Embed Size (px)
Citation preview

CUDA-Enabled Applications for Next-generation Sequencing
Bertil Schmidt
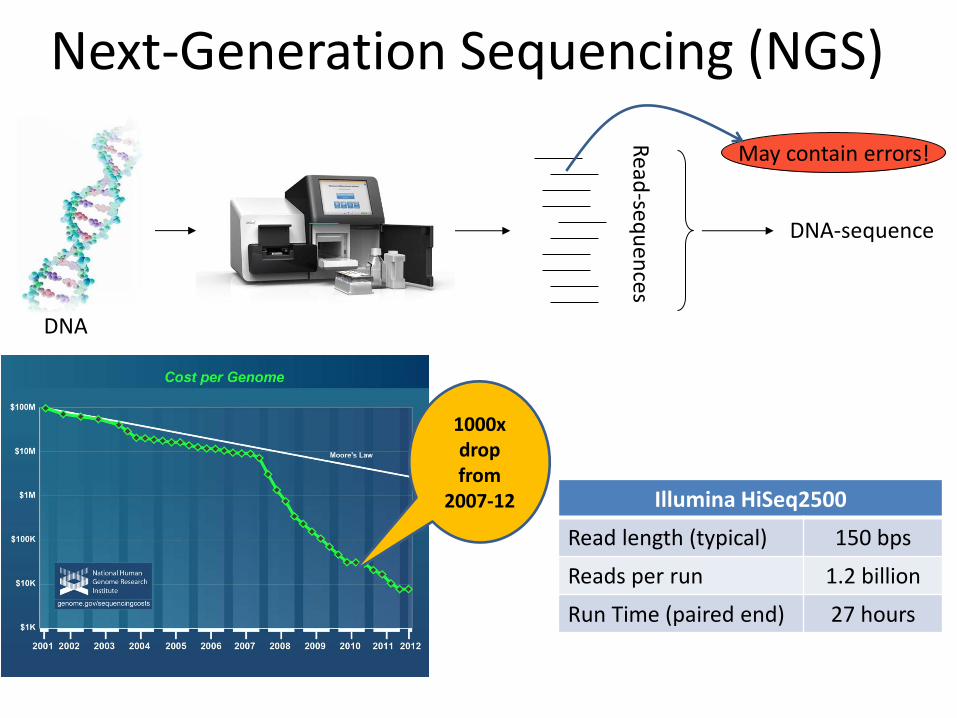
Next-Generation Sequencing (NGS)
Read
-sequ
ences
DNA-sequence
DNA
May contain errors!
Illumina HiSeq2500
Read length (typical) 150 bps
Reads per run 1.2 billion
Run Time (paired end) 27 hours
1000x drop from
2007-12

Short Read Mapping
• Problem definition – Given a huge set of reads and a reference genome, identify where
each read is from in the reference genome
• Computational challenges – Throughput (make it fast)
• BWA 0.5.9 with human reference genome – 7.5M reads/hour on quad-core CPU (4 threads, 2x100bps reads) – 1 CPU week for aligning a human genome at 40x coverage
– Sensitivity (make it accurate) • Sequencing errors • Genomic variation
taking many copies of a book, passing them all
through a shredder
find the location of each piece in a given
very similar book
Find alignments

Common Algorithmic Patterns in NGS Data Analysis
• Indexing and lookup – Hash tables – BWT and FM-Index – Bloom filter
• Dynamic programming – Smith-Waterman, Needleman-Wunsch
• NGS Bioinformatics Challenges – Scalability
• to deal with huge amounts of reads
– Algorithm design • to deal with short reads
– Parallelisation • Many Bioinformatics algorithms are irregular and therefore challenging to
map to parallel architectures
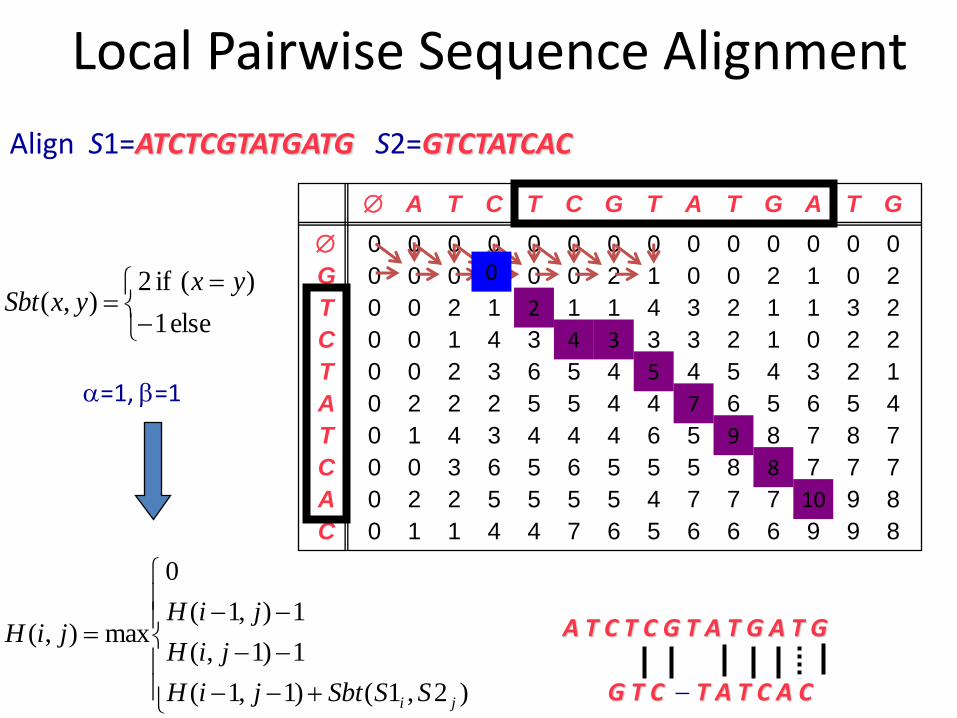
Local Pairwise Sequence Alignment
Align S1=ATCTCGTATGATG S2=GTCTATCAC
G
T
C
T
A
T
C
A
C
A T C T C G T A T G A T G
0 0 0 0 0 2 1 0 0 2 1 0
0
0
0
0
0
0
0
0
0
0
0 0 0 0 0 0 0 0 0 0 0 0 0
2
0 2 1 2 1 1 4 3 2 1 1 3 2
0
0
2
1
0
2
1
1
2
2
4
3
2
1
4
3
2
3
6
5
4
3
6
5
4
5
5
4
4
5
5
4
6
5
7
3
4
4
4
5
5
6
3
5
4
6
5
4
5
3
4
7
5
5
7
6
2
5
6
9
8
7
6
1
4
5
8
8
7
6
0
3
6
7
7
10
9
2
2
5
8
7
9
9
2
1
4
7
7
8
8
10
8
9
7
5
3 4
2
0
else 1
)( if 2),(
yxyxSbt
=1, =1
A T C T C G T A T G A T G
G T C T A T C A C
)2,1()1,1(
1)1,(
1),1(
0
max),(
ji SSSbtjiH
jiH
jiHjiH

Background on Short Read Mapping • Reference Genome and reads are too large for direct DP approach • „Seed-and-Extend“: Use an index data structure to rapidly find short
exact matches to seed longer in-exact alignments, e.g. – Build a hash table of all k-mers of genome – Segment query sequence into k-mer seeds
Ref Genome: …CAAACCAGCTCTTATGGTCAGAACTCTGAAAGACAACTGAGCTGCTG…
Read Seed: TGGTCAGAAC
k-mer positions

CUSHAW: CUDA-enabled short-read aligner to human genome based on BWT
• Indexing approaches (memory sizes for human genome) – Suffix tree (> 35 GB)
– Suffix array (> 12 GB)
– Hash tables (> 12 GB)
• CUSHAW: GPU-Approach – Index Reference Genome using BWT (Burrows Wheeler Transform)
– Needs 2.2 GB memory for Human Genome fits on Fermi C2050
– optimized for Fermi architecture using CUDA
– CUSHAW currently only supports a restrictive alignment models • exhaustive search with certain constraints
• allows few mismatches (default = 2) in seed
• no indels

BWT(cattattagga$)
Cyclic Rotations
c a t t a t t a g g a $
a t t a t t a g g a $ c
t t a t t a g g a $ c a
t a t t a g g a $ c a t
a t t a g g a $ c a t t
t t a g g a $ c a t t a
t a g g a $ c a t t a t
a g g a $ c a t t a t t
g g a $ c a t t a t t a
g a $ c a t t a t t a g
a $ c a t t a t t a g g
$ c a t t a t t a g g a
Sorting
$ c a t t a t t a g g a
a $ c a t t a t t a g g
a g g a $ c a t t a t t
a t t a g g a $ c a t t
a t t a t t a g g a $ c
c a t t a t t a g g a $
g a $ c a t t a t t a g
g g a $ c a t t a t t a
t a g g a $ c a t t a t
t a t t a g g a $ c a t
t t a g g a $ c a t t a
t t a t t a g g a $ c a
BWT
a
g
t
t
c
$
g
a
t
t
a
a

Backward search to calculate SA interval for “tta” in BWT(cattattagga$)
( ) ( [ ]) ( [ ], ( 1) 1) 1, 0
( ) ( [ ]) ( [ ], ( 1)), 0
a a
b b
I i C S i Occ S i I i i S
I i C S i Occ S i I i i S
t t a
$ c a t t a t t a g g a
a $ c a t t a t t a g g
a g g a $ c a t t a t t
a t t a g g a $ c a t t
a t t a t t a g g a $ c
c a t t a t t a g g a $
g a $ c a t t a t t a g
g g a $ c a t t a t t a
t a g g a $ c a t t a t
t a t t a g g a $ c a t
t t a g g a $ c a t t a
t t a t t a g g a $ c a
[1,4
]
t t a
$ c a t t a t t a g g a
a $ c a t t a t t a g g
a g g a $ c a t t a t t
a t t a g g a $ c a t t
a t t a t t a g g a $ c
c a t t a t t a g g a $
g a $ c a t t a t t a g
g g a $ c a t t a t t a
t a g g a $ c a t t a t
t a t t a g g a $ c a t
t t a g g a $ c a t t a
t t a t t a g g a $ c a
[8,9
]
t t a
$ c a t t a t t a g g a
a $ c a t t a t t a g g
a g g a $ c a t t a t t
a t t a g g a $ c a t t
a t t a t t a g g a $ c
c a t t a t t a g g a $
g a $ c a t t a t t a g
g g a $ c a t t a t t a
t a g g a $ c a t t a t
t a t t a g g a $ c a t
t t a g g a $ c a t t a
t t a t t a g g a $ c a
[10
,11
]

CUSHAW: short-read aligner to human genome based on BWT
• CUSHAW becomes less efficient with growing read length
SRR002273 8.5M Reads, 36bp
ERR000589 24.3M Reads, 51bp
ERR002273 20.4M Reads, 75bps
CUSHAW (Tesla M2090) 1.1 mins 5.5 mins 33.3 mins
BWA 0.5.9 (AMD quad-core CPU, 4 threads)
14.9 mins 67.6 mins 91.9 mins
Speedup 12.8 12.2 2.8
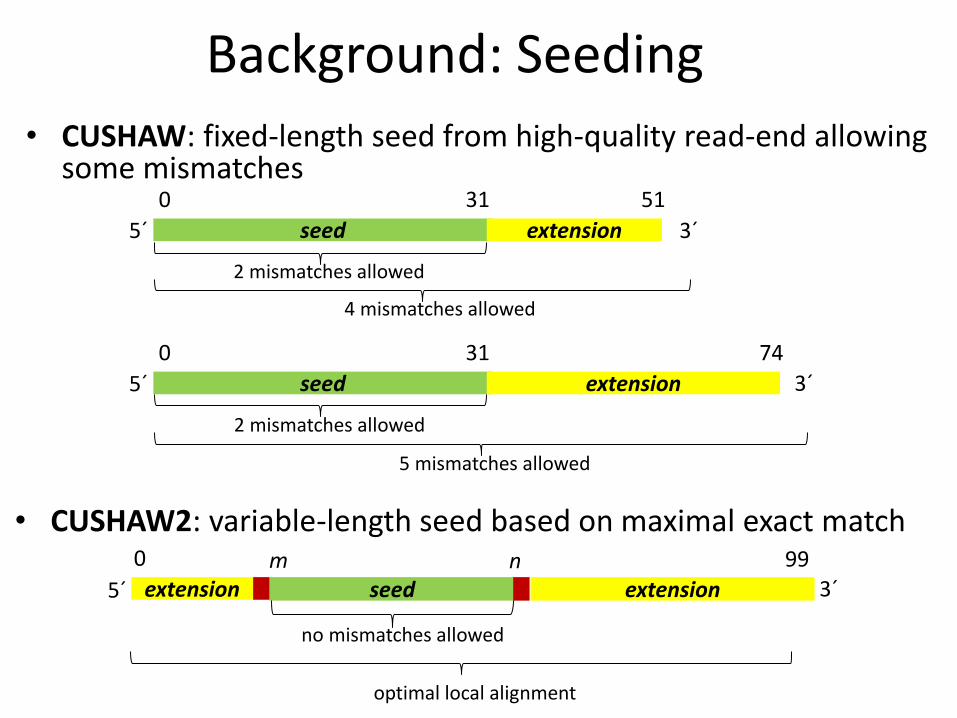
Background: Seeding
• CUSHAW: fixed-length seed from high-quality read-end allowing some mismatches
seed
0 51 31
extension
2 mismatches allowed
4 mismatches allowed
5´ 3´
seed
0 74 31
extension
2 mismatches allowed
5 mismatches allowed
5´ 3´
seed
0 99 m extension 5´ 3´ extension
n
no mismatches allowed
optimal local alignment
• CUSHAW2: variable-length seed based on maximal exact match
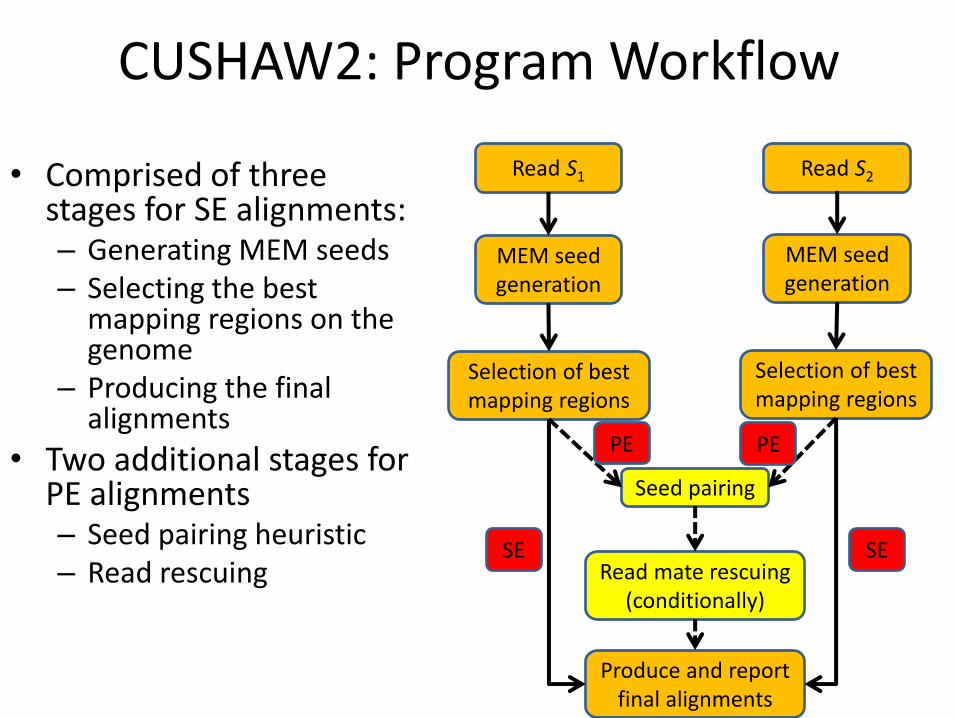
CUSHAW2: Program Workflow
• Comprised of three stages for SE alignments: – Generating MEM seeds – Selecting the best
mapping regions on the genome
– Producing the final alignments
• Two additional stages for PE alignments – Seed pairing heuristic – Read rescuing
Read S1 Read S2
MEM seed generation
Selection of best mapping regions
Seed pairing
Read mate rescuing (conditionally)
Produce and report final alignments
MEM seed generation
Selection of best mapping regions
PE PE
SE SE

Program Outline of CUSHAW2-GPU
Read batch
Seed generation
Top seeds selection
Alignment generation
Compute suffix array intervals of
seeds on GPU
Locate each seed on the reference on
GPU
Perform score-only Smith-
Waterman on GPU
Sort all seeds on GPU
Compute alignments of top seeds on
GPU
Read pairing and rescuing (for PE only)
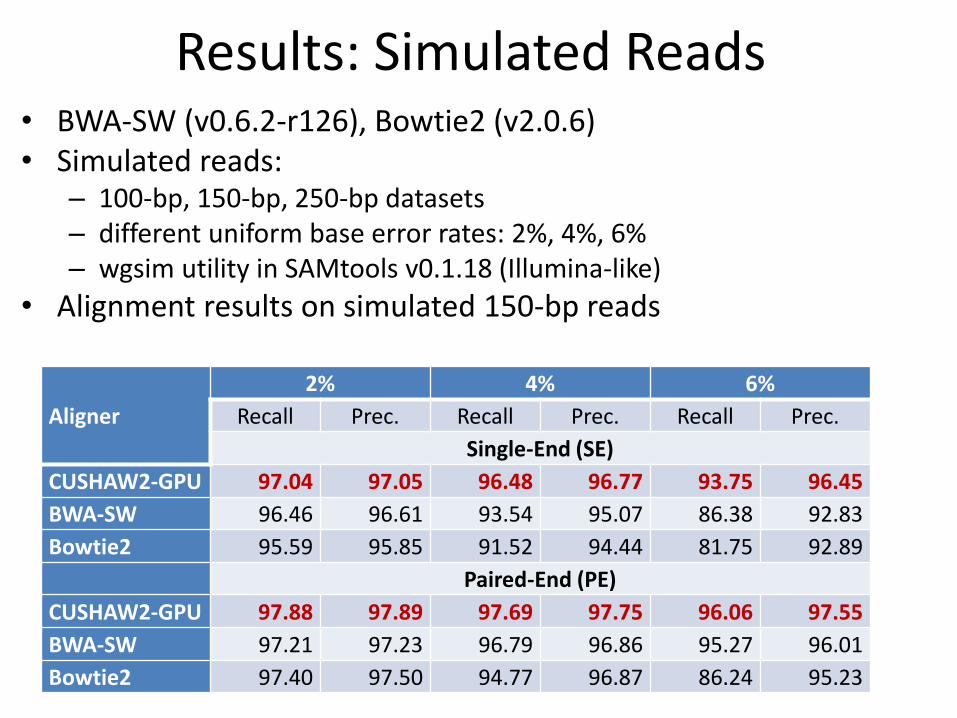
Results: Simulated Reads • BWA-SW (v0.6.2-r126), Bowtie2 (v2.0.6) • Simulated reads:
– 100-bp, 150-bp, 250-bp datasets – different uniform base error rates: 2%, 4%, 6% – wgsim utility in SAMtools v0.1.18 (Illumina-like)
• Alignment results on simulated 150-bp reads
Aligner
2% 4% 6%
Recall Prec. Recall Prec. Recall Prec.
Single-End (SE)
CUSHAW2-GPU 97.04 97.05 96.48 96.77 93.75 96.45
BWA-SW 96.46 96.61 93.54 95.07 86.38 92.83
Bowtie2 95.59 95.85 91.52 94.44 81.75 92.89
Paired-End (PE)
CUSHAW2-GPU 97.88 97.89 97.69 97.75 96.06 97.55
BWA-SW 97.21 97.23 96.79 96.86 95.27 96.01
Bowtie2 97.40 97.50 94.77 96.87 86.24 95.23

Results: Simulated Reads
• Simulated reads: – 454-like reads
– Mason simulator, default error mode settings
Aligner
100bps 150bps 250bps
Recall Prec. Recall Prec. Recall Prec.
SE
CUSHAW2-GPU 96.27 96.41 97.39 97.41 98.06 98.06
BWA-SW 93.96 95.72 96.89 97.06 97.91 97.91
Bowtie2 95.00 95.32 96.52 96.65 97.70 97.74

Effectiveness of mapping quality scores
• 100-bp dataset with 2% error rate • Cumulative recall and precision calculated from the
cumulative number of reported alignments and the cumulative number of correct alignments from high to low mapping quality scores

Runtime Evaluation
• 6-core Intel Xeon E5-2620 2.0 GHz CPU (using 12 threads), 16 GB RAM • K20 GPU • Runtimes in minutes
SRX00706 (454, 4M
Reads, 300bp)
SRX202788 (Ion Torrent, 4M Reads,
183bp)
SRX064195 (Illumina,
51M Reads, 100bp)
ERX009608 (Illumina,
53M Reads, 101bp)
CUSHAW2-GPU 9.0 4.8 31.1 34.2
CUSHAW2-CPU 25.3 11.1 55.9 61.4
BWA-SW 37.7 17.8 92.1 96.4
Bowtie2 13.3 8.0 48.2 51.3

Other CUDA-enabled HPC Bioinformatics Software developed by my group
• Sequence database searching – CUDASW++ (Smith-Waterman) – CUDA-BLASTP
• Multiple sequence alignment – MSA-CUDA
• Next-Generation Sequencing (NGS) – DecGPU (short-read error correction) – CUSHAW (short-read mapping) – CRiSPy-CUDA and CRiSPy-Embed (short-read clustering)
• Motif finding – CUDA-MEME
• Accessible via: http://hpc.informatik.uni-mainz.de/

Conclusion • CUSHAW is a CUDA-based short read aligner
– Open-source: http://cushaw.sourceforge.net/ – Speedup of one order-of-magnitude on a Tesla M2090 compared to
BWA on a Quad-core CPU for short reads – Less efficient for longer read length
• CUSHAW2 is a fast gapped NGS read aligner, employing MEMs as seeds – Open-source: http://cushaw2.sourceforge.net – Consistently among the highest-ranked aligners for both SE and PE in
terms of recall, precision, and speed – Partial Funding: NVIDIA Foundation (OpenGE)
• Publications – Y. Liu, B.Schmidt, D. Maskell: CUSHAW: a CUDA compatible short
read aligner to large genomes based on the Burrows-Wheeler transform, Bioinformatics 28(14), 1830-1837, 2012
– Y. Liu, B. Schmidt: Long read alignment based on maximal exact match seeds, Bioinformatics 28(18), i318-324, 2012

Conclusion
• NGS technologies establish the need for scalable Bioinformatics tools that can process massive amounts of short reads
• CUDA is a highly suitable technology to address this need
• NGS algorithms need to be adapted since throughput and read length continues to increase
• Website
– http://hpc.informatik.uni-mainz.de/





























