Embed Size (px)
Citation preview

I
CONJUGATED CYCLIC CHLOROCARBONS:
TRICHLOROCYCLOPROPENIUM ION,HEPTACHLOROTROPENIUM ION, AND
OCTACHLOROFULVALENE
ROBERT WEST
Department of Chemistry, University of Wisconsin, Madison,Wisconsin 53706, USA
ABSTRACTThe properties of the aromatic trichiorocyclopropenium ion (C3Cl) aredescribed. Evidence from vibrational spectroscopy indicates that the C—Cbonds in C3Cl are unusually short and strong. C3Cl undergoes Friedel—Crafts condensation reactions with aromatic hydrocarbons; with phenols,diaryiquinocyclopropenes are obtained, which upon oxidation yield noveltriquinocyclopropanes which show highly unusual electronic spectra Theseven-ring analogue to C3Cl, heptachiorotropylium ion (C7Cl), is obtainedfrom octachlorobicyclo[3,2,0]heptane and aluminium chloride at 1500 to 175°.C7Cl undergoes chemical reactions similar to those of C3Cl. Water convertsC7C Al2Cl to octachiorocycloheptatriene, which upon solvolysis withsulphuric acid is transformed to perchlorotropone.
The compound octachiorofulvalene, C10C18, and its bromo analogueC10Br8 have been investigated. Both compounds show strong bathochromicshifts of electronic bands, due to twisting of the rings about the central doublebond. C10C18 is a strongly electron-deficient molecule which serves as apowerful it-type charge-transfer acceptor toward aromatic hydrocarbons.
The charge-transfer spectra are discussed.
The chemistry of the chiorocarbons—fully or highly chlorinated organiccompounds—is now undergoing a renaissance, in which many of the mostinteresting findings centre about the cyclic conjugated perchloropolyenes.Monocyclic members of this family are shown in Figure 1. Of these speciesonly hexachlorobenzene was known before 1964. Within the last six yearsthe stable species C3Cl, C7Cl and C8C18 have been isolated (the cationsas salts) and the transient existence of C4C14, C5Cl and C5Cl has beenconvincingly demonstrated'.
The family of species in Figure 1, like the cyclic polyenes from which theyare derived, can be classified as aromatic if they contain 4n ± 2 and anti-aromatic if they contain 4n pi-electrons. Thus C3C1, C5C1, C6C16 andC7C1 can be considered aromatic and so are expected to be stabilized, butC4C14, C5Cl and C8C18 are antiaromatic and should be destabilized, atleast when planar. However, the chlorine substituents may perturb thecarbocyclic pi system either by withdrawing electronic charge inductively
379

ROBERT WEST
::cçci
®cE1
Figure 1. The monocyclic conjugated chiorocarbons.
through the C—Cl sigma bonds, or by direct participation of the non-bonding pairs on the chlorine in pi interaction with carbon. Fragmentaryevidence suggests that both effects may be significant in certain delocalizedchlorocarbons'.
This paper will discuss the chemistry of two of these species, C3Cl(I)and C7C14(II), both of which can be considered aromatic according to theusual rules. In addition, in the final section of the paper the chemistry of theinteresting conjugated molecule octachiorofulvalene will be described.
I. TRICHLOROCYCLOPROPENIUM IONThis simple aromatic species was first synthesized2 in 1964, by chloride
abstraction from tetrachiorocyclopropene (III), with aluminium chloride orother powerful Lewis acid-halide acceptors:
C3C14 + MC13 -+ C3Cl, MClIII M = Al,Ga,Fe; also SbCl5
The starting material LIII) is synthesized by dehydrohalogenation ofpentachiorocyclopropaneby warm concentrated aqueous potassium hydrox-ide solution in a two-phase system3. Pentachiorocyclopropane is obtainedby addition of dichiorocarbene to trichioroethylene; but the latter olefinis rather unreactive toward carbene addition, so most methods for carryingout this reaction give only very small yields. However, when sodium tn-chioroacetate is thermolysed in dimethoxyethane at 800 in the presence oftrichioroethylene, pentachiorocyclopropane is produced in 25 per centyield4.
H,Cl NaCCt3COO H\ J/ KOR Cl
c( ci (CH3OCH2)2, 8O ci' Kcici: ciIII
380

CONJUGATED CYCLIC CHLOROCARBONS
Tetrachiorocyclopropene prepared by this route is now available commer-cially in the USA. It shows promise as a vapour-phase fumigant as well asbeing useful as a chemical intermediate.
Substitution reactions on III normally take place with ring opening butwith boron tribromide, III gives BCI3 and tetrabromocyclopropene3. Thisreaction probably takes pktce through the formation of I as an intermediateand successive Br—Cl exchange reactions:
Cl Cl Cl Br Br Cl Cl Br Br Br/ \ /\1=== === 'BBBr3
Cl
jBBr3
Cl
BBr3
Br
11r3
r Br
Cl / Br / Br/ Br/1A1 CIAC1 Clr BrBrPartially fluorinated cyclopropenes can also be prepared from Ill, by treat-ment with SbF3 in the presence3 of SbC15, or with KF in tetramethylenesulphone5:
+
Cl Cl Cl
C3C14,III 4pFCl FF FFto
C1AC1+
C1ACI+
C1AFThese halogenated cyclopropenes can all serve as precursors to trihalo-cyclopropenium ions, when treated with appropriate halide acceptors,
C3Br4 A1Br3 C3Br, AlBrC3C13F AIcI3CC1F+ A1Cl
At the time it was prepared C3Cl was the simplest aromatic speciesknown, having just six atoms and D3h (triangular) symmetry6. The bondingin I was investigated using vibrational spectroscopy and normal coordinateanalysis. Observation of the infra-red spectrum presented no difficulty, butto determine the Raman spectrum measurement in solution in liquidsulphur dioxide was required. The resulting Raman spectrum is shown inFigure 2. Observed frequencies are given in Table j7
There are six constants in the Urey—Bradley force field used for the381

ROBERT WEST
m of C3C1ALC1 in SO2
I I I I I1000 800 600 400 200
SO2
c3ccm
1320
1800 1600J-
1/.00
Frequency, cm11200 1000
Figure 2. Raman spectrum of C3Cl A1C1 in liquid sulphur dioxide, showing polarization.
calculation and only five fundamental frequencies were observed, so one con-stant must be assumed; but fortunately the chlorine atoms are so far apartthat the nonbonded interaction constant can be taken as zero7. The resultsof a Urey—Bradley calculation are shown in Table 2, together with thosefrom a partial normal coordinate analysis for the corresponding brominatedion, C3Br. In Table 3, the C—C and C—Cl stretching force constants arecompared with those for other known aromatic species. Note that bothK and K1 are decidedly higher than for chlorobenzene; indeedfor C3Cl is markedly higher than [or any other known aromatic species.Consistent with this is the x-ray finding by Sundaralingam, that the C—Cdistance in triphenylcyclopropenium ion is shorter than that in benzene8.
Thble 1. Vibrational frequencies for C3Cl
cm1 Infra-red Raman Assignment
200 s — E459 — s,pol A1735 s vw E
1312 vs s,dep E1348 s — —1791 — m,pol A
382
Roman spectruC
cl C3CL AtCLC
u)
S02 T

CONJUGATED CYCLIC CHLOROCARBONS
Thble 2. Force constants for C3X, millidynes/A7
Species C3C1 C3Br
6.31 6.462.99 2.15
—0.248 (0.248)*0.385 (0.275)
F1 0.808 (0.674)Ct (0) (0)
* Values in parentheses are assumed.
Why should the C—C bond in cyclopropenium ions be so strong? SimpleHückel calculations predict a pi bond order of 0.667 for cyclopropenium,identical to that for benzene. The sigma bonding in cyclopropenium ionsis probably external to the ring and 'bent' even more strongly than in cyclo-propane or cyclopropene (Figure 3); if so the sigma bonds should be weaker
Table 3. C—C and C—Cl stretching force constants for aromaticspecies
Species p
C6H6 5.595.15
4.674.79
00.35
C5H 5.39 4.79 0.21C6C16 4.81 2.30 0.37C3Cl 6.32 2.99 0
p = Resonant constant.
than in benzene. But bending of the sigma bonds may bring the carbon atomscloser together, allowing for much increased overlap of p orbitals on adjacentcarbon atoms. According to this model the unprecedented bond strengthin C3Cl arises from the increased pi-bonding in cyclopropenium ions,which more than makes up for the decreased sigma bond strength.
In I, contributions from the chlorine atoms to the pi-bonding may alsoaid in raising both C—Cl and C—C bond strengths and force constants.
Figure 3. Schematic drawing of orbitals forming C—C bonds in cyclopropenium ions, indicatingoverlap external to the three-membered ring.
383

ROBERT WEST
Nuclear quadrupole resonance measurements of Lucken and Mazeline9give a value of 0.35 for the asymmetry parameter for the 35C1 nuclei hi I.This asymmetry parameter can be related directly to the partial doublebond character, which is found to be 0.16. Both the asymmetry parameterand the bond order are higher than for any other known carbon—chlorinecompound. The data suggest that about half of the positive charge on thering (3 >< 0.16) is delocalized through the pi system on to the chlorineatoms9.
The chemistry of I has been reviewed elsewhere1, so only those reactionswhich lead to compounds of some theoretical importance will be treatedhere. Hydrolysis of the tetrachioroaluminate salt of I, by flooding with water,leads to the regeneration of III in good yield7. Apparently water first attacksthe AlCl counterion rather than C3Cl, forming chloride which convertsI to C3C14. However, very slow solvolysis of I, preferably as the hepta-chiorodialuminate salt dissolved in dichioromethane, leads to the explosiveliquid, dichlorocyclopropenone10. The reaction is best carried out usingalcohol, and the first product is the A1CI3 complex of the cyclopropenone.Further alcoholysis of the complex converts it to an alkoxychiorocyclo-propenone' 1, but the last chlorine atom is removed simultaneously withring-opening. Upon cautious warming dichiorocyclopropenone is convertedto a spirolactone:
Cl O:A1CI 0
CIACICICIROH
R0C1However, this reaction may take place with mild detonation above 00, soquantities of dichlorocyclopropenone greater than ig should never be prepared.
The trichiorocyclopropenium ion undergoes a useful and importantFriedel—Crafts type reaction with aromatic hydrocarbons, leading to succes-sive replacement of chlorine atoms with aryl groups to form aryihalo- andeventually triaryl-cyclopropenium ions. (The final step to give Ar3Crequires, so far at least, an activated aromatic compound.) The arylatedcyclopropenium ions give a variety of products on hydrolysis. Thus ArC3C1yields the aryltrichiorocyclopropene or the arylhydroxycyclopropenone' 2This is by far the most convenient synthesis of arylhydroxycyclopropenones,which are strong protonic acids. Hydrolysis of Ar2C3Cl yields diary!-cyclopropenones; the aryl groups can be the same or different, so this is aflexible and general method for synthesis12.
384

CONJUGATED CYCLIC CHLOROCARBONS
Cl
Cl Cl
,AlC1
CIC1
Ar Cl
ArHjH20 H20
acetone
quench —
CI [ 0 I
ALLHAr CI LAr ci]
ArH
Cl CICI
H,O
acetone AOH
Ar Ar' Ar Ar' H2OSoc'2Ar Ar'
Ar'H
AAAr'H20
Ar
Especially interesting products are obtained when C3Cl is allowed toreact with phenols. If a diaryichlorocyclopropenium ion, for instance, istreated with a phenol, the product is a p-hydroxyaryldiphenylcyclopropeniumsalt, as expected. The latter, however, contains a phenolic proton easilyremoved by treatment with mild base, forming an orange or yellow diaryl-quinocyclopropene1 .
C6H,OH R3NAr2C3Cl — —
Diarylquinocyclopropenes are relatively little-known; at the time ourwork was carried out, only two examples were known, both obtained bymore laborious syntheses14' 15 Starting from C3Cl it is now possible toprepare these substances quite conveniently.
385
OH

Figure 4. Electronic spectrum of hexa-t-butyl triquinocyclopropane (V, R = t-butyl) indichioromethane.
386
ROBERT WEST
if all three aromatic groups allowed to react with I are phenols (preferably2,6-disubstituted), tris(p-hydroxyaryl)cyclopropenium ions are formed; thesereadily lose protons to form bis(hydroxyaryl)quinocyclopropenes. The latterundergo reversible oxidation to yield a remarkable new class of compounds,the triquinocyclopropanes (V)1 .
OH
R
RR
R
HO ED
oxdn redn
4)UC-a0-a
300 500 700
Waveength, m900

CONJUGATED CYCLIC CHLOROCARBONS
The triquinocyclopropanes are intensely violet coloured substances,remarkable in that their electronic absorption spectra extend throughoutthe visible and into the near infra-red; the strongest bond occurs at about
770 nm, with log e = 4.7. The electronic spectrum of V. R = tert-butyl,is shown in Figure 4. Compounds of this class are derivatives of 3-radialene,but are much more stable than normal 3-radialenes; some triquinocyclo-propanes will withstand heating to over 200°. The anion-radical and dianioncorresponding to V have also been prepared' 6• The e.s.r. spectrum of theanion-radical shows a symmetrical 7-line pattern (Figure 5) consistent withcomplete delocalization of the unpaired electron over the four-ring system.
Figure 5. Electron spin resonance spectrum of anion radical of V (R = t-butyl).
Both neutral triquinocyclopropanes and their dianions have singlet groundstates, in agreement with simple molecular orbital calculations.
II. HEPTACHLOROTROPYLLUM IONTropylium ion, C7H, is well known as a stable, six-it-electron cation.
Following isolation of C3Cl, attention was turned to synthesis of theseven-membered ring analogue, C7Cl. Starting material for this synthesiswas the bicyclic compound made from hexachiorocyclopentadiene, tn-chioroethylene, and aluminium chloride'1. Dehydrohalogenation leads tooctachlorobicyclo[3,2,0]beptadiene (VI), which reacts with aluminium
dci dC, dcl
Cç,Ci c=c2 ci_IIjI0G0KOH
,
ci_fIIjJ"
chloride on warming. Below 150° the bicyclic compound is regenerated onhydrolysis, and above 180° complete fragmentation occurs, but in the inter-mediate 30 deg. temperature range isomerization takes place to hepta-chiorocycloheptatrienium heptachiorodialuminate, which hydrolyses tooctachiorocycloheptatriene (VII)'8 This is a colourless crystalline com-pound, m.pt 86°, which reacts with Aid3 to form the yellowish tetrachioro-aluminate salt of II. Similar 1 :1 salts are formed between VII and SbC15,
387
Oe

ROBERT WEST
SnCJ4 or FeC13. The salts of II have essentially identical infra-red spectraabove 600 cm 1, consisting of just four bands at 1 252(w), 1180(s), 7 50(w) and700(s), in cm . At lower frequencies, bands characteristic of the halometallateions are observed (Figure 6). All of these salts regenerate VII upon treatmentwith water.
Models indicate that lone pairs on adjacent chlorine atoms would inter-fere strongly in the ion II if it were planar. The geometry of II is unknown,but it seems to have much of the aromatic character of tropylium ionsgenerally. It is at least as stable with respect to VII and Lewis acids as C3Clis with respect to its parent chiorocarbon C3C14. The chemical reactions of
II are as yet little known, but appear to resemble those of C3Cl. Forexample, II reacts with aromatic hydrocarbons to give Friedel—Crafts sub-stitution. With phenols, loss of a proton takes place to yield quinotropones.It seems probable that C7C1 will prove a valuable intermediate in thesynthesis of other novel delocalized structures, just at C3C1 is.
388
Figure 6. Infra-red spectrum of C7C1 Al2CI, as mull in petroleum oil.
Cl Cl Cl
Cl_
Cl ClCL ,ci
Cl1 Cl
VII
2A1C13
H20
H20
Cl Cl
Cl—'CICICl
155
ClCl Cl
C1ClCl Cl
II
,Al2Cl
Al2Cl

CONJUGATED CYCLIC CHLOROCARBONS
Halogen exchange reactions of Vii also take place, probably through theintermediate formation of 1119 When VII is warmed with boron tribromide,seven of the eight chlorine atoms are replaced by bromine, giving C7Br7C1(VIII). The remaining chlorine probably is in the geminal position and resistsreplacement for steric reasons. Solvolysis of VII with concentrated sulphuricacid yields perchiorotropone, which has also been synthesized by an inde-pendent photochemical method. Similarly, sulphuric acid treatment ofVIII yields perbromotropone, which can also be made'9 from perchioro-tropone and BBr3:
Cl Cl
ciX,ciJ/\C1\,/C1(
0ci }L C1
Cl
III. Octachlorofulvalene(X) and Its Charge-Transfer PropertiesThis fascinating chiorocarbon was first prepared by V. Mark2° via
dehalogenation of decachlorobi(2,4-cyclopentadien-l-yl), IX, which can besynthesized from bexachiorocyclopentadiene and copper or copper(i)chloride. Mark used triethyl phosphite in the final step, but a slightly betteryield can be obtained with stannous chloride dihydrate in acetone21:
389
VII
H2S04
B8r3
BBr3
Br ClBr Br11
VIII
BBr
Cl.

ROBERT WEST
Cl Clci ci
ci ci
dci dci(EtO)3p orCu-ligroin
ClLC1Cl Cl
SnCI2 2H20or CuCI-EtOHCl Cl acetone Cl \ / CIdci dci
Ix x
X has also been obtained by gas-phase dechlorination of hexachiorocyclo-pentadiene at high temperature22. Compared with other fulvalenes, X isremarkably inert and quite different in physical and chemical properties.Fulvalene itself is an orange-coloured material so unstable that it cannotbe isolated, but X is easily obtained as beautiful blue-violet crystals.
The chlorination of X has recently been investigated, and the severalproducts have been isolated and identified with the aid of nuclear quadrupoleresonance spectroscopy21:
clod8,x
Cl2
CHCI,
Ici ci ci
Ci
iiç1Cl Cl Cl Cl
clod!14
Cl Cl Cl CiClCl
C1)clCi Cl Cl Cl
clod12
Ci Cl Cl
+
Cl Cl
cinch0
Cl2
Cl2C10C114,m.pt23 169°
Br BrBr Br
Br.BrBr/ CuBr, MeOCH2CH2OMe—H20
3—5 mm,
Br Br Br BrBr Br
Clod10
Cl2
The perbromo analogue, octabromofulvalene (XI), has also been syn-thesized, directly from hexabromocyclopentadiene24.
570
XI
The perbromo compound XI is less stable thermally and more reactivechemically than X, but the two compounds show quite similar spectralproperties. The electronic spectra of these substances are shown in Figure 7.Both compounds show a strong band near 400 nm, and a broad band
390

CONJUGATED CYCLIC CHLOROCARBONS
between 600 and 650 nm. In contrast, fulvalene itself and simple substitutedfulvalenes show their longest wavelength absorption near 415 nm.
The striking bathochromic shift for X and XI compared to ordinaryfulvalenes may reflect in part contributions from nonbonded electrons onhalogen, but are probably due principally to twisting of the central C—Cdouble bond joining the rings. A two-dimensional x-ray study of X hasindicated a dihedral angle of 41° between the two rings25, which are twistedout of the preferred planar configuration by steric repulsions of the chlorineatoms at the 2, 2', 5 and 5' positions. Nonpianarity of the fulvalene rings has
5.0
L..0
3.0 -0
2.0 -
\1.0 I I I
200 400 600A,nm
Figure 7. Electronic spectra of octahalofulvalenes in dichioromethane solution.C10C18(X); ,C0Br8(XI).
previously been associated with bathochromic shifts of electronic transi-tions in fused-ring fulvalenes26. The molecular structure of XI is not yetknown, but the steric interactions should be even greater than in X, so theinter-ring angle will be of considerable interest.
Fulvalenes generally are electron-deficient molecules possessing an unfilledpi-orbital, and in octachiorofulvalene, according to liMO calculations, thisorbital is at even lower energy than in fulvalene itself (Figure 8)2 . Consistentwith this is the finding that X forms charge-transfer complexes, acting as api-acid. This property allows a spectacular demonstration of colour changesupon charge-transfer complex formation. Solutions of X in noninteractingsolvents such as dichioromethane are blue. Addition of benzene or toluenechanges the colour to green; mesitylene gives a yellow colour; phenanthrene,a yellow-brown; and with pyridines, red!
Spectral changes upon charge-transfer complex formation with X are subtle.The electronic spectrum of X contains bands at 390 nm (log = 4.60) and604 nm (log s = 2.40). In the presence of moderate pi-donors such as benzene
391
/ "/

ROBERT WEST
and toluene, these bands are unchanged except for some broadening anddetensification of the shorter wavelength band (Figure 9). In mesitylene,however, the 390 nm band of X is significantly broadened on its longerwavelength side. Such one-sided broadening must be due to the presence ofa new band in the 400—475 nm region of the spectrum (illustrated in Figure9). A set of similar curves is obtained when the spectra of X are plotted as oneincreases the concentration of an aromatic compound (Figure 10). In both ofthese cases the concurrent broadening of the 390 nm band of X and 'fillingin' of the absorption minimum near 475 nm shifts the )Lmin to longer wave-lengths and so gives rise to the observed colour changes27.
+1 -
>0)
LU
-1-
II—2 -
Figure 8. Schematic representation of the it molecular orbitals for fulvalene (left), twistedfulvalene (centre), and octachlorofulvalene(X) (right).
The spectral absorption in the 475 nm region was studied as a semi-quantitative measure of the effects of various donors on the spectra of X.A Log e (at 475 nm, relative to cyclohexane) was measured as a function ofdonor/acceptor mole ratio for a large number of donors. The results of thisstudy are summarized in Table 4 and Figure 11. In non-aromatic solvents,including dioxane and acetone, the spectrum of X gives no indication of strongcharge-transfer complex formation. The slight spectral changes which areobserved with dioxane probably arise from solvent shifts or n-donor charge-transfer. On the other hand, t-donors give rise to large values of A log e,
392

Figure 9. Variation in the electronic spectrum of C0C18(X) with solvent; methylenechloride; — — — — benzene; toluene; —. —. —. mesitylene. The position of the new bandappearing in the presence of excess mesitylene, and attributed to the charge-transfer absorption,
is also shown.
A, nm
Figure 10. Variations in the electronic spectrum of octachiorofulvalene with increasing donorconcentration. (C10C18/phenanthrene in CH2CI2; mole ratio 61 , 279 — — —. 552 — . —.).
393
CONJUGATED CYCLIC CHLOROCARBONS
300 1.00 500 600 700 800
A, nm
5.0
4.0
3.0
2.0300 400 500 600 700

ROBERT WEST
indicating the formation of strong molecular complexes. The data suggestthe following sequences of donor potency:
and
pentamethylbenzene, durene > hexamethylbenzene, mesitylene>xylene > toluene > t-butylbenzene > benzene, chlorobenzene
benzopyrene> phenanthrene> pyrene > naphthalene>acenaphthene.
In general this is the same order of donor strength which is observed in othercharge-transfer complex formation28' 29• Hexamethylbenzene and ace-naphthene are less effective donors than expected, probably because ofsteric hindrance to complexation.
0)0
L..O
Figure 11. Charge-transfer complexes of octachiorofulvalene. Variation in log e (at 475 nm)with mole ratio of donor to acceptor, for monocyclic aromatic donors.
Theoretically, increasing the concentration of a donor compound in adichioromethane solution should lead to gradual changes in the spectrumof X until essentially all of the C 10Cl8 is converted to the charge-transfercomplex; beyond this point no further spectral changes should occur. Thedata in Figure 11 show that no such levelling off of the A log versus moleratio curves is observed below donor/acceptor ratios of io and apparentlydoes not occur significantly even at mole ratios of 1O. This observationleads to the conclusion that the equilibrium constants for charge-transfercomplex formation,
KCT = (C10C18 donor)/(C1 0Cl8) (donor)
394
1.5 2.0 3.0Log (mole donor/mole acceptor)

CONJUGATED CYCLIC CHLOROCARBONS
cannot be greater than 10 . These values of Ka are approximately threeorders of magnitude smaller than those commonly observed in charge-transfer complex formation (for example, for the very stable tetracyano-ethylene—mesitylene complex KCT 17.3, and the relatively unstableiodine—benzene complex has KCT = 1.5 x 10 1)30.
Table 4. Effects of it-donors on the electronic spectrum of octachiorofulvalene
Donor Mole ratio3 log e(475 nm)
A loge(475 nm)
Cyclohexaneb 8.37 x io 2.03 0.00CHZC12bCHC131'
2.79 x io1.12 x io
2.102.05
+0.07+0.02
CCI4b 1.11 x io 2.03 0.001,4-Dioxan&' 2.28 x io 2.16 +0.13Ph—CE3t' 1.21 x tO4 2.31 +0.28PhHt' 9.60 x io 2.32 ± 0.29Ph—CU' 1.08 x io 2.31 ±0.28Ph—CH31' 9,04 x io 2.84 +0.81Ph—C(C113)31' 6.46 x io 2.64 + 0.61CH3C6H4CH3t'Mesitylen&'
7.6 x io6.29 >< iO
3.093.31
+ 1.06+ 1.28
p-C6H40CH3)f 2.12 x 102 2.11 +0.08pC6H4OCH3)2C 5.99 x 102 2.18 +0.15p-CH4OCH3)2t , 1.75 < lO 2.29 ±0.261,3,5-C6H3(OCH3)33 6.65 x 102 2.34 ±0.31Acridine3 5.72 x l0 2.08 ± 0.05Acridin& 3.84 x 102 2.30 ± 0.27
Donor/acceptor.Donor med at tht tolvent.Spectrum run at tolution in dichioromethanc.
Although a large number of pi-donors were employed with X, in no casewas it possible to observe the charge-transfer absorption band directly.With weak donors such as benzene and toluene this band apparently fallsalmost directly beneath the intense 390 nm band of X. With better donors(such as benzopyrene) absorption by the donor molecule (which must bepresent in large excess because of the low KCT) further obscures observa-tion of the charge-transfer band. However, for moderately strong pi-donorsthe charge-transfer band must lie between 400 and 475 nm, and this allowsa rough comparison of the acceptor strength of X with that of other pi-acids.Data for a number of acceptors with various donors are given in Table 5.From this table it appears that X is a slightly more powerful acceptor thanp-benzoquinone, and is comparable in pi-acid strength to chloranil or1,2,3-indanetrione27.
Although X is a relatively good pi-acceptor, it is not so strong as mightbe predicted from MO calculations. The energy of the charge-transfertransition, ECT, is commonly given34 as
EcT = JP — £4 + Cwhere JP is the ionization potential of the donor, EA is the electron affinityof the acceptor, and C is a constant denoting all other forms of energy
395

ROBERT WEST
(solvation, etc.) changing upon complex formation. The electron affinity ofX (LUMO at —0.413, see Figure 8) should, from MO theory, be even greaterthan for tetracyanoethylene (LUMO —03413)29. The reason why X is a poorerpi-acid than TCNE probably lies in the C term and reflects the nonpianarityof X which prevents close approach of the pi-system of the donor. The factthat the equilibrium constants for charge-transfer complex formation to Xare two or three orders of magnitude smaller than typically found for suchcomplexes is consistent with this explanation. Thus octachiorofulvalene isa unique example of a powerful but strongly sterically hindered p1-acid27.
Table 5. Charge-transfer absorption maxima for various complexes, nm
Substance Benzene Mesitylene NaphthalenePhenanthrene Pyrene Ref.
Tetracyanoethylene 385 — 560 540 720 31Chloranil
1,2,3-Indanetrione
339
—
427
—
390,480
—
473
400
435,599481
3229
X -350 400—475 400—475 400—475 — 27p-Benzoquinone 284 344 367 — 448 32Maleic anhydride — 325 354 384 33
Although the twisted molecular form of X apparently limits its effective-ness as a charge-transfer acceptor, the geometry of X should favour its out-right reduction to an anion radical or dianion. Imamura and Hoffmann havepointed out35 that while fulvalene and its derivatives should prefer a planarground state, the fulvalene dianion (isoelectronic with biphenylene) shouldprefer a nonpianar ground state. Hence the nonpianarity of X should adda driving force to the ability of Xto accept electrons in the ground state.
Treatment of octachiorofulvalene with Na—K alloy in tetrahydrofuranat room temperature in an e.s.r. tube gave rise to a purple solution showinga single, rather sharp e.s.r. signal (Aw = 2.4 G; Figure 12a) at g = 2.0048 ±0.0002. The intensity of this signal slowly decreased with time, and withina few hours the signal disappeared entirely. Electrolysis of a solution of Xin tetrahydrofuran with added tetra-n-butylammonium perchlorate at roomtemperature or at —50° and with 1—2 pA current led to a similar purplesolution and to the appearance of a similar e.s.r. signal (Lw = 2.5 G;Figure 12b) at g = 2.0043 ± 0.0001. Continued electrolysis of this sampleat somewhat higher currents (5—8 ptA) caused the signal to decay slowlyuntil it finally disappeared after about an hour. Numerous attempts toregenerate the radical species by reversing the leads to the electrolysis cellwere unsuccessful.
The radical observed in both of these experiments is believed to be theanion radical of X. Both of these species were generated under 'minimal'reaction conditions (short contact times and low currents), and the speciesobtained by alkali metal reduction is remarkably stable even at room tem-perature (ti. 1 h). The species obtained by electrolysis disappears easilyupon further reduction, presumably to give the dianion of X. Reductions ofboth X and its anion radical are apparently irreversible, however; both the
396
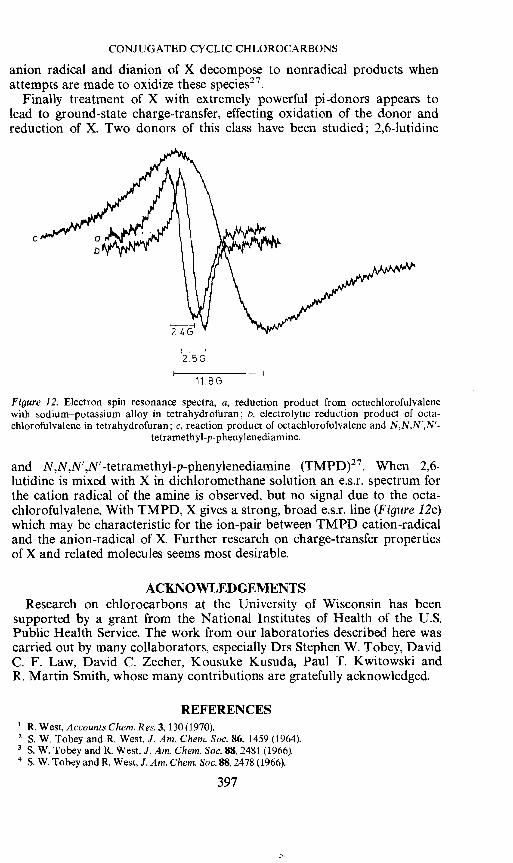
CONJUGATED CYCLIC CHLOROCARBONS
anion radical and dianion of X decompose to nonradical products whenattempts are made to oxidize these species27.
Finally treatment of X with extremely powerful pi-donors appears tolead to ground-state charge-transfer, effecting oxidation of the donor andreduction of X. Two donors of this class have been studied; 2,6-lutidine
2.5G
Figure 12. Electron spin resonance spectra, a, reduction product from octachiorofulvalenewith sodium—potassium alloy in tetrahydrofuran; b, electrolytic reduction product of octa-chiorofulvalene in tetrahydrofuran; c, reaction product of octachiorofulvalene and N,N,N',N'-
tetramethyl-p-phenylenediamine.
and N,N,N',N'-tetramethyl-p-phenylenediamine (TMPD)27. When 2,6-lutidine is mixed with X in dichioromethane solution an e.s.r. spectrum forthe cation radical of the amine is observed, but no signal due to the octa-chiorofulvalene. With TMPD, X gives a strong, broad e.s.r. line (Figure 12c)which may be characteristic for the ion-pair between TMPD cation-radicaland the anion-radical of X. Further research on charge-transfer propertiesof X and related molecules seems most desirable.
ACKNOWLEDGEMENTSResearch on chiorocarbons at the University of Wisconsin has been
supported by a grant from the National Institutes of Health of the U.S.Public Health Service. The work from our laboratories described here wascarried out by many collaborators, especially Drs Stephen W. Tobey, DavidC. F. Law, David C. Zecher, Kousuke Kusuda, Paul T. Kwitowski andR. Martin Smith, whose many contributions are gratefully acknowledged.
REFERENCES1 R.West, Accounts Chern. Res. 3, 130(1970).2 S. W. Tobey and R. West, J. Am. Chem. Soc. 86, 1459 (1964).
S. W. Tobey and R. West, J. Am. Chem. Soc. 88, 2481 (1966).S. W. Tobey and R. West, J. Am. Chem. Soc. 88,2478(1966).
397
b
II2.AG
11.8G

ROBERT WEST
D.C. F. Law and R. West, unpublished studies.6 More recently the cyclopropenium ion itself has been prepared: R. Breslow, J. T. Groves and
G. Ryan, J. Am. Chem. Soc. 89,5048 (1967).' R. West, A. Sadô and S. W. Tobey, J. Am. Chem. Soc. 88, 2488 (1966).
The C—C bond length in the three-membered ring in triphenylcyclopropenium perchlorateis 1.373 A, compared to 1.39 A for benzene. See M. Sundaralingam and L. H. Jenson, J. Am.Chem. Soc. 88, 198(1966).E. A. C. Lucken and C. Mazeline, J. Chem. Soc. A, 153(1968).
10 R. West, J. Chickos and E. Osawa, .J. Am. Chem. Soc. 90, 3885 (1968).J. Chickos and R. West, unpublished studies.
12 R. West, D. C. Zecher and S. W. Tobey, J. Am. Chem. Soc. 92, 168 (1970).13 R. West and D. C. Zecher, J. Am. Chetn. Soc. 92, 155 (1970).14 A. S. Kende, J. Am. Chem. Soc. 85, 1882(1963).15 B. Fohlisch and P. BUrgle, Tetrahedron Letters, 2661 (1965); Ann. Chem. 701,67(1967).16 R. West and D. C. Zecher, J. Am. Chem. Soc. 92, 161 (1970).17 A. Roedig and L. Horning, Ann. Chem. 598, 208 (1956), and references cited therein.
R. West and K. Kusuda, J. Am. Chem. Soc. 90, 7354 (1968).19 K. Kusuda, R. West and V. N. M. Rao, J. Am. Chem. Soc. 93, 3627 (1971).20 V. Mark, Tetrahedron Letters, 333 (1961).21 R. M. Smith and R. West, J. Org. Chem. 35, 2681 (1970).22 A. E. Ginsberg, R. Paatz and F. Korte, Tetrahedron Letters, 779(1962).23 E. T. McBee, J. D. Idol Jr and C. W. Roberts, U.S. Pat. Na 2911 448 (3 November 1959);
Chem. Abstr. 54,4527(1960).24 R. West and P. T. Kwitowski, J. Am. Chem. Soc. 90, 4697 (1968),25 P. J. Wheatley, .J. Chem. Soc. 4936(1961).26 E. D. Bergman, G. Berthier, A. Pullman and M. B. Pullman, Bull. Soc. Chim. Fr. 1079 (1950).27 R. M. Smith, Ph.D. thesis, University of Wisconsin (1970).28 L. J. Andrews and R. M. Keefer, Molecular Complexes in Organic Chemistry, p 21. Holden-
Day: San Francisco (1964).29 A. R. Lepley and J. P. Thelman, Tetrahedron, 22, 101 (1966).30 L. J. Andrews and R. M. Keefer, bc. cit. p28.31 M. J. S. Dewar and H. Rogers, J. Am. Chem. Soc. 84, 395 (1962).32 A. Kuboyama, Nippon Kagaku Zasshi, 81, 558 (1960).
M. Chowdhury, J. Phys. Chem. 66, 353 (1962).11. McConnell, J. S. Ham and J. R. Platt, J. Chem. Phys. 21, 66 (1953).A. Imamura and R. Hoffmann, J. Am. Chem. Soc. 90, 5379 (1968).
398





























