Embed Size (px)
Citation preview

Contribuição para o Estudo da Fase Nemática Biaxial
em Dendrímeros Líquido-Cristalinos Termotrópicos
Mariana Marcelino Belchior Cardoso
Dissertação para obtenção do Grau de Mestre em
Engenharia Física Tecnológica
Júri
Presidente: Prof. João Carlos Carvalho de Sá Seixas
Orientador: Prof. Carlos Manuel dos Santos Rodrigues da Cruz
Vogais: Prof. João Luís Maia Figueirinhas
Prof. Paulo Ivo Cortez Teixeira
Setembro 2007

i
Agradecimentos
Agradeço, antes de mais, ao Professor Carlos Cruz, orientador deste trabalho, por todo
o apoio, disponibilidade e paciência no decorrer deste trabalho.
Agradeço também ao Professor João Figueirinhas pelas várias sugestões e ideias, pela
preparação das amostras utilizadas, pelo auxilio no uso da consola de espectroscopia RMN, e
pela construção de um novo sistema de rotação. Agradeço, igualmente, a Paul Kouwer, pelos
dados de difracção de raio X e ao grupo do Professor Georg H. Mehl (Universiade de Hull,
Reino Unido) pela síntese dos compostos usados neste trabalho.
Gostaria ainda de agradecer à Fundação para a Ciência e Tecnologia, instituição
financiadora dos projectos POCTI/ISFL/261-516 e PTDC/FIS/65037/2006.
Gostaria também de agradecer a duas instituições, ao Instituto Superior Técnico e à
Fundação Gulbenkian pela atribuição do Prémio Professor António da Silveira.
Finalmente, agradeço ao Centro de Física da Matéria Condensada em particular ao
Grupo de Cristais Líquidos e Ressonância Magnética Nuclear pelo espaço e disponibilidade
cedidos para a realização deste trabalho.

ii
Resumo
Neste trabalho pretende-se contribuir para o estudo da fase nemática biaxial em
tetrapodes organosiloxanos, através da análise estrutural de fases nemáticas destes materiais
e de estudos de ordem molecular em fases nemáticas formadas pelos monómeros
constituintes dos mesmos.
Neste contexto, foram efectuados estudos estruturais por difracção de raios X (DRX)
em amostras alinhadas, nas mesofases formadas por dois tetrapodes organosiloxanos com
estruturas moleculares correlacionadas. Um dos compostos, Tas, apresenta uma fase nemática
biaxial, confirmada por técnicas ópticas e de espectroscopia RMN. No outro composto, Ts, que
apresenta uma fase nemática e uma fase esmética C, a biaxialidade na fase nemática foi
verificada opticamente. Com base nos resultados obtidos por DRX, são propostos modelos de
organização molecular nas mesofases destes compostos.
Com o objectivo de relacionar a ordem nemática biaxial com a estrutura das moléculas,
foram analisados dois monómeros de um dos tetrapodes por espectrocopia de RMN do
deutério de forma a avaliar se as fases nemáticas apresentadas pelos monómeros são
uniaxiais ou biaxiais. Para tal, foram preparadas misturas dos monómeros com sondas
nemáticas deuteradas. Da análise dos espectros resultou que, dentro da precisão permitida
pelo método experimental utilizado, as amostras dos monómeros não evidenciam ordem biaxial
nas respectivas fases nemáticas.
Estes resultados, conjuntamente com os estudos estruturais, sustentam a hipótese de
que a origem da ordem biaxial se relaciona com a especificidade da ligação entre os elementos
mesogénicos e o corpo do dendrímero e com o efeito desta ligação na estrutura peculiar da
fase nemática resultante.
Palavras Chave: Cristais Líquidos, Mesofase Nemática Biaxial, Espectroscopia RMN de
Deutério, Tetrapodes Organosiloxano, Dendrímeros

iii
Abstract
This work is a contribution to the study of the biaxial nematic phase of organosiloxane
tetrapodes based on structural studies of the nematic phases of these materials and molecular
order investigations by NMR in the nematic phases of the corresponding monomers.
In this framework, the structures of the mesophases exhibited by two organosiloxane
tetrapodes with related molecular structures were studied by means of X-ray diffraction (XRD) in
aligned samples. One of these compounds, Tas, presents a biaxial nematic phase confirmed by
optical techniques and NMR spectroscopy. The other compound, Ts, presents nematic and
smectic C phases. Packing models for the studied mesophases are proposed on the basis of
XRD results. Two monomers of the tetrapode Ts were analysed through deuterium NMR
spectroscopy in order to investigate the relationship between the molecular structure of the
tetrapodes and the biaxial ordering in the nematic phase. The key aim of this study was to verify
the existence (or inexistence) of biaxial nematic order in the monomers nematic phase. The
samples used in this analysis were mixtures of the monomers with a deuterated probe. From
this study, no evidence of biaxial nematic order was found, in the nematic phase of the
monomers.
The results obtained from NMR, together with the structural studies, support the
hypothesis that the biaxial order in the nematic phase is related to the structural specificity of
the linkage between mesogenic units and the dendritic core of the tetrapodes and its effect in
the peculiar structure of the resulting nematic phase.
Key Words: Liquid Crystals, Nematic Biaxial Mesophase, Deuterium NMR Spectroscopy,
Organosiloxane Tetrapodes, Dendrimers

iv
Apresentação do Trabalho
Este trabalho teve como objectivo contribuir para a compreensão da mesofase
nemática biaxial em tetrapodes organosiloxanos (dendrímeros de geração zero), através do
estudo experimental da estrutura destes compostos, por difracção de raios X, e ainda do
estudo por espectroscopia de RMN do deutério, nas mesofases nemáticas exibidas por
monómeros constituintes destes tetrapodes.
O trabalho encontra-se dividido em seis capítulos.
Inicialmente (capítulo 1) é feita uma introdução aos cristais líquidos e fases líquido
cristalinas, com particular incidência na fase nemática, atendendo aos objectivos dos estudos
aqui apresentados.
No capítulo 2, segue-se uma breve introdução às técnicas experimentais de difracção
de raios-X e ressonância magnética nuclear, com as quais se estudou a estrutura e ordem
molecular exibidas pelas mesofases destes compostos.
No capítulo 3 apresenta-se um resumo de estudos anteriormente publicados na
literatura sobre fases nemáticas biaxiais e abordagens que tem sido experimentadas na
procura de materiais potencialmente adequados à formação deste tipo de mesofase.
Após esta introdução e contextualização do trabalho segue-se, no capítulo 4, a
apresentação dos resultados de difracção de raios-X obtidos para os tetrapodes
organosiloxanos Ts e Tas. Nesta análise é dado particular ênfase à fase nemática dos dois
compostos e o seu comportamento particular. Os padrões bidimensionais de difracção de raios
X resultam de amostras alinhadas pela presença de um campo magnético. Este tipo de
estudos é particularmente útil para a análise estrutural das mesofases apresentadas por estes
compostos.
Segue-se, no capítulo 5, a apresentação dos resultados de espectroscopia de
ressonância magnética nuclear do deutério, para os monómeros Tm e Tm-NS. Para esta
análise foram preparadas amostras com a presença de uma sonda deuterada e foram obtidos
espectros com as amostras estáticas e em rotação. Da simulação destes espectros concluiu-se
que as amostras apresentam ordem uniaixial, em toda a extensão da gama de temperaturas
correspondente à fase nemática destes compostos.
Por fim, no capítulo 6, apresenta-se uma breve conclusão, após a discussão dos
resultados obtidos, onde se destaca a relação entre as características da ordem biaxial nos
tetrapodes, a respectiva organização molecular na fase nemática e os resultados de RMN
correspondentes aos seus monómeros constituintes.

v
Índice
Capítulo 1 – Generalidades Sobre Cristais Líquidos 1
Mesofases Nemáticas 5
Mesofase Nemática Quiral ou Colestérica 9
Mesofase Nemática Biaxial 10
Mesofases Esméticas 13
Mesofases Colunares 14
Capítulo 2 – Técnicas Experimentais 15
Microscopia Óptica Polarizante 15
Calorimetria Diferencial 16
Difracção de Raio X 17
Ressonância Magnética Nuclear 21
Capítulo 3 – Fase Nemática Biaxial 25
Capítulo 4 – Análise Estrutural dos Compostos Ts e Tas por Difracção de Raios–X 31
Composto Ts 34
Composto Tas 44
Capítulo 5 – Análise dos Compostos Tm e Tm NS por Espectroscopia RMN de Deutério 48
Capítulo 6 – Conclusão e Perspectivas de Trabalho Futuro 61
Bibliografia 65

1
Capítulo 1
Generalidades Sobre Cristais Líquidos
Os cristais líquidos assumiram, nos últimos anos, uma importância crescente devido às
suas múltiplas aplicações tecnológicas [ 1, 2]. Pode-se referir como exemplo disso: mostradores
electro-ópticos, ecrãs planos de computadores e televisão, indicadores de temperatura, janelas
de transparência controlável, válvulas de luz [ 3]. O nome cristal líquido está associado a com-
postos que apresentam formas de organização molecular com características comuns ao
estado sólido e ao estado líquido. Este estado intermédio apresenta geralmente a aparência de
um líquido translúcido, que o distingue dos líquidos “normais” (isótropos) opticamente
caracterizados pela sua transparência. As fases intermédias apresentadas por estes
compostos, ditas líquido-cristalinas são também designadas mesofases. Os compostos com
estas propriedades denominam-se mesogénicos. Estas mesofases conjugam características
macroscópicas típicas do estado sólido, como anisotropia óptica, eléctrica e magnética, com
características mecânicas dos líquidos (fluidez).
Os cristais líquidos podem ser de dois tipos – liotrópicos e termotrópicos. Nos primeiros,
a ocorrência das mesofases é controlada pela concentração de um composto com característi-
cas específicas (anfifílicas) num solvente apropriado, tipicamente, água.
No caso dos cristais líquidos termotrópicos o aparecimento das mesofases é deter-
minado pela temperatura. Estas mesofases surgem em gamas de temperatura, superiores à
temperatura de fusão (transição entre os estados cristalino e líquido-cristalino) e inferiores à
temperatura de clarificação (transição entre os estados líquido-cristalino e líquido isótropo).
A descoberta das fases líquido cristalinas ocorreu em 1888 aquando do estudo da fun-
ção do colesterol nas plantas, pelo botânico austríaco Friedrich Reinitzer. Os primeiros estudos
destes compostos (com aparência de líquidos) por microscopia óptica com luz polarizada,
foram efectuados pelo físico alemão Otto Lehmann. A observação de imagens de microscopia
semelhantes às dos cristais (sólidos) permitiram verificar a anisotropia óptica destes compostos,
tendo daí surgido a denominação de cristais líquidos, atribuída por Lehmann.
Esta introdução abordará essencialmente os cristais líquidos termotrópicos, uma vez
que os estudos apresentados neste trabalho irão incidir sobre um tipo particular destes mate-
riais.
Existem diferentes tipos de moléculas, normalmente compostos orgânicos, que exibem
comportamento líquido-cristalino termotrópico. Estes compostos são caracterizados por
apresentarem estruturas moleculares anisométricas, o que significa que as dimensões das
moléculas dependem fortemente da direcção espacial. Os exemplos mais importantes são as
moléculas calamíticas (em forma de bastonete) e discóticas (em forma de disco). Há no entanto

2
um grande conjunto de moléculas com diferentes geometrias (em geral estruturalmente mais
complexas) que também originam mesofases.
Na Figura 1 apresentam-se dois exemplos esquemáticos de moléculas mesogénicas,
respectivamente, calamíticas e discóticas, numa fase líquido cristalina. Nos cristais líquidos de
baixo peso molecular, (com massas moleculares tipicamente da ordem das centenas de unida-
des de massa atómica) as moléculas coincidem com as respectivas unidades mesogénicas
(elementos estruturais que determinam a simetria da mesofase).
Figura 1 – Representação esquemática de moléculas líquido cristalinas em forma de bastone-
tes (a) e de discos (b) (ambas numa fase líquida cristalina nemática).
Noutros casos, estas unidades megosénicas podem ser combinadas por ligação
química através de segmentos, geralmente flexíveis (por exemplo, cadeias alifáticas), dando
origem a estruturas moleculares de maior complexidade que apresentam ainda comportamento
líquido-cristalino. O exemplo mais comum deste tipo de estruturas são os cristais líquidos
poliméricos, cuja importância tem vindo a aumentar progressivamente nos últimos anos, devido
às suas aplicações tecnológicas, como fibras kevlar [ 5] e aplicações ópticas [ 6].
Os polímeros resultam da repetição de unidades elementares, formando cadeias muito
longas a que se associa grande flexibilidade. Os polímeros líquido cristalinos têm adicional-
mente unidades mesogénicas ao longo do polímero que lhe vão conferir comportamento e
fases características dos compostos líquido cristalinos. Os polímeros líquido cristalinos podem
ser divididos em dois grandes grupos, dependendo do posicionamento das unidades estrutu-
rais nas moléculas. No caso em que estas unidades se encontram ligadas lateralmente à ca-
deia polimérica, diz-se que se trata de polímeros de cadeia lateral. Alternativamente, no caso
em que as unidades mesogénicas estão incluídas ao longo da cadeia polimérica diz-se que se
trata de polímeros de cadeia principal [ 1]. A introdução destas unidades mesogénicas, de uma
ou outra forma, induz uma organização diferente da que o polímero tomaria na ausência das
mesmas e pode conferir diferentes estruturas moleculares que poderão dar origem a diferentes
mesofases.
As unidades mesogénicas incluídas nos polímeros líquidos-cristalinos podem ser cala-
míticas, discóticas ou de outro tipo. Podem obter-se diversos tipos de polímeros por combina-
ção de diferentes unidades mesogénicas ligadas em alternativa ou complementarmente em
cadeia lateral ou cadeia principal (ver Figura 2).
a) b)

3
Figura 2 – Representação esquemática de polímeros líquido-cristalinos de cadeia lateral (a) e
de cadeia principal (b).
Para além das estruturas lineares, representadas pelos polímeros, podem obter-se
moléculas de maior complexidade através, por exemplo, de substituição lateral de elementos
das cadeias poliméricas, reticulação (cross linking), ou ramificação (ver Figura 3). Neste último
caso, através de um processo de síntese sequencial, produzem-se moléculas com uma
estrutura em “árvore” designadas por dendrímeros [ 7]. Uma vez que, neste caso, o número de
camadas de ramificação (geração do dendrímero) pode ser controlado no processo de síntese,
obtém-se compostos com massa molecular bem determinada (monodispersos), contrariamente
ao que se passa com os polímeros. Os dendrímeros apresentam assim, maior organização
estrutural, contrariamente aos polímeros em que existe grande dispersão (associada a
distribuição estatística de massas moleculares) e desorganização dentro de uma amostra. Em
geral, devido à estrutura molecular característica, os dendrímeros de geração suficientemente
elevada tendem a adoptar estruturas globulares.
Figura 3 – Esquema de alguns dendrímeros líquido cristalinos de diferentes gerações.
Dendrímeros de segunda geração (a), terceira geração (b), geração zero (tetrapode) (c),
geração zero (octopode) (d).
À semelhança dos polímeros líquido cristalinos, os dendrímeros podem incluir na sua
estrutura unidades mesogénicas, que no caso mais comum se situam na camada periférica. A
presença destas unidades, associada à flexibilidade inerente ao corpo dendrimérico, confere a
estas moléculas a possibilidade de assumirem diversas formas. Daí resulta o aparecimento de
a) b)
a) b) c) d)

4
diferentes mesofases consoante a configuração molecular mais favorável, em função do ba-
lanço energia-entropia, para uma dada temperatura.
Os sistemas que serão abordados neste trabalho são o caso particular de dendrímeros
de baixa geração, tetrapodes (Figura 3), e os respectivos monómeros [ 9]. Estes sistemas são
especialmente interessantes pelo facto de apresentarem uma fase nemática biaxial a baixas
temperaturas. Esta fase, como será referido adiante, apresenta, para além do interesse em
termos de investigação fundamental, aspectos ligados a potenciais aplicações tecnológicas.
Os cristais líquidos apresentam propriedades intermédias relativamente às proprieda-
des dos líquidos isótropos e dos cristais. Estas propriedades relacionam-se com o tipo de
ordem que as diferentes mesofases apresentam. Numa classificação muito geral é possível
definir três tipos de mesofases: nemáticas, esméticas e colunares (Figura 4) [ 4, 8]. No caso das
fases nemáticas, mais próximas dos líquidos isótropos, verifica-se a existência de ordem
orientacional, reflectindo a tendência para o alinhamento das moléculas numa direcção média
comum. Numa fase esmética além de ordem orientacional existe ordem posicional a uma
dimensão. Neste caso, e à semelhança do que acontece num sólido há correlação (não local)
entre as posições das moléculas, mas apenas numa dimensão, o que significa que as
moléculas se organizam em camadas. E por fim, uma fase colunar apresenta ordem
orientacional, e ordem posicional a duas dimensões, o que significa que as moléculas (neste
caso, tipicamente em forma de disco) se dispõem em colunas paralelas, organizadas no plano
perpendicular ao eixo das mesmas. Nos extremos, estão, por um lado, os líquidos isótropos,
que não apresentam ordem orientacional nem ordem posicional, e por outro, os cristais, que
apresentam ordem posicional nas três dimensões. Segue-se uma apresentação destes
diferentes tipos de mesofases, dando particular enfase à fase nemática, uma vez que este
trabalho incide sobre o estudo da fase nemática biaxial, e à fase esmética, uma vez que um
dos compostos analisados também apresenta fases esméticas.
Figura 4 – Esquematização dos diferentes tipos de mesofases, nemáticas (a), esméticas (b) e
colunares (c).
a) b) c)

5
Mesofase Nemática
Uma fase nemática apresenta ordem orientacional, caracterizada pela existência de um
vector que define a orientação média das moléculas, designado por vector director ou director
(ver Figura 5). A ordem posicional que as moléculas experimentam restringe-se a uma
vizinhança local. Diz-se desta mesofase que apresenta ordem posicional de curto alcance.
Considerando cristais líquidos com origem em moléculas calamíticas, é possível definir um eixo
molecular principal que indica a orientação natural das moléculas numa fase nemática, como
se ilustra na Figura 5.
O facto da ordem posicional ser apenas de curto alcance permite que, à semelhança
do que acontece nas fases líquidas isótropas, os centros de massa das moléculas estejam
aleatoriamente distribuídos. A ordem orientacional garante que as moléculas fiquem orientadas
preferencialmente numa direcção. As posições dos centros de massa variam no tempo, à
semelhança do que acontece num líquido isótropo, devido a fenómenos de difusão e agitação
térmica. A densidade desta mesofase, assim como acontece num líquido isótropo, não depen-
de das coordenadas, é uniforme no espaço (ρ(r)=ρ0).
Figura 5 – Esquema representativo de uma fase nemática e da direcção média de orientação
das moléculas e eixo molecular principal.
A ordem orientacional pode ser definida por um tensor de segunda ordem simétrico de
traço nulo designado tensor de ordem de Saupe. Considere-se o referencial cartesiano do
laboratório (x,y,z) e um referencial solidário com uma molécula calamítica definido pelas
coordenadas de Euler, (ξ,η,ζ), Figura 6. No sistema de coordenadas do laboratório, o eixo z
coincide com o director, n.

6
Figura 6 – Definição dos ângulos de Euler para uma molécula calamítica.
O tensor de ordem de Saupe vem então dado por:
(1)
com α, β = ξ, η, ζ, onde os vectores k são as projecções do director n (orientado segundo o
eixo z) em cada um dos eixos ξ, η, ζ associados à molécula e onde ⟨.⟩ denota a média num
domínio nemático. Uma vez que se trata de um tensor simétrico de traço nulo, pode ser
diagonalizado obtendo-se os parâmetros S e D a partir dos valores próprios:
ψθηη
θ
ξξ
ζζ
2cossin2
3
1cos32
1
2
2
=−=
−==
SSD
SS
(2)
O parâmetro S mede a ordem do eixo longitudinal da molécula, ou seja, mede o desvio
do eixo molecular em relação ao director. Constata-se que o parâmetro de ordem orientacional,
S, é dado pela média sobre as moléculas da amostra do segundo polinómio de Legendre
P2(cosθ) (onde P2(x)= ½(3x2-1)). O parâmetro D mede o grau de anisotropia transversal (acha-
tamento) da molécula.
A definição para o parâmetro de ordem orientacional ou parâmetro de ordem nemático
é conveniente uma vez que toma o valor zero para uma amostra isótropa, organizada aleatoria-
mente, e o valor 1 para uma amostra perfeitamente alinhada. Em geral, o parâmetro de ordem
diminui com o aumento da temperatura. Verifica-se uma abrupta diminuição no parâmetro de
αβ β α αβ δ θ θ − = cos cos 3 2
1 S
θ
ψ
Z
Y
X
ζ
n
ξ
η
⊥ ao plano zζ φ

7
ordem para zero, quando a amostra experimenta uma transição de uma fase líquido-cristalina
para a fase isótropa (transição de fase de primeira ordem). Este parâmetro de ordem pode ser
determinado experimentalmente, por exemplo através de RMN. Em geral, para cristais líquidos
na mesofase nemática, S toma valores que se situam tipicamente entre 0.4 e 0.7 sendo a sua
variação principalmente controlada pela temperatura.
Os compostos nemáticos não assumem uma orientação preferencial, pelo que os dois
sentidos que a direcção média permite são equivalentes. O sentido do vector director não é
importante, apenas a sua direcção importa. Se um composto contém dipolos permanentes,
com componente não nula segundo o director, então numa qualquer amostra haverá tantos
dipolos num sentido como no sentido inverso e o sistema não será ferroeléctrico.
O alinhamento segundo uma direcção bem definida das moléculas de um cristal líquido
nemático dá origem a uma anisotropia óptica. Ou seja, uma onda polarizada segundo n propa-
ga-se com um velocidade diferente de uma onda polarizada perpendicularmente a n. Isto
significa que existe uma diferença entre os índices de refracção paralelo e perpendicular (∆n =
n ⁄ ⁄ - n⊥) que evidencia a birrefringência dos cristais líquidos, resultante principalmente da
anisotropia dieléctrica ∆ε = ε ⁄ ⁄ - ε⊥ (para as frequências ópticas).
O facto de existir anisotropia nas mesofases nemáticas indica a presença de forças
intermoleculares, que têm a propriedade de conferir orientação às moléculas, sem impedir a
sua mobilidade, como num líquido isótropo.
A teoria contínua elástica descreve o que se passa com as distorções que uma amos-
tra nemática experimenta. Esta teoria considera perturbações numa amostra que se impõe
orientada, mas cujo alinhamento pode não ser uniforme ao longo de toda a amostra. Há então
três constantes elásticas que importa definir K1, K2, K3 – constantes elásticas de Frank – que
descrevem os três modos básicos de distorção, que são, respectivamente: afunilamento, torção
e flexão (ver Figura 7) . A energia livre de Frank é dada por:
(3)
Figura 7 – Definição das geometrias de distorção fundamentais para amostras confinadas: afu-
nilamento (a), torção (b) e flexão (c).
A distorção relativamente à orientação uniforme é resultado da adaptação do composto
às condições a que se encontra sujeito, ou seja, à presença de defeitos (por exemplo
( ) ( ) ( ) ( ) dv n n K n n K n K F vol d ∫ × ∇ × + ∇ ⋅ + ⋅ ∇ =
2 3
2 2
2 1
2
1 ×
a) b) c)

8
originados por impurezas), às condições fronteira das superfícies com que contacta ou à
presença de um campo externo, e resultam de uma condição de minimização de energia. A
resposta das amostras a diferentes constrangimentos pode ser decomposta nos três modos de
distorção fundamentais caracterizadas pelas respectivas constantes elásticas.
Considerando uma amostra nemática depositada sobre uma superfície plana, as molé-
culas do composto tenderão, nos dois casos mais comuns, a alinhar-se paralelamente à super-
fície, ou perpendicularmente a esta: Estes dois tipos de alinhamentos designam-se, respectiva-
mente, alinhamento planar e alinhamento homeotrópico, Figura 8.
Figura 8 – Amostra nemática depositada numa superfície; alinhamento planar (a), alinhamento
homeotrópico (b).
Um amostra, em geral, pode estar confinada entre diferentes superfícies. O alinhamen-
to das moléculas junto a cada superfície, pode ser planar ou homeotrópico, daí resultando
diferentes configurações. Os tipos de alinhamentos fundamentais, mais simples, que as
moléculas podem assumir quando confinadas entre duas superfícies planas (casos mais
relevantes no estudo de cristais líquidos) estão resumidos na Figura 9. Configurações mais
complexas resultam de o director ter tendência a assumir uma configuração uniforme, mesmo
quando se registam alinhamento distintos junto a cada superfície. A configuração uniforme que
o director tende a assumir indica a presença de forças internas, na mesofase, as quais são de
natureza elástica, descritas pela teoria contínua elástica.
Figura 9 – Tipos de alinhamento fundamentais que as moléculas tomam quando confinadas
entre duas placas paralelas. A: planar uniforme. B: homeotrópica. C: planar helicoidal.
Uma amostra nemática pode alinhar-se não só devido ao confinamento entre superfí-
cies, mas também pela aplicação de campos (magnético ou eléctrico).
Na presença de um campo exterior, uma amostra nemática tem tendência a alinhar-se
com o campo aplicado (se o alinhamento é paralelo ou perpendicular ao campo aplicado é
função da anisotropia, da susceptibilidade magnética ou eléctrica, ou seja, se esta for positiva o
a) b)
a) b) c)

9
alinhamento será paralelo, e será perpendicular se a anisotropia for negativa). A equação
seguinte, válida para uma amostra nemática (caracterizada por um director n), dá a relação
entre o vector magnetização, o campo aplicado e as componentes da susceptibilidade
magnética do material relativamente ao director nemático (na zona de resposta linear ao cam-
po aplicado):
(4)
Como os cristais-líquidos, em particular a fase nemática têm como característica serem
fase anisótropas, quando uma amostra é sujeita a um campo magnético, há uma interacção
entre o campo introduzido e a orientação do nemático que origina um termo adicional na densi-
dade de energia livre.
( )2
0
2
0
B
0mag nB
2
1B
2
1dBMF ⋅χ∆
µ−χ
µ−=⋅= ⊥∫
(5)
Uma situação análoga verifica-se na presença de um campo eléctrico exterior, no
entanto a situação onde há um campo magnético exterior será mais relevante no contexto
deste trabalho, uma vez que este foi o utilizado para promover o alinhamento das amostras.
Quando se verifica a competição entre o alinhamento induzido pela presença de cam-
pos exteriores e efeitos de superfície dá-se uma transição entre um e outro alinhamento, em
função da intensidade do campo, que é descrita no quadro das chamadas transições de
Fredericksz. Esta competição está na origem dos ecrãs de cristal líquido nemático, que são
uma importante aplicação tecnológica dos cristais líquidos.
Para além da fase nemática uniaxial (isto é definida por um único eixo de simetria)
existem outras variantes de fases nemáticas que importa referir e que serão apresentadas de
seguida.
Mesofase nemática quiral ou colestérica
Uma destas mesofases é a mesofase colestérica ou nemática quiral. A origem deste
nome, colestérico, vem da família dos primeiros compostos líquido cristalinos identificados,
derivados do colesterol, que são constituídos por moléculas quirais. Entenda-se por moléculas
quirais: moléculas que não são idênticas à sua imagem num espelho, ou seja, estas moléculas
não são invariantes perante uma reflexão.
Um composto que exiba uma mesofase colestérica pode ser descrito como um nemá-
tico com uma torção natural uniforme em torno de um eixo normal ao director local, pelo que
) n ( n B 1
B 1
M 0 0
⋅ χ ∆ µ
+ χ µ
= ⊥

10
nestes compostos o director local roda ao longo da amostra, de forma periódica. Esta torção
natural demonstrada por estes compostos deve-se ao facto de as moléculas não se alinharem
numa determinada direcção de forma exactamente paralela. Procuram antes uma forma de
emparelhamento aproximadamente paralelo que acaba por induzir rotação, com um ângulo
residual, umas relativamente às outras. Não se criam no entanto camadas de moléculas onde o
director toma estas diferentes direcções, Figura 10. Na direcção normal à evolução helicoidal
do director verifica-se uma evolução gradual da orientação das moléculas. Assim ao avançar
no eixo perpendicular à torção, percorrendo um passo p, conhecido por passo da hélice,
chega-se a uma situação de alinhamento idêntica à inicial. De notar que os dois sentidos que o
director pode tomar são equivalentes, pelo que o passo da hélice correspondente a uma
rotação de 360º na orientação do director define no espaço uma distância que é o dobro do
período de repetição da estrutura molecular.
Figura 10 – Estrutura de uma fase colestérica, variação especial do respectivo director local e
definição do passo da hélice (trata-se de uma fase nemática, as camadas indicadas são
apenas para facilitar a compreensão do esquema, não existem num sistema real),.
A anisotropia óptica dos colestéricos é negativa. Outra particularidade é a ordem de
grandeza deste passo, em geral é da ordem das centenas de nanómetros, ou seja, na escala
do visível. Estas duas propriedades, anisotropia óptica e passo com comprimento na ordem
dos comprimentos de onda da luz visível, são interessantes para a aplicação a dispositivos de
cristais líquidos.
Por fim, uma fase nemática não quiral pode ser entendida como um caso limite de uma
fase nemática quiral com um passo infinito.
Mesofase nemática biaxial
As fases nemáticas uniaxias são definidas por um único eixo relativamente ao qual se
dá o alinhamento preferencial das moléculas, sendo os dois outros eixos equivalentes (neste
caso as moléculas podem considerar-se em média como objectos com simetria cilíndrica). No
entanto, é possível definir, num caso mais geral, a chamada fase nemática biaxial. Neste caso
p/2

11
as moléculas para além de se orientarem segundo o seu eixo maior, ou principal, também o
podem fazer adicionalmente em relação a um eixo secundário, o que não acontecia para os
nemáticos uniaxiais (Figura 11).
Figura 11 – Exemplo de uma molécula calamítica e respectivo director, e uma molécula tipo
“tábua” e os directores principal (n1) e secundários (n2 e n3).
Um domínio de um composto que exiba uma fase biaxial continua a ser homogéneo no
espaço, no entanto apresenta três eixos ópticos distintos, ao contrário de uma fase uniaxial que
só apresenta um eixo preferencial em torno do qual o sistema é simétrico para rotações e um
eixo ortogonal a este, desfavorável à propagação de luz. O grupo de simetria pontual para um
nemático biaxial é D2h.
A biaxialidade presente numa fase nemática biaxial, pode ser discutida em função da
estrutura dos compostos e da estrutura local da fase nemática resultante.
A ordem biaxial pode ser descrita com base na matriz de ordem (6).
(6)
Onde i,j = X,Y,Z, referente aos eixos do referencial da fase (Z coincide com o director principal,
em cada domínio nemático), α,β = ξ,η,ζ, referentes aos eixos do referencial molecular, θiα,θjβ
ângulos entre o referencial da fase e o referencial molecular.
A existência de ordem nemática biaxial reflectir-se-à na medição de qualquer grandeza
física tensorial de segunda ordem relacionada com a estrutura da fase. Enquanto numa fase
uniaxial uma grandeza deste tipo poderá ser definida no seu referencial principal apenas por
duas quantidades, Q|| e Q⊥, numa fase biaxial são necessárias três quantidades (associados
aos três eixos do referencial principal) Qxx, Qyy e Qzz para definir essa grandeza. Um exemplo
de uma grandeza com estas características é o tensor gradiente de campo eléctrico na
vizinhança de uma ligação Carbono-Deutério (C-D) numa molécula deuterada.
Segundo a formulação de Straley [ 10], os elementos do tensor gradiente de campo
eléctrico médio (AFG, do inglês, averaged field gradient) associados a cada domínio, no
referencial da fase, podem ser relacionados com os valores das componentes principais do
n n1
n2
n3
( ) αβ β α αβ δ ij θ θ − =
j i ij S cos cos 3
2 1

12
tensor gradiente de campo eléctrico médio (Vii) associado à ligação C-D, no referencial
molecular, utilizando quatro parâmetros de ordem: S, D, P e C. Estas componentes
determinam o parâmetro de assimetria, que caracteriza uma fase nemática biaxial:
ZZ
YYXX
V
VV −=η
(7)
As componentes do tensor gradiente de campo médio, no referencial da fase, podem
ser determinadas em função de quatro parâmetros de ordem, S, D, P e C, e das componentes
do tensor AFG no referencial molecular:
(8a)
(8b)
Os parâmetros de ordem são dados por:
(9a)
(9b)
(9c)
(9d)
Os ângulos indicados reportam uma vez mais a Figura 6. Adicionalmente, podem agora
obter-se os parâmetros P e C, que não apareciam no caso de fases uniaxiais. O parâmetro S
continua a representar a ordem orientacional nemática e mede a tendência do eixo molecular
se alinhar com o director, no domínio em que a molécula se encontra. O parâmetro D,
anteriormente introduzido, relaciona-se com o achatamento das moléculas numa fase uniaixial,
e mede a diferença na tendência da projecção do director principal no plano ξη se alinhar com
os eixos ξ η moleculares. O parâmetro P é um parâmetro biaxial que mede a diferença na
2
1 cos
2
3 2 − = ≡ θ Z ζζ S S
ψ θ 2 cos sin 2
3 2 = − ≡ Z ηη
Z ξξ S S D
γ θ 2 cos sin 2
3 2 = − ≡ Y ζζ
X ζζ S S P
( ) ( )
γ ψ θ γ ψ
θ 2 sin 2 sin cos 3 2 cos 2 cos 2
3 cos
2
3 2 −
+ =
= − − − ≡ Y ηη
Y ξξ
X ηη
X ξξ S S S S C
( ) ζζ ηη ξξ
YY XX V P V V C
V V . 3
+ − = −
( ) ζζ ηη ξξ ZZ V S V V
D V .
3 + − =

13
tendência do alinhamento da projecção do eixo molecular no plano XY em alinhar-se numa
direcção, X ou Y. E C é um parâmetro intrínseco de biaxialidade molecular para uma fase
biaxial. Numa experiência de espectroscopia RMN de deutério estes parâmetros de ordem não
são determinados directamente. O parâmetro relevante numa experiência deste tipo é o
parâmetro de assimetria, que se relaciona com os parâmetros de ordem através das
expressões (8) e (9).
Mesofases Esméticas
As mesofases esméticas caracterizam-se pela organização das moléculas em cama-
das, sobrepostas de espessura idêntica. Nesta mesofase (para além de ordem orientacional,
que neste caso é em geral superior ao que se verificava na mesofase nemática), os cristais
líquidos exibem ordem posicional, numa determinada direcção.
As camadas podem ser desordenadas (por exemplo: fases esméticas A, SmA ou C,
SmC) ou ordenadas (por exempo: fase esmética B, SmB).
Nas mesofases esméticas de camadas desordenadas, a distribuição dos centros de
massa das moléculas faz-se, dentro de cada camada, de forma aleatória, estabelecendo-se
uma periodicidade espacial da densidade na direcção normal às camadas, Figura 12.
Figura 12 – Estrutura de fases esméticas, para o caso de moléculas desordenadas, SmA (a) e
para o caso de uma fase de moléculas inclinadas, SmC (b).
Há dois tipos de fases esméticas de camadas desordenadas, as ortogonais (em que as
de moléculas se dispõem perpendicularmente às camadas), SmA, e as de moléculas inclinadas,
SmC (Figura 12) (em que as moléculas apresentam um média uma inclinação em relação à
perpendicular às camadas). O ângulo médio que dá a inclinação das moléculas na fase SmC, f,
evolui na temperatura, diminuindo, geralmente, com o aumento da mesma. No caso mais
comum (fases esméticas de monocamada) o comprimento das moléculas é semelhante à
espessura das camadas esméticas, ℓ, no caso da fase SmA, e dado por ℓ cosf, para a SmC,.
a) b)
ℓ ℓ
φ

14
As fases esméticas de moléculas inclinadas, ou fase esméticas inclinadas são biaxiais.
A direcção média de inclinação das moléculas definida, no plano das camadas, permite definir
uma ordem de orientação molecular transversa.
As mesofases esméticas de camadas ordenadas, como a fase esmética B, onde se
observa ordem posicional de longa distância nas três dimensões, devem, em rigor, ser consi-
deradas cristalinas. Verifica-se geralmente num processo de aumento da temperatura, as fases
mais ordenadas precedem as menos ordenadas.
Mesofases Colunares
As mesofases colunares são, nos casos mais comuns, formadas por moléculas em
forma de disco (discóticas). As moléculas discóticas organizam-se preferencialmente com os
planos dos discos aproximadamente paralelos. A direcção importante nestas unidades
mesogénicas é a direcção normal ao plano molecular. Nas fases nemáticas de moléculas
discóticas a orientação média comum, a que se associa o director, será definida pelos versores
normais aos planos dos discos.
Relativamente à estrutura das mesofases colunares, esta caracteriza-se pelo empi-
lhamento dos discos moleculares formando colunas. A organização espacial das colunas no
plano transversal ao plano onde se encontra o director permite ainda classificar esta mesofase
em termos das redes cristalinas bidimensionais formadas pelas colunas; a considerar fases
colunares hexagonal, rectangulares ou oblíquas, Figura 13. Nas fases discóticas desordenas, a
posição e movimento relativo da terceira dimensão são livres, o que confere carácter líquido ao
composto, ou seja, os discos podem deslocar-se entre colunas. As fases colunares ordendas,
uma vez mais, têm comportamento e estrutura que se aproximam mais nos cristais.
Figura 13 – Fases colunares de moléculas desordenadas.

15
Capítulo 2
Técnicas Experimentais
Das diversas técnicas experimentais utilizadas para o estudo de fases de compostos
líquido cristalinos termotrópicos aquelas que importa evidenciar, no contexto deste trabalho,
são: microscopia óptica polarizante, calorimetria diferencial, difracção de raios X e ressonância
magnética nuclear. A microscopia óptica polarizante e a calorimetria diferencial serão
brevemente referidas, uma vez que foram aplicadas na determinação da sequência de fases e
respectivas temperaturas de transição dos compostos utilizados neste trabalho. Esses
resultados foram obtidos na sequência da síntese dos compostos e preparação das amostras e
antecederam a realização deste trabalho. A difracção de raios-X e ressonância magnética
nuclear foram utilizadas de forma sistemática ao longo deste trabalho e por isso serão referidas
de forma mais pormenorizada.
Microscopia Óptica Polarizante
A microscopia óptica polarizante (MOP) é uma técnica fundamental para a determina-
ção da sequência de fases em compostos líquido cristalinos. Esta técnica consiste em observar
num microscópio uma pequena amostra do composto através de polarizadores cruzados,
controlando a temperatura a que a amostra se encontra. Os compostos líquido-cristalinos,
como tinha sido já referido, caracterizam-se por apresentarem anisotropia óptica. Ao sujeitar
uma amostra no estado sólido a aquecimento então esta transitará para uma fase líquida (ou
líquido-cristalina). A utilização dos polarizadores cruzados permite fazer a distinção entre uma
fase líquida cristalina e uma fase isótropa. No caso de uma fase isótropa, como as moléculas
podem tomar todas as orientações possíveis, não há uma direcção preferencial para a
propagação de luz na amostra. Então, uma amostra isótropa colocada entre polarizadores
cruzados não permite a passagem de luz, e o que se observa é simplesmente um padrão
negro uniforme.
No caso de amostras cristalinas ou líquido-cristalinas, o que se obtém com os polariza-
dores cruzados é um padrão que depende da organização molecular do material constituinte
da amostra. Estes padrões (texturas) são caracteristicamente diferentes para as diferentes
fases. Assim, quando no processo de evolução da temperatura (quer aquecimento, ou
arrefecimento) se atinge uma transição de fase, há uma variação significativa nos padrões
apresentados. Este processo pode ser executado tanto no caso de subida ou descida de
temperatura mas é de ter em conta que a transição de fases é mais nitidamente verificada
quando há diminuição da temperatura a partir da fase isótropa. Nota-se ainda que é frequente

16
nestes compostos verificar-se sobre-arrefecimento das amostra quando se transita das fases
líquido-cristalinas para a fase cristalina, ou seja, a cristalização dá-se a uma temperatura
inferior àquela a que se dá a transição de fase no sentido ascendente da temperatura.
O facto das texturas observadas por este método apresentarem padrões característi-
cos para as diferentes mesofases permite não só determinar as temperaturas para as quais se
dão as transições de fase, mas também (com algumas precauções associadas à interpretação
das texturas) identificar as fases envolvidas neste processo.
Calorimetria diferencial
Outro método experimental utilizado na determinação de transições de fase é calorime-
tria diferencial de varrimento (DSC, do inglês, differential scanning calorimetry). Na aplicação
desta técnica, as amostras são aquecidas e arrefecidas, progressivamente, medindo-se
durante este processo, o calor latente associado às diferentes transições de fase. Nestas
experiências a diferença na quantidade de calor necessária para variar a temperatura da
amostra em estudo e de uma amostra de referência, são medidas, como função da
temperatura. A referência tem uma capacidade térmica bem determinada na gama de
temperaturas que se está a inspeccionar, o que permite avaliar a quantidade de calor envolvida
no varrimento em temperatura da amostra. Desta variação da quantidade de calor é possível
detectar as temperaturas a que se dão eventuais transições de fase.
No caso de o processo que a amostra experimenta requerer maior ou menor quanti-
dade de calor do que a referência para atingir a mesma temperatura, assim vai ser maior ou
menor o fluxo de calor que é necessário transmitir à amostra em relação à referência. Por
exemplo, quando uma amostra transita de uma fase sólida para líquida (isótropa) esse
processo vai requerer maior fluxo de calor a fluir para a amostra para aumentar a temperatura
à mesma taxa do que acontece na referência. Isto acontece devido à absorção de calor por
parte da amostra quando atravessa uma transição de fase endotérmica, de sólido para líquido.
Da mesma forma, se a amostra experimentar uma transição exotérmica, como um processo de
cristalização, é necessária uma menor variação da quantidade de calor para diminuir a
temperatura da amostra. Ao observar a diferença entre o fluxo de calor da amostra e referência,
então é possível determinar a quantidade de calor absorvida e libertada durante as transições.
A DSC pode também detectar transições de fase mais subtis, como as transições para fases
líquido-cristalinas ou a transição vítrea. Estas transições envolvem variações de energia mais
pequenas, quer na transição de sólido para cristal líquido quer de cristal líquido para a fase
isótropa. Esta última situação é muito relevante no contexto do estudo de compostos líquido-
-cristalinos.
Trata-se de uma técnica complementar para a determinação das temperaturas a que
ocorrem as diferentes fases. Mais uma vez os fenómenos de sobre-arrefecimento podem

17
conduzir a resultados que não são simétricos nas situações de subida e descida da temperatu-
ra. Esta situação é recorrente e é necessário tê-la em conta, na análise dos dados.
Difracção de raios-X
Enquanto, nos casos anteriormente descritos, da MOP e da DSC, o principal objectivo
é a caracterização das temperaturas de transição de fases e identificação das respectivas
mesofases, no caso da difracção de raios-X (DRX) o objectivo é a caracterização da estrutura
das mesofases presentes.
Um experiência de DRX necessita de uma fonte de raios X a qual radia sobre a
amostra. A interacção da radiação com os átomos da amostra provoca um padrão de difracção
em função da dispersão que a radiação incidente sofre na interacção com a amostra. O padrão
de difracção resulta da combinação das várias ondas difractadas, provenientes de diferentes
átomos que constituem a amostra, as quais interferem construtiva ou destrutivamente
(consoante as respectivas fases). Este padrão tem contribuições de todos os átomos
espacialmente distribuídos por toda a amostra e, por isso, reflecte a estrutura da mesma. Uma
imagem típica de DRX corresponde a zonas maior e menor intensidade em função da
distribuição da radiação electromagnética difractada segundo cada direcção.
No caso de haver interferência construtiva, resultante de sucessivas direcções
preferenciais na amostra (no caso cristalino, corresponde a planos da rede cristalina) os
máximos de intensidade verificados relacionam-se com a estrutura da amostra pela lei de
Bragg (da diferença de percurso óptico nos diferentes planos, Figura 14), 2 d sin θ = n λ,
Figura 14 – Esquema ilustrativo da difracção de Bragg.
onde d corresponde às distâncias entre planos definidos pela organização molecular (no caso
cristalino, corresponde à distância entre planos cristalinos), θ é o ângulo de incidência, λ o
comprimento de onda da radiação incidente, e n é um inteiro (correspondente às diferentes
ordens de difracção.
No caso de uma rede cristalina, o padrão de difracção, idealmente, terá infinitas ordens
de difracção em n 2π/d. A intensidade destes picos, no caso de interferência construtiva, pode
d
θ
θ
d.sinθ
θ
θ

18
ser matematicamente descrita por um impulso δ de Dirac, na situação de interferência
destrutiva a intensidade será zero. No entanto, nos cristais líquidos a ordem que se estabelece
nunca se estende a tão longa distância como num cristal. No caso da fase nemática, em que a
ordem é a curta distância, os picos de Bragg dão lugar, em geral, a picos difusos e não é
possível detectar mais do que uma ordem de difracção. Nos cristais líquidos em que existe
ordem posicional (esméticos e colunares) o padrão de difracção inclui pseudo picos de Bragg,
picos de grande intensidade (contudo inferior ao observado no caso dos cristais, propriamente
ditos) que reflectem a ordem posicional a quase longa distância.
O padrão de difracção de raios-X pode ser registado com recurso a detectores unidi-
mensionais ou bidimensionais, sendo que nestes últimos o difractograma fornece mais informa-
ção sobre a estrutura. Os dados de DRX analisados neste trabalho provêm de um detector
bidimensional, pelo que a apresentação que se segue descreve o que acontece nesta situação.
Para uma amostra nemática, idealmente orientada (S=1), constituída por um único
domínio, o padrão observado, num detector bidimensional, é formado por quatro picos difusos,
distribuídos como se indica na Figura 15. Note-se que a orientação das amostras se consegue
com recurso a um campo magnético ou eléctrico. Este padrão reflecte a difracção
correspondente às distâncias entre moléculas consecutivas (d) (associada a uma distância 2π/d,
na região dos pequenos ângulos), e às distâncias laterais entre moléculas (a) (associadas a
uma distância 2π/a, na região dos grandes ângulos). Os picos, na região dos pequenos e
grandes ângulos estão desfasados no espaço de π/2 (difracções em direcções perpendicula-
res).
Figura 15 – Padrão de DRX de uma amostra nemática alinhada, com parâmetro de ordem S=1,
e as dimensões características no espaço real e recíproco.
No caso de uma amostra com parâmetro de ordem, S, diferente da unidade, as bandas
difusas alargam angularmente indicando variações na orientação que as moléculas assumem,
na amostra, como se exemplifica na Figura 16.
a
d
2π/d
2π/a

19
Figura 16 – Padrão de DRX de uma amostra nemática alinhada, com S<1, e as dimensões
características no espaço real e recíproco.
Para amostras esméticas, em que há ordem posicional, o padrão que antes era
composto por bandas difusas na região dos pequenos ângulos passa a dar lugar a pseudo
picos de Bragg, em n×2π/ℓ (n dá a ordem de difracção), resultantes da difracção nos planos das
camadas esméticas. Na região dos grandes ângulos continuam a estar presentes as bandas
difusas (em 2π/a), ver Figura 17.
Figura 17 – Padrão de DRX de uma amostra esmética alinhada e as dimensões características
no espaço real e recíproco.
Para fases esméticas de moléculas inclinadas, situação que terá interesse para a
análise dos dados apresentados neste trabalho, os padrões encontrados são parcialmente
semelhantes aos encontrados para a fase esmética A, obsevando-se o padrão rodado de um
ângulo (que em geral corresponde ao ângulo de inclinação da fase) na região dos pequenos ou
dos grandes ângulos consoante o tipo de organização dos domínios esméticos da amostra. No
caso em que as moléculas se encontram inclinadas, mas com as camadas orientadas
perpendicularmente ao campo usado para alinhar a amostra, esta orientação da amostra
esmética designa-se bookshelf. Neste caso a inclinação detecta-se na região dos grandes
ângulos (Figura 18 a) . Para uma orientação chevron, em que as moléculas ficam alinhadas
d
a
n 2π/d
2π/a
a 2π/a
ℓ
2π/ℓ 2π/ℓ

20
com o campo aplicado, mas as camadas inclinadas, o ângulo de inclinação da fase é detectado
na região dos pequenos ângulos. Na Figura 18 estão esquematizados os padrões de DRX para
amostras na fase SmC totalmente alinhadas, assumindo as orientações bookshelf e chevron,
respectivamente.
Figura 18 – Padrões característicos de DRX para amostras na fase SmC para a orientação
bookshelf (a) e chevron (b) e as respectivas organizações moleculares.
No caso de uma experiência de DRX é frequente as moléculas ou camadas, para as
orientações bookshelf e chevron, respectivamente, tomarem mais que uma orientação
preferencial, resultando os novos padrões e organizações moleculares indicados na Figura 19.
Figura 19 – Padrões característicos de DRX para amostras na fase SmC para a orientação
bookshelf (a) e chevron (b) e as respectivas organizações moleculares, numa experiência típica
de DRX.
No caso em que os padrões de DRX resultam de amostras policristalinas (pós), deixam
de se detectar picos e bandas, passando o padrão a ser composto por imagens circulares de
2πℓ
2πa
ψ
2πℓ
2πa
a) b)
d
a
d
a ψ
n
2π/a
ψ n
2π/d d
a
2π/d
2π/a
a) b)

21
raio correspondente às distâncias anteriormente indicadas. Neste caso, deixa de haver
informação sobre a inclinação nas fases esméticas de moléculas inclinadas. A informação que
é possível recolher de uma imagem de difracção de um pó, é idêntica à que se pode obter com
um detector unidimensional (em que se usam amostras de pós, ou se colocam as amostras em
rotação, para percorrer todos os diferentes ângulos possíveis).
Nos resultados analisados, no contexto deste trabalho, as amostras encontram-se
alinhadas pela presença de um campo magnético.
Ressonância Magnética Nuclear
A ressonância magnética nuclear (RMN) é uma outra técnica experimental, que repre-
senta um importante contributo para o estudo de fases líquido-cristalinas, uma vez que permite
obter informação sobre um conjunto grande de parâmetros [ 11]. Para além da sua importância
nos estudos de ordem molecular em fases líquido-cristalinas, é uma das técnicas mais
utilizadas no estudo da dinâmica molecular através da determinação dos tempos
característicos associados ao diferentes tipos de movimentos observados.
Trata-se de uma técnica experimental que se baseia no facto de alguns núcleos atómi-
cos exibirem propriedades magnéticas intrínsecas, apresentando momento magnético intrínse-
co, mesmo que a amostra não apresente magnetização natural. Esta técnica baseia-se no ali-
nhamento dos momentos magnéticos nucleares (o que acontece devido à presença de uma
campo magnético estático muito forte, B0) e posterior análise da resposta do sistema a
perturbações a este alinhamento.
Para o caso mais simples de um sistema com spins 1/2, são possíveis dois
alinhamentos com o campo, paralelo e anti-paralelo. Entre as duas situação há uma diferença
energética, o nível orientado paralelamente (α – spin up) ao campo tem menor energia
enquanto que o outro ocupará um estado mais energético (β – spin down). Assim, o nível
menos energético será o mais ocupado, na situação de equilíbrio – minimização da energia,
havendo uma diferença de energia, ∆E= h ν = ω, entre estes dois níveis. O desequilíbrio
energético na ocupação dos dois níveis é função da intensidade do campo aplicado (ν = γ
B0/2π), resultando ∆E = γ B0 (γ é a relação giromagnética). Esta diferença energética surge
como consequência da interacção de Zeeman associada ao campo B0. A diferença de energia
entre os dois níveis, considerados provoca uma diferença entre a ocupação dos mesmos, que
em equilíbrio termodinâmico a uma dada temperatura é descrita pela distribuição de Boltzmann.
Existem então dois níveis energicamente afastados de ∆E com diferentes ocupações.
Uma experiência RMN supõe então que é aplicada uma radiação de frequência
adequada, a qual terá exactamente o valor da frequência que separa os dois níveis α e β (ωL =
γ B0 esta é a frequência de precessão de Larmor no campo estático, B0). Esta radiação é
aplicada com recuso a um campo electromagnético oscilante, B1, com frequência ωL, induzindo

22
transições entre os dois níveis. Assim haverá uma reocupação dos níveis, com aumento da
população no nível mais energético, quando a amostra já se encontra sujeita ao campo estático.
Nesta situação a magnetização total da amostra toma um valor diferente do valor que tomava
quando sujeita apenas ao campo B0. No limite, a situação em que as ocupações dos dois
níveis se igualam corresponde à situação de saturação. Assim, para analisar o sistema será
necessário perturbá-lo relativamente à condição de equilíbrio inicial. Na ausência do campo, B1,
os momentos magnéticos têm tendência a voltar a alinhar-se com o campo estático, B0,. Numa
experiência de RMN, a frequência típica encontra-se na gama das radiofrequências.
Em termos macroscópicos esta circunstância corresponde a rodar a magnetização de
um ângulo, θ (θ = γ B1 ∆t), em que ∆t representa o intervalo de tempo durante o qual o impulso
é aplicado . Em particular, para experiências típicas importa considerar rotações de 90 e 180º,
correspondentes a impulsos designados π/2 e π. No entanto ao retirar a frequência excitadora
dá-se um processo de relaxação dos spins nucleares que conduz o sistema de novo para o
equilíbrio.
Os processos de relaxação não são um fenómeno de emissão espontânea. Ou seja, a
relaxação dá-se através de um fenómeno de recuperação de um estado de menor energia
após um fenómeno de ressonância. Não é fruto ocasional dos processos associados à emissão
espontânea. As transições típicas em RMN tem origem em transições que são induzidas pela
rede, a qual funciona como reservatório térmico ideal, pelo que terá facilidade em absorver o
excedente de energia do sistema de spins quando este entrou em ressonância.
A análise dos processos de perda de energia para retornar à configuração mais estável
são caracterizados por uma relaxação spin-spin (relaxação transversal, com tempo característi-
co: T2) e relaxação spin-rede (relaxação longitudinal, com tempo característico: T1). À relaxação
spin-spin não está associada uma transferência energética, enquanto que na outra se supõe
uma perda de energia para a rede. A relaxação spin-spin não implica trocas de energia, basta
que o excesso de spins responsável pela magnetização fora do equilíbrio passe a ter
distribuição aleatória. No entanto a relaxação spin-rede, como já foi referido implica uma
transição de energia do sistema de spins para a rede a qual funciona como reservatório térmico
ideal. Assim os tempos de relaxação característicos tanto spin-spin como spin-rede são impor-
tantes, embora se refiram ao processo de relaxação permitem extrair informações diferentes. A
análise deste tempo de relaxação spin-rede característico vai ser importante para a compre-
ensão de fenómenos associados à dinâmica molecular. Uma análise RMN para além de um
espectro em frequências com informação sobre a ordem das moléculas na amostra permite-
-nos saber mais, em particular sobre movimentos moleculares.
A evolução da magnetização para o equilíbrio é que vai permitir determinar os tempos
característicos dos fenómenos de relaxação. A evolução da magnetização é dada pelas equa-
ções de Bloch, estas equações são equações diferenciais que descrevem o comportamento da
magnetização na presença dos campos magnéticos B0 e B1.

23
(10)
Numa experiência de espectroscopia de RMN detecta-se um espectro de absorção de
energia por parte da amostra que contém informação sobre a ordem orientacional e a estrutura
da amostra. O hamiltoniano do sistema, é dado por:
H = Hz + Hdq + Hd + Hi + Hq (11)
Onde o termo Hz do hamiltoniano é a interacção de Zeeman (reflecte a interacção dos spins nu-
cleares com o campo magnético B0); Hdq é o desvio químico (representa a interacção dos spins
nucleares com o campo magnético local criado pela nuvem electrónica que se cria em torno
deste); Hd dá o acoplamento dipolar entre pares de spins (reflecte a interacção magnética entre
spins); Hi dá o acoplamento dipolar indirecto entre spins (reflecte a perturbação induzida por
cada spin na nuvem electrónica em seu redor); finalmente, Hq é a interacção quadrupolar, para
núcleos com spins pelo menos iguais a 1 (representa a interacção do momento quadrupolar
eléctrico nuclear com o gradiente de campo eléctrico que existe na posição do núcleo criado
pela nuvem electrónica que o rodeia).
No contexto deste trabalho, em que foi utilizado deutério (I = 1), a perturbação ao termo
de Zeeman relevante é a da interacção quadrupolar. O hamiltoneano de Zeeman é dado por:
HZ = – γ B0 Iz (12)
Onde IZ é a componente de spin segundo a direcção do campo B0 (definida como coincidente
com o eixo dos ZZ). O termo da interacção quadrupolar é dado por:
Hq=„i
3
4 e Qi XVZZ
i\Ii H2 Ii -1L BIZ
i2-
1
3 Ii IIi +1MF
(13)
Onde ⟨ViZZ⟩ é a componente ZZ do gradiente de campo eléctrico médio no referencial do
laboratório.
Numa experiência de espectroscopia de RMN, na presença de deutério resulta um
espectro de absorção, que é função das transições entre estados diferentes pela aplicação dos
campos magnéticos adequados na amostra. As energias associadas a estas transições podem
ser determinadas calculando, pela a teoria das perturbações, os níveis de energia associados
ao hamiltoneano da interacção quadrupolar (13), como perturbações à interacção de Zeeman.
dMx
dt=g @My B0+ Mz B1 sin Hw tLD- Mx
T2
dMy
dt=g @Mz B1cos Hw tL - MxB0D- My
T2
dMz
dt=-g @MxB1 sin Hw tL+MyB1 sin Hw tLD- Mz-M0
T1

24
Num espectro de RMN de deutério (no caso em que a molécula contém um único núcleo de
deutério) vai haver um padrão de riscas, duas para cada domínio nemático que vão ficar
afastadas, em frequência, de uma distância que depende do parâmetro de assimetria (de onde
se pode avaliar, experimentalmente, a biaxialidade). O afastamento quadrupolar resultante da
perturbação de primeira ordem ao hamiltoneano de Zeeman devida à interacção quadupolar, é
dado por (consultar Figura 6, para ver definição dos ângulos):
ϕθ
η+
−θν=δν 2cossin
22
1cos
2
3 22Q2
3
(14)
No caso de uma amostra com a presença de deutério ligado a um núcleo de carbono, θ
e ϕ são respectivamente os ângulos polar e azimutal que definem a orientação do campo B0 no
referencial principal do tensor gradiente de campo eléctrico associado à ligação C-D.
O afastamento quadrupolar tem dependência no parâmetro de assimetria, η, que é
função dos parâmetros de ordem (resultantes das matrizes de ordem de Saupe), já indicado no
Capítulo 1. Pelo que se pode avaliar a biaxialidade de uma fase nemática recorrendo a
espectroscopia RMN de deutério.
A forma que o espectro assume relaciona-se com a ordem da fase subjacente à
amostra. Mais uma vez, este estudo é feito para diferentes temperaturas e em geral requer já
um conhecimento relativo do esquema de transições de fase que o composto apresenta.

25
Capítulo 3
Fase Nemática Biaxial
O tema da biaxialidade em fases nemáticas embora introduzido teoricamente há várias
décadas (1970) por Freiser [ 12], é um tema que experimentalmente se encontra ainda envolto
em controvérsia. A primeira evidência experimental da existência de uma fase nemática biaxial
em compostos líquido cristalinos aconteceu em 1980, por Saupe [ 13], para um sistema
liotrópico. Os resultados apresentados suscitaram grande interesse e desde logo foram
iniciados estudos em sistemas termotrópicos, a fim de investigar a possível existência de fases
nemáticas biaxias, neste tipo de compostos líquido-cristalinos. Se no caso dos sistemas
liotrópicos as experiências eram claras, para os sistemas termotrópicos o processo tem-se
desenrolado mais lentamente e com resultados menos evidentes.
Uma fase nemática biaxial caracteriza-se pelo facto das três componentes principias de
qualquer propriedade tensorial de segunda ordem serem diferentes entre si. As técnicas
experimentais que se usam (a maioria são provenientes de resultados de dispersão) são, por
exemplo: difracção de raios X, de electrões, de luz (por exemplo com recurso a laseres), de
neutrões, difusão dinâmica de luz, espectroscopia RMN de deutério, conoscopia.
O índice de refracção, por exemplo, permite avaliar a simetria da fase, uma vez que
pequenas diferenças no índice de refracção são mensuráveis. A técnica experimental que
permite avaliar a biaxialidade através de variações no índice de refracção denomina-se
conoscopia. Para tal é necessária a preparação de um monodomínio nemático. Que pode
conseguir-se sujeitando uma pequena camada de amostra na fase nemática ao efeito de um
campo eléctrico que alinhe o director principal. As tensões superficiais são então usadas para
alinhar o director secundário, das fases nemáticas biaxiais. A diferença entre os índices de
refracção ao longo das direcções no plano ortogonal ao director principal, ou seja ao longo das
direcções dos directores secundários, pode ser determinada por técnicas de conoscopia. As
imagens obtidas para fases uniaxiais e biaxiais são distintas. Para uma fase nemática uniaxial
o padrão de interferência é dado por duas riscas escuras, conhecidas por isogiras, que se
cruzam, formando uma cruz no centro da imagem. No caso de uma fase biaxial estas isogiras
abrem, deixando de se cruzar no centro da imagem, como indicado na Figura 20. A
biaxialidade será tanto maior quanto maior for o desvio que as isogiras experimentem.

26
Figura 20 – A imagem a) indica o padrão esperado para uma amostra uniaxial, em b) está o
assinatura biaxial, com a abertura das isogiras.
Há no entanto um problema associado a esta forma de identificar fases biaxiais, para
uma amostra de pequena espessura é possível induzir biaxialidade óptica na amostra uniaxial,
através da interacção com as superfícies.
No contexto das transições de Fredericks, é possível partindo de um monodomínio de
uma amostra, conjugado com a aplicação de uma campo que alinha a amostra, induzir
transições, ditas transições de Fredericks. Nesta situação pode acontecer que o director
principal esteja inclinado em relação à normal à superfície. Neste caso o perfil obtido seria
característico de uma fase biaxial e não uniaxial, mesmo que a amostra seja uniaixial. Para
verificar inequivocamente que uma fase é biaxial é necessário que os resultados verificados
por conoscopia sejam validados por outra técnica experimental [ 14].
Uma outra técnica experimental que permite avaliar a biaxialidade de uma fase
nemática, é a espectrocopia RMN de deutério. Como foi já referido, o afastamento quadrupolar
tem dependência explícita no parâmetro de assimetria, η, que caracteriza uma fase biaxial.
Numa experiência para avaliar a biaxialidade por espectroscopia RMN de deutério,
podem seguir-se dois métodos diferentes para tentar detectar se existem, ou não, mais do que
um director segundo os quais as moléculas se alinham. Os dois métodos apresentados em
seguida, não podem ser utilizados indiferentemente para qualquer amostra, a viscosidade das
amostras condiciona as experiências, como será adiante indicado.
Um dos métodos de verificar, por RMN do deutério, a existência (ou inexistência) de
ordem biaxial numa amostra nemática consiste na comparação de dois espectros de RMN
adquiridos respectivamente antes e após a aplicação de uma rotação de um ângulo fixo a uma
amostra alinhada, em torno de um eixo perpendicular a B0. Para uma amostra nemática que
esteja alinhada com o campo aplicado, B0, o espectro apresenta duas riscas (por cada átomo
de deutério presente na molécula em estudo) referentes à orientação do director principal com
o campo. No caso de a amostra ser rodada de 90º (ortogonal ao alinhamento inicial), ao ser
adquirido um novo espectro, se não houver um outro director secundário (fase nemática
uniaxial) os picos estarão agora afastados de metade do afastamento verificado para a
situação inicial, 0º. Pelo contrário se for detectado um par de picos com um afastamento
diferente de metade do afastamento para o ângulo 0º, estes reflectem uma orientação
preferencial na direcção ortogonal ao director principal. Considerando a equação para o
a) b)

27
afastamento quadrupolar (14), para θ=0º e ϕ=0º (amostra alinhada), resulta: ∆νQ=(3/2) νQ, para
θ=90º e ϕ=0º (amostra rodada de 90º) agora resulta ∆νQ=(3/4) νQ. Se a viscosidade for muito
baixa, acontece que a amostra ao ser rodada do ângulo fixo, reorienta-se muito depressa com
o campo. Neste caso o espectro obtido reflecte o realinhamento do director principal com a
amostra, não sendo possível avaliar a biaxialidade.
Alternativamente, pode tomar-se uma amostra policristalina (pó). No caso de um pó o
espectro obtido passa a ser mais complexo, reflectindo todas as orientações do director,
correspondentes aos diferentes domínios nemáticos presentes na amostra. Estes espectros
resultam como indicado na Figura 21.
Figura 21 – Espectro RMN de deutério típico de uma amostra policristalina bidimensional, a
preto: amostra alinhada (a); a verde: uniaxial (b); a vermelho: biaxial (c).
Estes espectros resultam da sobreposição de pares de riscas, cada uma das quais
associada a uma direcção específica assumida pelo director no conjunto dos domínios
nemáticos existentes na amostra. No caso de uma amostra uniaxial ou biaxial, em rotação
contínua em torno de um eixo perpendicular ao campo magnético estático, B0, o tipo de perfil
que se espera encontrar apresenta, em ambos os casos (se no caso biaxial um dos directores
secundários se alinhar com o eixo de rotação), dois pares de picos (exteriores e interiores).
Correspondentes, respectivamente, a ângulos de 0º e 90º entre o director principal e o campo
magnético estático, B0. No caso de uma amostra uniaxial o afastamento entre picos interiores é
metade do dos exteriores e no caso de uma amostra biaxial esta largura será diferente de
metade dos exteriores. Trata-se de uma assinatura clara da biaxialidade. A viscosidade da
amostra condiciona a experiência. É necessário que a amostra rode com velocidade suficiente
para que o director não tenha tempo de se alinhar com o campo, B0.
A espectroscopia RMN de deutério é uma forma eficaz de avaliar a biaxialidade. A
verificação da biaxialidade por técnicas de conoscopia está associada a alguma incerteza, uma
vez que numa experiência de conoscopia pode ser induzido carácter biaxial a uma amostra
uniaxial.
Os cristais líquidos termotrópicos mais simples são constituídos por pequenas molé-
culas, de baixo peso molecular e que têm comportamento líquido cristalino intrínseco numa
gama de temperaturas bem determinada. A biaxialidade tem sido investigada para estas
a) b) c)

28
moléculas. Os estudos nesta área, com moléculas de baixo peso molecular, têm recorrido a
compostos com estrutura molecular biaxial, no sentido de investigar a existência, ou não, de
fases biaxias, nestes sistemas. Os compostos com estrutura molecular biaxial apresentam forte
anisotropia molecular, apresentando conformações com dimensões distintas nas diferentes
direcções.
Dos sistemas de baixo peso molecular, mais simples, destacam-se as moléculas em
forma de tábua. Nestes sistemas, em 1988, Chandrasekhar et al [ 15], identificaram uma fase
nemática biaxial, com recurso a técnicas de conoscopia. O sistema apontado é estruturalmente
biaxial, no entanto, esta condição não garante a existência de uma fase biaxial, tinham sido já
testadas, anteriormente, outras moléculas, com a mesma forma, mas sem resultados, no que
respeita à biaxialidade das fases nemáticas.
Um dos tipos de estruturas que têm sido igualmente investigado são os compostos em
forma de banana ou boomerang, cuja ordem nemática biaxial foi prevista teoricamente [ 16] .
Em 2004, Madsen et al [ 17], publicaram os resultados do estudo com este tipo de moléculas
em forma de boomerang exibindo simetria biaxial. Neste artigo ficou experimentalmente
provada a existência de uma fase nemática biaxial para estes compostos. As técnicas
experimentais utilizadas foram a conoscopia e espectroscopia de RMN com a presença de uma
sonda deuterada, estes resultados são consistentes entre si e demostram a existência de uma
fase nemática biaxial.
Nesse trabalho, [ 17], foram preparadas amostras para a análise por técnicas de
conoscopia, as quais foram testadas para diferentes temperaturas, tendo sido verificada uma
possível fase nemática biaxial. No entanto estas observações conoscópicas, não são uma
confirmação unívoca da biaxialidade, assim em [ 17] apresentam-se também os resultados
referentes à espectroscopia RMN. O parâmetro de assimetria encontrado por RMN do deutério
para este composto é não nulo (aproximadamente igual a 0.11), o que indica tratar-se de uma
fase nemática biaxial. A origem desta biaxialidade, de acordo com [ 17], tem origem na forma de
empacotamento que as moléculas assumem na fase nemática. Pelo facto de o ângulo entre os
dois “ramos” da molécula ser de 140º (superior ao que, em geral, se verifica para moléculas em
forma de banana, ~120º) permitindo melhor empacotamento das moléculas. Este empacota-
mento conduzirá a uma fase nemática mais organizada, com duas orientações preferenciais
distintas, ou seja propicía o aparecimento da fase nemática biaxial, como se verifica.
No entanto existem diferentes abordagens, em sistemas mais complexos, que os sis-
temas de baixo peso molecular. A primeira alteração que se pode pensar para estes sistemas é
obter misturas [ 18] e compostos uniaixiais, com estruturas e orientações preferenciais distintas,
de modo a propiciar diferentes organizações moleculares. Em particular, a mistura de cristais
líquidos em forma de bastonetes com cristais líquidos discóticos, uma vez que estas moléculas
são mais simples, e têm os directores orientados de forma distinta, em relação ao corpo das
moléculas.
A forma destes dois tipos de moléculas e a orientação que o director assume
relativamente a cada um deles (paralelamente aos bastonetes e perpendicularmente aos

29
discos) permite antever a possibilidade de obtenção de misturas onde coexistam dois directo-
res mutuamente perpendiculares, potenciando, deste modo, a existência de ordem biaxial
numa fase nemática. Esta hipótese tem sido explorada com resultados até à data inconclusivos.
Um dos principais condicionantes que se têm verificado para a obtenção destas fases é a
dificuldade em obter misturas entre os compostos de uma e outra espécie. As moléculas
tendem a separar-se, conduzindo à segregação entre os componentes, resultando numa bifase
e não numa mistura, como se pretenderia. Ao contrário dos resultados teóricos e dos que se
têm obtido por simulação [ 19, 20, 21, 22], que são promissores, os resultados experimentais
requerem esforços adicionais, no sentido de produzir sistemas miscíveis. Resultados recentes
de estudos de miscibilidade entre compostos calamíticos e discóticos apontam uma via
promissora neste sentido [ 23]. Atendendo aos problemas de miscibilidade suscitados por estes
sistemas, torna-se pertinente estudar amostras em que os componentes calamíticos e
discóticos estão quimicamente ligados entre si, originando amostras onde a segregação fica
limitada, esse é o princípio agora estudado por G. H. Mehl [ 23].
Paralelamente às misturas e aos compostos de baixo peso molecular a biaxialiadade
têm sido investigada em sistemas mais complexos como polímeros. Em 1987, Hessel et al [ 24],
encontraram, experimentalmente (conoscopia), uma fase nemática biaxial num composto
polimérico líquido cristalino. O polímero onde esta fase foi encontrada apresenta cadeias
ligadas lateralmente ao corpo central dos polímeros, polímero de cadeia lateral. Estas cadeias
laterais correspondem a grupos mesogénicos, com comportamento, intrinsecamente, líquido
cristalino. Neste tipo de polímeros os movimentos de rotação das unidades mesogénicas em
torno do seu eixo principal encontram-se condicionados, pela forma como estas unidades estão
ligadas à cadeia principal do polímero (ligação lateral através de espaçadores). A explicação
sugerida para a fase nemática biaxial nestes composto baseia-se na forma como as cadeias
estão ligadas ao corpo central (lateralmente) surgindo limitações nas rotações que estarão na
origem da fase nemática biaxial [ 24]. Pelo facto de os movimentos de rotação das unidades
mesogénicas em torno do seu eixo maior estarem limitados proporcionam-se condições para o
aparecimento de ordem biaxial.
Para um sistema idêntico ao apresentado por Hessel et al [ 24] foram publicados, em
2004, por Severing e Saalwächter [ 25] os resultados do afastamento quadrupolar resultante de
espectroscopia RMN na presença de uma sonda deuterada. Os resultados verificaram a
biaxialidade do sistema. O trabalho [ 25] é de grande importância no contexto da verificação,
inequívoca da biaxialidade em polímeros nemáticos. Foram estudados dois sistemas, um dos
quais muito próximo do sistema [ 24], mas com as cadeias centrais quimicamente diferentes. O
outro sistema trata-se de também um polímero, em que as cadeias mesogénicas estão ligadas
à cadeia central, não lateralmente, mas terminalmente; designando-se polímeros de cadeia
principal. Estes dois sistemas comportam-se globalmente de forma diferente, no que respeita
ao possível comportamento biaxial das fases nemáticas. Por RMN de deutério, concluiu-se que
apenas o polímero de cadeia lateral tem comportamento biaxial, apresentando um parâmetro
de assimetria não nulo (~0.18, no ensaio a temperatura mais baixa dentro da fase nemática).

30
A origem da biaxialidade resultante deste tipo de estruturas é fundamentalmente
diferente da que resulta para cristais líquidos de baixo pelo molecular. Neste caso a
biaxialidade não está relacionada com unidades mesogénicas com formas particulares, ou
achatas e longas, ou em forma de banana, ou outras variantes, mas relaciona-se antes com a
forma como os constrangimentos, na ligação das unidades mesogénicas à cadeia central,
condicionam a organização que diferentes partes do polímero assumem.
Além dos polímeros, também em dendrímeros, a biaxialidade tem sido explorada. Em
particular tem sido explorados tetrapodes (dendrímeros de geração zero).
Em 2004, K. Merkel et al [ 26], apresentaram um estudo de espectroscopia dos infra
vermelhos polarizada, para um sistema formado por tetrapodes organosiloxanos. Neste estudo
foram avaliados os parâmetros de ordem orientacional (S), molecular (D) e da biaxialidade da
fase (P). Os resultados indicam uma fase nemática com parâmetro de ordem P não nulo, o que
indica, promissoramente, uma possível fase biaxial. Neste contexto, em 2005, Figueirinhas et al
[ 27] estudaram um sistema semelhante, constituído por uma mistura de um tetrapode
organosiloxano com uma sonda deuterada, por RMN de deutério. O resultado obtido confirma a
existência de uma fase nemática biaxial, com um parâmetro de assimetria de aproximadamente
0.8, para a temperatura mais baixa ensaiada, na fase nemática. Trata-se do primeiro resultado
com cristais líquidos termotrópicos em que o parâmetro de assimetria assuem um valor tão
elevado, reforçando a validade dos resultados obtidos. Uma vez mais, volta a ser defendido
que a forma de organização que as moléculas assumem na fase nemática é condição
fundamental para o aparecimento da ordem biaxial. Corroborando estes resultados de
espectroscopia RMN do deutério estão outros estudos dos mesmos autores em que são
adoptadas técnicas de RMN complementares (rotação contínua com aquisições síncronas e
assíncronas) C. Cruz et al [ 28] e de K. Neupane et al [ 29] com estudos de difusão dinâmica de
luz.

31
Capítulo 4
Análise Estrutural dos Compostos Ts e Tas por
Difracção de Raios–X
Neste capítulo são apresentados dados de difracção de raios-X para os compostos Ts
e Tas. O composto Tas apresenta uma fase nemática biaxial, que foi verificada por
espectroscopia por RMN do deutério. O outro composto, Ts, é estruturalmente semelhante a
este, Tas. A nomenclatura Ts e Tas, relaciona-se com a estrutura dos corpos aromáticos, que é
simétrica (s) para o composto Ts, e assimétrica (as) para o outro composto, Tas. Neste
contexto, é importante caracterizar de forma detalhada a estrutura das mesofases exibidas por
estes compostos, com o objectivo de procurar estabelecer uma correlação entre as proprieda-
des estruturais e a ordem biaxial.
No decorrer deste trabalho foram analisados dados de difracção de raios-X (DRX)
obtidos para amostras alinhadas num difractrómetro MAR345 com um detector bidimensional,
plano. A ampola de raios-X utilizada é de cobre (Cu), emite radiação Ka, o monocromador é de
grafite e o comprimento de onda da radiação X é de 1.54 Å.
A estrutura química dos compostos, Ts e Tas, bem como os esquemas de transições
de fase são apresentados na Figura 22. As sequências de fases dos compostos foram anterior-
mente identificada por DSC e MOP. Os dois compostos agora apresentados correspondem a
dendrímeros de geração zero. A síntese respectiva encontra-se descrita em [ 30, 31, 32, 26].
Si
Si
O
Si
Si
O
C8H17O OC8H17
O
O
O
4
O
O
Si
Si
O
Si
Si
O
C8H17O OC8H17
O
O
O
4
O
O
O
O
C 70ºC SmC 89ºC N 132ºC IC 70ºC SmC 89ºC N 132ºC I

32
Si
Si
O
Si
Si
O
C8H17OOC11H23O
O
O
4
Si
Si
O
Si
Si
O
C8H17OOC11H23O
O
O
4
Tg -30ºC N 47ºC ITg -30ºC N 47ºC I
Figura 22 – Estrutura química dos compostos analisados e respectivas sequências de fases.
Em cima: Ts, em baixo Tas.
O composto Ts tem massa molecular 3910.06 gmol-1. Trata-se de um tetrapode orga-
nosiloxano baseado num núcleo de siloxano ligado a quatro unidades mesogénicas. Os espa-
çadores flexíveis que ligam o núcleo mesogénico ao núcleo de siloxano são constituídos por
cinco grupos metileno, e grupos tetrametildisiloxano. O composto Tas, de massa molecular de
3597.96 gmol-1, difere do anterior no número de grupos aromáricos nas unidades mesogénicas.
O núcleo é formado por três anéis benzénicos (ao contrário dos quatro que caracterizavam o
composto Ts) e, adicionalmente, onze grupos metileno (no composto Ts eram oito) formam as
cadeias alifáticas de cada unidade mesogénica. O comprimento total das unidades mesogéni-
cas, na configuração mais distendida é aproximadamente o mesmo nos dois compostos. Na
Figura 23 apresenta-se uma imagem esquemática da estrutura molecular dos compostos
estudados em que se indicam os comprimentos característicos médios dos diferentes
segmentos moleculares.

33
lm≅ 4 nm
lc≅ 2 nm
lch≅ 1 nm
lch≅ 1 nm
ls≅1.4 nm
lp≅ 1.4 nm
ls≅1.4 nm
lc≅ 1.6 nm
lch2≅ 1.4 nm
lch1≅ 1 nm
lp≅ 1.4 nm
lm≅ 4 nm
lc≅ 2 nm
lch≅ 1 nm
lch≅ 1 nm
ls≅1.4 nm
lp≅ 1.4 nm
ls≅1.4 nm
lc≅ 1.6 nm
lch2≅ 1.4 nm
lch1≅ 1 nm
lp≅ 1.4 nm
Figura 23 – Representação esquemática das moléculas e os comprimentos caracterísiticos
médios dos diferentes segmentos moleculares. A molécula da esquerda tem os corpos
aromáticos simétricos (Ts) e a da direita, os corpos assimétricos (Tas).
Convém notar que, embora no esquema apresentado na Figura 23, as moléculas
estejam representadas no plano, a respectiva estrutura é, efectivamente tridimensional,
partindo do grupo siloxano central com simetria tetraédrica. Do esquema apresentado é
possível também verificar a semelhança química entre as duas moléculas. Os dados de raios-X
que em seguida se apresentam, confirmam as sequências de fases determinadas anterior-
mente por DSC e MOP.
Nos resultados obtidos utilizaram-se integrações azimutais e radiais (na gama dos
pequenos e grandes ângulos) com recurso ao programa Fit2D 12.077. Os ajustes foram reali-
zados recorrendo a um processo de regressão não-linear de minimização do c2 reduzido. Na
Figura 24 estão indicados os ângulos segundo os quais foram efectuadas as integrações,
angulares radiais (ângulos y e c) e azimutais (ângulo q) necessárias à análise de dados.

34
Figura 24 – Esquema da forma de integração adoptada no programa Fit2D. Para estudos de
variação angular azimutal nos pequenos ângulos, os resultados são integrados em θ
(radialmente), na coroa circular interior indicada, e a variável angular é y. Nos grandes ângulos
procede-se analogamente para a coroa circular exterior, sendo a variável angular χ. Para obter
um perfil em função de θ, correspondente ao que seria obtido numa amostra não alinhada, os
dados são integrados azimutalmente (num domínio de 360º).
Os resultados de integrações azimutais, dependentes do ângulo q, são apresentadas
em função da variável q que se relaciona com θ pela Lei de Bragg (2d sinθ=nλ), tendo em
conta que q=2π/d, resulta:
λ
θπ=
nsin4
q (15)
Os compostos estudados exibem as fases já indicadas na Figura 22. A apresentação
dos resultados é feita respectivamente para cada uma das fases líquido cristalinas de cada um
dos compostos, começando pela fase esmética C do composto Ts, seguindo-se a fase
nemática do mesmo composto e por fim a fase nemática do composto Tas.
Composto Ts
Na Figura 25 encontra-se registado o padrão bidimensional de difracção de raios-X
numa temperatura incluída na gama correspondente à fase esmética C para uma amostra do
composto Ts parcialmente alinhada, pela presença de um campo magnético. Os resultados de
integração azimutal destes dados são apresentados na Figura 26.
0
χχχχ
ψψψψ q

35
Figura 25 – Padrão de difracção bidimensional para uma amostra alinhada do composto Ts, na
fase esmética C, a 78ºC.
2 4 6 8 10 12 14 16100
1000
Lo
g (
I)
q (nm -1)
Figura 26 – Ajuste da integração azimutal do padrão de difracção na fase esmética C (78ºC).
A análise dos resultados apresentados nas Figura 25 e Figura 26 evidencia claramente
tratar-se de uma fase esmética de camadas desordenadas. Os dois picos, na região dos
pequenos ângulos, correspondem a pseudo picos de Bragg de primeira e segunda ordem,
associados a uma organização lamelar, com camadas de espessura, ℓ. O valor de ℓ é apro-
ximadamente 3.1 nm, a 78ºC. De acordo com as dimensões indicadas na Figura 23, o
comprimento das unidades mesogénicas é de aproximadamente 4 nm, ou seja superior à
distância entre camadas esméticas. Pelo facto de a distância entre camadas esméticas ser

36
inferior ao comprimento molecular das unidades mesogénicas, ℓm, indica que se trata de uma
fase de moléculas inclinadas. A identificação da fase esmética C tinha também sido já confir-
mada por MOP.
Na região dos grandes ângulos é possível detectar duas bandas difusas, que cor-
respondem a distâncias latereais entre segmentos moleculares. A 78ºC as bandas detectadas
estão centradas em 0.44 nm e 0.68 nm, respectivamente. A primeira reflexão corresponde à
distância lateral entre os corpos aromáticos e entre as cadeias alifáticas, a segunda,
corresponde à distância lateral média no plano das camadas entre as cadeias siloxano
desordenadas. Aos pseudo picos de Bragg na região dos pequenos ângulos foram ajustadas
curvas lorentzianas e as bandas difusas detectadas na região dos grande ângulos foram
ajustadas funções gaussianas. Na Tabela 1 estão resumidos os dados obtidos para as
diferentes distâncias, resultantes da integração azimutal do padrão de difraccção.
Picos de difracção q (nm-1) Distância (nm) (2p/q)
001
siloxano
Aromática-alifática
2,03
9,17
14,05
3,1
0,68
0,44
Tabela 1– Resultado das distâncias obtidas por integração azimutal do padrão de difracção do
composto Ts a 78ºC, fase esmética C.
Relativamente à fase nemática evidenciada por esta amostra, é de notar que se
formam grupos cibotáticos em que se observa ordem local semelhante à de uma fase esmética
C. Na Figura 27 está apresentado o padrão de difracção, para a amostra alinhada, caracterís-
tico desta fase nemática.

37
Figura 27 – Padrão de difração de raios-X bidimensional para uma amostra alinhada do
composto Ts, na fase nemática, a 104ºC.
Figura 28 – Ajuste da integração azimutal do padrão de difracção na fase nemática a 104ºC.
Na Figura 28 apresentam-se os resultados de integração azimutal dos dados experi-
mentais. Uma vez que os resultados indicam a presença de domínios cibotáticos com estrutura
esmética C, o tratamento adoptado na análise de dados correspondente à fase esmética C foi
igualmente adoptado para fase nemática. No caso da fase nemática verifica-se a existência de
uma banda difusa (x), que, a 104ºC, corresponde a uma distância de 1.31 nm. Uma explicação
2 4 6 8 10 12 14 16120
140
160
180
200
220
I (u
ni.
arb
.)
q (nm -1 )
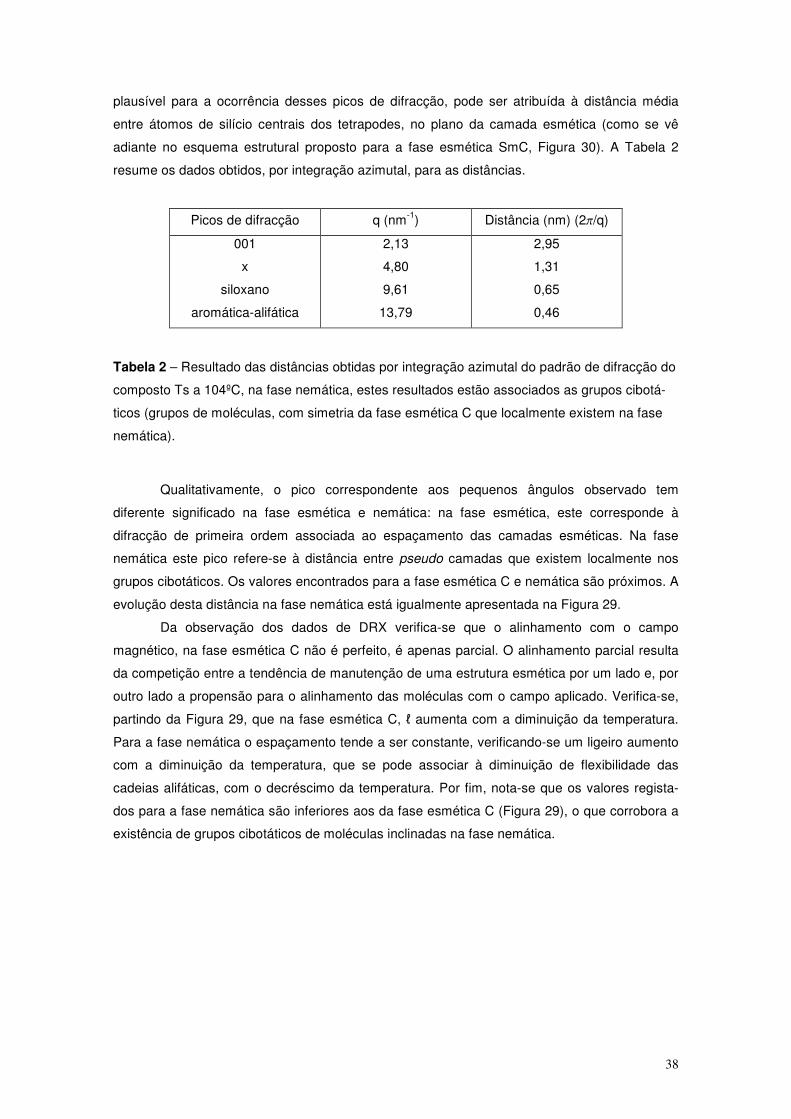
38
plausível para a ocorrência desses picos de difracção, pode ser atribuída à distância média
entre átomos de silício centrais dos tetrapodes, no plano da camada esmética (como se vê
adiante no esquema estrutural proposto para a fase esmética SmC, Figura 30). A Tabela 2
resume os dados obtidos, por integração azimutal, para as distâncias.
Picos de difracção q (nm-1) Distância (nm) (2p/q)
001
x
siloxano
aromática-alifática
2,13
4,80
9,61
13,79
2,95
1,31
0,65
0,46
Tabela 2 – Resultado das distâncias obtidas por integração azimutal do padrão de difracção do
composto Ts a 104ºC, na fase nemática, estes resultados estão associados as grupos cibotá-
ticos (grupos de moléculas, com simetria da fase esmética C que localmente existem na fase
nemática).
Qualitativamente, o pico correspondente aos pequenos ângulos observado tem
diferente significado na fase esmética e nemática: na fase esmética, este corresponde à
difracção de primeira ordem associada ao espaçamento das camadas esméticas. Na fase
nemática este pico refere-se à distância entre pseudo camadas que existem localmente nos
grupos cibotáticos. Os valores encontrados para a fase esmética C e nemática são próximos. A
evolução desta distância na fase nemática está igualmente apresentada na Figura 29.
Da observação dos dados de DRX verifica-se que o alinhamento com o campo
magnético, na fase esmética C não é perfeito, é apenas parcial. O alinhamento parcial resulta
da competição entre a tendência de manutenção de uma estrutura esmética por um lado e, por
outro lado a propensão para o alinhamento das moléculas com o campo aplicado. Verifica-se,
partindo da Figura 29, que na fase esmética C, ℓ aumenta com a diminuição da temperatura.
Para a fase nemática o espaçamento tende a ser constante, verificando-se um ligeiro aumento
com a diminuição da temperatura, que se pode associar à diminuição de flexibilidade das
cadeias alifáticas, com o decréscimo da temperatura. Por fim, nota-se que os valores regista-
dos para a fase nemática são inferiores aos da fase esmética C (Figura 29), o que corrobora a
existência de grupos cibotáticos de moléculas inclinadas na fase nemática.
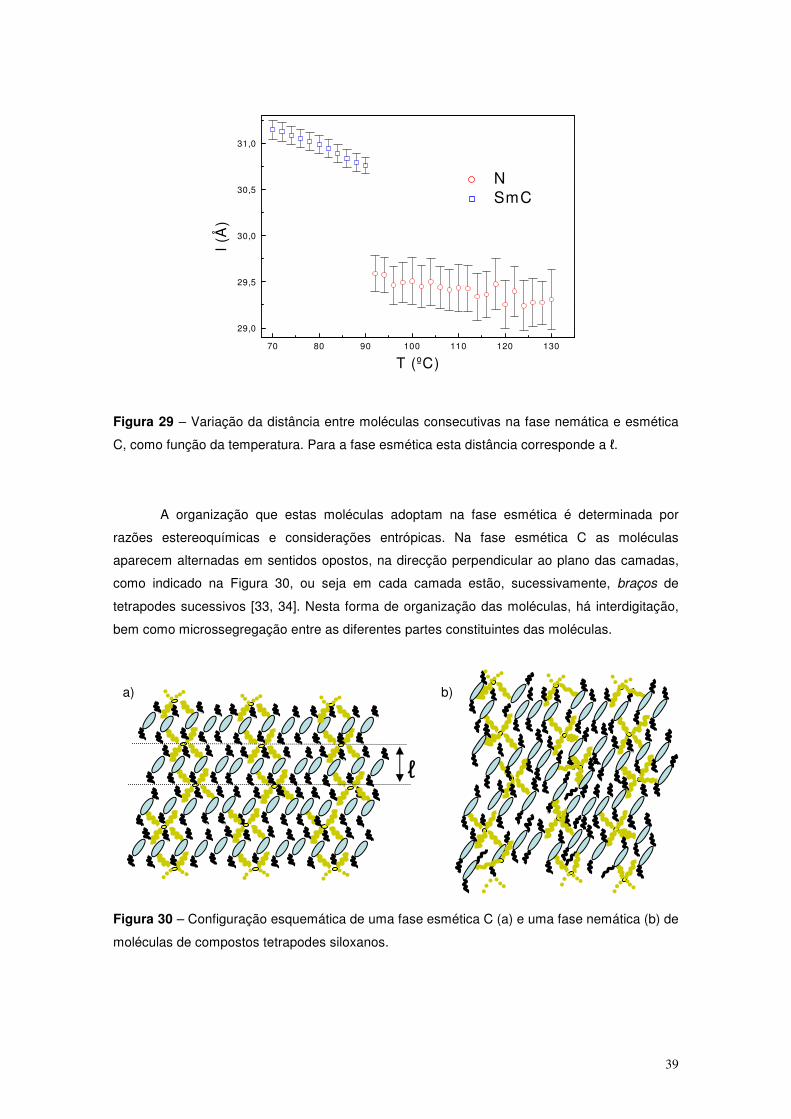
39
70 80 90 100 110 120 130
29,0
29,5
30,0
30,5
31,0
N SmC
l (Å
)
T (ºC)
Figura 29 – Variação da distância entre moléculas consecutivas na fase nemática e esmética
C, como função da temperatura. Para a fase esmética esta distância corresponde a ℓ.
A organização que estas moléculas adoptam na fase esmética é determinada por
razões estereoquímicas e considerações entrópicas. Na fase esmética C as moléculas
aparecem alternadas em sentidos opostos, na direcção perpendicular ao plano das camadas,
como indicado na Figura 30, ou seja em cada camada estão, sucessivamente, braços de
tetrapodes sucessivos [ 33, 34]. Nesta forma de organização das moléculas, há interdigitação,
bem como microssegregação entre as diferentes partes constituintes das moléculas.
Figura 30 – Configuração esquemática de uma fase esmética C (a) e uma fase nemática (b) de
moléculas de compostos tetrapodes siloxanos.
ℓ
a) b)

40
Os resultados das integrações radiais, no caso dos pequenos ângulos (para a fase
esmética C, ver Figura 31, e para a fase nemática, consultar Figura 32) reforçam a hipótese do
tipo de estrutura atrás proposta. No caso dos resultados integrados radialmente, foram
ajustadas funções gaussianas e lotentzianas, dependendo do perfil mais adequado a cada
caso. Na Figura 32 é possível verificar a presença de quatro picos, como resultado dos grupos
cibotáticos presentes na amostra. Inversamente, na fase esmética C detectam-se apenas dois
picos, devido à competição do alinhamento molecular entre as camadas esméticas e o campo
aplicado. É importante reforçar que o perfil de difracção dos picos na região dos pequenos
ângulos mostra que se trata efectivamente de uma fase nemática, embora apresente uma
estrutura com grande organização local e padrões de difracção não comuns numa fase
nemática, para temperaturas afastadas na temperatura de transição. Os resultados de DSC e
MOP assim reforçam esta conclusão. Este efeito de ordem local foi verificado em toda a gama
de temperaturas da fase nemática, tratando-se de uma ordem local suficientemente forte para
não ser, significativamente, alterada na gama de temperaturas correspondentes a esta fase.
0 45 90 135 180 225 270 315 3600
200
400
600
800
1000
I (u
ni.
arb
.)
ψ (º)
Figura 31 – Ajuste do padrão radialmente integrado na região dos pequenos ângulos, na fase
esmética C, a 78ºC.

41
0 45 90 135 180 225 270 315 360
130
140
150
160
170
180
190
I (u
ni.
arb
.)
ψ (º)
Figura 32 – Ajuste do padrão radialmente integrado na região dos pequenos ângulos, na fase
nemática, a 104ºC.
Estes dados mostram que a fase nemática presente nesta amostra tende a assumir
localmente uma estrutura cibotática como uma fase esmética C, numa estrutura em chevron,
conforme é discutido, em termos gerais no capítulo 2 e exemplificado nas Figura 18 e Figura 19.
Da presença destes quatro pseudo picos (Figura 32) foi possível determinar a evolução do
ângulo de inclinação nos núcleos moleculares em relação à ordem local apresentada pela fase
nemética, nos domínios cibotáticos. Tratando-se de uma fase nemática não existem camadas,
no entanto é conservada alguma memória da organização da fase esmética e este ângulo que
se determina está relacionado com esta reminiscência da estrutura da fase esmética nos
grupos cibotáticos da fase nemática. Na Figura 33 apresenta-se a evolução do ângulo de
inclinação dos corpos aromáticos em relação ao director nemático que se verifica nos grupos
cibotáticos (este ângulo corresponde ao ângulo em y definido na Figura 24).

42
90 95 100 105 110 115 120 125 130 135
60,0
60,5
61,0
61,5
62,0
62,5
ψ (
º)
T (ºC)
Figura 33 – Evolução, na temperatura, do ângulo de inclinação presente nos grupos cibotáticos
da fase nemática.
Em complemento da discussão até aqui apresentada pode estimar-se analiticamente o
ângulo de inclinação da fase esmética C. A área molecular é definida como Vm/ℓ, onde Vm é o
volume molecular e ℓ é a espessura das camadas. O volume molecular pode ser estimado
partindo da massa molecular e da densidade (considera-se que a densidade de compostos
deste tipo é próxima de 1 gcm-3). O valor do volume molecular a 78ºC é de aproximadamente
6.493 nm3, resultando a área molecular ~2.09 nm2.
A estrutura da fase esmética apresentada (Figura 30 a) é importante para relacionar a
área molecular com o ângulo de inclinação dos corpos aromáticos na fase esmética C. Nota-se
que a área molecular nas diferentes subcamadas é necessariamente constante por razões
geométricas. Assim, a área molecular associada à subcamada na qual os corpos mesogénicos
tendem a alinhar-se é a mesma área da subcamada onde coexistem as cadeias alifáticas e
siloxanos. Paralelamente, cada molécula é composta por quatro unidades mesogénicas.
Da microssegregação que se verifica na fase esmética C resulta uma subcamada,
composta pelos corpos aromáticos, disposta entre duas subcamadas onde se encontram as
cadeias alifáticas e siloxano, Figura 34. Considerando uma camada esmética, para cada
elemento mesogénico (contido numa subcamada) haverá em média uma cadeia alifática e
apenas meia cadeia siloxano (Figura 34).

43
Figura 34 – Esquematização de uma fase esmética de moléculas inclinadas de tetrapodes
organosiloxanos.
A área molecular correspondente a cada uma das unidades mesogénicas é dada por
Σ=Vm/4ℓ, que é de aproximadamente 0.52 nm2, para 78ºC. A relação entre as áreas
moleculares do corpo mesogénico (c), das cadeias alifáticas (ch) e das cadeias siloxano (s) é
Σ=Σc=Σch+Σs/2. Para a determinação do ângulo de inclinação da fase esmética, é ainda
necessário considerar a secção transversal molecular. Para cada segmento molecular a
secção transversal, σ, dada por: Σcosψ, onde ψ é o ângulo entre o eixo do segmento
considerado e a normal às camadas esméticas. Considerando este argumento para todos os
segmentos moleculares, o ângulo entre os corpos aromáticos e a normal às camadas esméti-
cas resulta em (16).
s
s
ch
ch
c
c
ψ
σ
ψ
σ
σψ
cos2
1
cos
cos
+
=
(16)
O ângulo ψc é o ângulo que dá inclinação da fase esmética, uma vez que os corpos
aromáticos, são as unidades que apresentam uma rigidez significativa, e que a conferem à fase
esmética. Esta rigidez é relativa ao que se passa com as cadeias alifática e siloxano, que têm
grande mobilidade permitindo grandes oscilações em relação a uma posição média, com-
portando-se como cadeias quase líquidas. Minimizando em ordem a ψc, resulta
cosψc=σc/(σch+1/2σs). Este ângulo define a inclinação da fase esmética. Para o cálculo do valor
deste ângulo são necessários os valores típicos para as secções transversais de cada uma das
secções. Para os núcleos aromáticos o valor é σc~0.22-0.24 nm2, para as cadeias alifáticas
σch~0.2-0.4 nm2 e para as cadeias siloxano σc~0.43 nm2. Resultando o ângulo da fase esmética
~60º.
ψc
ψch
Σ σ
ℓ
ψs

44
Por haver competição entre o alinhamento do director nas camadas esméticas e no
campo aplicado, a partir dos resultados experimentais resultantes da difracção de raios X, não
é possível verificar directamente este valor a partir dos dados de DRX da fase SmC. No
entanto, uma vez que a fase nemática apresenta ordem local, assumindo uma organização
local cibotática idêntica à fase esmética C, dos resultados associados à fase nemática resulta
um ângulo para a inclinação das moléculas em relação à ordem local que se verifica. Verifica-
se que os ângulos encontrados experimentalmente (consultar Figura 33) estão próximo do
determinado teoricamente, reforçando a hipótese relativa ao tipo de estrutura adoptada por
esta amostra.
As integrações radiais nos grandes ângulos são também relevantes neste contexto. Os
perfis apresentados para a fase esmética e nemática, na região dos grandes ângulos, são
semelhantes, e caracterizados por dois picos. Na Figura 35 está indicada a evolução, na tem-
peratura, da largura a meia altura (full width at half maximum, FWHM) dos picos referentes às
integrações radiais, nos grande ângulos. Verifica-se que a largura destes picos é significativa-
mente superior na fase esmética C do que na fase nemática. Verifica-se que na fase esmética
os picos registados na integração radial para os grandes ângulos são mais difusos do que para
a fase nemática. Este dado corrobora a apontada competição entre o campo magnético aplica-
do no alinhamento no director das moléculas, e a formação das camadas esméticas.
70 80 90 100 110 120 130 14040
60
80
100
120
140
160
180
200
N SmC
FW
HM
(º)
T (ºC)
Figura 35 – Evolução da largura a meia altura (full width at half maximum, FWHM), com a
temperatura, para os picos de difracção na região dos grandes ângulos – ângulo c.
O gráfico indica que os corpos moleculares estão mais alinhados com o campo mag-
nético na fase nemática do que na fase esmética C. Energeticamente, na fase esmética C
verifica-se a competição entre o termo magnético, que tende a alinhar o director no campo
aplicado, e a energia associada à formação das camadas esméticas. Uma estrutura esmética
apenas permite deformações de flexão. Ao submeter a amostra a um campo magnético, os

45
corpos moleculares tendem a alinhar-se na direcção desse campo, no entanto a estrutura em
camadas esméticas tende a inibir esse alinhamento, dando origem a uma estrutura em chevron.
Na Figura 36 pode notar-se que a distância lateral entre os corpos aromáticos e os
grupos alifáticos aumenta com o aumento de temperatura.
70 80 90 100 110 120 130
4,45
4,50
4,55
4,60
4,65
N SmC
d (
Å)
T (ºC)
Figura 36 – Evolução da distância lateral entre moléculas com a temperatura, nas fases
nemática e esmética.
Composto Tas
O composto Tas apresenta apenas uma fase liquido cristalina nemática, Figura 22. O
padrão de difracção para 25ºC é apresentado na Figura 37.

46
Figura 37 – Padrão de difração de raios-X bidimensional para uma amostra alinhada do
composto Tas, na fase nemática, a 25ºC.
A Figura 37, correspondente ao composto Tas, é bastante semelhante à Figura 27, cor-
respondente ao composto, Ts. Ambas as figuras correspondem a padrões de difracção na fase
nemática. Este perfil indica uma vez mais a presença de grupos cibotáticos na fase nemática
onde as moléculas adoptam uma estrutura semelhante a uma estrutura chevron de uma fase
esmética de moléculas inclinadas, com camadas líquidas. Os dados de DRX agora obtidos
para o composto Tas foram tratados de forma semelhante à do composto anterior, os
resultados seguem nas Figuras: Figura 38 (integração azimutal, os valores obtidos estão
agrupados na Tabela 3), Figura 39 (integração radial nos pequenos ângulos) e Figura 40
(integração radial nos grandes ângulos).

47
2,5 5,0 7,5 10,0 12,5 15,0
75
150
225
300
375
450
525
I (u
ni.
arb
.)
q (nm -1 )
Figura 38 – Ajuste da integração azimutal do padrão de difracção na fase nemática, a 25ºC.
0 45 90 135 180 225 270 315 360
220
240
260
280
300
320
340
360
380
400
I (u
ni.
arb
.)
ψ (º)
Figura 39 – Ajuste do padrão radialmente integrado na região dos pequenos ângulos, na fase
nemática.

48
Figura 40 – Ajuste do padrão radialmente integrado na região dos grandes ângulos, na fase
nemática, a 25ºC.
O perfil agora determinado para este composto é similar aos resultados já obtidos para
o composto Ts. Os valores obtidos pela integração azimutal determinam a distância típica entre
moléculas adjacentes, na região dos grandes ângulos e o comprimento molecular, na região
dos pequenos ângulos.
Picos de difracção q (nm-1) Distância (nm) (2p/q)
001
x
siloxano
aromática-alifática
2,31
4,46
12,42
14,09
2,72
1,41
0,51
0,45
Tabela 3 – Resultado das distâncias obtidas por integração azimutal do padrão de difracção do
composto Tas a 25ºC, na fase nemática, estes resultados estão associados aos grupos
cibotáticos que localmente existem na fase nemática.
Na região dos pequenos ângulos, é possível, uma vez mais, verificar a existência dos
quatro picos semelhantes aos observados numa fase esmética C , Figura 39. Uma vez mais é
possível determinar um ângulo associado à inclinação que se detecta nos grupos cibotáticos
que coexistem na fase nemática. A evolução deste ângulo vem dada na Figura 41.
0 45 90 135 180 225 270 315 360200
250
300
350
400
450
500
I (u
ni.
arb
.)
χ (º)

49
25 30 35 40 45 5040
42
44
46
48
50
52
54
56
58
ψ (
º)
T (ºC)
Figura 41 – Evolução, na temperatura, do ângulo de inclinação presente nos grupos cibotáticos
da fase nemática.
É possível notar uma ligeira diferença, de alguns graus, entre este ângulo para os dois
compostos, o ângulo é superior para o composto Ts. O composto Tas tem cadeias alifáticas
mais longas e corpos aromáticos menos extensos, em relação a Tas. As cadeias alifáticas
conferem flexibilidade à fase, trata-se do segmento mais flexível da molécula. Assim as
moléculas do composto Ts conseguem inserir-se em modelos de empacotamento menos
inclinados resultando num menor valor do ângulo de inclinação detectado.

50
Capítulo 5
Análise dos Compostos Tm e Tm NS por
Espectroscopia RMN de Deutério
No decurso deste trabalho foram estudados dois monómeros do tetrapode, Tas
(anteriormente estudado por Figueirinhas et al. [ 27], por espectrocopia RMN de deutério) os
resultados de difracção de raios X deste tetrapode foram apresentados no capítulo anterior. No
capítulo 3 são apresentados alguns sistemas de cristais líquidos termotrópicos onde tem sido
investigada a existência de fases nemáticas biaxiais.
Neste trabalho procura-se contribuir para a investigação da origem da ordem nemática
biaxial, anteriormente observada num composto tetrapode organosiloxano [ 27]. A questão
central que será colocada prende-se com a comparação entre dois efeitos previamente
associados à origem da fase nemática biaxial em cristais líquidos termotrópicos. Por um lado
uma contribuição da forma das moléculas no sentido em que estas possam assumir uma
biaxialidade molecular, resposável pela estrutura biaxial da fase, como no caso dos compostos
em forma de boomerang [ 17] e, por outro, o constrangimento das rotações dos elementos
mesogénicos como foi observado nos polímeros líquido cristalinos de cadeia lateral [ 25]. Na
abordagem da questão da origem da fase nemática biaxial, será tida em conta a estrutura dos
tetrapodes detalhadamente analisada por DRX no capítulo 4. No sentido de contribuir para a
solução deste problema, foram também estudados, por RMN de deutério, monómeros dos
tetrapodes, anteriormente investigados.
O estudo a que estes compostos foram sujeitos teve como objectivo determinar se
misturas (com a mesma sonda e em semelhante concentração (percentagem de peso) à que
tinha sido já realizada com o tetrapode) agora com os monómeros, apresentam comportamento
biaxial na fase nemática. Assim, utilizou-se a mesma sonda que em [ 27], 7CB, deuterada na
posição a. A utilização de uma sonda deuterada, prende-se com o tipo de análise a que estas
moléculas vão ser submetidas (RMN do deutério), uma vez que as amostras dos compostos
puros não são deuteradas. Na escolha das sondas teve-se em conta que as mesmas deverão
possuir uma estrutura molecular tal que as leve a assumir o tipo de ordem apresentada pelo
composto em que se encontram dissolvidas. A sonda escolhida apresenta uma fase nemática
(à semelhança dos monómeros). Deste modo, uma condição crucial será a forma como a
sonda se posiciona em relação às moléculas do composto. Neste caso, pretendia-se que a
sonda se dispusesse junto dos corpos aromáticos, uma vez que se supõe serem estes os
responsáveis pela ordem nemática biaxial.
As misturas são constituídas por 15% (em peso) de sonda e 85% (em peso) do
monómero considerado. Como é normal, a presença da sonda provocou alterações nas
temperaturas de transição dos monómeros puros, para as misturas (dados obtidos por DSC). A

51
estrutura química das moléculas, bem como o esquema de transições de fase das respectivas
misturas com a sonda utilizada, estão indicados na Figura 42.
a)
Si
Si
O
Si
Si
O
C8H17O OC11H23 O
O
O
4\
I N T C C
g → → − º 37 º 40
b)
CN D D
c)
I N T C C g → → º 33.6 º 5.5
d)
C8H17O
OC11H23 O
O
O
I N T C C
g → → º 72.6 º 51.6
Figura 42 – Estruturas moleculares dos compostos considerados neste capítulo e sequência
de fases para as respectivas misturas com a sonda deuterada, 7CBαD2. (a) O tetrapode, Tas;
(b) a sonda deuterada, 7CBαD2; (c) o monómero, Tm e (d) o monómero não substituído, Tm
NS (d).
Para avaliar se estas misturas apresentam fases nemáticas biaxiais foram obtidos
espectros de RMN do deutério, cuja importância foi já referida no capítulo 3, uma vez que o
afastamento quadrupolar resultante inclui um termo dependente do parâmetro de assimetria, η,
que se não for nulo indica estarmos em presença de uma fase biaxial. Conforme foi referido no
capítulo 3, a expressão do afastamento quadrupolar é dada por:
ϕθ
η+
−θν=δν 2cossin
22
1cos
2
3 22Q2
3
(17)
Os parâmetros dos quais depende o afastamento quadrupolar são νQ, que corresponde
ao valor médio da constante de acoplamento quadrupolar do deutério (abreviadamente, cons-
tante quadrupolar) e η, o parâmetro de assimetria. Este afastamento é também dependente
dos ângulos θ e ϕ. O ângulo θ mede o desvio da orientação do director principal em relação ao
campo magnético estático, B0. O ângulo ϕ dá, por seu lado, o desvio do alinhamento do direc-
tor secundário em relação ao campo estático. No caso em que se tratam de amostras uniaxiais
o segundo termo é nulo, uma vez que η=0 (Figura 11).
Si
O
Si
C8H17O
OC11H23 O
O
O

52
Os espectros que se podem obter por RMN do deutério, no caso mais simples, em que
estamos na presença de uma amostra estática e alinhada com o campo estático aplicado, cor-
respondem a duas riscas centradas em zero e separadas por um intervalo de frequências
correspondente a 3/2 da constante quadrupolar, νQ (corresponde à situação em que θ é zero,
ou seja o director principal está completamente alinhado com o campo estático). Esta situação
mais simples corresponde ao que se obtém para uma amostra nemática totalmente alinhada
(monodomínio). No caso de espectros policristalinos, a análise é mais complexa. Para estas
amostras, policristalinas, os diferentes ângulos entre as orientações do director em cada
domínio e o campo estático vão ser ponderados no afastamento quadrupolar, originando um
espectro que precisa de ser analisado caso a caso. O afastamento máximo volta a ser dado
por 3/2νQ (situação de alinhamento total).
Dependendo das condições experimentais, uma amostra pode apresentar-se como um
pó cristalino em duas ou três dimensões. Os espectros obtidos para policristais bidimensionais
ou tridimensionais têm características diferentes. Em ambos os casos, para amostras uniaxiais
e biaxiais os espectros apresentam características, intrinsecamente diferentes. Dado que os
espectros para os pós são distintos no caso biaxial ou uniaxial, a existência de ordem nemática
biaxial poderá ser avaliada utilizando amostras com estas características.
Uma amostra policristalina nas três dimensões resulta de uma distribuição uniforme de
dos directores principais no espaço tridimensional. Já um policristal bidimensional resulta de se
ter uma amostra, sujeita ao campo magnético estático, em rotação em torno de um eixo
perpendicular ao campo aplicado, B0. Daí decorre uma distribuição uniforme do director
principal no plano perpendicular ao eixo de rotação, resultante da minimização da energia na
presença do campo magnético. Neste trabalho foram obtidas amostras policristalinas a duas
dimensões (pós bidimensionais).
No caso de um pó bidimensional uniaxial o espectro apresenta dois picos, correspon-
dentes às situações em que θ = 0 e 90º, das quais resultam, respectivamente, afastamentos
δνQ=3/2νQ e 3/4νQ. O espectro é caracterizado por quatro picos, em que os picos interiores se
encontram afastadas de metade do intervalo associado às das exteriores. No espectro de uma
amostra biaxial, contribui também o termo associado ao parâmetro de assimetria, e neste caso,
os picos exteriores encontram-se na mesma posição (afastadas da mesma distância). Mas as
riscas interiores têm um afastamento que é diferente de metade do afastamento entre as riscas
exteriores (portanto diferente do que se observava no caso uniaxial, consultar Figura 21).
Um outro processo possível para investigar a possível existência de ordem biaxial
consiste em rodar uma amostra totalmente alinhada de um ângulo fixo e obter espectros
associados a esta nova direcção, fixa, como descrito no capítulo 3. Este procedimento não é
adequado a este problema com os monómeros (no caso do tetrapode é um bom método), por
questões que se prendem com a viscosidade das amostras. A viscosidade das amostras
condiciona os procedimentos, na medida em que para viscosidades suficientemente baixas
(como é o caso dos monómeros) o tempo de reorientação é da mesma ordem de grandeza que
o tempo de observação, o que impossibilita a observação do sistema por este método. Partindo

53
de uma amostra líquido-cristalina alinhada, se a esta for aplicada uma rotação uniforme, em
torno de um eixo perpendicular ao campo magnético estático, com velocidade superior à
velocidade crítica (abaixo da velocidade crítica a amostra mantém-se alinhada com o director
principal desviado de um ângulo fixo em relação ao campo estático), a amostra assumirá uma
distribuição do director correspondente a um pó bidimensional no plano perpendicular ao eixo
de rotação. Nestas condições, se se obtiver um espectro com aquisições não sincronizadas
poder-se-á averiguar da existência (ou inexistência) da assinatura associada à ordem nemática
biaxial.
No trabalho em causa, para avaliar a velocidade crítica das amostras, estas foram
colocadas em rotação contínua com diferentes velocidades e foram registados os respectivos
espectros. No caso de a velocidade ser inferior à velocidade crítica, o espectro resultante
apresenta um par de riscas, à semelhança do que acontecia na rotação por um ângulo fixo, ou
na situação totalmente alinhada. Para velocidades crescentes, verifica-se que o director deixa
de estar alinhado paralelamente ao campo aplicado passa a ter um ângulo entre o director e o
campo aplicado. No espectro resulta uma diminuição do afastamento quadrupolar. Já no caso
em que a amostra roda com velocidade superior à velocidade crítica o espectro passa a ser
distinto, apresentando dois pares picos, correspondentes a uma amostra policristalina. Foi
inspeccionada a velocidade a partir da qual a amostra perdia o alinhamento total com o campo
magnético, passando a exibir um espectro de um pó. Verificou-se que a velocidade crítica
surge, para o composto Tm para uma velocidade superior ao que acontecia para Tm NS (todos
os espectros de amostras policristalinas apresentados foram obtidos para velocidades
superiores à velocidade crítica).
Os dados experimentais foram obtidos num espectrómetro BRUKER AVANCE II 300
equipado com um electromagneto BRUKER B-E30 operando a um campo magnético com
intensidade de 2.3T, correspondendo a uma frequência de ressonância do deutério de 15.1
MHz.
Verificou-se no circuito a existência de ringing acústico. A intensidade do sinal com
origem no ringing, sem significado para a física do problema, introduz ruído nas medições.
Para melhorar a relação sinal ruído foi necessário recorrer a um programa de impulsos em que
se tentasse compensar o ruído proveniente do ringing. A sequência utilizada inclui uma
composição de duas sequências de eco sólido (solid echo), em que uma das sequências foi
antecedida de um impulso–p que inverte a magnetização do sistema. Numa sequência de eco
sólido são aplicados dois impulsos π/2, (com fases relativas deslocadas de 90º, ver Figura 43)
que rodam a magnetização de π/2. Estes impulsos estão separados de um intervalo de tempo τ.
Quando se dá finalmente a relaxação do sistema para a situação inicial, o máximo de
intensidade é registado, um tempo, τ, após o segundo impulso. A combinação destas duas
sequências de impulsos resulta na soma do sinal de precessão livre que se pretendia medir
resultante da presença do deutério (uma vez que os sinais de precessão livre de uma e outra
situação são absolutamente equivalentes). No caso do sinal resultante do ringing pela inversão
que o sistema sofreu o resultado é a subtracção dos sinais de ringing das duas situações. Na

54
Figura 43 apresenta-se o esquema de impulsos adoptado. A sequência de impulsos indicada
foi a utilizada para a obtenção de todos os espectros adiante apresentados.
Figura 43 – Esquema dos impulsos adoptado.
O esquema de montagem utilizado nestes ensaios é apresentado na Figura 44.
Figura 44 – Esquema da montagem experimental.
Foram adquiridos espectros para as duas misturas (dos monómeros com a sonda) dos
dois diferentes monómeros do tetrapode. Foram tomadas aquisições para diferentes
temperaturas, dentro da fase nemática dos compostos. No início de cada experiência, as
amostras foram submetidas ao campo magnético estático e foram aquecidas, com recurso ao
sistema de controlo da temperatura, até se dar a transição para a fase isótropa, posteriormente
foram arrefecidas, uma vez mais na presença do campo, até à temperatura pretendida. A
subida da temperatura aconteceu a uma taxa máxima de 5ºC/min, já na descida a velocidade
máxima foi de -3ºC/min. Neste processo as amostras estiveram sempre estáticas, ou seja, nas
amostras, o director ficou uniformemente alinhado com o campo estático. Para cada
temperatura foi obtido um espectro em que a amostra estática estava alinhada com o campo
p p/2x p/2y p/2x p/2y
FID FID Solid echo Solid echo
τ τ τ τ
Motor M
AG
NE
TE
MA
GN
ET
E
Amostra Cabeça de medida

55
aplicado. A amostra foi seguidamente submetida a uma rotação contínua e foram adquiridos
novos espectros, utilizando aquisições assíncronas.
Foram feitas aquisições entre as temperaturas 8 e 24ºC, e 20 e 65 ºC, para os
compostos Tm e Tm NS, respectivamente. Às temperaturas analisadas os compostos
apresentam sempre fases nemáticas. Esta condição manteve-se válida quando se desce a
temperatura para valores inferiores à temperatura de transição para o estado sólido, devido ao
fenómeno de sobre-arrefecimento [ 15], que neste caso ocorre numa larga gama de temperatu-
ras.
A única mesofase exibida pelos monómeros investigados, à semelhança do que já
acontecia para o tetrapode, correspondente, é uma fase nemática. Uma fase biaxial é uma fase
mais ordenada que uma fase uniaxial. Em geral fases mais ordenadas surgem para
temperaturas inferiores às das fases mais desordenadas. Será então de esperar que, para
temperaturas mais baixas, a possibilidade de existir uma fase nemática biaxial seja maior do
que para uma temperatura mais elevada. Assim o fenómeno de sobre-arrefecimento pode ser
útil no sentido de propiciar eventualmente o aparecimento de fases nemáticas biaxiais.
Pela simulação dos espectros pretende-se averiguar, se existe ou não um parâmetro
de assimetria, h, não nulo, dentro do erro experimental. Os espectros obtidos foram simulados
tendo como critério de ajuste a minimização do c2 reduzido. O erro admitido para os pontos dos
espectros segue uma distribuição estatística normal em que o erro é, naturalmente, dado pela
raíz do número de contagens. Este erro não pondera o facto de as riscas terem mais relevância
no ajuste dos espectros. Os ajustes parecem, no entanto, suficientemente fieis aos espectros
experimentais para se considerar que os modelos utilizados descrevem adequadamente o
problema.
Para o caso dos espectros de amostras estáticas, em que se verifica o alinhamento do
director com o campo estático aplicado, o que é evidenciado pela presença de duas únicas
riscas, o espectro experimental pode ser simulado pela expressão:
( ) ( ) ( )2 / L 2 / L G δν + ν + δν − ν = ν (18)
( )( )22 2/
2/Lδν−ν+λ
λ=δν−ν
(19)
No caso dos espectros obtidos para amostras em rotação, como há contribuição de
muitos domínios nemáticos, a situação verificada é equivalente a ter uma distribuição de
domínios no plano perpendicular ao eixo de rotação da amostra. Esta situação é equivalente à
distribuição de pares de riscas como na situação em repouso (orientação de 0º em relação ao
campo aplicado) para as diferentes orientações possíveis, no plano ortogonal ao eixo de
rotação da amostra. Os espectros resultantes desta nova situação são descritos pela
expressão (20):

56
( ) ( ) ( )[ ] ( ) ΩΩδν+ν+δν−ν=ω ∫Ω
dP2/L2/LG
(20)
Se se tratar de um pó bidimensional P(Ω) corresponde a uma distribuição uniforme no
plano ortogonal ao eixo de rotação da amostra. No caso da amostra em rotação verificou-se
que a descrição anterior não era suficientemente precisa para o bom ajuste dos espectros.
Dada a baixa viscosidade destes compostos, foi necessário considerar que a distribuição dos
directores P(Ω) apresentava uma contribuição não uniforme resultante de reorientações
parciais devido ao acoplamento com o campo B0. Esta correcção pode ser descrita por uma
expansão em série de Fourier onde só aparecem os termos pares em coseno, uma vez que o
alinhamento do director é indistinguível para θ=0 e θ=π. O facto de aparecerem apenas termos
em coseno e não em seno resulta da simetria física do problema. A expansão foi desenvolvida
até à terceira ordem, (21).
( ) ( ) ( ) ( )[ ]θ+θ+θ+=Ω 6cosC4cosC2cosC1P 321 (21)
Verifica-se que a correcção introduzida tem um papel significativo na simulação dos
espectros. Esta correcção é tanto maior quanto menor a velocidade, para velocidades acima da
velocidade crítica. Trata-se de uma situação esperada uma vez que quanto menor for a
velocidade de rotação maior a influência do campo no alinhamento das moléculas, que para
ângulos diferentes resulta na aceleração ou desaceleração dos directores ao tentarem alinhar-
se com o campo. Para todas as velocidades experimentadas, dentro da gama de velocidades
possível para o sistema motor–suporte da amostra não foi possível chegar a nenhuma
velocidade para a qual não houvesse um realinhamento parcial dos directores com o campo
magnético estático.
Para os diferentes ensaios foram obtidos sempre os espectros estáticos antecedendo
os espectros de amostra em rotação. Os parâmetros νQ e λ (obtidos para os espectros
estáticos) foram usados como valores iniciais na simulação dos espectros de amostras em
rotação.
Em seguida estão apresentados alguns dos resultados obtidos para os dois compostos
estudados, para diferentes temperaturas e velocidades de rotação da amostra.
Para o monómero Tm NS os espectros obtidos para amostras estáticas são da forma
indicada na Figura 45.

57
-40 -20 0 20 40-50
0
50
100
150
200
250
300
I (u
ni. a
rb.)
ν (kHz)
Figura 45 – Espectro RMN do deutério, para amostra estática do composto Tm NS a 25ºC.
Aos espectros das amostras estáticas foi ajustada a função (18). Os mesmo parâme-
tros referentes à forma das riscas foram utilizados subsequentemente, para os espectros em
rotação. O que se passa na situação de rotação, em relação ao caso estático, é que em rota-
ção não haverá apenas a contribuição, para o espectro final, de um par de riscas, mas sim de
uma distribuição de riscas que será ponderada pelo ângulo entre o campo e o director. Nesta
situação foi ainda ponderada uma possível contribuição referente ao parâmetro de assimetria.
Assim as características de cada par de riscas em relação à situação estática mantêm-se, e daí
a importância dos resultados do ajuste relativo aos espectros de amostras estáticas aquando
do ajuste aos espectros correspondentes a amostras em rotação.
O resultado da simulação do espectro obtido em condições estáticas em causa vem
indicado na Figura 46. À mesma temperatura o espectro resultante para a amostra em rotação
vem dado na Figura 47.
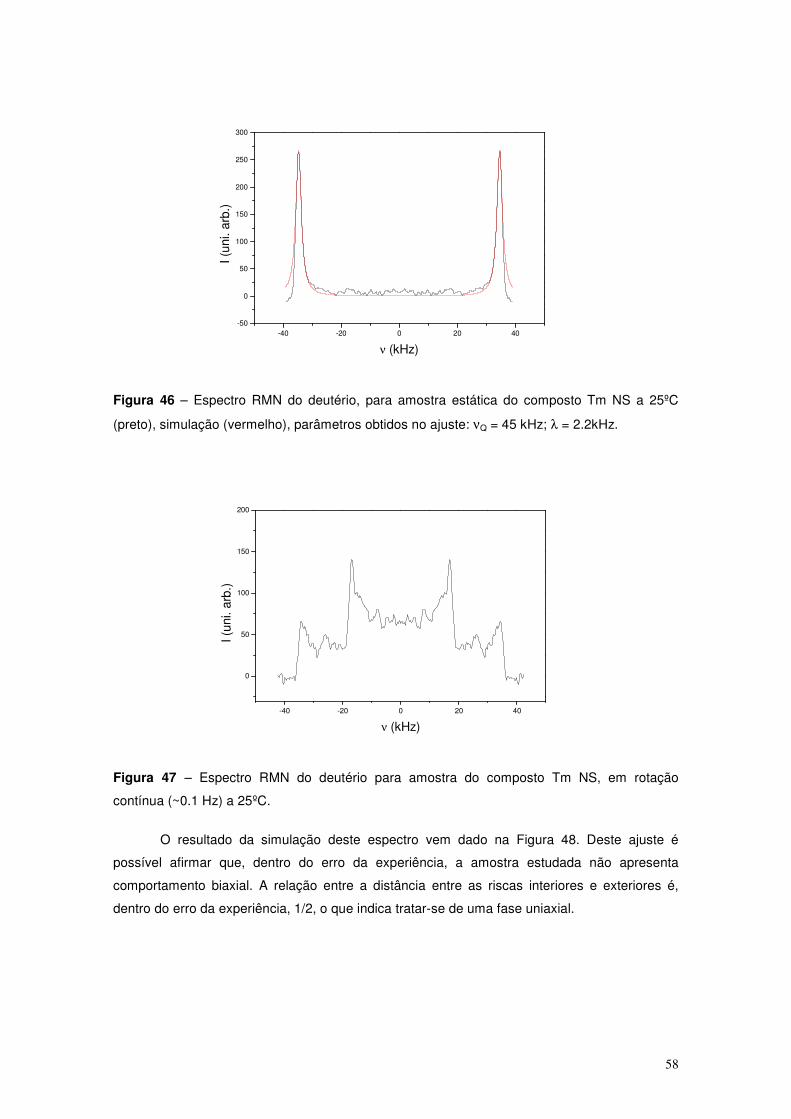
58
-40 -20 0 20 40-50
0
50
100
150
200
250
300
I (u
ni. a
rb.)
ν (kHz)
Figura 46 – Espectro RMN do deutério, para amostra estática do composto Tm NS a 25ºC
(preto), simulação (vermelho), parâmetros obtidos no ajuste: νQ = 45 kHz; λ = 2.2kHz.
-40 -20 0 20 40
0
50
100
150
200
I (un
i. ar
b.)
ν (kHz)
Figura 47 – Espectro RMN do deutério para amostra do composto Tm NS, em rotação
contínua (~0.1 Hz) a 25ºC.
O resultado da simulação deste espectro vem dado na Figura 48. Deste ajuste é
possível afirmar que, dentro do erro da experiência, a amostra estudada não apresenta
comportamento biaxial. A relação entre a distância entre as riscas interiores e exteriores é,
dentro do erro da experiência, 1/2, o que indica tratar-se de uma fase uniaxial.

59
-40 -20 0 20 40
0
50
100
150
200
I (un
i. ar
b.)
ν (kHz)
Figura 48 – Espectro RMN do deutério, para amostra em rotação contínua (~0.1 Hz) do
composto Tm NS a 25ºC (preto), simulação do espectro na ausência da correcção à
distribuição do director (verde).
Como tinha já sido referido, foi considerada uma correcção adicional na simulação dos
espectros associados a amostras em rotação. Com esta correcção é tida em conta a diferença
entre as velocidades efectivas que o director nemático toma na amostra e a velocidade a que a
amostra é posta em rotação. No caso das amostras em estudo, os dados mostram que esta
correcção não é desprezável. Na Figura 49 é apresentado, a verde, o ajuste na ausência deste
termo correctivo e é possível verificar um desvio significativo entre a situação com e sem
correcção.
-40 -20 0 20 40
0
50
100
150
200
I (un
i. ar
b.)
ν (kHz)
Figura 49 – Espectro RMN do deutério, para amostra em rotação contínua (~0.1 Hz) do
composto Tm NS a 25ºC (preto), simulação do espectro na ausência da correcção à

60
distribuição do director (verde), simulação do espectro com correcção à distribuição do director
(vermelho) segundo a expressão (21), parâmetro de assimetria obtido do ajuste: η =
0.003±0.012.
A correcção angular utilizada para a distribuição do ângulo θ, segundo a expressão
(21), para o ensaio apresentado, é representada na Figura 50, pela curva a vermelho. Na
ausência desta correcção, a distribuição angular seria uniforme, dando o mesmo peso a todos
os ângulos, como indicado na Figura 50 (curva a verde).
0,0 0,5 1,0 1,5 2,0 2,5 3,00,0
0,2
0,4
0,6
0,8
1,0
1,2
P(θ
)
θ (rad)
Figura 50 – Correcção angular (segundo a expressão (21)) para ensaio ao composto Tm NS à
temperatura de 25ºC e velocidade de rotação ~ 0.1 Hz (curva a vermelho). A linha a verde
corresponde à distribuição uniforme do director em θ.
A correcção angular encontrada reflecte o que se esperava: uma valorização dos
ângulos correspondentes a 45º, situação em que é favorecida a reorientação do director pelo
campo magnético. Há então um decréscimo da distribuição do director para os ângulos 0 e 90º,
que se reflecte nos espectros através de uma diminuição nas intensidades dos picos interiores
e exteriores. No resultado apresentado a verde, no ajuste, para espectro simulado sem a
correcção, está já patente esta situação. Ou seja, em geral, esperar-se-ia que a contribuição
angular em 0 e 90º fosse superior ao efectivamente registado.
Para o outro composto, Tm, os resultados são semelhantes, aqui a viscosidade do
composto, em relação ao anterior, é superior, mas verificam-se o mesmo tipo de fenómenos.
As figuras que se seguem apresentam um estudo semelhante ao anterior, de onde resulta uma
vez mais que a amostra não apresenta comportamento nemático biaxial. A Figura 51
apresenta-se os resultados para o espectro relativo à amostra estática, na Figura 52 encontra-
se o espectro correspondente à amostra em rotação e, por fim, na Figura 53 temos o resultado
da correcção da distribuição angular.

61
-40 -20 0 20 40
0
50
100
150
200
I (un
i. ar
b.)
ν (kHz)
Figura 51 – Espectro RMN do deutério, para amostra estática do composto Tm a 8ºC (preto),
ajuste do espectro (vermelho), parâmetros obtidos dos ajuste: νQ = 42 kHz; λ = 2.7 kHz.
-40 -20 0 20 40
0
75
150
225
300
375
450
I (un
i. ar
b.)
ν (kHz)
Figura 52 – Espectro RMN do deutério, para amostra em rotação contínua (~1.3 Hz) do
composto Tm a 8ºC (preto), ajuste do espectro na ausência da correcção à distribuição do
director (verde), ajuste do espectro (vermelho), parâmetro de assimetria obtido do ajuste: η =
0.015±0.016.

62
0,0 0,5 1,0 1,5 2,0 2,5 3,00,00
0,25
0,50
0,75
1,00
1,25
P(θ
)
θ (rad)
Figura 53 – Correcção angular (segundo a expressão (21)) para ensaio ao composto Tm à
temperatura de 8ºC e velocidade de rotação ~ 1.3 Hz (curva a vermelho). A linha a verde
corresponde à distribuição uniforme do director em θ.
Os dados apontados foram obtidos para condições especificas de temperatura e
velocidade de rotação das amostras. Verificou-se, dentro da gama de temperaturas da fase
nemática de cada uma das amostras, que estas não apresentam comportamento biaxial, ao
contrário do que foi verificado para o tetrapode correspondente. Estes resultados indicam que a
origem da fase nemática biaxial no tetrapode não surge associada ao comportamento dos
monómeros considerados individualmente. Para diferentes velocidades de rotação da amostra
é de notar que a correcção na distribuição angular é tanto mais significativa quanto menor for a
velocidade de rotação, se acima da velocidade crítica.

63
Capítulo 6
Conclusão e Pespectivas de Trabalho Futuro
O trabalho que deu origem a esta tese teve como objectivo o estudo da fase nemática
biaxial em dendrímeros líquido cristalinos (de geração zero). Os estudos na fase nemática
biaxial, de sistemas termotrópicos, têm sido alvo de controvérsia [ 20]. Neste trabalho preten-
deu-se estudar a origem da fase nemática biaxial, do composto tetrapode organosiloxano Tas,
verificada por técnicas ópticas [ 35] e de espectroscopia RMN de deutério [ 27] (a verificação da
presença de biaxialidade numa fase nemática através destas duas técnicas experimentais,
indica tratar-se de uma fase nemática biaxial). A ordem biaxial que se observa na fase
nemática encontra-se geralmente associada à forma das moléculas ou elementos mesogénicos
que constituem esta fase. É, então, considerado expectável que uma fase nemática formada
por moléculas cuja estrutura química se afaste das convencionais moléculas em forma de
bastonete ou de disco (ambas com simetria de rotação em torno de um eixo) tenda a
apresentar ordem biaxial. Este é o caso das fases nemáticas de moléculas em forma de
boomerang [ 17]. Por outro lado, foi observada, tanto em polímeros líquido cristalinos
termotrópicos [ 25], como em tetrapodes organosiloxanos [ 27], a existência de fases nemáticas
biaxiais em sistemas cujas unidades mesogénicas são relativamente próximas das que
constituem as fases nemáticas uniaxiais. Neste caso, a ordem biaxial surge associada à
complexidade dos sistemas, no sentido em que a quebra de simetria de rotação em torno do
eixo associado ao director principal resulta dos constrangimentos impostos pela estrutura dos
polímeros ou dendrímeros em causa e não pela forma dos elementos mesogénicos. Nos casos
conhecidos, este tipo de fase aparece em sistemas formados por ligação lateral dos elementos
mesogénicos a uma cadeia polimérica ou ao corpo central de um dendrímero, respectivamente.
No sentido de averiguar qual o factor que contribui, de forma mais determinante, para a
fase nemática biaxial, o tetrapode Tas bem como Ts foram estudados por DRX para
determinação das suas estruturas moleculares. Por outro lado, dois monómeros do tetrapode
Tas foram submetidos a espectroscopia RMN de deutério, para averiguar da existência (ou
inexistência) da assinatura biaxial na fase nemática destes compostos. A realização deste
trabalho permitiu concluir que a biaxialidade observada em mesofases nemáticas para o
tetrapode organosiloxano, Tas, não tem origem nos monómeros, individualmente, mas antes
na forma como estes estão ligados nas moléculas e como a estrutura molecular condiciona a
organização das fases.
Em termos estruturais, verifica-se que os compostos Ts e Tas, que têm dimensões
moleculares idênticas (ver Figura 23), no entanto, os aparentes detalhes que diferenciam estas
duas moléculas, determinam que o composto cujos elementos mesogénicos são simétricos
apresenta uma fase esmética C, para além da fase nemática, enquanto o composto de

64
elementos mesogénicos assimétricos apresenta apenas uma fase nemática. Os padrões de
DRX mostram claramente que ambos os compostos apresentam fases nemáticas com uma
estrutura peculiar associada à presença de grupos cibotáticos, do tipo esmético C, cuja
existência prevalece em toda a extensão do domínio térmico da fase. É significativo notar que,
em trabalhos relativos ao estudo da fase nemática de um composto de moléculas em forma de
boomerang, este tipo de padrões de DRX aparece associado à presença de ordem biaxial [ 17].
Por outro lado, os dois compostos apresentam espessuras diferentes para as pseudo-camadas
presentes nos grupos cibotáticos das respectivas fases nemáticas, sendo inferiores para o
composto Tas que para o composto Ts (consultar Tabela 2 e Tabela 3). Esta diferença significa
que a agregação molecular é mais eficaz para o composto Tas relativamente a Ts. Este ponto
pode ser significativo para a discussão da origem da fase nemática biaxial. Em termos da
constituição dos compostos, as diferenças entre Ts e Tas manifestam-se na extensão dos
corpos aromáticos e das cadeias alifáticas. No composto Tas os corpos aromáticos são mais
longos, e as cadeias alifáticas menos extensas. Os corpos aromáticos estão associadas a
maior rigidez que as cadeias alifáticas. Os detalhes da forma de agregação molecular, que são
diferentes entre Ts e Tas poderão estar relacionados com esta diferença de rigidez e de
comprimentos entre corpos e cadeias, assegurando um empacotamento molecular mais eficaz
na fase nemática para o composto Tas. Esta poderá ser uma condição importante da
biaxialidade em fase nemáticas.
A espessura das pseudo-camadas, ℓ, que se formam localmente nos grupos cibotáticos
da fase nemática, sofre uma evolução na temperatura. No composto Ts que apresenta uma
fase nemática e uma fase esmética C a evolução de é ℓ registada na Figura 29. Neste gráfico
fica indicado que ℓ assume valores superiores na fase esmética C do que na fase nemática.
Esta variação de ℓ indicía um alinhamento mais fácil nas camadas esméticas para temperaturas
mais baixas (onde há menos mobilidade de o sistema e consequentemente o alinhamento com
o campo fica prejudicado), ou seja, o alinhamento local é facilitado na fase nemática em
relação à fase esmética. Fica evidenciada a crescente mobilidade da fase nemática em relação
à fase esmética. Esta mobilidade na fase nemática tem efeito no sentido de originar uma
estrutura para a fase nemática mais compacta do que para a fase esmética C. Uma vez mais
esta compactação pode propiciar o aparecimento de uma fase nemática biaxial. Experimental-
mente a biaxialidade na fase nemática do composto Ts foi verificada por técnicas ópticas e de
espectroscopia do infravermelho [ 26].
Da análise estrutural dos dois tetrapodes pode concluir-se, pela permanência dos
grupos cibotáticos, que ambos apresentam fases nemáticas localmente mais organizadas do
que as fases nemáticas mais comuns. Este tipo de comportamento existe em toda a gama de
temperaturas ensaiadas da fase nemática. Para o composto Ts foram tomados dados em toda
a gama de temperaturas de existência da fase nemática (as temperaturas de transição estão
indicadas Figura 22). Relativamente ao composto Tas foram efectuados ensaios na fase
nemática entre as temperaturas de 47ºC e 25ºC, não tendo sido realizadas medidas abaixo da
temperatura ambiente, no entanto o composto apresenta a transição vítrea a -30ºC (Figura 23).

65
Em estudos de biaxialidade em fases nemáticas em polímeros líquido-cristalinos
publicados na literatura [ 24, 25] concluiu-se que nos polímeros de cadeia lateral era possível a
existência de fases nemáticas biaxiais, enquanto nos polímeros de cadeia principal, as fases
nemáticas apresentadas eram invariavelmente uniaxais. A explicação para esta diferença de
comportamento prende-se com constrangimentos nos movimentos de rotação das unidade
mesogénicas nos polímeros de cadeia lateral (que não existe nos polímeros de cadeia
principal). No caso dos tetrapodes estudados, e embora a sua estrutura seja idêntica,
possivelmente, no composto Tas os corpos aromáticos e as cadeias alifáticas adoptam uma
estrutura mais compacta, com movimento de rotação mais restringidos, propiciando uma fase
nemática biaxial mais inequívoca. Este efeito é especialmente importante para as baixas
temperaturas incluídas no domínio térmico de existência da fase nemática do composto Tas.
Para avaliar a importância da estrutura molecular dos tetrapodes, no que respeita à
presença da ordem biaxial na fase nemática, foram estudados dois monómeros do tetrapode
Tas. Os monómeros, à semelhança do tetrapode, foram estudados por espectroscopia RMN de
deutério. Os monómeros estudados não têm deutério na sua composição, pelo que foram
preparadas misturas destes compostos para os ensaios de espectroscopia. A sonda utilizada
(7CBαD2), é um cristal líquido nemático com uma estrutura molecular que, previsivelmente, o
levará a assumir o tipo de ordem apresentada pelo composto em que se encontra dissolvido.
Da análise dos resultados experimentais obtidos por RMN do deutério para as misturas
dos monómeros do tetrapode Tas conclui-se que as amostras estudadas não apresentam uma
fase nemática biaxial. A assinatura nos espectros de pós bidimensionais (resultantes de
rotação da amostra segundo um eixo ortogonal ao campo estático B0 aplicado) é a de uma fase
nemática uniaxial (a relação entre o afastamento entre riscas exteriores e interiores é de 1/2).
Por seu lado, a mistura do tetrapode [ 27] tem uma fase nemática biaxial. Comparando os
resultados obtidos para monómeros e tetrapode pode-se dizer que a fase nemática biaxial que
se observa no tetrapode não é resultado de uma característica intrínseca dos monómeros.
O facto de não ter sido verificada biaxialidade na fase nemática dos monómeros, indica
que, assim como nos polímeros [ 24, 25], a estrutura dos elementos mesogénicos considerados
individualmente não é necessariamente determinante para o comportamento biaxial ou não das
fases nemáticas que exibem. Esta conclusão já tinha sido também inferida para sistemas de
baixo pelo molecular [ 20]. Por outro lado, os resultados estruturais para os tetrapodes apontam
para organizações compactas onde se pode presumir inibição de determinados movimentos de
rotação na fase nemática (biaxial). As fases nemáticas que os tetrapodes exibem são,
localmente, muito organizadas, mais organizadas do que em geral se verifica para uma fase
nemática convencional. Resumindo: o facto das fases nemáticas formadas pelos monómeros
apresentarem apenas ordem uniaxial (contrariamente ao que acontece com os tetrapodes) é
compatível com a hipótese de que a ordem nemática biaxial resulta de constrangimentos
orientacionais das unidades mesogénicas provocados pela ligação ao núcleo central dos
dendrímeros.

66
Em termos de possíveis análises futuras de espectroscopia RMN de deutério, seria
interessante considerar os tetrapodes deuterados, em vez de se recorrer a uma sonda, para
confirmar o carácter biaxial dos compostos, separadamente das misturas. Seria igualmente
interessante fazer a caracterização dos espectros obtidos para os tetrapodes em função da
velocidade de rotação da amostra, recorrendo a equações para a dinâmica do director (para
amostras biaxiais). Os estudos da caracterização dos espectros em função da velocidade
seriam também interessantes para os monómeros, uma vez que estes têm movimentos muito
rápidos o que pode ser útil em termos do estudo comparativo com os tetrapodes.

67
Bibliografia
1. A. F. Martins, Os Cristais Líquidos, Colóquios Ciência, Ed. Gulbenkian (1991)
2. P. G. De Gennes, J. Prost, The Physics of Liquid Crystals, 2ª edição, Clarendon Press,
Oxford (1974)
3. P. J Collings and M. Hird, Introduction to Liquid Crystals, Taylor and Francis (1998)
4. S. Chandrasekhar, Liquid Crystals, 2nd editon, Cambriedge University Press (1992)
5. Sítio: http://www2.dupont.com/Kevlar/en_US/
6. M. Moritsugu, S. N. Kim, T. Ogata, T. Nonaka, S. Kurihara, S. Kubo H. Segawa and O. Sato,
Appl. Phys. Lett. 89, 153131 (2006)
7. R. M. Richardson, S. A. Ponomarenko, N. I. Boiko and V. P. Shibaev, Liquid Crystals 26, 1,
101 (1999)
8. C. Cruz, Estudo por RMN da Ordem e Dinâmica Molecular em Mesofases com Característi-
cas Singulares, Tese de Doutoramento, Universidade Técnica de Lisboa (1994)
9. G. R. Newkome, C. N. Moorefield and F. Vögtle, Dendrimers and Dendrons, Wiley-VCH
(2001)
10. J.P. Straley, Phys. Rev. A 10, 1881 (1974)
11. J. L. Figueirinhas, A Deuterium Nuclear Magnetic Resonance Study of the Smectic F Phase,
Ph. D. Thesis, Kent University (1987)
12. M. J. Freiser, Phys. Rev. Lett. 24, 1041 (1970).
13. L. J. Yu and A. Saupe, Phys. Rev. Lett. 45, 1000 (1980)
14. Y. Galerne, Mol. Cryst. Liq. Cryst. 323 (1998)
15. S. Chandrasekhar, B. K. Sadashiva, B. R. Ratna and V. N. Raja, Pramana 30, L-491 (1988)
16. P. I. C. Teixeira, A. J. Masters and B. M. Mulder, Mol. Cryst. Liq. Cryst. 323, 167 (1997)
17. L. A. Madsen, T. J. Dingemans, M. Nakata and E. T. Samulski, Phys. Rev. Lett. 92,
145505-1 (2004)
18. D. W. Bruce, The Chemical Record 4, 10-22 (2004)
19. A. Stroobants and H. N.W. Lekkerkerker, J. Phys. Chem. 88, 3669 (1984)
20. G. R. Luckhurst, Thin Solid Films 393, 40 (2001)
21. H. H. Wensink, G. J. Vroege and H. N.W. Lekkerkerker, Phys. Rev. E 66, 041704 (2002)
22. Y. Martínez-Raton and J. A. Cuesta, Phys. Rev. Lett. 89, 185701 (2002)
23. D. Apreutesei and G. H. Mehl, Chem. Commun. (2006)
24. F. Hessel, R.-P. Herr and H. Finkelmann, Makromol. Chem. 188 (1987)
25. K. Severing and K. Saalwächter, Phys. Rev. Lett. 92, 125501 (2004)
26. K. Merkel, A. Kocot, J. K. Vij, G. H. Mehl and T. Meyer, J. Chem. Phys., 121, 5012 (2004);
P. Kouwer, G. H. Mehl, European Conference Liquid Crystals, Jaca, Spain (2004)
27. J. L. Figueirinhas, C. Cruz, D. Filip, G. Feio, A. C. Ribeiro, Y. Frère, T. Meyer and G. H.
Mehl, Phys. Rev. Lett. 94, 107802 (2005)
28. C. Cruz, J. L. Figueirinhas, D. Filip, G. Feio, A. C. Ribeiro, Y. Frère, T. Meyer and G. H.
Mehl (a publicar)

68
29. K. Neupane, S.W. Kang, S. Sharma, D. Carney, T. Meyer, G. H. Mehl, D.W. Allender, S.
Kumar and S. Sprunt, Phys. Rev. Lett. 97, 207802 (2006)
30. S. Diez, D. Dunmur, M. R. De La Fuente, P. Karahaliou, G. H. Mehl, T. Meyer, M. A. Perèz
Jubindo and D. J. Photinos, Liq. Cryst., 30, 1021(2003)
31. R. Elsassar, G. H. Mehl, J. W. Goodby and D. J. Photinos, Chem. Commun. 851, (2000)
32. T. Meyer and G.H. Mehl, The synthesis and the liquid crystal properties of Germanium
containing multipodes, 19th International Liquid Crystal Conference, 30.6-5.7, Edinburgh, Uk
(2002)
33. I. M. Saez and J. W. Goodby, Liq. Cryst., 26, 1101 (1999)
34. J. Barberá, M. Marcos, A. Omenat, J-L. Serrano, J. I. Martínez and P. J. Alonso, Liq. Cryst.,
27, 255 (2000)
35. K. Merkel, A. Kocot, J. K. Vij, R. Korlacki, G. H. Mehl, and T. Meyer, Phys. Rev. Lett. 93,
237801 (2004)
36. B. R. Acharya , A. Primak and S. Kumar, Pramana 61, 231 (2003); reprinted in Phys. Rev.
Lett. 92, 145506 (2004)





























