the myc gene. EMBO J. 13:2124–2130.
6. Bortner, D. M., and M. P. Rosenberg. 1997. Induction of mammary glandhyperplasia and carcinomas in transgenic mice expressing human cyclin E.Mol. Cell. Biol. 17:453–459.
7. Bresnahan, W. A., I. Boldogh, T. Ma, T. Albrecht, and E. A. Thompson. 1996.Cyclin E/dk2 activity is controlled by different mechanisms in the G0 and G1phases of the cell cycle. Cell Growth Differ. 7:1283–1290.
8. Broekhuyse, R. M., E. D. Kuhlmann, and A. L. Stols. 1976. Lens membranes.II. Isolation and characterization of the main intrinsic polypeptide (MIP) ofbovine lens fiber membranes. Exp. Eye Res. 23:365–371.
9. Chepelinsky, A. B., C. R. King, P. S. Zelenka, and J. Piatigorsky. 1985.Lens-specific expression of the chloramphenicol acetyltransferase gene pro-moted by 59 flanking sequences of the murine alpha A-crystallin gene inexplanted chicken lens epithelia. Proc. Natl. Acad. Sci. USA 82:2334–2338.
10. Connell-Crowley, L., J. W. Harper, and D. W. Goodrich. 1997. Cyclin D1/CDK4 regulates retinoblastoma protein-mediated cell cycle arrest by site-specific phosphorylation. Mol. Biol. Cell 8:287–301.
11. Duronio, R. J., and P. H. O’Farrell. 1995. Developmental control of the G1to S transition in Drosophila: cyclin E is a limiting downstream target of E2F.Genes Dev. 9:1456–1468.
12. Duronio, R. J., A. Brook, N. Dyson, and P. H. O’Farrell. 1996. E2F-inducedS phase requires cyclin E. Genes Dev. 10:2513–2525.
13. Elledge, S. J., and J. W. Harper. 1994. CDK inhibitors: on the threshold ofcheckpoints and development. Curr. Opin. Cell Biol. 6:847–852.
14. Ewen, M. E., H. K. Sluss, C. J. Sherr, H. Matsushime, J. Kato, and D. M.Livingston. 1993. Functional interactions of the retinoblastoma protein withmammalian D-type cyclins. Cell 73:487–497.
15. Fantl, V., G. Stamp, A. Andrews, I. Rosewell, and C. Dickson. 1995. Micelacking cyclin D1 are small and show defects in eye and mammary glanddevelopment. Genes Dev. 9:2364–2372.
16. Finley, R. L., Jr., B. J. Thomas, S. L. Zipursky, and R. Brent. 1996. Isolationof Drosophila cyclin D, a protein expressed in the morphogenetic furrowbefore entry into S phase. Proc. Natl. Acad. Sci USA 93:3011–3015.
17. Fromm, L., and P. A. Overbeek. 1996. Regulation of cyclin and cyclin-dependent kinase gene expression during lens differentiation requires theretinoblastoma protein. Oncogene 12:69–75.
18. Furuya, Y., S. Ohta, K. Yasuda, and H. Ito. 1997. Enhanced expression ofcyclin-dependent kinase apoinhibitor in apoptosis of androgen-independentprostatic cancer cell line induced by calcium ionophore. Anticancer Res.17:2391–2394.
19. Gao, C. Y., Z. Zakeri, Y. Zhu, H. He, and P. S. Zelenka. 1997. Expression ofCdk5, p35, and Cdk5-associated kinase activity in the developing rat lens.Dev. Genet. 20:267–275.
20. Gavrieli, Y., Y. Sherman, and S. A. Ben-Sasson. 1992. Identification ofprogrammed cell death in situ via specific labeling of nuclear DNA fragmen-tation. J. Cell Biol 119:493–501.
21. Gorman, C. M., L. F. Moffat, and B. H. Howard. 1982. Recombinant ge-nomes which express chloramphenicol acetyltransferase in mammalian cells.Mol. Cell. Biol. 2:1044–1051.
22. Guan, K. L., C. W. Jenkins, Y. Li, M. A. Nichols, X. Wu, C. L. O’Keefe, A. G.Matera, and Y. Xiong. 1994. Growth suppression by p18, p16INK4a/MTS1-and p14INK4B/MTS2-related CDK6 inhibitor correlates with wild-type pRbfunction. Genes Dev. 8:2939–2952.
23. Hatakeyama, M., J. A. Brill, G. R. Fink, and R. A. Weinberg. 1994. Collab-oration of G1 cyclins in the functional inactivation of the retinoblastomaprotein. Genes Dev. 8:1759–1771.
24. Kato, J., and C. J. Sherr. 1993. Inhibition of granulocyte differentiation byG1 cyclins D2 and D3 but not D1. Proc. Natl. Acad. Sci. USA 90:11513–11517.
25. Kato, J., M. Matsuoka, K. Polyak, J. Massague, and C. J. Sherr. 1994. CyclicAMP-induced G1 phase arrest mediated by an inhibitor (p27Kip1) of cyclin-dependent kinase-4 activation. Cell 79:487–496.
26. Koh, J., G. H. Enders, B. D. Dynlacht, and E. Harlow. 1995. Tumor-derived
TABLE 2. Average number of positive nuclei per lens 6 standarddeviation in E13.5 transgenic, pRb2, and/or p57KIP2-
deficient embryos
Transgene No. of BrdU-positive nuclei No. of TUNEL-positive nuclei
p572/2 2.5 6 0.5 3.9 6 0.7D3/p572/2 25.9 6 1.1 9.1 6 0.7pRb2/2 25.5 6 1.3 9.3 6 0.7D1/p572/2 10.5 6 1.3 4.2 6 0.6E/p572/2 2.0 6 0.6 4.0 6 0.7
362 LAHOZ ET AL. MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Nov
embe
r 20
21 b
y 39
.110
.32.
138.

p16 alleles encoding proteins defective in cell-cycle inhibition. Nature 375:506–510.
27. Lovec, H., A. Grzeschiczek, M. B. Kowalski, and T. Moroy. 1994. CyclinD1/bcl-1 cooperates with myc genes in the generation of B-cell lymphoma intransgenic mice. EMBO J. 13:3487–3495.
28. Lukas, J., M. Pagano, Z. Staskova, G. Draetta, and J. Bartek. 1994. CyclinD1 protein oscillates and is essential for cell cycle progression in humantumour cell lines. Oncogene 9:707–718.
29. Lukas, J., J. Bartkova, M. Rohde, M. Strauss, and J. Bartek. 1995. Cyclin D1is dispensable for G1 control in retinoblastoma gene-deficient cells indepen-dently of cdk4 activity. Mol. Cell. Biol. 15:2600–2611.
30. Lukas, J., T. Herzinger, K. Hansen, M. C. Moroni, D. Resnitzky, K. Helin,S. I. Reed, and J. Bartek. 1997. Cyclin E-induced S phase without activationof the pRb/E2F pathway. Genes Dev. 11:1479–1492.
31. Matsushime, H., D. E. Quelle, S. A. Shurtleff, M. Shibuya, C. J. Sherr, andJ. Kato. 1994. D-type cyclin-dependent kinase activity in mammalian cells.Mol. Cell. Biol. 14:2066–2076.
32. McAvoy, J. W. 1980. Beta- and gamma-crystallin synthesis in rat lens epithe-lium explanted with neural retina. Differentiation 17:85–91.
33. Medema, R. H., R. E. Herrera, F. Lam, and R. A. Weinberg. 1995. Growthsuppression by p16ink4a requires functional retinoblastoma protein. Proc.Natl. Acad. Sci. USA 92:6289–6293.
34. Meyerson, M., and E. Harlow. 1994. Identification of G1 kinase activity forcdk6, a novel cyclin D partner. Mol. Cell. Biol. 14:2077–2086.
35. Miller, M. W., and R. S. Nowakowski. 1988. Use of bromodeoxyuridineimmunohistochemistry to examine the proliferation, migration and time oforigin of cells in the central nervous system. Brain Res. 457:44–52.
36. Morgenbesser, S. D., B. O. Williams, T. Jacks, and R. A. DePinho. 1994.p53-dependent apoptosis produced by Rb-deficiency in the developingmouse lens. Nature 371:72–74.
37. Morgenbesser, S. D., N. Schreiber-Agus, M. Bidder, K. A. Mahon, P. A.Overbeek, J. Horner, and R. A. DePinho. 1995. Contrasting roles for c-Mycand L-Myc in the regulation of cellular growth and differentiation in vivo.EMBO J. 14:743–756.
38. Murata, Y., T. Kudoh, H. Sugiyama, K. Toyoshima, and T. Akiyama. 1997.The Wilms tumor suppressor gene WT1 induces G1 arrest and apoptosis inmyeloblastic leukemia M1 cells. FEBS Lett. 409:41–45.
39. Nourse, J., E. Firpo, W. M. Flanagan, S. Coats, K. Polyak, M. H. Lee, J.Massague, G. Crabtree, and J. M. Roberts. 1994. Interleukin-2-mediatedelimination of p27Kip1 cyclin-dependent kinase inhibitor prevented by rapa-mycin. Nature 372:570–573.
40. Ohtsubo, M., and J. M. Roberts. 1993. Cyclin-dependent regulation of G1 inmammalian fibroblasts. Science 259:1908–1912.
41. Ohtsubo, M., A. M. Theodoras, J. Schumacher, J. M. Roberts, and M.Pagano. 1995. Human cyclin E, a nuclear protein essential for the G1-to-Sphase transition. Mol. Cell. Biol. 15:2612–2624.
42. Overbeek, P. A., A. B. Chepelinsky, J. S. Khillan, J. Piatigorsky, and H.Westphal. 1985. Lens-specific expression and developmental regulation ofthe bacterial chloramphenicol acetyltransferase gene driven by the murinealpha A-crystallin promoter in transgenic mice. Proc. Natl. Acad. Sci. USA82:7815–7819.
43. Pagano, M., S. W. Tam, A. M. Theodoras, P. Beer-Romano, G. DalSal, V.Chau, P. R. Yew, G. F. Draetta, and M. Rolfe. 1995. Role of the ubiquitin-proteosome pathway in regulating abundance of the cyclin-dependent kinaseinhibitor p27. Science 269:682–685.
44. Park, D. S., B. Levine, G. Ferrari, and L. A. Greene. 1997. Cyclin dependent
kinase inhibitors and dominant negative cyclin dependent kinase 4 and 6promote survival of NGF-deprived sympathetic neurons. J. Neurosci. 17:8975–8983.
45. Piatigorsky, J. 1981. Lens differentiation in vertebrates. A review of cellularand molecular features. Differentiation 19:134–153.
46. Quelle, D. E., R. A. Ashmun, S. A. Shurtleff, J. Kato, D. Bar-Sagi, M. F.Roussel, and C. J. Sherr. 1993. Overexpression of mouse D-type cyclinsaccelerates G1 phase in rodent fibroblasts. Genes Dev. 7:1559–1571.
47. Rao, S. S., and D. S. Kohtz. 1995. Positive and negative regulation of D-typecyclin expression in skeletal myoblasts by basic fibroblast growth factor andtransforming growth factor beta. A role for cyclin D1 in control of myoblastdifferentiation. J. Biol. Chem. 270:4093–4100.
48. Resnitzky, D., and S. I. Reed. 1995. Different roles for cyclins D1 and E inregulation of the G1-to-S transition. Mol. Cell. Biol. 15:3463–3469.
49. Resnitzky, D., M. Gossen, H. Bujard, and S. I. Reed. 1994. Acceleration ofthe G1/S phase transition by expression of cyclins D1 and E with an induciblesystem. Mol. Cell. Biol. 14:1669–1679.
50. Robles, A. I., F. Larcher, R. B. Whalin, R. Murillas, E. Richie, I. B. Gimenez-Conti, J. L. Jorcano, and C. J. Conti. 1996. Expression of cyclin D1 inepithelial tissues of transgenic mice results in epidermal hyperproliferationand severe thymic hyperplasia. Proc. Natl. Acad. Sci. USA 93:7634–7638.
51. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: alaboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, ColdSpring Harbor, N.Y.
52. Sherr, C. J. 1994. G1 phase progression: cycling on cue. Cell 79:551–555.53. Sherr, C. J. 1996. Cancer cell cycles. Science 274:1672–1677.54. Sherr, C. J., and J. M. Roberts. 1995. Inhibitors of mammalian G1 cyclin-
dependent kinases. Genes Dev. 9:1149–1163.55. Sicinski, P., J. L. Donaher, S. B. Parker, T. Li, A. Fazeli, H. Gardner, S. Z.
Haslam, R. T. Bronson, S. J. Elledge, and R. A. Weinberg. 1995. Cyclin D1provides a link between development and oncogenesis in the retina andbreast. Cell 82:621–630.
56. Sicinski, P., J. L. Donaher, Y. Geng, S. B. Parker, H. Gardner, M. Y. Park,R. L. Robker, J. S. Richards, L. K. McGinnis, J. D. Biggers, J. J. Eppig, R. T.Bronson, S. J. Elledge, and R. A. Weinberg. 1996. Cyclin D2 is an FSH-responsive gene involved in gonadal cell proliferation and oncogenesis. Na-ture 384:470–474.
57. Takemoto, L., J. Kuck, and K. Kuck. 1988. Changes in the major intrinsicpolypeptide (MIP26K) during opacification of the Emory mouse lens. Exp.Eye Res. 47:329–336.
58. Terui, Y., Y. Furukawa, J. Kikuchi, and M. Saito. 1995. Apoptosis duringHL-60 cell differentiation is closely related to a G0/G1 cell cycle arrest.J. Cell. Physiol. 164:74–84.
59. Vallance, S. J., H. M. Lee, M. F. Roussel, S. A. Shurtleff, J. Kato, D. K.Strom, and C. J. Sherr. 1994. Monoclonal antibodies to mammalian D-typeG1 cyclins. Hybridoma 13:37–44.
60. Wang, T. C., R. D. Cardiff, L. Zukerberg, E. Lees, A. Arnold, and E. V.Schmidt. 1994. Mammary hyperplasia and carcinoma in MMTV-cyclin D1transgenic mice. Nature. 369:669–671.
61. Weinberg, R. A. 1995. The retinoblastoma protein and cell cycle control. Cell81:323–330.
62. Zhang, P., N. J. Liegeois, C. Wong, M. Finegold, H. Hou, J. C. Thompson, A.Silverman, J. W. Harper, R. A. DePinho, and S. J. Elledge. 1997. Altered celldifferentiation and proliferation in mice lacking p57KIP2 indicates a role inBeckwith-Wiedemenn syndrome. Nature 387:151–158.
VOL. 19, 1999 CONTROL OF G1 EXIT IN VIVO 363
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Nov
embe
r 20
21 b
y 39
.110
.32.
138.




![The regulation of SIRT2 function by cyclin-dependent kinases ......916JCB • VOLUME 180 • NUMBER 5 • 2008 with recombinant baculoviral cyclin E – Cdk2 and -[ 32 P]ATP ( Fig](https://img.dokumen.tips/doc/110x75/60d8933f6f7c6259ee7c52cd/the-regulation-of-sirt2-function-by-cyclin-dependent-kinases-916jcb-a.jpg)




![RESEARCH Open Access P276-00, a cyclin-dependent ......leukins viz; IL-6 and IL-10 [7]. P276-00, a novel small molecule inhibitor of cyclin-dependent kinases (Cdks), is currently in](https://img.dokumen.tips/doc/110x75/60d7f116f079b4414742a5cb/research-open-access-p276-00-a-cyclin-dependent-leukins-viz-il-6-and-il-10.jpg)



















