Embed Size (px)
Citation preview

CSP02 Uncommon Gastrointestinal Disorders: Ignorance is Not Bliss
Rhonda Yantiss MD Sarah Dry MD Dhanpat Jain
Alexandros Polydorides Jeffrey Goldsmith David Lewin MD
2011 Annual Meeting – Las Vegas, NV
AMERICAN SOCIETY FOR CLINICAL PATHOLOGY 33 W. Monroe, Ste. 1600
Chicago, IL 60603

CSP02 Uncommon Gastrointestinal Disorders: Ignorance is Not Bliss Members of the Rodger C. Haggitt Gastrointestinal Pathology Society Education Committee felt that a discussion of potential mimics of common gastrointestinal disorders may be helpful to practicing surgical pathologists. Recent advances in our understanding of inflammatory gastrointestinal conditions have improved our recognition of subtle differences between these disorders and generated novel schemes for their classification. Pathologists should be aware of the spectrum of disorders that may mimic common gastrointestinal diseases, since many require specific therapeutic interventions. Our proposed program includes a panel of speakers who will address these issues in a case-based format. Each speaker will discuss common pitfalls, diagnostic considerations, and the use of ancillary testing in the establishment of a diagnosis.
• Develop an algorithmic approach to the interpretation of medical biopsies of the gastrointestinal tract. • Discriminate between common gastrointestinal illnesses and their potential mimics. • Apply ancillary techniques to the classification of gastrointestinal diseases and understand the biologic
implications of the results. FACULTY: Rhonda Yantiss MD Sarah Dry MD Dhanpat Jain Alexandros Polydorides Jeffrey Goldsmith David Lewin MD Practicing Pathologists Surgical Pathology Gastrointestinal 3.0 CME/CMLE Credits Accreditation Statement: The American Society for Clinical Pathology (ASCP) is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education (CME) for physicians. This activity has been planned and implemented in accordance with the Essential Areas and Policies of the Accreditation Council for Continuing Medical Education (ACCME). Credit Designation: The ASCP designates this enduring material for a maximum of 3 AMA PRA Category 1 Credits™. Physicians should only claim credit commensurate with the extent of their participation in the activity. ASCP continuing education activities are accepted by California, Florida, and many other states for relicensure of clinical laboratory personnel. ASCP designates these activities for the indicated number of Continuing Medical Laboratory Education (CMLE) credit hours. ASCP CMLE credit hours are acceptable to meet the continuing education requirements for the ASCP Board of Registry Certification Maintenance Program. All ASCP CMLE programs are conducted at intermediate to advanced levels of learning. Continuing medical education (CME) activities offered by ASCP are acceptable for the American Board of Pathology’s Maintenance of Certification Program.

10/8/2011
1
Do you remember life before GISTs?A brief review of other
mesenchymal lesions of the gut
Sarah Dry, MD
UCLA Department of Pathology
ASCP 2011 Annual Meeting
Disclosures
• None
Objectives
• Review benign and malignant non‐GIST gut mesenchymal lesions most commonly encountered or considered in DDx
• Review one uncommon mesenchymal lesion• Review one uncommon mesenchymal lesion of the gut with unique presentation

10/8/2011
2
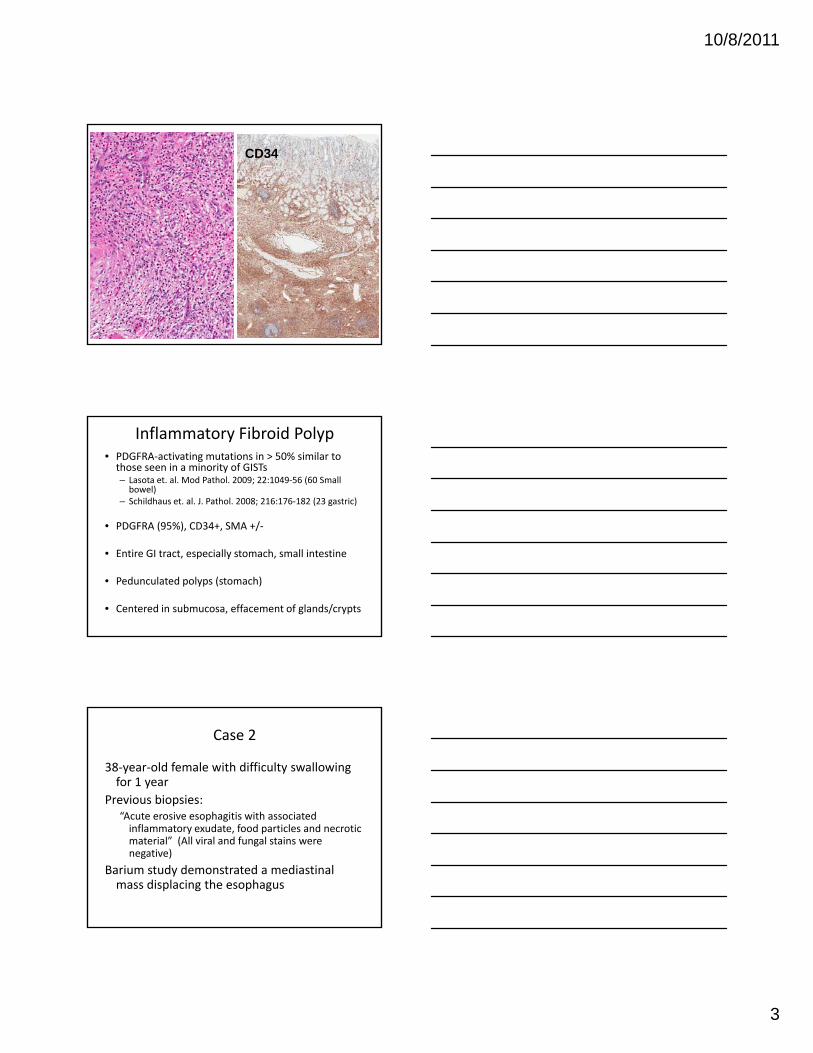
Case 1: 77 y.o. male, gastric polyp
Submucosal centered
Delicate blood vessels, inflammation (eosinophils)
10/8/2011
3
CD34
Inflammatory Fibroid Polyp• PDGFRA‐activating mutations in > 50% similar to those seen in a minority of GISTs – Lasota et. al. Mod Pathol. 2009; 22:1049‐56 (60 Small bowel)
– Schildhaus et. al. J. Pathol. 2008; 216:176‐182 (23 gastric)
• PDGFRA (95%) CD34+ SMA +/• PDGFRA (95%), CD34+, SMA +/‐
• Entire GI tract, especially stomach, small intestine
• Pedunculated polyps (stomach)
• Centered in submucosa, effacement of glands/crypts
Case 2
38‐year‐old female with difficulty swallowing for 1 year
Previous biopsies:“Acute erosive esophagitis with associatedAcute erosive esophagitis with associated inflammatory exudate, food particles and necrotic material” (All viral and fungal stains were negative)
Barium study demonstrated a mediastinalmass displacing the esophagus

10/8/2011
4
Key featurePeripheral lymphoid aggregates with or without germinal center formation
Hypocellular areas
Hypo/Hypercellular Areas
10/8/2011
5
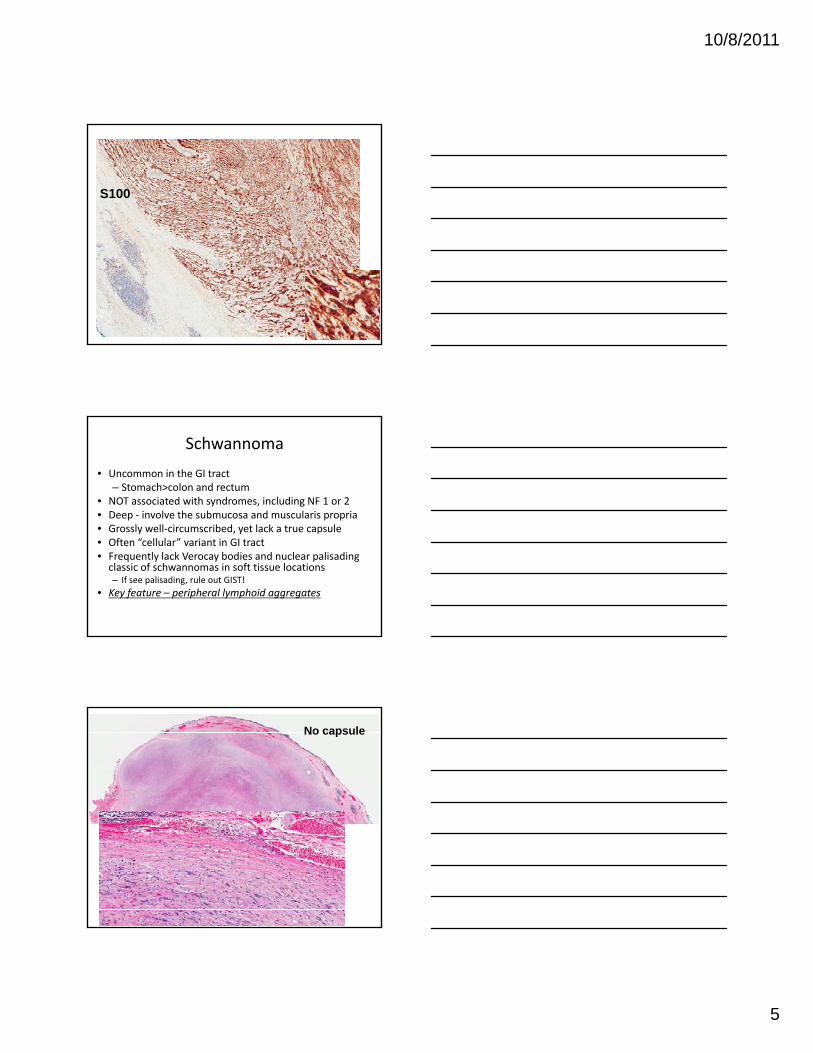
S100
Schwannoma
• Uncommon in the GI tract– Stomach>colon and rectum
• NOT associated with syndromes, including NF 1 or 2• Deep ‐ involve the submucosa and muscularis propriaG l ll i ib d t l k t l• Grossly well‐circumscribed, yet lack a true capsule
• Often “cellular” variant in GI tract• Frequently lack Verocay bodies and nuclear palisading classic of schwannomas in soft tissue locations– If see palisading, rule out GIST!
• Key feature – peripheral lymphoid aggregates
No capsule

10/8/2011
6
GI Schwannoma with Verocay bodies
GISTs with neural features
Case 3• 50 y.o. male
• Screening colonoscopy
• “Thickened fold” with diminutive polyp in d di ldescending colon
• Separate TA in cecum

10/8/2011
7
Histology
S100
Schwann cell hamartoma vs. Schwannoma
• Schwannoma– Mural based (SM and MP)
– Well‐circumscribed
• Schwann cell hamartoma– Mucosal based (LP, not MM)
– Poorly circumscribed
– Spindle cell variant (most common) and epithelioid variant
– Verocay bodies rare
– Strong diffuse S100 positive
• Entraps adjacent crypts
– Uniform bland spindle cells with indistinct cell borders
– Verocay bodies rare
– Strong diffuse S100 positive

10/8/2011
8
DDx: Ganglioneuroma
Retroperitoneum, mediastinumRare in GI tract (colon and rectum)3 main groups
Solitary polypoidGanglioneuromatouspolyposis**Diffuse ganglioneuromatosis**** associated with Cowden’s syndrome, MEN 2b and NF1
Case 431 y.o. male, abd pain
13 cm mass
Centered in smallCentered in small bowel mesentery
At surgery, involved jejunum, distal duodenum, colon, stomach
Infiltrative borders

10/8/2011
9
Regularly distributed blood vessels
Short intersecting fasciclesUniform nucleiMitoses inconspicuous
Mesenteric fibromatosis
• 8% of all desmoids
• Sporadic (most) ‐ some associated w/trauma
• Inherited (FAP or Gardner’s)– Gardner’s: FAP plus extra‐abdominal tumors (skin kertinous cysts, osteomas, desmoid fibromatosis)
• Associated with APC, beta‐catenin gene mutations
• Prominent myxoid change in some
• DDx: sclerosing mesenteritis (fibrosis, chronic inflammation, fat necrosis), IGG4 sclerosinglesions (often storiform, inflammation present)

10/8/2011
10
Case 563‐year‐old female with bowel obstruction. PMH: Breast cancer and non‐small cell lung cancer On follow‐up PET‐CT scan, 8.3 x 7.7 cm mass noted within the pelvis.
10/8/2011
11
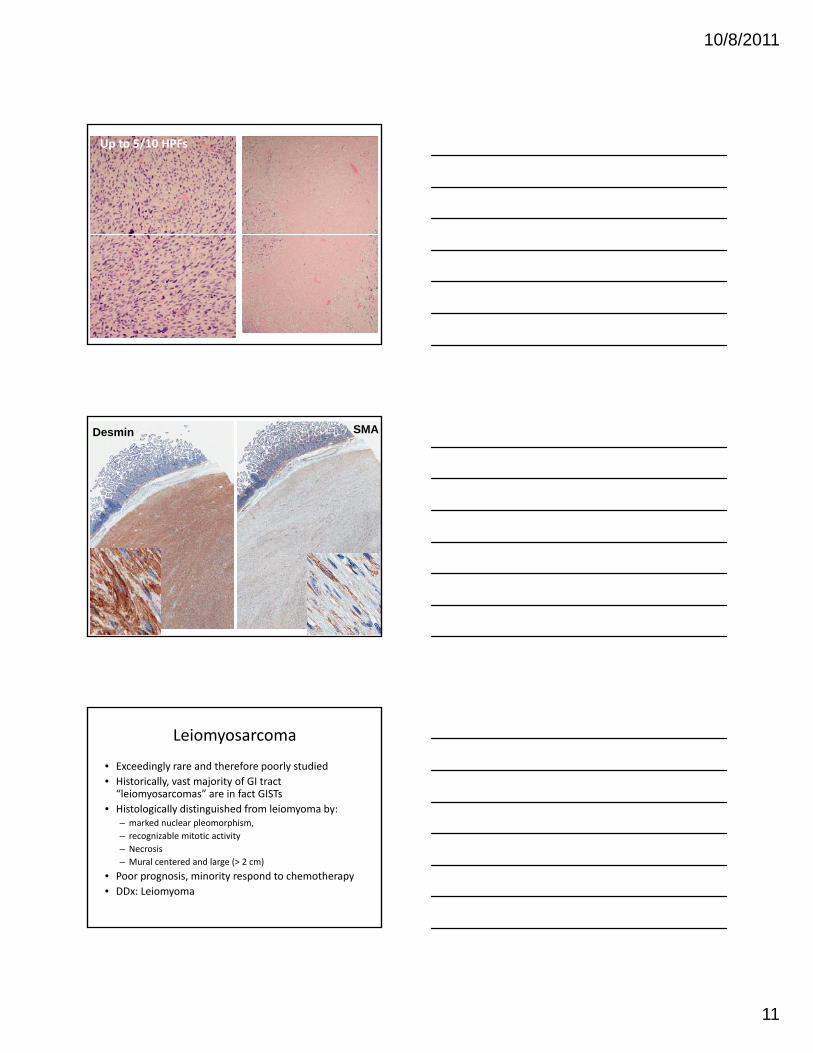
Up to 5/10 HPFs
Desmin SMA
Leiomyosarcoma
• Exceedingly rare and therefore poorly studied• Historically, vast majority of GI tract “leiomyosarcomas” are in fact GISTs
• Histologically distinguished from leiomyoma by: – marked nuclear pleomorphism, – recognizable mitotic activity– Necrosis– Mural centered and large (> 2 cm)
• Poor prognosis, minority respond to chemotherapy• DDx: Leiomyoma

10/8/2011
12
DDx: Leiomyoma
Small, asymptomaticMuscularis mucosaEsophagus, colon
Rare in stomach, SIRare presentations
Intramural rectal massIntramural rectal massCan resemble uterine LM+/‐ attached to external rectal wall+/‐ ER and PR positive
Esophageal leiomyomatosis
Case 6 – unusual tumor
31 year old male, presented with vomiting and abdominal pain
Small bowel lesion #1 ‐ cellular

10/8/2011
13
Small bowel lesion #2

10/8/2011
14
Colon
Both hypo-and hypercellularareas
SMA SMA
Desmin

10/8/2011
15
Small bowel #2
Same SMA
Patchy DesminPatchy Desmin
Summary
• All positive for smooth muscle actin
• Some positive for desmin
• Negative for:– CD117
– CD34
– S100
– AE1/AE3
• Is the patient immunosuppressed?
EBV EBER

10/8/2011
16
EBV‐associated smooth muscle tumors• Rare, occur in immunocompromised individuals, adults and children– Renal transplant, AIDS, steroid users
• Often multiple synchronous tumors• Diverse locations – soft tissue, liver, gut, gallbladder, spleen tonsil vocal cords nasopharynx bonespleen, tonsil, vocal cords, nasopharynx , bone, extradural, spinal cord, bladder, adrenal
• Histology: Well‐differentiated smooth muscle with primitive cellular round cells in about 50% of cases– Mitoses low to high (up to 18/10 HPFs)– SMA in all, desmin in half
• EBV infection was confirmed in all cases through in situ hybridization for EBV early RNAs (EBER)
EBV‐associated smooth muscle tumors
• Deyrup et. al. (Am. J. Surg. Pathol. 2006; 30:75‐82) studied EBV genome in patients with multiple tumors (19 patients, 29 tumors)
• Each tumor clonally distinct – From tumor to tumor within the same patientp– From patient to patient
• Multiple tumors appear to be multiple infection events, not metastasis
• Follow up (mean 15 months; 1‐105 months) – Persistent disease (11/18)– Dead of disease (1/18)
Conclusions
• Inflammatory fibroid polyps
• Schwann cell hamartoma (ganglioneuroma)
• Schwannoma
• Leiomyosarcoma (leiomyoma)
• Mesenteric fibromatosis
• EBV‐associated smooth muscle tumors

10/8/2011
1
NonNon--reflux Esophagitides:reflux Esophagitides:Knowledge is PowerKnowledge is Power
(New and Weird Stuff)(New and Weird Stuff)
Jeffrey Goldsmith MDJeffrey Goldsmith MDDirector of Surgical PathologyDirector of Surgical Pathology
Beth Israel Deaconess Medical CenterBeth Israel Deaconess Medical Center
Consultant in Gastrointestinal PathologyConsultant in Gastrointestinal PathologyChildren’s Hospital BostonChildren’s Hospital Boston
Harvard Medical SchoolHarvard Medical SchoolBoston, MABoston, MA
New(ish)New(ish)
Allergic / eosinophilic esophagitisAllergic / eosinophilic esophagitis‘Lymphocytic esophagitis’‘Lymphocytic esophagitis’
Weird (but important)Weird (but important)
Esophagitis dessicans superficialisEsophagitis dessicans superficialisSkin diseases that can affect the esophagusSkin diseases that can affect the esophagus

10/8/2011
2
80100120140160
“PubMed = (“Allergic” or “Eosinophilic”) + “Esophagitis”
Man
usc
ripts
0204060
1998
1999
2000
2001
2002
2003
2004
2005
2006
2007
2008
2009
2010
Num
ber
of

10/8/2011
3

10/8/2011
4
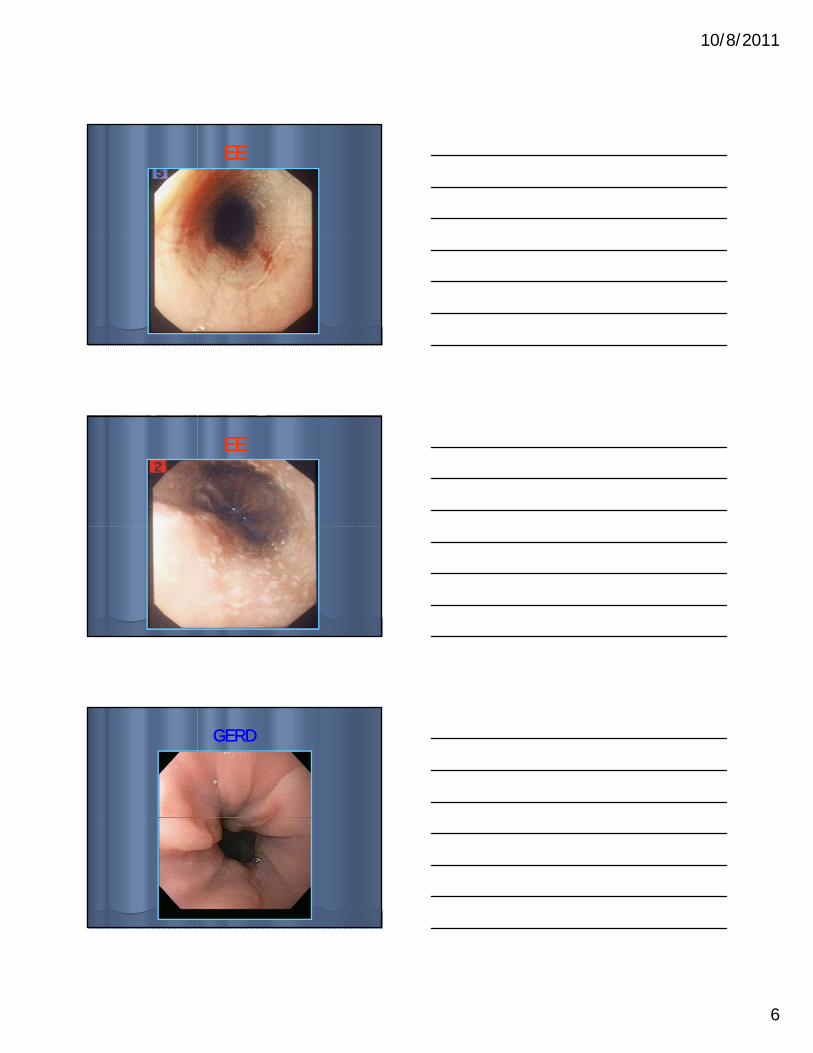
BackgroundBackgroundEEEE
•• M >> FM >> F
•• Usually young.Usually young.
•• Incidence 1.7Incidence 1.7--3 /3 /
GERDGERD•• M = FM = F•• Any age; common in Any age; common in
infantsinfants•• 7% of pediatrician 7% of pediatrician
visitsvisits Incidence 1.7Incidence 1.7 3 / 3 / 10,00010,000
•• Increasing prevalenceIncreasing prevalence
•• GERD like symptomsGERD like symptoms•• DysphagiaDysphagia
•• Food impactionFood impaction
visits.visits.•• Adults: Adults:
•• Epigastric pain with Epigastric pain with radiation to back.radiation to back.
•• Infants:Infants:•• RegurgitationRegurgitation•• IrritabilityIrritability•• ApneaApnea

10/8/2011
5
SignificanceSignificanceEEEE
•• Tx:Tx:•• SteroidsSteroids•• Diet manipulationDiet manipulation
GERDGERD•• Tx:Tx:
•• PPIsPPIs•• SurgerySurgery
•• Sequelae:Sequelae:•• StrictureStricture
•• Sequelae:Sequelae:•• StrictureStricture•• Barrett’s EsophagusBarrett’s Esophagus•• AdenocarcinomaAdenocarcinoma
PathogenesisPathogenesisEEEE
•• AllergyAllergy•• TH2 lymphocyte TH2 lymphocyte
responseresponse•• Interleukin secretionInterleukin secretion
GERDGERD•• LESLES
•• Transient relaxationTransient relaxation•• Hiatal herniaHiatal hernia•• FoodsFoods
•• EotaxinEotaxin•• EosinophilsEosinophils
•• SalivaSaliva
EEEE
10/8/2011
6
EEEE
EEEE
GERDGERD

10/8/2011
7
GERDGERD vs. vs. EEEEFew EosFew Eos Lots of EosLots of Eos
GERDGERD vs. vs. EEEE
Increased numbers distallyIncreased numbers distally Uniform distributionUniform distribution
GERDGERD vs. vs. EEEE-- Eosinophilic MicroabscessesEosinophilic Microabscesses ++

10/8/2011
8
GERDGERD vs. vs. EEEEMinimal degranulationMinimal degranulation Lots of degranulationLots of degranulation
GERDGERD vs. vs. EEEEUniform Distribution Uniform Distribution Increased Surface DensityIncreased Surface Density
GERDGERD vs. vs. EEEE-- Eosinophilic CrustEosinophilic Crust ++

10/8/2011
9
GERD EE
BUT …
GERD EE
Histology Histology –– rules of thumbrules of thumbEEEE
•• >15 eos / hpf>15 eos / hpf•• Increased lumenal Increased lumenal
density of eosdensity of eos•• Eosinophilic Eosinophilic
microabscessesmicroabscesses
GERDGERD•• <15 eos / hpf<15 eos / hpf•• Uniform Uniform
distribution of distribution of eosinophils eosinophils
microabscessesmicroabscesses•• Eosinophilic Eosinophilic
degranulationdegranulation•• Surface Surface
eosinophilic eosinophilic exudateexudate
•• Reactive epithelial Reactive epithelial changeschanges
ppthroughout throughout epitheliumepithelium
•• Reactive epithelial Reactive epithelial changeschanges
Allergic / Eosinophilic Esophagitis Allergic / Eosinophilic Esophagitis
Signout: Descriptive.Signout: Descriptive.Mention highest number of eosinophils / Mention highest number of eosinophils / hpf.hpf.N t h ld ti diff ti l di iN t h ld ti diff ti l di iNote should mention differential diagnosis.Note should mention differential diagnosis.

10/8/2011
10
‘Lymphocytic Esophagitis’‘Lymphocytic Esophagitis’(as it were)(as it were)
Defined as “increased” intraepithelial Defined as “increased” intraepithelial lymphocytes with or without epithelial lymphocytes with or without epithelial edema.edema.No or rare neutrophils / eosinophilsNo or rare neutrophils / eosinophilsNo or rare neutrophils / eosinophils.No or rare neutrophils / eosinophils.
‘Lymphocytic Esophagitis’‘Lymphocytic Esophagitis’(as it were)(as it were)
‘Lymphocytic Esophagitis’‘Lymphocytic Esophagitis’(as it were)(as it were)

10/8/2011
11
‘Lymphocytic Esophagitis’‘Lymphocytic Esophagitis’(as it were)(as it were)
Rubio et al (AJCP 2006; 125:432).Rubio et al (AJCP 2006; 125:432).81 patients, retrospective.81 patients, retrospective.
20 pts had ‘lymphocytic esophagitis’ = 20 pts had ‘lymphocytic esophagitis’ = lymphocytes only.lymphocytes only.y p y yy p y y61 controls = lymphocytes + granulocytes 61 controls = lymphocytes + granulocytes
Controls had predetermined diagnoses (50 Controls had predetermined diagnoses (50 GERD, 6 post radiation esophagitis, 5 Candida GERD, 6 post radiation esophagitis, 5 Candida esophagitis)esophagitis)Diagnosis in LE group were variable: 40% had Diagnosis in LE group were variable: 40% had Crohn's disease.Crohn's disease.
‘Lymphocytic Esophagitis’‘Lymphocytic Esophagitis’(as it were)(as it were)
Purdy et al (AJCP 2008; 130:508).Purdy et al (AJCP 2008; 130:508).42 pts., retrospective, with control group 42 pts., retrospective, with control group (n=34; non(n=34; non--BE surveillance).BE surveillance).
LE group differ from controls with respectLE group differ from controls with respectLE group differ from controls with respectLE group differ from controls with respectDemographicsDemographicsEndoscopyEndoscopyMedical history (inc. allergies, Crohn’s disease, Medical history (inc. allergies, Crohn’s disease, H. pylori, H. pylori, celiac disease)celiac disease)
Pediatric subset analysis donePediatric subset analysis done
‘Lymphocytic Esophagitis’‘Lymphocytic Esophagitis’(as it were)(as it were)
We see cases of ‘lymphocytic esophagitis.’We see cases of ‘lymphocytic esophagitis.’Significance? Who knows.Significance? Who knows.Should we mention it?Should we mention it?
Lichen planusLichen planus

10/8/2011
12
Esophagitis Dissecans Superficialis Esophagitis Dissecans Superficialis
AKA: Esophagitis dessicans, sloughing AKA: Esophagitis dessicans, sloughing esophagitis.esophagitis.Approximately 100 cases in literature.Approximately 100 cases in literature.A 60A 60 7070Average age 60Average age 60--70 years70 yearsSometimes extreme clinical presentationSometimes extreme clinical presentationTypical symptoms are dysphagia and Typical symptoms are dysphagia and melena.melena.
Esophagitis Dissecans Superficialis Esophagitis Dissecans Superficialis
EndoscopyEndoscopy
Esophagitis Dissecans Superficialis Esophagitis Dissecans Superficialis
PhotoPhoto

10/8/2011
13
Esophagitis Dissecans Superficialis Esophagitis Dissecans Superficialis
PhotoPhoto
Esophagitis Dissecans Superficialis Esophagitis Dissecans Superficialis
Typically treated with highTypically treated with high--dose PPIs.dose PPIs.Complete resolution seems to occur in a Complete resolution seems to occur in a subset of patients.subset of patients.
Major histologic DDx: Vesiculobullous Major histologic DDx: Vesiculobullous dermatoses involving the esophagus.dermatoses involving the esophagus.
Esophageal Vesicobullous DiseaseEsophageal Vesicobullous Disease
Can rarely occur in the absence of skin Can rarely occur in the absence of skin involvement.involvement.Pemphigus (vulgaris, foliaceus, vegitans).Pemphigus (vulgaris, foliaceus, vegitans).
Antibodies against various desmogleins.Antibodies against various desmogleins.Antibodies against various desmogleins.Antibodies against various desmogleins.Vulgaris: Suprabasilar blister.Vulgaris: Suprabasilar blister.Foliaceus: Subcorneal blister.Foliaceus: Subcorneal blister.Vegitans: Acanthosis with inflammation Vegitans: Acanthosis with inflammation (including eosinophils).(including eosinophils).Most (~95%) have positive IF in an Most (~95%) have positive IF in an intercellular pattern.intercellular pattern.

10/8/2011
14
Esophageal Vesicobullous DiseaseEsophageal Vesicobullous Disease
Esophageal Vesicobullous DiseaseEsophageal Vesicobullous Disease
Esophageal Vesicobullous DiseaseEsophageal Vesicobullous Disease
IgG

10/8/2011
15
Take Home PointsTake Home Points
•• EoE: Diagnosis requires clinical EoE: Diagnosis requires clinical correlation. Do not make a definitive correlation. Do not make a definitive diagnosis using only pathologic diagnosis using only pathologic information.information.
•• “Lymphocytic Esophagitis:” We see it; “Lymphocytic Esophagitis:” We see it; who knows what it means.who knows what it means.
•• Esophagitis dissecans superficialis: Rare, Esophagitis dissecans superficialis: Rare, seems to be a real entity.seems to be a real entity.
•• Vesicobullous diseases: Also rare, can Vesicobullous diseases: Also rare, can be limited to the esophagus.be limited to the esophagus.

10/8/2011
1
Non-IBD Chronic Colitis
(malicious mimics and important impostors)
Alexandros D. Polydorides, MD, PhDMount Sinai School of Medicine, NY, NY
Disclosure
I do not have conflicts of interest to disclose for this learning session.
I affirm that all discussions of drug and device use will be consistent with either FDA or compendia (i.e., medical textbook, published medical literature, professional society guidelines) approved indications.
Case presentation
• 67 year old male
• Rare blood in stool, diarrhea
• Occasional abdominal discomfort
• PMH: MVP, aortic aneurysm, thyroid ca
• Rx: Toprol, Synthroid
• Colonoscopy:
- Diminutive polyp (transverse colon)
- Internal hemorrhoids
- Inflammatory stenosis (5 cm long)
stricture at 20 cm
inflammation

10/8/2011
2
sigmoid colon biopsy

10/8/2011
3
Case presentation (additional findings)
• Diverticulosis (descending, sigmoid)
• Stenosis next to extensive area of:
- Erythema, edema, ↓ vascular pattern
- Moderately sized diverticula
rectum
inflammation near diverticula• No evidence of colitis in remaining colon
- Rectum
- Proximal colon
rectum biopsy
Segmental colitis in diverticulosis
• Diverticular disease-associated (segmental) colitis (DAC)
• Diverticular disease-associated chronic colitis
• Diverticular disease-related colitis
• Diverticular colitis
• Segmental colitis associated with diverticular disease (SCAD)
• (Chronic) Segmental colitis associated with diverticula
• Segmental colitis complicating diverticular disease
• Crescentic colitis
• Isolated sigmoiditis

10/8/2011
4
sigmoid colon biopsy
Diverticular colitis – Key points
• Prevalence: 0.3% (all comers), 3-15% (in diverticulosis)
• Presentation: older (>60), male, rectal bleeding, LLQ pain
• Endoscopy: diverticula, erythema, granularity, friability
• Histology: variable inflammation/chronicity, IBD-like
• Association with IBD (“overlap hypothesis”)
- Pathogenesis: luminal antigens, mucosal permeability
- Treatment: mesalazine, infliximab (anti-TNFα), surgery
- Progression: up to 10% → IBD (“blind pouch effect”)
• Diagnosis requires clinicopathologic correlation!

10/8/2011
5
Segmental Colitis Associated With Diverticular Disease and Other IBD Look-alikes.
Harpaz, Noam; Sachar, David.
Journal of Clinical Gastroenterology. 40 Supplement 3:S132-S135, August 2006.
Prevalence and Significance of Inflammatory Bowel Disease-Like Morphologic Features in Collagenous and Lymphocytic Colitis.
Ayata, Gamze; Ithamukkala, Sarathehandra; Sapp, Heidi; Shaz, Beth; Brien, Tom; Wang, Helen;Antonioli, Donald; Farraye, Francis; Odze, Robert.
American Journal of Surgical Pathology. 26(11):1414-1423, November 2002.
TABLE 2. Summary of histologic features in patients with collagenous or lymphocytic colitis.
Case presentation (continued)
• Diverticulitis
• Hartmann’s procedure:
- Sigmoidectomy
- Colostomy
- Rectal stump (“Hartmann’s pouch”)
• Rectal discomfort, mucoid discharge
• Proctoscopy of diverted segment:
- Numerous small (2-3 mm) polyps
- Smooth, but friable mucosa
small polyps
smooth friable mucosa

10/8/2011
6
rectum (diverted) biopsy

10/8/2011
7
Diversion colitis – Key points
• Prevalence: up to 90% (in diverted), 3 mos. to 3 yrs. postop
• Presentation: asymptomatic, tenesmus, discharge
• Endoscopy: stump, erythema, friability, nodularity
• Histology: follicular lymphoid hyperplasia, IBD-like
• Colonic bacteria: ferment dietary starch into short-chain FAs
- Pathogenesis: nutritional deficiency → cell injury
- Treatment: short-chain FA enemas
- Regression: within 3 months after re-anastomosis
• Can coexist with IBD (impossible to discern effect of each)
Case presentation (still going…)
• Reanastomosis (descending colon-rectum)
• Aortic aneurysm repair, severe blood loss
• Post-op: hypotension, shock
- Acute onset abdominal pain
- Hematochezia, vomiting
• Colonoscopy:
- Abnormal segment 35-55 cm
- Erosions, exudate, friability
- Patchy, normal above and below
erosions, exudate
normal patch in-between
left colon biopsy

10/8/2011
8
Colonic ischemia – Pathogenesis
ARTERIAL OCCLUSIONThrombosisEmbolismAtherosclerosis/aneurysm
VASCULAR/INTRINSIC EXTERNAL/EXTRINSIC
CENTRAL CAUSESHypotension/ShockHypoxemia
yRadiation injury
VENOUS OCCLUSIONThrombosis(Mesenteric Veno-occlusive disease)
Hypercoagulable states
VASCULITISArteritisPhlebitisSmall vessel disease
COMPRESSIONVolvulus, intussusceptionAdhesions, tumor
OBSTRUCTIONMotility disorders, strictureTumor, prolapse
MEDICATIONS/DRUGSVasospasmLocalized vasoconstriction

10/8/2011
9

10/8/2011
10
Ischemic colitis – Key points
• Prevalence: increasing incidence, 0.1% hospitalizations
• Presentation: >60, multiple problems, pain, hematochezia
• Endoscopy: edema, “geographic” ulcers, pseudomembranes
• Left colon: splenic flexure, descending, sigmoid
• Histology (early): withered crypts, hyalin, little inflammation
• Chronic/late changes: stricture, crypt distortion, inflammation
• Prognosis: nonocclusive > mesenteric artery thrombosis
• Often significant regenerative atypia
• Spectrum of disorders with common histologic endpoint
Non-IBD chronic colitis – Differential
Structural/mechanical(anatomic/histologic changes)
DIVERTICULAR, MICROSCOPIC, SRUS
Infectious(chronic/granulomatous)C. DIFF, TB, SALMONELLA
Ischemic/vascular(protracted injury)
NSAIDs, BEHCET’S, MVOD, NEC
Immune-mediated(suppression, autoimmunity)
GVHD, CVID, CMV
Treatment-related(prior medical/surgical therapy)
DIVERSION, RADIATION, CELLCEPT, FIBROSING COLOPATHY
References
• Rickert RR. J Clin Gastroenterol. 1984; 6:153.
• Harpaz N, Sachar DB. J Clin Gastroenterol. 2006; 40:S132.
• Yantiss RK, Odze RD. Histopathology. 2006; 48:116.
• Lamps LW, Knapple WL. Clin Gastroenterol Hepatol. 2007; 5:27.
• Odze RD, et al. Saunders, Philadelphia, 2004.
• Fenoglio-Preiser CM, et al. 3rd ed. LWW, Philadelphia, 2008.
• Lauwers GY. Int J Surg Pathol. 2010; 18:48S.
• Greenson JK, et al. LWW, Philadelphia, 2009.
• Nielsen OH, et al. Nat Clin Pract Gastroenterol Hepatol. 2008; 5:28.

Approach To Pathologic Diagnosis Of Motility Disorders Of The Bowel
Dhanpat Jain Associate Professor
Director Program in GI pathology Yale University School of Medicine, New Haven, CT
Introduction: The neuro-muscular organization of the bowel is very similar throughout the gastrointestinal(GI) tract, with some minor variations(1) and the knowledge of the basic organization of the neuromuscular apparatus of the bowel is essential if its motility disorders are to be understood. Normal bowel motility depends on the interplay of smooth muscle, ICC, the intrinsic and extrinsic nerve supply and various neuroendocrine peptides. Abnormality in any of these components may result in bowel dysmotility. The clinical manifestations of the disorders eventually depend on the extent and localization of the abnormality. Some of these disorders present with distinctive clinical features (e.g. idiopathic hypertrophic pyloric stenosis, Hirschprung disease, achalasia); others have non-specific manifestations. Pathogenesis of many of these conditions is still very poorly understood, and many disorders lack specific diagnostic histomorphology. Pathogenesis of some of the rare and familial forms is beginning to be understood and underlying genes that play an important role are being recognized. Currently, the work-up of bowel motility disorders remains a challenge, both for clinicians and pathologists. A variety of primary and secondary motility disorders can involve the GI tract (table 1)A complete discussion of all GI motility disorders is beyond the scope of this review and only selected disorders that involve the large bowel are discussed. Various developmental disorders and conditions seen in children are also not discussed here. Chronic Intestinal Pseudo-obstruction Chronic intestinal pseudo-obstruction (CIPO) is caused by a variety of disorders that may affect different components of the bowel neuromuscular apparatus(2, 3) It most commonly involves the small intestine and/or colon. It may primarily involve the bowel (idiopathic) (Table 2) or be part of a generalized or systemic disorder (secondary) (Table 3). Among the idiopathic/ primary cases, four major categories have been recognized; those with abnormalities of the smooth muscle (myopathic form), those with abnormalities of the neural system (neuropathic form), those with ICC abnormalities, and those with abnormalities of neurohormonal peptides. Clinical features: Many of the clinical features of CIPO are common to the various subtypes. In most cases, particularly the familial ones, symptoms begin in childhood. Some patients remain asymptomatic until middle age, and others are entirely asymptomatic. The diagnosis is often delayed for many years (median 8 years), and repeated exploratory laparotomy or surgery is not uncommon in the clinical history of these patients(4) Symptoms are typical of intestinal obstruction with abdominal distention, pain and vomiting. Distention may be gradual, but may also become severe, especially when both the small intestine and colon are involved. Generally, these patients have alternating diarrhea and constipation, rather than obstipation. Diarrhea is generally

secondary to bacterial overgrowth due to stasis and may result in substantial weight loss. Perforation occurs rarely. Table 1: Chronic Idiopathic Intestinal Pseudo-obstruction (Priamary/Idiopathic) ------------------------------------------------------------------------------------------------------------ Myopathic Forms A. Familial visceral myopathy 1. Autosomal dominant (Type I) 2. Autosomal recessive (Type II ) 3. Autosomal recessive (Type III) 4. Autosomal recessive (Type IV) B. Sporadic visceral myopathy
1. Degenerative, non-inflammatory leiomyopathy i) Sporadic degenerative, non-inflammatory leiomyopathy ii) Non-familial South African leiomyopathy 2. Degenerative, inflammatory leiomyopathy
Neuropathic Forms A. Familial visceral neuropathies
1. Autosomal recessive 2. Autosomal recessive with neuronal intranuclear inclusions
3. Autosomal dominant 4. X-linked B. Sporadic visceral neuropathies 1. Degenerative, non-inflammatory 2. Degenerative, inflammatory C. Developmental abnormalities 1. Hirschsprung disease i). Classic or short segment disease ii) Ultra-short segment disease
iii) Long segment disease iv)Total bowel aganglionosis iv) Zonal aganglionosis
3. Hirschsprung disease allied disorders (Pseudo- Hirschsprung disease) i) Hypognaglionosis ii) Hyperganglionosis
- Gangiloneuromatosis - Intestinal neuronal dysplasia (IND-B)
Disorders of interstitial cells of Cajal 1. With Visceral Myopathy-type changes 2. Without other significant pathologic changes
Disorders of Neurohormonal peptides - --------------------------------------------------------------------------------------------------------------------
A. CIPO-Primary/ idiopathic forms a. CIPO- Myopathic forms Both sporadic and familial myopathic forms are recognized(2) Care must be exercised before considering a case to be sporadic since involved family members may be asymptomatic and a reliable family history may be difficult to elicit. Familial visceral myopathy may also involve other organs (urinary bladder or biliary tract) and has also

been called “hollow visceral myopathy” (5, 6). Type I is the most common, and is characterized by redundant colon, esophageal dilatation, megaduodenum, megacystis, and sometimes uterine inertia. Type II tends to show gastric dilatation, slight small intestinal dilatation often with diverticuli formation, ptosis and external opthalmoplegia. In type III, the entire GI tract, from esophagus to rectum, may be involved and show marked dilatation. Type IV is characterized by gastroparesis, a tubular (narrow) small intestine, normal esophagus and normal colon(7) In general, sporadic cases resemble autosomal recessive familial type III visceral myopathy. Other rare forms, partially resembling type I and III ,with autosomal recessive mode of transmission, and esophageal and cardiac abnormalities have also been described (8). Although smooth muscle degeneration is thought to be responsible for bowel dysmotility, the etiopathogenesis for most of these cases remains obscure. Rare cases with actin or desmin abnormalities have been described(9, 10). In cases of “desmin myopathy,” systemic skeletal and cardiac muscle involvement is also commonly noted. Rare cases show a T-cell rich inflammatory leiomyositis, and are possibly autoimmune in nature(11) A distinctive type of non-familial visceral myopathy has been described in young children from southern, central and eastern Africa (African visceral leiomyopathy)(12) In some cases, absence of c-kit-positive ICC has been thought to be the underlying mechanism(13) (see later). It is likely that this is a heterogenous group of disorders, and many of the histologic changes likely represent end-stage disease. Many cases probably go unrecognized. Thus the spectrum of these disorders may be even wider than is currently known. Pathologic features: The involved segment is often dilated and the bowel wall may appear thick, normal or thin depending on the degree of distention. Although initially no mucosal pathology is often noted, inflammation, ulceration, and ischemia may supervene secondary to stasis and extensive dilatation (2). Microscopy reveals degeneration and fibrous replacement of the smooth muscle. Degenerative changes are most prominent in the muscularis propria, but they also affect the muscularis mucose and, thus, may be identified in mucosal biopsy specimens (14) The longitudinal layer tends to be more severely involved. However, rarely only the inner circular layer is involved (2, 6) In these cases, distinction from scleroderma may be difficult. Muscle degeneration results in fibrosis, cytoplasmic vacuolation, variation in muscle fiber size and thinning of the bowel wall. Fibrosis may be subtle and may require a trichrome stain to be fully appreciated (2). Other changes include nuclear atypia, increased mitotic activity, and PAS positive intracytoplasmic inclusions (15). Ultrastructurally, cytoplasmic inclusions represent aggregates of degenerated myofibrils (15). However, one must be aware of potential artifactual changes in the muscle not uncommonly seen in surgical specimens. Rare cases with deficient smooth muscle α-actin show an absence of staining with smooth muscle actin antibodies, particularly in the inner circular muscle layer (10, 16) however, the significance of this finding has recently been questioned and caution is warranted in interpreting this finding in clinical practice(17). Electron microscopy shows nonspecific degenerative changes, which include mitochondrial vacuolation and may be the only diagnostic evidence of myopathy when light microscopy is normal (2, 6, 12, 18). b. CIPO-Neuropathic Forms

This group includes abnormalities of the intrinsic or extrinsic neural network of the bowel, in both sporadic and familial forms (2, 19) The mode of inheritance may be autosomal dominant, autosomal recessive, or, rarely, X-linked. Autosomal recessive cases tend to show intranuclear inclusions in ganglion cells; some are characterized by mental retardation and basal ganglia calcification. The autosomal dominant variant does not show extraintestinal manifestations. The X-linked form is associated with short small intestine, malrotation and pyloric hypertrophy(20) The etiopathogensis of the neurodegenerative changes remains obscure. Several pathogenetic mechanisms may be involved which include altered calcium signaling, mitochondrial dysfunction, and free radical injury(21) Rare autosomal recessive cases present with a progressive multisystem neurodegenerative disorder and reveal abnormalities in mitochondrial DNA(22) Many genes have been identified that are responsible for the syndromic forms of intestinal pseudo-obstruction that include thymidine phosphorylase (also known as endothelial cell growth factor-1 or ECGF-1), DNA polymerase gamma gene (POLG) and the transcription factor SOX10(23). Mutations in the thymidine phosphorylase have been shown to be responsible for familial cases of Mitochondrial neurogastrointestinal encephalpomyopathy (MNGIE), a disorder characterized by intestinal pseudo-obstruction, progressive external opthalmoplegia, ptosis, polyneuropathy and leukoencephalopathy (24, 25) Decreased ganglion cell survival may be a factor in some cases, as suggested by decreased Bcl-2 gene product in the enteric ganglion cells(26) Some cases reveal inflammatory neuronal degeneration, which suggests an autoimmune or infectious etiology(27); neuronal autoantibodies are detected in some patients(28). Some of these cases represent a para-neoplastic manifestation, while some remain idiopathic (29). Pathologic features: Gross findings are similar to other forms of intestinal pseudo-obstruction and do not help differentiate the various subtypes. Examination of routine sections is often unrevealing except in cases where neurons are markedly decreased in number or when cytomegalovirus-like (CMV-like) intranuclear inclusions can be identified in neurons(19, 30, 31) Electron microscopy has revealed these inclusions to be proteinaceous material composed of curving filaments, and not viral particles. Some cases tend to show lymphocytic or eosinophilic inflammation of the ganglions and myenteric plexus (29). In addition, subtle degenerative changes in neurons and abnormal dendritic processes are also identified (2). These changes are best appreciated with a silver stain on thick en-face/tangential embedded sections of the bowel, or whole mount preparations. Silver stains also help identify abnormalities of argyrophobic and argyrophilic ganglion cell populations however, these stains are obsolete in current practice. A variety of immunohsitochemical markers have been used that include vasoactive intestinal polypeptide (VIP), substance P/ related tachykinis, nitric oxide synthase, neuropeptide Y, calcitonin gene-related peptide and Bcl-2 that show abnormal expression in the enteric nervous system in the neuropathic forms, but lack disease specificity and fail to differentiate primary from secondary changes (4, 29) Of these Bcl-2 has been more widely used as a marker of increased neuronal apoptosis and supports neuropathic changes (26). Limited experience with these conditions, necessity of employing fastidious neuron counting techniques, and tedious silver stains has limited the study of such cases to only a few highly specialized centers. One must also be aware of

artifactual changes in ganglion cells that are frequently encountered in clinical practice, and do not imply neuropathic changes. c. CIPO-ICC abnormalities (Mesenchymopahic form) Recent insight into the role of ICC in bowel motility, and their putative role as the pacemaker cells of the bowel, has led to speculation that they may play a role in chronic idiopathic intestinal pseudo-obstruction. Steel mutant mice which lack c-kit-positive ICCs show marked constipation and features suggestive of chronic intestinal pseudo-obstruction(32, 33). Also, blockade of the c-kit receptor results in severe disturbance of bowel motility(34). Piebaldism in humans, a condition associated with inactivating c-kit mutations, is associated with life-long constipation(35, 36) It has recently been shown that some cases of intestinal pseudo-obstruction show near total, to total, loss of c-kit-positive ICCs(13, 37-39). Rare case of ICC hyperplasia without underlying germline c-kit mutation and presenting as chronic idiopathic intestinal pseudo-obstruction in a pediatric patient has also been reported (40). Pathologic features: Routine H&E stains may show changes typical of visceral myopathy, or appear normal however, immunohistochemistry reveals near total, to total, loss of c-kit-positive ICCs in the involved segment (small bowel and/ or colon). Some cases may show the presence of ICCs but their network may be abnormal, or only a subset of ICCs (submucosal plexus) may be lacking, however, these abnormalities are difficult to appreciate on routine formalin fixed tissues (41). In rare cases with ICC hyperplasia, distinct band like proliferation of benign spindle cells between the two layers of muscularis propria can be appreciated even on H&E stains. These cells stain strongly with C-kit antibody, which makes their recognition as ICC easier (40). d. CIPO- Neurohormonal peptide abnormalities This ill-defined group includes cases of neuroblastoma and ganglioneuroblastoma associated with chronic intestinal pseudo-obstruction(42, 43). Tumor resection results in resolution of the pseudo-obstruction. Vasoactive intestinal polypeptide (VIP) produced by tumor has been implicated as causing intestinal dysmotility. A rare case of pancreatic polypepide cell hyperplasia associated with intestinal pseudo-obstruction has also been reported(44). B. CIPO-Secondary forms Table 2: Chronic Intestinal Pseudo-obstruction (Secondary) -------------------------------------------------------------------------------------------------------------------- A. Associated with Systemic Disorders 1. Progressive systemic sclerosis/polymyositis. 2. Systemic Lupus Erythematosus 3. Progressive muscular dystrophy 4. Myotonic dystrophy 5. Fabry’s disease 6. Parkinson’s disease 7. Multiple sclerosis B. Endocrine and Metabolic Disorders 1. Diabetes Mellitus 2. Hypothyroidism

3. Hypoparathyroidism 4. Pheochromocytoma 5. Acute intermittent porphyria C. Infiltrative Disorders
1. Amyloidosis 2. Diffuse lymphoid infiltration 3. Eosinophilic gastroenteritis D. Paraneoplastic 1. Small cell carcinoma 2. Others F. Infections
1. Trypanosoma Cruizi (Chagas’ disease) 2. Herpes virus 3. Cytomegalovirus 4. Epstein Barr virus 5. Lyme disease 6. JC virus
D. Miscellaneous conditions 1. Ceroidosis (Brown Bowel Syndrome) 2. Small intestinal diverticulosis 3. Radiation enteritis 4. Jejunoileal bypass
H. Toxins and Pharmacologic Agents 1. Tricyclic antidepressants 2. Phenothiazines 3. Ganglionic blockers 4. Clonidine 5. Antiparkinsonism medication 6. Opiates (Narcotic bowel syndrome) 7. Amanita Phalloides toxin -------------------------------------------------------------------------------------------------------------------- a. Systemic disorders Patients with scleroderma or progressive systemic sclerosis may have significant involvement of the bowel; the result of this is a severe motility disorder that often requires surgical resection(45). Clinically, esophageal involvement usually predominates. The inner circular layer is often preferentially involved, in contrast to primary visceral myopathy which involves the outer longitudinal muscle layer preferentially. (2, 46) In scleroderma, collagenous replacement of the muscle layer tends to be nearly complete, unlike the delicate form of interstitial fibrosis characteristic of primary visceral myopathy. Fibrosis may cause muscle weakness resulting in diverticuli with the formation of squared-mouth ostia. Mucosal changes are non-specific and secondary to the underlying motility problem (e.g., reflux esophagitis and villous blunting due to bacterial overgrowth in the small bowel). Pseudo-obstruction, with muscle damage, may occur in patients with dermatomyositis/polymyositis, systemic lupus erythematosus, myotonic dystrophy and progressive muscular dystrophy(45, 47-49) Amyloid deposition in the muscularis propria (myopathy) or myenteric plexus (neuropathy) may, uncommonly, present with intestinal pseudo-obstruction. AA-type amyloid is often deposited in the myenteric plexus while AL-type amyloid is more often deposited in the muscularis propria(50).

Parkinson’s disease, Familial autonomic dysfunction and Shy-Drager syndrome may be associated with dysmotility, but no specific pathologic changes are identified in these conditions. Diffuse polyclonal lymphoid infiltration of the small intestine is another rare condition of uncertain etiopathogenesis(51) Intestinal pseudo-obstruction may also occur in patients with hypoparathyroidism, hypothyroidism, and pheochromocytoma. However, diabetes is by far the commonest endocrine disorder associated with bowel dysmotility and this may result from autonomic dysfunction, electrolyte abnormalities, and vasculopathy. Eosinophilic gastroenteritis and radiation enteritis may also result in intestinal pseudo-obstruction. Destruction of ganglion cells, as a paraneoplastic syndrome, has been well described in patients with small cell carcinoma of the lung and rarely with other tumors as well (52-54) In such cases, neuronal autoantibodies have been detected, and ganglionic destruction is likely to be immune-mediated. b. Drugs and Toxins: A variety of pharmacologic agents (e.g., phenothiazines, tricyclic antidepressants, ganglionic blockers, clonidine and antiparkinsonian medication) have a marked effect on bowel motility, and use or ingestion of naturally occurring toxins such as Amanita phalloides may result in intestinal pseudo-obstruction. c. Infections: Viral infection, particularly with infection with herpes group of viruses, has been associated with systemic autoimmune disturbances and bowel dysmotility. Visceral involvement concurrent with Varicella zoster cutaneous involvement has been shown to result in dysmotility of the stomach, small intestine, colon and anus(55, 56). Bowel dysfunction resolves with improvement of the cutaneous diease. CMV infection has also been implicated in intestinal pseudo-obstruction, especially in immunocompromised individuals(57, 58). In some cases, evidence of EBV infection has been demonstrated by PCR and in-situ hybridization studies of the myenteric plexus(59) Histologically, the only clues may be the presence of inflammatory cells surrounding ganglia and myenteric plexus, or typical viral inclusions in the ganglion cells. Lyme disease and Chagas’ disease may involve the small and/or large intestine resulting in intestinal pseudo-obstruction(60-62). d. Miscellaneous Conditions i. Ceroidosis (Brown Bowel Syndrome): This condition is characterized by deposition of light brown, granular, lipofuscin-like pigment within the smooth muscle cells of the muscularis mucosae and/or muscularis propria of any bowel segment (63, 64). Ultrastructurally, the granular electron-dense material contains myelin figures and abnormal distorted mitochondria. Ceroidosis has been seen in many processes associated with malabsorption, including celiac disease, Whipple’s disease and chronic pancreatitis; vitamin E deficiency have also been implicated as underlying factors. It is unclear whether this is a purely non-specific morphologic marker of a systemic disease or represents a primary smooth muscle disorder. ii. Irritable Bowel Syndrome: Irritable bowel syndrome (IBS) is a common disorder of uncertain etiopathogenesis most commonly affecting adult females. Symptoms include a combination of diarrhea, constipation, bloating and abdominal pain. Disturbance of bowel motility and enhanced visceral sensitivity have been implicated as etiologic factors. Colonoscopy is normal and routine examination of mucosal biopsy specimens does not normally show any pathologic abnormalities. However, quantitative histologic studies, immunohistochemical analysis and ultrastructural studies may show subtle alterations that include an increase in the number of lymphocytes, mast cells, and

enterochromaffin cells(65) These changes point to activation of the enteric immune system and neuro-immune interactions, but have little value in routine diagnostic evaluation of biopsy specimen from IBS patients. Biopsies are often performed merely to rule out other potential causes of the patients symptoms. Diagnostic approach and work-up of cases with intestinal dysmotility Diagnosis of motility disorders remains a challenge for both clinicians and pathologists as any of the disorders lack specific diagnostic pathologic features. A good diagnostic approach to patients with intestinal derangements requires a careful evaluation of the clinical presentation, family history, history of medications, exposure to toxins, imaging and physical findings, and pathologic features (66) Early onset of symptoms, in childhood or in the neonatal period, suggests a developmental or congenital etiology, whereas the majority of motility disorders diagnosed in adults are acquired, or secondary. Many disorders, particularly CIPO have an insidious onset, and the chronic nature of the disease may not be overtly obvious. A family history is often false-negative since the diseases may be mild or sub-clinical, and many affected individuals do not seek clinical attention. The presence of other associated abnormalities (e.g., external opthalmoplegia), dilatation of other segments of GI tract or other viscera (e.g., duodenum, gallbladder or urinary bladder) may point towards an inherited form of visceral myopathy. Careful evaluation of associated symptoms or signs can often lead to the primary cause of bowel dysmotility. Occasionally, the underlying systemic disorder may be diagnosed only subsequent to pathologic evaluation of bowel specimen, as in some collagen-vascular disorders, such as scleroderma. A positive history of medication use, or exposure to toxin, is often difficult to evaluate since many patients consume multiple drugs, and the impact of the drugs on bowel motility may not be well known, or previously reported. A positive history of a preceding viral illness should always be evaluated for a possible infectious/ post-infectious cause of pseudo-obstruction(55, 56) In select cases, serology for circulating anti-neuronal and anti-smooth muscle antibodies may be helpful (11, 23) Endoscopy, laparotomy and radiology may help exclude mechanical causes of intestinal obstruction. GI manometry, although not essential, also helps differentiate mechanical from function obstruction (23, 66) It also helps to differentiate neuropathic from myopathic causes of dysmotility. Other investigations, such as neurologic and autonomic tests, also play a role in the diagnostic work-up. The majority of patients with intestinal obstruction in routine practice are secondary to mechanical causes (e.g., adhesions, extrinsic compression or internal hernia). At present, molecular and genetic tests play a very limited role in the diagnostic work up of motility disorders.
From a pathologists point of view, a careful evaluation of the gross findings and utilization of a systematic approach to the histologic examination of tissue specimen are essential. Mucosal biopsies often show nonspecific findings. An appropriate diagnostic work-up often requires a full-thickness biopsy, combined with electron microscopy and special stains (Table 4). Whenever feasible, some tissue should be frozen, and some immediately fixed in gluteraldehyde for possible electron-microscopy. By light microscopy, careful examination of the mucosal changes and the neuromuscular apparatus should be undertaken. Particular attention should be given to the thickness of the muscle layers, myocyte morphology, pattern of fibrosis, number and morphology of ganglion cells, number and distribution of ICC, presence or absence of neural plexus

hypertrophy or atrophy, and presence of absence of inflammation involving the neuromuscular apparatus. Inflammation surrounding the neural plexus, and ganglionitis, may point towards an infectious or para-neoplastic/ autoimmune neuropathy, whereas dense lymphocytic inflammation limited to the muscular layers may suggest autoimmune leiomyositis(11, 27) However, one should be cautious when evaluating inflammation within the neuromuscular apparatus, since secondary involvement with inflammatory disorders (e.g., inflammatory bowel disease) is more common than primary involvement. Neural hypertrophy and atrophy, although non-specific, may indicate involvement of the neuromuscular apparatus and an underlying motility disorder. Artifactual cytoplasmic vacoulation and nuclear pyknosis in muscle and ganglion cells should be separated from true pathology (Figures 7-10 & 7-13). A panel of histochemical stains (trichrome, congo red and PAS) and immunohistochemical stains (S100, c-Kit, smooth muscle actin and desmin) is helpful in evaluating cases in which routine histological examination is either normal, non-specific, or non-diagnostic. Interstitial cells of cajal are difficult to appreciate on H&E stained tissue sections, and need immunohistochemical analysis with c-kit antibody. Intestinal cells of cajal abnormalities always need to be considered in the differential diagnosis, particularly when routine histology is unremarkable. Although the presence or absence of ICC is easily appreciated on routine tissue sections with immunohistochemical, subtle abnormalities of the deep muscular/submucosal ICC plexus are better evaluated on frozen tissue (41). Quantiation of ICC, and evaluation of their network, is also difficult on routine tissue sections (67, 68). A more detailed evaluation of the enteric nervous system with an elaborate immunohistochemical antibody panel (vasoactive intestinal polypeptide (VIP), substance P/ related tachykinins, nitric oxide synthase, neuropeptide Y, calcitonin gene-related peptide and Bcl-2) should be limited to select cases. Electron microscopy is extremely valuable in some cases, particularly when light microscopy is non-diagnostic. Many degenerative changes in the muscle, neuronal cells or mitochondria can only be detected by ultrastructural examination. Unfortunately, despite extensive work-up, many cases of primary intestinal dysmotility remain of unclear etiology and are a source of frustration for both pathologists and clinicians. Table 3. Diagnostic approach and work-up of Motility Disorders of GI Tract . H&E stain 1
Look for number of ganglion cells, morphology of ganglion cells,
thickness of muscle layers, histology of muscle fibers.
Look for inflammation in the muscle layers, around the ganglion
cells or neural plexuses, and the nature of inflammatory
infiltrate
2 Histochemical stains
a. Trichrome stain
.
Look for pattern fibrosis

b. PAS stain
c. Congo Red
Look for cytoplasmic i
ule out Amyloidosis
nclusion in smooth muscle fibers
R
3 Im histoch. muno emical stains
a. S/100
b. C‐Kit (CD117)
muscle actin c. Smooth
in d. Desm
e. Bcl‐2
Look for neural plexuses
Look for interstitial cells of Cajal
Decreased /absent staining in myopathic cases
cases Decreased /absent staining in myopathic
Decreased staining in neuropathic cases
4. Electron Microscopy
Look for degenerative changes in muscle fibers and ganglion
ondria. cells, or abnormal mitoch
Look for viral infections
Look for interstitial cells of Cajal.

References: 1. Sternberg S. Histology for pathologists, 2nd ed. New York, Lippincott‐Raven. 1997:461‐571.
intestine d colo
2. Krishnamurthy S, Schuffler MD. Pathology of neuromuscular disorders of the small an n. Gastroenterology. 1987 Sep;93(3):610‐39. 3. Coulie B, Camilleri M. Intestinal pseudo‐obstruction. Annu Rev Med. 1999;50:37‐55. 4. Stanghellini V, Cogliandro RF, De Giorgio R, Barbara G, Morselli‐Labate AM, Cogliandro L, et Natural. al history of chronic idiopathic intestinal pseudo‐obstruction in adults: a single center study.
Clin Gastroenterol Hepatol. 2005 May;3(5):449‐58. M dies o t p t5. Schuffler D, Pope CE, 2nd. Stu f idiopa hic intestinal seudoobs ruction. II. Hereditary
hollow visceral myopathy: family studies. Gastroenterology. 1977 Aug;73(2):339‐44. 6. Schuffler MD, Lowe MC, Bill AH. Studies of idiopathic intestinal pseudoobstruction. I. Hereditary hollow visceral myopathy: clinical and pathological studies. Gastroenterology. 1977 Aug;73(2):327‐38. 7. Kansu A, Ensari A, Kalayci AG, Girgin N. A very rare cause of intestinal pseudoobstruction: familial visceral myopathy type IV. Acta Paediatr. 2000 Jun;89(6):733‐6. 8. Mungan Z, Akyuz F, Bugra Z, Yonall O, Ozturk S, Acar A, et al. Familial visceral myopathy with eudo‐obstps ruction, megaduodenum, Barrett's esophagus, and cardiac abnormalities. Am J
Gastroenterol. 2003 Nov;98(11):2556‐60. 9. Ariza A, Coll J, Fernandez‐Figueras MT, Lopez MD, Mate JL, Garcia O, et al. Desmin myopathy:
ultis ol. a m ystem disorder involving skeletal, cardiac, and smooth muscle. Hum Path 1995 Sep;26(9):1032‐7.
d10. Smith VV, Lake BD, Kamm MA, Nicholls RJ. Intestinal pseudo‐obstruction with eficient smooth muscle alpha‐actin. Histopathology. 1992 Dec;21(6):535‐42. 11. Ruuska TH, Karikoski R, Smith VV, Milla PJ. Acquired myopathic intestinal pseudo‐
truct a b o u e yobs ion m y e due t a toimmun enteric leiom ositis. Gastroenterology. 2002 Apr;122(4):1133‐9. 12. Moore SW, Schneider JW, Kaschula RD. Non‐familial visceral myopathy: clinical and pathologic features of degenerative leiomyopathy. Pediatr Surg Int. 2002 Jan;18(1):6‐12. 13. Jain D, Moussa K, Tandon M, Culpepper‐Morgan J, Proctor DD. Role of interstitial cells of Cajal in motility disorders of the bowel. Am J Gastroenterol. 2003 Mar;98(3):618‐24.
t c14. Alstead EM, Murphy MN, Flanagan AM, Bishop AE, Hodgson HJ. Familial au onomic vis eral myopathy with degeneration of muscularis mucosae. J Clin Pathol. 1988 Apr;41(4):424‐9. 15. Fogel SP, DeTar MW, Shimada H, Chandrasoma PT. Sporadic visceral myopathy with inclusion bodies. A light‐microscopic and ultrastructural study. Am J Surg Pathol. 1993 May;17(5):473‐81. 16. Knowles CH, Silk DB, Darzi A, Veress B, Feakins R, Raimundo AH, et al. Deranged smooth
cle amus lpha‐actin as a biomarker of intestinal pseudo‐obstruction: a controlled multinational case series. Gut. 2004 Nov;53(11):1583‐9.
E n a17. Gamba , Carr NJ, Bateman AC. Deficient alpha smooth muscle acti expression as cause of intestinal pseudo‐obstruction: fact or fiction? J Clin Pathol. 2004 Nov;57(11):1168‐71. 18. Wedel T, Van Eys GJ, Waltregny D, Glenisson W, Castronovo V, Vanderwinden JM. Novel
oth smo muscle markers reveal abnormalities of the intestinal musculature in severe colorectal motility disorders. Neurogastroenterol Motil. 2006 Jul;18(7):526‐38. 19. Roper EC, Gibson A, McAlindon ME, Williams LH, Cook JA, Kandler RH, et al. Familial visceral neuropathy: a defined entity? Am J Med Genet A. 2005 Sep 1;137(3):249‐54. 20. Auricchio A, Brancolini V, Casari G, Milla PJ, Smith VV, Devoto M, et al. The locus for a novel
romsynd ic form of neuronal intestinal pseudoobstruction maps to Xq28. Am J Hum Genet. 1996 Apr;58(4):743‐8. 21. Hall KE, Wiley JW. Neural injury, repair and adaptation in the GI tract I. New insights into neuronal injury: a cautionary tail. Am J Physiol. 1998;274:G978‐83. 22. Haftel LT, Lev D, Barash V, Gutman A, Bujanover Y, Lerman‐Sagie T. Familial mitochondrial intestinal pseudo‐obstruction and neurogenic bladder. J Child Neurol. 2000 Jun;15(6):386‐9.

23. De Giorgio R, Sarnelli G, Corinaldesi R, Stanghellini V. Advances in our understanding of the pathology of chronic intestinal pseudo‐obstruction. Gut. 2004 Nov;53(11):1549‐52. 24. Hirano M, Silvestri G, Blake DM, Lombes A, Minetti C, Bonilla E, et al. Mitochondrial
roganeu strointestinal encephalomyopathy (MNGIE): clinical, biochemical, and genetic features of an autosomal recessive mitochondrial disorder. Neurology. 1994 Apr;44(4):721‐7. 25. Nishino I, Spinazzola A, Papadimitriou A, Hammans S, Steiner I, Hahn CD, et al. Mitochondrial
roga nneu strointestinal encephalomyopathy: a autosomal recessive disorder due to thymidine phosphorylase mutations. Ann Neurol. 2000 Jun;47(6):792‐800. 26. De Giorgio R, Santini D, Ceccarelli C. Defective expression of Bcl‐2 in the enteric nervous
em ( l u a f c tsyst ENS): a new potentia ly seful m rker for severe un tional bowel disorders (abstract). I al J Gastroenterol. 1996;28:100. 27. Schobinger‐Clement S, Gerber HA, Stallmach T. Autoaggressive inflammation of the myenteric plexus resulting in intestinal pseudoobstruction. Am J Surg Pathol. 1999 May;23(5):602‐6. 28. Smith VV, Gregson N, Foggensteiner L, Neale G, Milla PJ. Acquired intestinal aganglionosis
circand ulating autoantibodies without neoplasia or other neural involvement. Gastroenterology. 1997 Apr;112(4):1366‐71. 29. De Giorgio R, Barbara G, Stanghellini V, De Ponti F, Salvioli B, Tonini M, et al. Clinical and
phofunmor ctional features of idiopathic myenteric ganglionitis underlying severe intestinal motor dysfunction: a study of three cases. Am J Gastroenterol. 2002 Sep;97(9):2454‐9.
m i30. Schuffler MD, Bird TD, Sumi SM, Cook A. A fa ilial neuronal disease presenting as ntestinal pseudoobstruction. Gastroenterology. 1978 Nov;75(5):889‐98. 31. El‐Rifai N, Daoud N, Tayyarah K, Baydoun A, Jaubert F. Neuronal intranuclear inclusion
ase prese ting a d u t i dise n s chronic intestinal pseu o‐obstr ction in the neona al period n the absence of neurologic symptoms. J Pediatr Gastroenterol Nutr. 2006 Mar;42(3):321‐3. 32. Ward SM, Burns AJ, Torihashi S, Harney SC, Sanders KM. Impaired development of
rstitiinte al cells and intestinal electrical rhythmicity in steel mutants. Am J Physiol. 1995 Dec;269(6 Pt 1):C1577‐85. 33. Isozaki K, Hirota S, Nakama A, Miyagawa J, Shinomura Y, Xu Z, et al. Disturbed intestinal movement, bile reflux to the stomach, and deficiency of c‐kit‐expressing cells in Ws/Ws mutant rats. Gastroenterology. 1995 Aug;109(2):456‐64. 34. Torihashi S, Ward SM, Nishikawa S, Nishi K, Kobayashi S, Sanders KM. c‐kit‐dependent
ldevelopment of interstitial cells and electrical activity in the murine gastrointestina tract. Cell Tissue Res. 1995 Apr;280(1):97‐111. 35. Giebel LB, Spritz RA. Mutation of the KIT (mast/stem cell growth factor receptor) protooncogene in human piebaldism. Proc Natl Acad Sci U S A. 1991 Oct 1;88(19):8696‐9. 36. Fleischman RA, Saltman DL, Stastny V, Zneimer S. Deletion of the c‐kit protooncogene in the human developmental defect piebald trait. Proc Natl Acad Sci U S A. 1991 Dec 1;88(23):10885‐9. 37. Isozaki K, Hirota S, Miyagawa J, Taniguchi M, Shinomura Y, Matsuzawa Y. Deficiency of c‐kit+
in o ticells patients with a myopathic form of chr nic idiopathic intestinal pseudo‐obstruc on. Am J Gastroenterol. 1997 Feb;92(2):332‐4. 38. Yamataka A, Ohshiro K, Kobayashi H, Lane GJ, Yamataka T, Fujiwara T, et al. Abnormal
ribut o i s a hdist ion of intestinal pacemaker (C‐KIT‐p sit ve) cell in an inf nt with chronic idiopat ic intestinal pseudoobstruction. J Pediatr Surg. 1998 Jun;33(6):859‐62. 39. Kenny SE, Vanderwinden JM, Rintala RJ, Connell MG, Lloyd DA, Vanderhaegen JJ, et al.
yed Dela maturation of the interstitial cells of Cajal: a new diagnosis for transient neonatal pseudoobstruction. Report of two cases. J Pediatr Surg. 1998 Jan;33(1):94‐8. 40. Jeng YM, Mao TL, Hsu WM, Huang SF, Hsu HC. Congenital interstitial cell of cajal hyperplasia with neuronal intestinal dysplasia. Am J Surg Pathol. 2000 Nov;24(11):1568‐72. 41. Feldstein AE, Miller SM, El‐Youssef M, Rodeberg D, Lindor NM, Burgart LJ, et al. Chronic
tina l Jintes l pseudoobstruction associated with altered interstitial cells of caja networks. ournal of Pediatric Gastroenterology & Nutrition. 2003;36(4):492‐7. 42. Gohil A, Croffie JM, Fitzgerald JF, Gupta SK, Del Rosario MA. Reversible intestinal pseudoobstruction associated with neural crest tumors. J Pediatr Gastroenterol Nutr. 2001 Jul;33(1):86‐8.

43. Malik M, Connors R, Schwarz KB, O'Dorisio TM. Hormone‐producing ganglioneuroblastoma simulating intestinal pseudoobstruction. J Pediatr. 1990 Mar;116(3):406‐8.
44. Albazaz R, Da Costa PE, Verbeke CS. Pancreatic polypeptide cell hyperplasia of the pancreas. J Clin Pathol. 2006 Oct;59(10):1087‐90. 45. Rohrmann CA, Jr., Ricci MT, Krishnamurthy S, Schuffler MD. Radiologic and histologic
disdifferentiation of neuromuscular orders of the gastrointestinal tract: visceral myopathies, visceral neuropathies, and progressive systemic sclerosis. AJR Am J Roentgenol. 1984 Nov;143(5):933‐41. 46. Venizelos ID, Shousha S, Bull TB, Parkins RA. Chronic intestinal pseudo‐obstruction in two
ents. f s o l tpati Overlap of eatures of sy temic scler sis and viscera myopathy. His opathology. 1988 May;12(5):533‐40. 47. Nowak TV, Ionasescu V, Anuras S. Gastrointestinal manifestations of the muscular dystrophies. Gastroenterology. 1982 Apr;82(4):800‐10. 48. Leon SH, Schuffler MD, Kettler M, Rohrmann CA. Chronic intestinal pseudoobstruction as a complication of Duchenne's muscular dystrophy. Gastroenterology. 1986 Feb;90(2):455‐9. 49. Perlemuter G, Chaussade S, Wechsler B, Cacoub P, Dapoigny M, Kahan A, et al. Chronic intestinal pseudo‐obstruction in systemic lupus erythematosus. Gut. 1998 Jul;43(1):117‐22. 50. Tada S, Iida M, Yao T, Kitamoto T, Fujishima M. Intestinal pseudo‐obstruction in patients
amywith loidosis: clinicopathologic differences between chemical types of amyloid protein. Gut. 1993 Oct;34(10):1412‐7.
huf d a51. McDonald GB, Sc fler MD, Ka in ME, Tytg t GN. Intestinal pseudoobstruction caused by diffuse lymphoid infiltration of the small intestine. Gastroenterology. 1985 Oct;89(4):882‐9. 52. Lennon VA, Sas DF, Busk MF, Scheithauer B, Malagelada JR, Camilleri M, et al. Enteric neuronal autoantibodies in pseudoobstruction with small‐cell lung carcinoma. Gastroenterology. 1991 Jan;100(1):137‐42.
in , E r t n53. Kull g D Reed C , Ve ne GN, Co ton PB, Tar asky PR. Intestinal pseudo‐obstruction as a paraneoplastic manifestation of malignant thymoma. Am J Gastroenterol. 1997 Sep;92(9):1564‐6.
her 54. Lee HR, Lennon VA, Camilleri M, Prat CM. Paraneoplastic gastrointestinal motor dysfunction: clinical and laboratory characteristics. Am J Gastroenterol. 2001 Feb;96(2):373‐9. 55. Kebede D, Barthel JS, Singh A. Transient gastroparesis associated with cutaneous herpes zoster. Dig Dis Sci. 1987 Mar;32(3):318‐22. 56. Pai NB, Murthy RS, Kumar HT, Gerst PH. Association of acute colonic pseudo‐obstruction (Ogilvie's syndrome) with herpes zoster. Am Surg. 1990 Nov;56(11):691‐4. 57. Mathias JR, Baskin GS, Reeves‐Darby VG, Clench MH, Smith LL, Calhoon JH. Chronic intestinal
doo ie t‐lung tpseu bstruction in a pat nt with hear transplant. Therapeu ic effect of leuprolide acetate. Dig Dis Sci. 1992 Nov;37(11):1761‐8. 58. Sonsino E, Mouy R, Foucaud P, Cezard JP, Aigrain Y, Bocquet L, et al. Intestinal
doo Epseu bstruction related to cytomegalovirus infection of myenteric plexus. N ngl J Med. 1984 Jul 19;311(3):196‐7. 59. Debinski HS, Kamm MA, Talbot IC, Khan G, Kangro HO, Jeffries DJ. DNA viruses in the pathogenesis of sporadic chronic idiopathic intestinal pseudo‐obstruction. Gut. 1997 Jul;41(1):100‐6. 60. Pinotti HW, Felix VN, Zilberstein B, Cecconello I. Surgical complications of Chagas' disease:
aesomeg phagus, achalasia of the pylorus, and cholelithiasis. World J Surg. 1991 Mar‐Apr;15(2):198‐204. 61. Chatila R, Kapadia CR. Intestinal pseudoobstruction in acute Lyme disease: a case report. Am J Gastroenterol. 1998 Jul;93(7):1179‐80.
t L C M n e
62. Iantorno G, Basso ti G, Kogan Z, umi CM, abanne A , Fisogni S, et al. The e teric n rvous system in chagasic and idiopathic megacolon. Am J Surg Pathol. 2007 Mar;31(3):460‐8.
t i63. Ruchti C, Eisele S, Kaufmann M. Fa al ntestinal pseudo‐obstruction in brown bowel syndrome. Arch Pathol Lab Med. 1990 Jan;114(1):76‐80. 64. Hitzman JL, Weiland LH, Oftedahl GL, Lie JT. Ceroidosis in the "brown bowel syndrome". Mayo Clin Proc. 1979 Apr;54(4):251‐7.
a 65. Kirsch R, Riddell RH. Histopathological lterations in irritable bowel syndrome. Mod Pathol. 2006 Dec;19(12):1638‐45. 66. Cogliandro RF, De Giorgio R, Barbara G, Cogliandro L, Concordia A, Corinaldesi R, et al. Chronic intestinal pseudo‐obstruction. Best Pract Res Clin Gastroenterol. 2007;21(4):657‐69.

67. Nemeth L, Yoneda A, Kader M, Devaney D, Puri P. Three‐dimensional morphology of gut rvat nal Pediatr inne ion in total intesti aganglionosis using whole‐mount preparation. J Surg. 2001
Feb;36(2):291‐5. 68. He CL, Burgart L, Wang L, Pemberton J, Young‐Fadok T, Szurszewski J, et al. Decreased interstitial cell of cajal volume in patients with slow‐transit constipation. Gastroenterology. 2000 Jan;118(1):14‐21.

Mimics of Celiac Disease David Lewin MD Professor of Pathology Medical University of South Carolina

Nothing to disclose

In non-neoplastic disease of the small intestine, biopsy specimens are typically
obtained from the proximal small intestine in patients with malabsorption and diarrhea. The small intestine biopsy is indispensable in the work-up of mucosal disorders.1, 2 The three most common biopsy abnormalities observed in the evaluation of patients with malabsorption or diarrhea are an unexplained (presumably postinfectious) mild lesion of villous architecture, celiac sprue, and to clinically unsuspected giardiasis. The basic approach is to categorize lesions as specific (diagnostic) or nonspecific (nondiagnostic). In addition, the severity of mucosal injury is subjectively expressed as degrees of abnormality in villous architecture.
NORMAL VILLOUS ARCHITECTURE: A diagnosis of "normal" requires the examination of a number of sections. The villous-to-crypt ratio should be at least 3:1. To feel secure in calling a biopsy normal, three or four villi in a row should be observed. The following can cause artifacts in small intestine biopsy specimens: regional variations in mucosal biopsy appearances, geographic variation, tangential artifact, and endoscopic trauma.
EVALUATION OF A SMALL INTESTINE BIOPSY SPECIMEN: In evaluating jejunal or duodenal biopsy specimens, the initial screening at lower magnification gives an impression of whether obvious abnormalities are present. The overall grading of villous architectural change is usually done at this magnification. Next, a higher magnification is used to look for specific (diagnostic) features and to assess the mucosal components (the villi, crypts, surface mucus, and lamina propria inflammation) systematically in a number of adjoining oriented villi. When an injury occurs, there is typically shortening of the villi, often with hyperplasia of the crypts.
ABNORMAL SMALL INTESTINE BIOPSY APPEARANCE: The severity of the villous abnormality are expressed as mild, moderate, and severe ("flat") lesions. The term variably severe refers to abnormalities in villous architecture that are patchily distributed and of variable severity.
The Mild Lesion: The main abnormalities here are in the surface epithelium and in the lamina propria. Villous height is normal to decreased by a third. The surface epithelium commonly exhibits a disordered nuclear polarity and an increase in intraepithelial lymphocytes. There may be an apparent increase in mitotic figures, with mitotic figures extending higher in the crypts. The lamina propria exhibits an apparent increase in mononuclear cells. Designation of a mild abnormality in a biopsy specimen generally refers to changes in numerous villi.
The Moderate Lesion: The main difference between this grade and the mild lesion is that there is an unequivocal shortening and broadening of villi with more exaggerated surface epithelial and lamina propria round cell changes, usually one-third to two-thirds shortening. The surface epithelium may exhibit both increased numbers of intraepithelial lymphocytes and a cuboid appearance of the enterocytes.
The Severe Flat Lesion: Here the villi are virtually absent. The most common disorder accounting for this appearance is celiac sprue. Others describe the severe flat lesion as "subtotal villous atrophy."
The Thin Flat Mucosa.: This appearance is seen most typically in familial enteropathy (microvillus inclusion disease) and in conditions associated with marked reductions in cell turnover (radiomimetic injury), such as irradiation, chemotherapy, and

severe folate and vitamin B12 deficiency. There are exceptions to the usual severe lesion, namely, the mucosa may be very thin, the usual striking increase in lamina propria inflammatory cells may be minimal or absent, and numbers of mitotic figures may be reduced or at least not increased as they usually are in moderate or severely abnormal biopsy specimens.
Severe Flat Mucosa, Nonspecific and Diagnostic: Celiac Sprue (Celiac Disease, Gluten-Sensitive Enteropathy, Nontropical
Sprue).Pathogenesis: Celiac sprue is a small intestine disease, most common in Ireland and northern Europe, characterized by damage to the small intestine mucosa and malabsorption of a variety of nutrients. The intestinal mucosal injury is due to gluten, a group of proteins found in wheat, rye, barley, and oats.3 The mechanism whereby the gluten causes the mucosal injury is not well understood but involves interactions between environmental, genetic, and immunologic factors. Cell-mediated immune mechanisms appear to play an important role in the tissue injury that occurs in celiac sprue.4 There is strong evidence of genetic predisposition in celiac sprue. For example, the disease is clearly linked with certain HLA antigens, particularly HLA-DQ2 and DQ85 There are two criteria for the diagnosis of celiac sprue. One is demonstration of the characteristic severe flat lesion. Although the lesion is in the nonspecific category, it virtually always indicates celiac sprue in North America. The second criterion for diagnosis is demonstration that malabsorption disappears when gluten is removed from the diet. Serum antigliadin, antiendomysial (EMA), and antitissue transglutaminase (TTG) antibodies are helpful in establishing the diagnosis of celiac disease.6 There sensitivity and specificity depend on the population of patients tested. Most studies have shown that EMA and TTG antibody tests are more sensitive and specific (typically greater than 95%); however, these are IgA antibodies, and response is negative in patients with selective IgA deficiency (10 times higher frequency in celiac patients). The mucosal lesion of celiac sprue is most severe in the duodenum and proximal jejunum. The ileum is usually spared or less severely affected with a mild lesion characterized primarily by an increased number of intraepithelial lymphocytes. In general, the degree of malabsorption is proportional to the length of involved small intestine. Biopsy in Untreated Celiac Sprue.Endoscopic Appearance: Scalloping of the valvulae conniventes with a mosaic pattern or a loss of the valvulae results in a smooth tubular appearance.7 Biopsy specimens from the region of the ligament of Treitz or more proximal in the duodenum show a diffuse flat lesion in symptomatic patients. Traditional Microscopic Findings.The typical lesion shows absence of villi and an abnormal surface epithelium. The surface epithelial cells are histologically often but not invariably cuboid, demonstrate enhanced basophilia, and characteristically contain numerous intraepithelial lymphocytes (cytotoxic T cells)8,9 . Crypts are elongated and contain increased numbers of mitoses, some of which may almost reach the surface epithelium. There is a marked increase in the numbers of lamina propria round cells, predominantly plasma cells. In addition, scattered increased numbers of lamina propria eosinophils and free neutrophils and mast cells may also be observed. More rarely seen are crypt abscesses and sparsely distributed neutrophils in the surface epithelium, but they disappear quickly after gluten withdrawal. Paneth cell numbers may appear reduced. The typical flat mucosa represents one end of the spectrum of pathologic

abnormalities in gluten-sensitive individuals. The "infiltrative lesion" of latent sprue characterizes the earliest recognizable pathologic change. This lesion is characterized by an increase in intraepithelial T cells in the surface epithelium (more than 40 lymphocytes per 100 epithelial cells)9 but no alteration in villous or crypt morphology. This lesion is identified in biopsy specimens from first-degree relatives of celiac sprue patients (latent sprue) and patients with dermatitis herpetiformis. A Marsh classification scheme has been adopted to describe the progression of abnormalities in the celiac mucosa. 10 Some recent studies have suggested using CD3 immunohistochemical stains to help highlight the increased IELs in Marsh I and II lesions. 11
Refractory Celiac Sprue: This refers to individuals who have an initial partial and
laboratory response to a gluten-free diet or have a symptom relapse after an initial excellent response to the diet. It requires documentation that the persistent malabsorption and abnormal biopsy appearance in the face of a strict gluten-free diet are not associated with other causes outlined previously. These patients are usually observed for several years, with repeated evaluations for complications such as lymphoma and collagenous sprue before a final "wastebasket" diagnosis of refractory celiac sprue is made.12
Unclassified Sprue. This is defined as a malabsorption disorder associated with a severe form of diffuse mucosal injury, presumably throughout most of the small intestine including ileum, as evidenced by vitamin B12 malabsorption; it often involves a gastritis and colitis as well. In protracted diarrhea of infancy associated with small intestine mucosal lesions, abnormal immune markers have been identified; autoantibodies to enterocytes and a polyclonal T cell surface expression defect were found in another study.
Collagenous Sprue. This is a rare variant or complication of celiac sprue with approximately 50 cases described in the literature.13 The finding of collagenous sprue usually portends a gloomy prognosis, either a progressive downhill course and death or the requirement for corticosteroid maintenance therapy.14 The mucosa exhibits a severe flat lesion similar to that of untreated celiac sprue, but in addition, there is a unique diagnostic feature of a striking broad band of collagen below the surface epithelial cells that may occupy about one third of the superficial part of the lamina propria. The deposition of collagen may be patchy, especially early in the disease.
Autoimmune Enteropathy: This is characterized by onset of chronic severe secretory diarrhea in early infancy (however adult cases have been reported) that typically does not resolve with the use of elimination diets.15 The diagnostic serologic marker is the presence of circulating autoantibodies directed against gut enterocytes.16, 17 Many patients also have a self or family history of other autoimmune-mediated disorders. The pathologic changes in the small intestine mucosa vary, but there is usually villous atrophy with crypt hyperplasia. There is also an increased mononuclear inflammation in the lamina propria, and neutrophilic infiltration with crypt abscesses may be present in severe cases. The main differential diagnosis is celiac disease; however, autoimmune enteropathy does not have the increase in surface intraepithelial lymphocytes seen in celiac disease. Treatment is with total parenteral nutrition and immunosuppression. Successful therapy and clinical response can be verified with decreasing circulating antienterocyte antibodies.

Other Protein Injury (Soy and Milk Protein), Food Hypersensitivity: Most of the patients in whom food hypersensitivities are identified are children, and the only two documented (in terms of gut abnormalities) are milk and soy hypersensitivities.18, 19 Diarrhea and vomiting are prominent symptoms.
Morphologic Reactions in Food Hypersensitivity. Eosinophils are not a prominent part of the mucosal injury in the first 6 months of life, whereas they may be after 6 months. These older infants, who have delayed reactions, may represent part of the spectrum of eosinophilic gastroenteritis rather than a food hypersensitivity. Some infants have nonspecific mucosal lesions of variable severity with ingestion of soy or milk protein. Prominent eosinophils may be seen in children with allergic proctitis and gastroenteritis with food sensitivities mainly to wheat, soy, and milk proteins.
Tropical Sprue Syndrome. Tropical sprue is defined as a malabsorption syndrome (often with prominent vitamin B12 and folate deficiency) associated with small intestine abnormalities and occurring in parts of the world considered to be tropical.20,21 Tropical sprue has been documented in the Caribbean, India, Malay Peninsula, and Philippines but not in Africa south of the Sahara. The pathogenesis is unclear, although most favor environmental influences, specifically in relation to the microbial flora and enterotoxin production. Biopsy, however, does not separate symptomatic from asymptomatic cohorts in these parts of the world. Many asymptomatic individuals who live in these regions may have mucosal lesions in the proximal small intestine indistinguishable from those of the symptomatic patients. The usual pattern in multiple biopsy specimens is that of variable severity, with lesions ranging from mild to severe. Unlike in celiac sprue, in which the mucosal injury is most severe proximally, the mucosal injury in tropical sprue has been reported to be of similar severity throughout the small intestine.
The usual approach is to treat any suspected tropical sprue patient with a broad-spectrum antibiotic such as tetracycline, vitamin B12, and folic acid for 6 months. Tropical sprue patients improve with this treatment, and the intestinal mucosa usually reverts toward normal. If there is no improvement, a presumptive diagnosis of celiac sprue is made if there is a diffuse flat mucosa.
Childhood Kwashiorkor (Protein-Calorie Malnutrition). This disorder may result in a severe mucosal lesion indistinguishable from that of celiac sprue, except that mitoses may be slightly reduced.22 The mucosa will be fully restored if a normal diet is instituted.
Microvillus Inclusion Disease, Familial Enteropathy (Hypoplastic Villus Syndrome, Congenital Microvillus Atrophy). This severe malabsorption syndrome begins at birth with severe watery diarrhea; it is associated with a thin and translucent intestine wall and a moderate to severe flat lesion.23 The definitive diagnosis is made with electron microscopy. Characteristic microvillus inclusions are more intensely PAS positive than goblet cells, consist of intracytoplasmic vacuoles lined by microvilli, and are contained within the cytosol of enterocytes. Surface microvilli are reduced and irregular.
The etiology of this syndrome is unknown, and the refractoriness of both the mucosal lesion and the malabsorption syndrome to therapy was illustrated by the lack of response to either a gluten-free diet or prolonged total parenteral nutrition. The prognosis is extremely poor because infants are completely dependent on parenteral alimentation for nutritional support and maintenance of hydration and usually die as a result of liver failure caused by therapy.

Variable Severity Lesions: Nonspecific The differential diagnosis in this group is broad. Also, not all patients with these
disorders will necessarily exhibit any abnormality in villous architecture. Variable severity in this group refers to lesions that usually fall into the mild to moderate category, but within any group of three or four biopsy specimens, there may be one that shows a severe flat lesion. The clinical presentation and differential diagnosis of this group of disorders often arise in the evaluation of a new patient with protracted diarrhea or steatorrhea. An underlying infection and postinfectious enteropathy including bacterial overgrowth are by far the most common causes.
Immunodeficiency Syndromes.The three major adult immunodeficiency syndromes24 are 1) common variable immunodeficiency; 2) selective IgA deficiency; and 3) AIDS.
Common Variable Immunodeficiency25, 26 This is a heterogeneous group of immunologic disorders, often familial and characterized by panhypogammaglobulinemia, recurrent infections (especially of the respiratory tree) and malabsorption.27 The mucosa in patients with common variable immunodeficiency may range from normal or only mildly abnormal to completely flat. Accompanying each pattern of villous architectural change, there may be concomitant diffuse NLH. Histologic patterns similar to graft-versus-host disease, inflammatory bowel disease, and Whipple disease have all been described. When infections are treated in these individuals, the severity of the villous architectural lesions generally improves, but usually not to normal. Thus, these individuals may continue to have "intrinsic" mucosal lesions. Although reduced or absent plasma cells may be a specific clue to the diagnosis, they may not be reduced at all. When immunoglobulin staining is done in such individuals, it is found that most of the plasma cells stain with IgG, an intermediate number with IgM, and few or none with IgA. The main challenge for the pathologist is to find evidence of parasitic infestation in reviewing biopsy specimens from these patients.
Selective IgA Deficiency.28 This disorder occurs in approximately 1 in 700 persons, and most of them have no apparent clinical abnormalities. A few, especially if they have an accompanying deficiency in certain IgG subclasses, may have signs of pulmonary disease and malabsorption. Biopsy specimens from these individuals are thus not commonly obtained, and if they are, they do not reveal an absence of plasma cells. However, with immunostaining, the IgA-staining plasma cells are seen to be markedly reduced in number.
AIDS. The main challenge for the pathologist in reviewing biopsy specimens from these patients is to attempt to find evidence of specific infections. When diarrhea originates from a small intestine source in AIDS, it may be due to parasitic infestation, either with organisms known to be more prevalent in the homosexual population at large, such as G. lamblia, or with organisms that tend to produce more protracted illness in this syndrome, namely, Cryptosporidium and Isospara belli. Smears are often more sensitive for organisms. Much more rarely, there is diffuse mucosal involvement with Mycobacterium avium-intracellulare, but acid-fast stains may be needed to detect occasional positive macrophages. In both normal-appearing and abnormal-appearing biopsy specimens, there may be evidence of cytomegalovirus infection, as represented by typical cytomegalic cells with large intranuclear inclusions. Often, in the evaluation of

patients with AIDS and chronic diarrhea, no infection is found, and yet the mucosa may be abnormal, with lesions in the mild or moderate category. This has been termed AIDS enteropathy.29
Cryptosporidium is a coccidial organism first described in humans in 1976. Transmissions of cryptosporidiosis can occur in a variety of ways: from animals to humans, from person to person, and from a common source.30,31 At histologic examination, the organism may be patchily distributed. They have a characteristic appearance with medium- or high-power objectives, namely, pinpoint blue dots primarily concentrated right at the villus tips or the upper sides of the villi. This is in contrast to their location in the rectal mucosa, where they tend to cluster in the rectal crypts. The mucosal changes associated with cryptosporidiosis are focally distributed and variable in severity. Even in patients with AIDS who have massive secretory diarrhea, there may be only mild mucosal injury, although some patients do have more severe lesions. There is sometimes an intense neutrophilic infiltrate of the surface epithelium. Diagnosis of cryptosporidiosis can be made most readily by examination of stool concentrates with use of acid-fast stains and searching for oocytes.32
Cytomegalovirus. Evidence for this infection is usually based on the finding of cytomegalic cells with their typical intranuclear inclusions. In the small intestine, it appears that Brunner glands are more commonly affected. Evidence of the typical cytomegalovirus cells may be found in immunocompromised patients.
Infectious Gastroenteritis. Bacterial Overgrowth Syndrome (Stasis Syndromes, Contaminated Intestine Syndrome). Bacterial overgrowth occurs in diseases that contribute to small intestine stasis.33 The small intestine becomes colonized predominantly by anaerobes and resembles the colon in terms of flora. The most common causes are diabetes mellitus and scleroderma. Others include surgical blind loops as in the afferent limb of a Billroth II anastomosis, jejunal diverticula, localized obstruction, complication of radiation therapy or Crohn's disease, and idiopathic pseudo-obstruction. Although diagnostic maneuvers may suggest the diagnosis (vitamin B12 malabsorption, breath tests),34 culture of small intestine, the ultimate "gold standard," demonstrates an unequivocal clinical and laboratory response to empirical treatment with antibiotics. Some patients have completely normal villous architecture and others have lesions of variable severity, usually in the mild or moderate category. Only rarely is there a severe flat lesion.
Zollinger-Ellison Syndrome (Gastrinoma), Acid Dumping. There are generally only mild to moderate changes in the epithelium, but focal flat biopsy specimens are occasionally encountered as a direct effect of acid on the mucosa.35 Certain clues may suggest a relationship with hyperacidity. If the mucosa is flat, the surface enterocytes are tall and columnar without intraepithelial lymphocytes, in marked contrast to celiac sprue, nor is there the heavy plasmacytic infiltrate in the lamina propria. Neutrophilic infiltrates may be more prominent both in the surface epithelium and free in the lamina propria and may be accompanied by surface microulcerations and gastric mucous cell metaplasia.
Variable Severity Lesions: Diagnostic Eosinophilic Gastroenteritis36 The criteria for the diagnosis should be 1) presence of
gastrointestinal symptoms, 2) histologic evidence of masses of eosinophilic infiltrates in one or more areas of the gastrointestinal tract, and 3) no evidence of obvious causes such

as parasites or systemic disease. The pathogenesis is unclear; some patients have an atopic history and may have high circulating levels of serum IgE. Biopsy diagnosis may not be possible because the dominant infiltrate may be in the muscular or subserosal layers. The disease may dominate in one of three general patterns in decreasing order of frequency: mucosal layer, muscle layer, and subserosal layer.37 Endoscopic examination reveals a spectrum of changes, including an edematous, friable, granular, or nodular mucosa.Histologic Features.The most common sites of involvement are the distal stomach and the small intestine, less commonly the large intestine. By far the greatest remediable cause is an infection with a worm such as Strongyloides, which may not always be easy to detect on repeated examinations of the stool. The distribution of the disease suggests a dietary allergen. In infants, apart from parasites, milk protein or soy protein enterocolitis is frequently associated with an increase in eosinophils, including eosinophilic crypt abscesses in the large intestine. The hypereosinophilic syndrome, which is primarily associated with endomyocardial fibrosis and thromboembolic phenomena, has gastrointestinal involvement with an eosinophilic infiltrate in about half. Many inflammatory lesions of the intestine (e.g., ulcerative colitis, Crohn disease, and reflux esophagitis) contain an increased number of eosinophilis as a component of the inflammatory infiltrate. However, in eosinophilic gastroenteritis, the eosinophils dominate the inflammatory infiltrate, and other inflammatory cells are usually sparse. Polyarteritis nodosa and other vasculitides (e.g., Churg-Strauss syndrome) may show a marked tissue eosinophilia. However, this is typically associated with tissue necrosis and an arteritis.
Crohn Disease: In the small bowel, the terminal ileum is the most commonly affected site, however upper gastrointestinal tract involvement of the distal antrum and proximal duodenum is not uncommon. The gross appearances are discrete and longitudinal ulcers, strictures, serositis, thickened intestine wall, edema, fistula, and fat wrapping. The histologic features are granulomas, apthoid ulcers, transmural lymphoid aggregates, granulomas, neuronal hyperplasia, patchy thickening of the muscularis mucosae, focal architectural distortion, pyloric metaplasia, lymphangiectasia, and submucosal edema38. When a patient has focal active enteritis of the distal antrum and duodenum (usually first and second portions), Crohn disease may be considered even if typical granulomas cannot be found.39
Graft versus Host Disease: This disease is seen after bone marrow transplantation and affects the skin, liver, and gastrointestinal tract. The histologic features are similar to that seen in small bowel transplant rejection biopsies. The earliest changes consist of individual crypt cell necrosis40. More progressive changes include crypt abscesses and subsequent dropout of crypts. Relative to the severity of crypt and epithelial cell injury, the lamina propria may actually appear empty. The most severe lesion is total denudation of the epithelium and an autolyzed appearance of the mucosa similar to that described in detail in the rectal mucosa. A chronic graft-versus-host disease syndrome may develop in a delayed fashion (4 to 12 months) after transplantation. When intestinal involvement occurs, it is manifested by intractable diarrhea, malabsorption, and abdominal pain. Autopsy specimens in these individuals may reveal focal fibrosis, especially in the submucosa and serosa.41

1. Rubin CE, Dobbins WO III: Peroral biopsy of the small intestine. A review of its diagnostic usefulness. Gastroenterology 49:676–697, 1965.
2. Dandalides SM, Carey WD, Petras R, et al: Endoscopic small bowel mucosal biopsy: a controlled trial evaluating forceps size and biopsy location in the diagnosis of normal and abnormal mucosal architecture. Gastrointest Endosc 35:197–200, 1989.
3. Kolberg J, Sollid L: Lectin activity of gluten identified as wheat germ agglutinin. Biochem Biphys Res Commun 130:867–872, 1985.
4. Kagnoff MF: Immunopathogenesis of celiac disease. Immunol Invest 18:449–508, 1989.
5. Liu E, Rewers M, Eisenbarth GS. Genetic testing: who should do the testing and what is the role of genetic testing in the setting of celiac disease? Gastroenterology 128:S33-S37, 2005.
6. Hill ID. What are the sensitivity and specificity of serologic tests for celiac disease? Do sensitivity and specificity vary in different populations? Gastroenterology 128:S25-S32, 2005.
7. Jabbari M, Wild G, Goresky CA, et al: Scalloped valvulae conniventes: an endoscopic marker of celiac sprue. Gastroenterology 95:1518–1522, 1988.
8. Guix M, Skinner JM, Whitehead R: Measuring intraepithelial lymphocytes, surface area, and volume of lamina propria in jejunal mucosa of coeliac patients. Gut 20:275–278, 1979.
9. Marsh MN: Studies of intestinal lymphoid tissue. III. Quantitative analysis of epithelial lymphocytes in the small intestine of human control subjects and of patients with celiac sprue. Gastroenterology 79:481–492, 1980.
10. Marsh MN. Gluen, major histocompatibility complex, and the small intestine. a molecular and immunobiological approach to the spectrum of gluten sensitivity. Gastroenterology 102:330-354, 1992.
11. Mino M, Lauwers GY. Role of lymphocytic immunophenotyping in the diagnosis of gluten-sensitive enteropathy with preserved villous architecture. Am J Surg Pathol. 27:1237-1242, 2003.
12. Collin P, Rennala T, Pukkala E, et al: Coeliac disease—associated disorders and survival. Gut 35:1215–1218, 1994.
13. Weinstein WM, Saunders DR, Tytgat GN, et al: Collagenous sprue—an unrecognized type of malabsorption. N Engl J Med 283:1297–1301, 1970.
14. Holdstock DJ, Olesky S: Successful treatment of collagenous sprue with combination of prednisolone and gluten-free diet. Postgrad Med 49:664–667, 1973.
15. Russo PA, Brochu P, Seidman EG, et al. Autoimmune enteropathy. Ped Devel Pathol 2(1):65-71, 1999.
16. Colletti RB, Guillot AP, Rosen S, et al: Autoimmune enteropathy and nephropathy with circulating anti-epithelial cell antibodies. J Pediatr 118:858–864, 1991.
17. Akram S, Murray JA, Pardi DS, et al. Adult Autoimmune enteropathy: Mayo clinic Rochester experience. Clin Gastroenterol Hepatol 5:12282-90, 2007.

18. Anderson JA: The establishment of a common language concerning adverse reactions to foods and food additives. J Allergy Clin Immunol 78:140–144, 1986.
19. Metcalfe DD: Diseases of food hypersensitivity (editorial). N Engl J Med 321:255–257, 1989.
20. O'Brien W: Tropical sprue. A review. J R Soc Med 72:916–920, 1979. 21. Swanson VL, Thomasson RW: Pathology of the jejunal mucosa in tropical
sprue. Am J Pathol 46:511–551, 1965. 22. Cook GC, Lee FD: The jejunum after kwashiorkor. Lancet 2:1263–1267,
1966. 23. Cutz E, Rhoads JM, Drumm B, et al: Microvillus inclusion disease: an
inherited defect of brush-border assembly and differentiation. N Engl J Med 320:646–651, 1989.
24. Rosen FS, Copper MD, Wedgewood RJ: The primary immunodeficiencies. N Engl J Med 333:431–440, 1995.
25. Cunningham-Rundles C: Clinical and immunologic analyses of 103 patients with common variable immunodeficiency. J Clin Immunol 9:22–33, 1989.
26. Washington K, Stenzel TT, Buckley RH, et al. Gastrointestinal pathology in patients with common variable immunodeficiency and x-linked agammaglobulinemia. Am J Surg Pathol 20(10):1240-1252, 1996.
27. Rosen FS, Wedgwood RJ, Aiuti F, et al: Primary immunodeficiency diseases: report prepared for the WHO by a scientific group on immunodeficiency. Clin Immunol Immunopathol 28:450–475, 1983.
28. Cunningham-Rundles C: Selective IgA deficiency. J Pediatr Gastroenterol Nutr 7:482–484, 1988.
29. Batman PA, Miller ARO, Foster SM, et al: Jejunal enteropathy associated with human immunodeficiency virus infection: quantitative histology. J Clin Pathol 42:275–281, 1989.
30. Hayes EB, Matte TD, O'Brien TR, et al: Large community outbreak of cryptosporidiosis due to contamination of a filtered public water supply. N Engl J Med 320:1372–1376, 1989.
31. D'Antonio RG, Winn RE, Taylor JP, et al: A waterborne outbreak of cryptosporidiosis in normal hosts. Ann Intern Med 103:886–888, 1985.
32. Garcia LS: Fluorescence detection of cryptosporidium oocysts in human fecal specimens by using monoclonal antibodies. J Clin Microbiol 25:119–121, 1987.
33. King CE, Toskes PP: Small intestine bacterial overgrowth. Gastroenterology 76:1035–1055, 1979.
34. Kerlin P, Wong L: Breath hydrogen testing in bacterial overgrowth of the small intestine. Gastroenterology 95:982–988, 1988.
35. Fang M, Ginsburg AL, Glassman L, et al: Zollinger-Ellison syndrome with diarrhea as the predominant clinical feature. Gastroenterology 76:378–387, 1979.
36. Lee M, Hodges WG, Huggins TL, et al: Eosinophilic gastroenteritis. South Med J 89:189–194, 1996.

37. Talley NJ, Shorter RG, Phillips SF, et al: Eosinophilic gastroenteritis: a clinicopathological study of patients with disease of the mucosa, muscle layer and subserosal tissues. Gut 31:54–58, 1990.
38. Kramer K, Kotanogi H, Fazio V, et al: Do microscopic abnormalities at resection margins correlate with increase of anastomotic recurrence in Crohn's disease? A retrospective analysis of 100 cases. Dis Colon Rectum 34:909–916, 1991.
39. Schuffler MD, Chaffee RG: Small intestinal biopsy in a patient with Crohn's disease of the duodenum: the spectrum of abnormal findings in the absence of granulomas. Gastroenterology 76:1009–1014, 1979.
40. Snover DC: Graft-versus-host disease of the gastrointestinal tract. Am J Surg Pathol 14(suppl 1):101–108, 1990.
41. McDonald GB, Shulman HM, Sullivan KN, et al: Intestinal and hepatic complications of human bone marrow transplantation. Parts I and II. Gastroenterology 90:460–477, 770–784, 1986.





























