Embed Size (px)
Citation preview

Comparison of NADHand NADPHOxidase Activities
in Granules Isolated from Human Polymorphonuclear
Leukocytes with a Fluorometric Assay
DAVID IVERSON, LAWRENCER. DECHATELET,JOHNK. SPITZNAGEL, and PATSYWANG
From The Department of Biochemistry, BowmanGray School of Medicine, Winston-Salem,North Carolina 27103, and The Department of Bacteriology and Immunology, University of NorthCarolina Medical School, Chapel Hill, North Carolina 27514
A B S T RA C T A fluorometric method for the determi-nation of pyridine nucleotides has been adapted for usein studying the reduced pyridine nucleotide oxidases inhuman polymorphonuclear leukocytes. In the presenceof strong base the oxidized forms of the pyridinenucleotides form a highly fluorescent product. Thesmall amounts of NAD(P) formed by the oxidasereactions can be determined with great sensitivity.This method has been compared to the radioisotopicassay for NADPH oxidation. Both methods gaveessentially the same results in terms of nanomolesNADPproduced by control, resting, and phagocytizingsamples.
Both NADPHand NADHoxidase activities wereinsensitive to cyanide. NADPHoxidation had a pHoptimum of 5.5, while that for NADHappeared to be6.0. Granules isolated from phagocytizing cells rou-tinely showed more activity toward both substrates(two to threefold) than granules from resting cells.Both activities were located primarily in a granulefraction prepared by differential centrifugation. Oxida-tion of NADPHwas routinely four to five times that ofNADHat all except very high substrate levels. Measur-able NADHoxidation was rarely seen below 0.80 mMNADH, while NADPHoxidation was easily measur-able at 0.20 mM. One patient with chronic granulo-matous disease was studied. At low substrate levels,there was no activity toward either substrate in granulesisolated from either resting or phagocytizing cells ofthis patient, while granules isolated from normal controlcells showed substantial activity at these substratelevels.
Purification of the activities has been initiated withlinear sucrose gradients. Both activities co-sediment to
Received for publication 19 July 1976 and in revised form20 October 1976.
a very dense region of the gradient, a region differentfrom that in which membrane or azurophil granulesusually equilibrate. The peak gradient fractions show a10-30-fold increase in specific activity over comparablegranule fractions.
These data suggest that the oxidase activities areassociated with one enzyme that has different affinitiesfor the two substrates and support the contention thatthe oxidation of NADPHis responsible for the metabolicburst accompanying phagocytosis in human PMNL.
INTRODUCTION
A reduced pyridine nucleotide oxidase is thought to bethe enzyme that initiates the respiratory burst ac-companying phagocytosis in polymorphonuclear leu-kocytes (PMNL).1 This enzyme catalyzes the reaction ofthe reduced nucleotide with molecular oxygen pro-ducing the oxidized nucleotide and hydrogen peroxide.There has been some controversy over the substratespecificity of the oxidase; some work has implicatedan NADHoxidase (1-3) while other studies have sug-gested NADPHoxidase (4, 5) as the critical enzyme.Both oxidase reactions can directly explain the increasesin oxygen consumption and H202 production thatneutrophils exhibit upon phagocytosis. Both can beused to explain the increase in the hexose mono-phosphate shunt activity: NADPHoxidase can directlygenerate NADP, the rate limiting substance for thehexose monophosphate shunt activity; NADHoxidasemight indirectly generate NADPby means of a trans-hydrogenase (6) or by the glutathione peroxidasesystem (7). Implicit in these arguments seems to be the
IAbbreviations used in this paper: CGD, chronic gran-ulomatous disease; MPO, myeloperoxidase; PBS, phosphatebuffered saline; PMNL, polymorphonuclear leukocytes.
The Journal of Clinical Investigation Volume 59 February 1977-282-290282

assumption that there are two oxidase enzymes, onethat uses NADPHand another that uses NADH; thishas not been rigorously demonstrated. Although it ispossible that there are two separate enzymes, it isalso possible that there is a single enzyme which canoxidize either reduced pyridine nucleotide.
Work on the oxidases in general has been plaguedby the lack of a sensitive, specific assay. A typicalassay involves measuring oxygen uptake with a Clarkelectrode. This method is fairly nonspecific as it wouldmeasure any and all metabolic activities of the cellsrequiring oxygen, not necessarily just the oxidase, andvariable nonenzymatic rates of oxygen consumption areseen from day to day (8). In addition, the assay is notvery sensitive, requiring relatively large, nonphysio-logical amounts of nucleotide to observe activity inhuman cells. A spectrophotometric method whichmeasures a decrease in absorbance at 340 nmwith timehas also been used. This method, though specific, alsosuffers from a lack of sensitivity. Since the pyridinenucleotides absorb so strongly, it is necessary to uselow concentrations of the substrate or a 1-mm pathlength to insure that the measurement is kept withinthe linear range of the spectrophotometer accordingto Beer's law. Such a concentration is lower thanoptimal for measuring the enzyme in subcellularfractions of human PMNL. Also, regardless of whatconcentration is used, one is forced by this method tomeasure a relatively small decrease in a relativelylarge initial absorbance reading. This is not a veryaccurate procedure as instrument noise becomes moreand more important.
In 1975, DeChatelet and his co-workers publishedan isotopic procedure for assaying NADPHoxidase (8).This assay eliminated the problems of the others bybeing both absolutely specific and very sensitive. Itmeasures the amount of NADPformed by coupling theoxidase reaction to the 6-phosphogluconic dehy-drogenase reaction in a two-step incubation procedure.The major problem with this assay is its inability tomeasure the oxidation of NADHdue to the absolutespecificity of the dehydrogenase enzyme for NADPand thus its inability to answer the question of whichoxidase is the important one in the respiratory burstof the human neutrophil.
Lowry et al. (9), expanding on the work of Kaplanet al. (10), devised a very sensitive assay system forthe pyridine nucleotides relying on the fact that thereduced forms are destroyed by acid, while the oxidizedforms are destroyed by base. Further, when theoxidized form of either nucleotide is incubated instrong base (i.e., 6 N) the destruction yields a highlyfluorescent product. Such a system allows measure-ment of a small amount of NAD(P) that is formedin the presence of a relatively large amount of NAD(P)Hin a typical oxidase assay. In addition, nanomolar
amounts of the nucleotides can be measured and theassay has a 2,000-fold useful range (9). These factorsmake this system a very powerful one, capable ofdealing with most questions related to the oxidaseproblem.
METHODSPreparation of crude granule fraction. Venous blood was
obtained from normal, healthy volunteers. The erythrocyteswere sedimented with plasmagel (HTI Corp., Buffalo, N. Y.)as previously described (8). The leukocytes were collected bycentrifugation at 160 g for 8 min at 4°C, washed once withDulbecco's phosphate buffered saline (PBS), and contaminat-ing erythrocytes removed by hypotonic lysis. The leukocyteswere suspended to a final concentration of 1.5 x 108PMNL/ml. Total and differential counts were performed. Thesepreparations routinely yielded 70-80% neutrophils, 1-7%eosinophils, and 10-20% lymphocytes and monocytes.
Zymosan (ICN, Nutritional Biochemicals Division, Inter-national Chemical & Nuclear Corp., Cleveland, Ohio) wassuspended in PBS at a concentration of 50 mg/ml. 2 vol ofhuman pooled serum was added to 1 vol of the zymosan sus-pension and incubated at 37°C for 30 min. At the end of theincubation, the opsonized zymosan was sedimented by cen-trifugation at 17,000 g for 10 min. The supemate was dis-carded and the zymosan was resuspended in 3 vol of PBS.
1 ml of cell suspension was incubated with 2 ml of PBSto give resting cells or with 2 ml of opsonized zymosan togive phagocytizing cells. The PBS and zymosan were pre-incubated at 37°C for 5 min, the cells were added, and theincubation continued for another 3 min. At the end of this time3.0 ml of cold 0.68 M sucrose was added and the cells wereput immediately on ice. They were then homogenized togreater than 90%Yo breakage in a Potter-Elvehjem homogenizerwith a motor-driven Teflon pestle run at 12,000 rpm (Tri-RStirrer model S-63C operated at maximum speed; Tri-RInstruments, Inc., Jamaica, N.Y.). The whole homogenatewas centrifuged at 500g to remove unbroken cells, largedebris, and zymosan particles. The 500 g supemate wasthen centrifuged at 27,000 g, and the resulting granule pelletwas resuspended in 0.34 M sucrose with a Douncehomogenizer. The procedure of Lowry et al. was usedto estimate protein (11). The granule fractions generally hadgreater than 1.0 mg/ml protein. For ease in comparing day-to-day results with different preparations, all preparations wereroutinely diluted to 1 mg/ml with 0.34 Msucrose and 0. 10-mlsamples were used for assay in the 1-ml incubations.
Isopycnic centrifugation of PMNLgranules. Venous bloodwas obtained, the erythrocytes sedimented, and the leukocytescollected as before. After the initial collection of theleukocytes, the cells were suspended in PBSto approximately107 cells/ml. 4 ml of this solution was layered on 3 ml ofFicoll-Hypaque solution (final Ficoll/Hypaque concentrationsequal to 6.06%/10.0%) and centrifuged at 350 g for 35 min at20°C. This procedure effectively removed lymphocyte andmonocyte contamination and routinely yielded preparationsof 90-95% neutrophils, 5-7% eosinophils and basophils, and1-3% lymphocytes and monocytes. After Ficoll-Hypaque treat-ment the cell pellet was resuspended and washed in PBS. Hypo-tonic lysis removed erythrocyte contamination, and the PMNLwere suspended to a final concentration of 1.5 x 108/ml. Thezymosan preparation and cell incubation were performed asdescribed above.
At the end of the 3-min incubation the cell suspension wascentrifuged and the cell pellet resuspended to a concentrationof 3 x 108 cells/ml in 0.34 Msucrose. This was done to facilitate
NADPHand NADHOxidase Activities in Human Polymorphonuclear Leukocytes 283
00)
'I.
qJ
80
60
40
20
40 60 80
(nmol) NADP
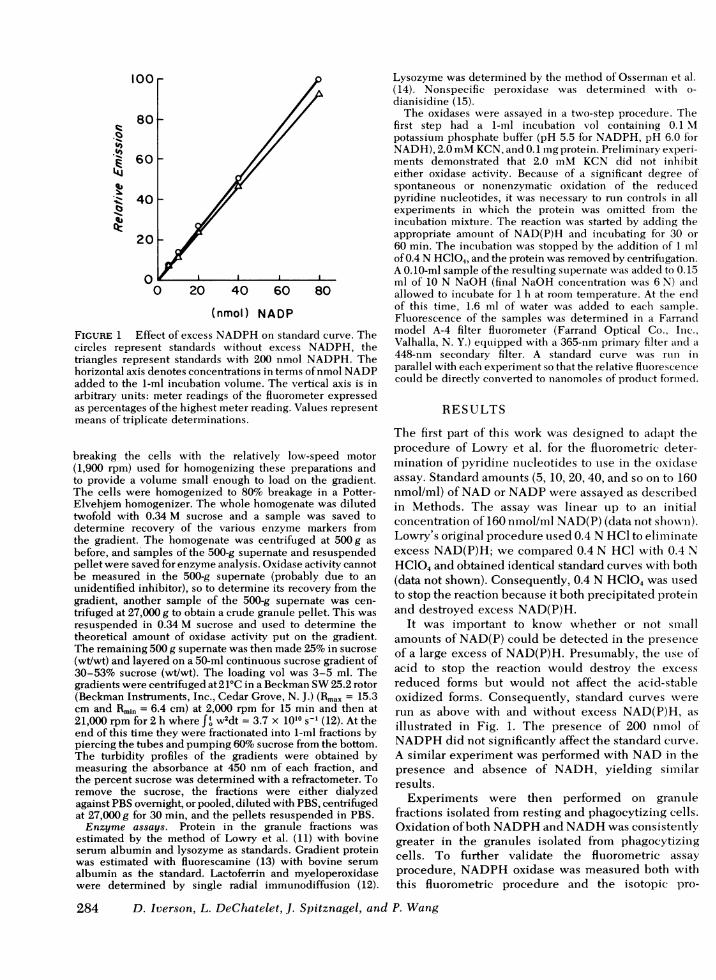
FIGURE 1 Effect of excess NADPHon standard curve. Thecircles represent standards without excess NADPH, thetriangles represent standards with 200 nmol NADPH. Thehorizontal axis denotes concentrations in terms of nmol NADPadded to the 1-ml incubation volume. The vertical axis is inarbitrary units: meter readings of the fluorometer expressedas percentages of the highest meter reading. Values representmeans of triplicate determinations.
breaking the cells with the relatively low-speed motor(1,900 rpm) used for homogenizing these preparations andto provide a volume small enough to load on the gradient.The cells were homogenized to 80% breakage in a Potter-Elvehjem homogenizer. The whole homogenate was dilutedtwofold with 0.34 M sucrose and a sample was saved todetermine recovery of the various enzyme markers fromthe gradient. The homogenate was centrifuged at 500g asbefore, and samples of the 500-g supemate and resuspendedpellet were saved for enzyme analysis. Oxidase activity cannotbe measured in the 500-g supernate (probably due to anunidentified inhibitor), so to determine its recovery from thegradient, another sample of the 500-g supernate was cen-trifuged at 27,000 g to obtain a crude granule pellet. This wasresuspended in 0.34 M sucrose and used to determine thetheoretical amount of oxidase activity put on the gradient.The remaining 500 g supernate was then made 25% in sucrose(wt/wt) and layered on a 50-ml continuous sucrose gradient of30-53% sucrose (wt/wt). The loading vol was 3-5 ml. Thegradients were centrifuged at 21°C in a Beckman SW25.2 rotor(Beckman Instruments, Inc., Cedar Grove, N. J.) (Rmax = 15.3cm and Rmin = 6.4 cm) at 2,000 rpm for 15 min and then at21,000 rpm for 2 h where ft w2dt = 3.7 x 1010 s-' (12). At theend of this time they were fractionated into 1-ml fractions bypiercing the tubes and pumping 60'% sucrose from the bottom.The turbidity profiles of the gradients were obtained bymeasuring the absorbance at 450 nm of each fraction, andthe percent sucrose was determined with a refractometer. Toremove the sucrose, the fractions were either dialyzedagainst PBSovernight, or pooled, diluted with PBS, centrifugedat 27,000 g for 30 min, and the pellets resuspended in PBS.
Enzyme assays. Protein in the granule fractions wasestimated by the method of Lowry et al. (11) with bovineserum albumin and lysozyme as standards. Gradient proteinwas estimated with fluorescamine (13) with bovine serumalbumin as the standard. Lactoferrin and myeloperoxidasewere determined by single radial immunodiffusion (12).
Lysozyme was determined by the method of Osserman et al.(14). Nonspecific peroxidase was determined wvith o-dianisidine (15).
The oxidases were assayed in a two-step procedure. Thefirst step had a 1-ml incubation vol containing 0.1 Mpotassium phosphate buffer (pH 5.5 for NADPH, pH 6.0 forNADH), 2.0 mMKCN, and 0.1 mgprotein. Preliminary experi-ments demonstrated that 2.0 mMKCN did not inhibiteither oxidase activity. Because of a significant degree ofspontaneous or nonenzymatic oxidation of the redtucedpyridine nucleotides, it was necessary to run controls in allexperiments in which the protein was omitted from theincubation mixture. The reaction was started by adding theappropriate amount of NAD(P)H and incubating for 30 or60 min. The incubation was stopped by the addition of 1 mlof 0.4 N HClO4, and the protein was removed by centrifugation.A 0.10-ml sample of the resulting supernate was added to 0.15ml of 10 N NaOH (final NaOHconcentration was 6 N) andallowed to incubate for 1 h at room temperature. At the en-dof this time, 1.6 ml of water was added to each sample.Fluorescence of the samples was determined in a Farrandmodel A-4 filter fluorometer (Farrand Optical Co., Inc.,Valhalla, N. Y.) equipped with a 365-nm primary filter anid a448-nm secondary filter. A standard curve was run inparallel with each experiment so that the relative fluorescenlcecould be directly converted to nanomoles of product formed.
RESULTS
The first part of this work was designed to adapt theprocedure of Lowry et al. for the fluiorometric deter-mination of pyridine nucleotides to uise in the oxidlaseassay. Standard amounts (5, 10, 20, 40, and so on to 160nmol/ml) of NADor NADPwere assayed as describedin Methods. The assay was linear up to an initialconcentration of 160 nmol/ml NAD(P) (data not shownvi).Lowry's original procedure used 0.4 N HCI to eliminateexcess NAD(P)H; we compared 0.4 N HCI with 0.4 NHC104 and obtained identical standard curves with both(data not shown). Consequently, 0.4 N HC104 was usedto stop the reaction because it both precipitated proteinand destroyed excess NAD(P)H.
It was important to know whether or not smnallamounts of NAD(P) could be detected in the presenceof a large excess of NAD(P)H. Presumably, the uise ofacid to stop the reaction would destroy the excessreduced forms but would not affect the acid-stableoxidized forms. Consequently, standard curves wererun as above with and without excess NAD(P)H, asillustrated in Fig. 1. The presence of 200 nmol ofNADPHdid not significantly affect the standard cturve.A similar experiment was performed with NADin thepresence and absence of NADH, yielding similarresults.
Experiments were then performed on grantulefractions isolated from resting and phagocytiziuig cells.Oxidation of both NADPHand NADHwas consistentlygreater in the granules isolated from phagocytizingcells. To further validate the fluorometric assayprocedure, NADPHoxidase was measured both withthis fluorometric procedure and the isotopic pro-
284 D. Iverson, L. DeChatelet, J. Spitznagel, and P. Wang
60 Phogocytizing
40 -
20- rF60r Resting
40 t
201-
060
40
20
0
r
Control
0.2mM 0.4mM 0.8mM
NADPH
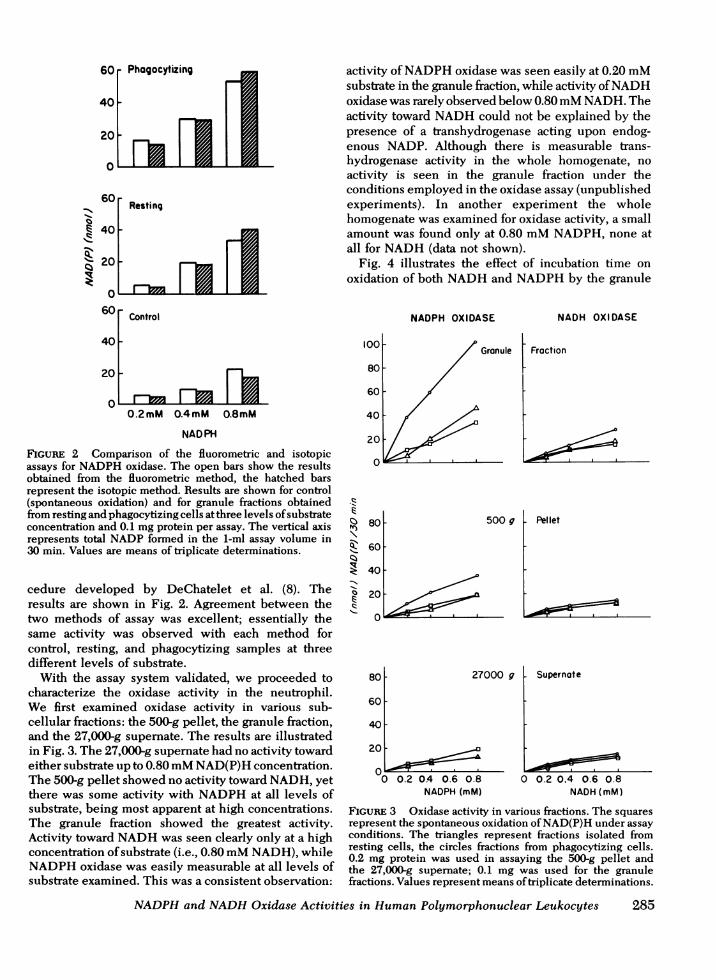
FIGURE 2 Comparison of the fluorometric and isotopicassays for NADPHoxidase. The open bars show the resultsobtained from the fluorometric method, the hatched barsrepresent the isotopic method. Results are shown for control(spontaneous oxidation) and for granule fractions obtainedfrom resting and phagocytizing cells at three levels of substrateconcentration and 0.1 mgprotein per assay. The vertical axisrepresents total NADPformed in the 1-ml assay volume in30 min. Values are means of triplicate determinations.
cedure developed by DeChatelet et al. (8). Theresults are shown in Fig. 2. Agreement between thetwo methods of assay was excellent; essentially thesame activity was observed with each method forcontrol, resting, and phagocytizing samples at threedifferent levels of substrate.
With the assay system validated, we proceeded tocharacterize the oxidase activity in the neutrophil.We first examined oxidase activity in various sub-cellular fractions: the 500-g pellet, the granule fraction,and the 27,000-g supernate. The results are illustratedin Fig. 3. The 27,000-g supernate had no activity towardeither substrate up to 0.80 mMNAD(P)H concentration.The 500-g pellet showed no activity toward NADH, yetthere was some activity with NADPHat all levels ofsubstrate, being most apparent at high concentrations.The granule fraction showed the greatest activity.Activity toward NADHwas seen clearly only at a highconcentration of substrate (i.e., 0.80 mMNADH), whileNADPHoxidase was easily measurable at all levels ofsubstrate examined. This was a consistent observation:
activity of NADPHoxidase was seen easily at 0.20 mMsubstrate in the granule fraction, while activity of NADHoxidase was rarely observed below 0.80 mMNADH.Theactivity toward NADHcould not be explained by thepresence of a transhydrogenase acting upon endog-enous NADP. Although there is measurable trans-hydrogenase activity in the whole homogenate, noactivity is seen in the granule fraction under theconditions employed in the oxidase assay (unpublishedexperiments). In another experiment the wholehomogenate was examined for oxidase activity, a smallamount was found only at 0.80 mMNADPH, none atall for NADH(data not shown).
Fig. 4 illustrates the effect of incubation time onoxidation of both NADHand NADPHby the granule
101
81
6
4
2
(11
zlz.N)
.q-C.)z
11
Q1.r,
1--
NADPH OXIDASE
0 Granule
'0 f0O
0o0--
80 500 g
60 F
40
20
0
80 F 27000 g
60 [
40[
20
w0 0.2 04 0.6 0.8NADPH(mM)
NADH OXIDASE
Fraction
Pellet
Supernate
0 0.2 0.4 0.6 0.8NADH(mM)
FIGURE 3 Oxidase activity in various fractions. The squaresrepresent the spontaneous oxidation of NAD(P)H under assayconditions. The triangles represent fractions isolated fromresting cells, the circles fractions from phagocytizing cells.0.2 mg protein was used in assaying the 500-g pellet andthe 27,000-g supemate; 0.1 mg was used for the granulefractions. Values represent means of triplicate determinations.
NADPHand NADHOxidase Activities in Human Polymorphonuclear Leukocytes 285

i10t -
0 15 30 45 60Time (min)
FIGURE 4. Effect of time of incubation on oxidase activity.The squares show results from NADPHoxidase activity; thecircles show NADHoxidase activity. 0.1 mg granule proteinisolated from phagocytizing cells was used with 0.20 mMNADPHor 0.80 mMNADH. The vertical axis gives totalnumber of nmol of NAD(P) formed in the 1-ml incubationvolume. Values are means of triplicate determinations cor-rected for spontaneous oxidation.
fraction from phagocytizing cells. Oxidation increasedprogressively up to at least 60 min in a fairlylinear fashion. Fig. 5 shows a pH curve for bothactivities; oxidation of NADPH had an apparentoptimum pH of 5.5 as reported earlier (8); NADHoxidation had an apparent optimum of 6.0. It shouldbe noted in these two figures that there was asubstantial difference in the activities toward the twosubstrates reflected in the fact that the concentrationof NADPHin the assay mixture was 0.20 mMwhilethat of NADHwas four times higher (0.80 mM). Theeffect of protein concentration was likewise deter-
I0
4.5 5.0 5.5 6.0 6.5 7.0pH
FIGURE 5 Effect of pH on oxidase activities. The squaresshow NADPHoxidase activity with 0.20 mMNADPH; thecircles show NADHoxidase activity at 0.80 mMNADH. 0.1mg granule protein isolated from phagocytizing cells wasused. The horizontal axis represents the pH of the 0.1-Mpotassium phosphate buffers used. The vertical axis repre-sents total nano moles NAD(P) formed in the 1-ml incubationvolume in 30 min. Values are means of triplicate determina-tions corrected for spontaneous oxidation.
mined for both substrates. Both activities were linearup to approximately 0.2 mg protein per assay whenthe incubation time was 30 min (data not shown).Table I shows the compilation of data from severalexperiments assayed at 0.20 mMNADPHand 0.80 mMNADH. As can be seen, the phagocytizing samplesshowed significantly more activity than either controlor resting samples. Also, in spite of the fourfoldsubstrate concentration difference, there is more oxida-tion of NADPHthan NADH in the phagocytizingsamples.
Chronic granulomatous disease (CGD) is a childhoodsyndrome in which the PMNLfail to kill invadingbacteria, and they do not show a normal increase inmetabolic activity with phagocytosis (16). A defect inthe oxidase could explain that observation and, in fact,previous reports showed there was no oxidation ofNADPHwith granules from patients with CGD(5, 17).Fig. 6 shows the effect of substrate concentrationat three levels of NADPHand NADHon the oxidaseactivities of granules obtained from a patient withCGDand granules from normal cells prepared inparallel. The granules from the patient with CGDshowed no significant activity toward NADPHat thesubstrate levels examined. The normal control showedactivity at all levels with resting activity becomingequal to phagocytizing activity at and above 0.80 mM.The same pattern holds for NADH; again no activitywas observed with granules isolated from cells ofthe patient with CGDwhile activity was observedwith granules isolated from normal cells at 0.40 and0.80 mMNADH. Granules from CGDcells did showsome activity toward both substrates, but only atvery high substrate levels (6-12 mMNAD(P)H) andno difference was seen between resting and phago-cytizing samples (data not shown). The control cellsused in this experiment were not typical of other
TABLE INAD(P)H Oxidation in Control, Resting, and
Phagocytizing Samples
NAD(P)H Control Resting Phagocytizingoxidation
nmol/30 min
NADP 8.2+0.8 (10)* 4.6±1.0 (8) 24.4+1.6 (10)NAD 10.1+0.7 (11) 10.9+1.2 (9) 17.7+1.6 (11)
Values are averages+SEM for the number of experiments.Values in each experiment were determined in triplicate.Assay concentration of NADPH was 0.20 mM, NADHconcentration was 0.80 mM. Protein concentration was 0.1mg/ml. Control is the spontaneous oxidation of NAD(P)Hunder assay conditions. Resting and phagocytizing representgranule fractions isolated from resting and phagocytizingcells.* Number of experiments.
286 D. Iverson, L. DeChatelet, J. Spitznagel, and P. Wang

routine preparations as the oxidase activities, es-pecially from the resting cells, were unusually high(see Fig. 3 and Table I). This preparation contained15% eosinophils, an unusually high percentage.Unpublished data from this laboratory showed thateosinophils have three to five times more oxidaseactivity than do neutrophils, consequently the elevatedactivity shown here may be at least partially due tothe elevated eosinophil count. That, however, does notchange the observation that the CGDcells showed noactivity toward either substrate, even if they arecompared to the data in Fig. 3 or Table I.
Because of the relatively higher activity in theeosinophil than the neutrophil, the possibility must beconsidered that the measured activities reside entirelyin the eosinophilic contamination and not in the neutro-phil itself. This does not seem to be the case asevidenced by the following facts: (1) in one prepara-tion from a normal subject, the isolated cells con-tained only 1% eosinophils, yet the specific activityof NADPHoxidase in this preparation was 32.3 nmol/30
NADPHOXIDASE
ft
120
100
80
60
40
20
100
80
60
40
20
CGD
NADH OXIDASE
CGD
0.2 04 0.6 0.8 0.2 0.4 0.6 0.8NAD(P)H (mM)
FIGURE 6 Effect of NAD(P)H concentration on NAD(P)Hoxidase activities of granules isolated from CGDand normalPMNL. The top two panels show normal and CGDactivitytoward NADPH; the bottom panels show normal andCGDactivity toward NADH. The squares show the spon-taneous oxidation of NAD(P)H under assay conditions. Theopen circles represent granules obtained from resting cells;the closed circles represent granules from phagocytizingcells. Protein concentration was 0.1 mg. Each point repre-sents the mean of triplicate determinations.
40t 30
2010
16ta 12q 8
4
Z 200
I 100
4 200
Q 100
0.8t o.6
0.40.2
I - IWWgwi
l~~~~~~ I~~~~~ 0
1- NADH1 OxidoseA
Protein
;r Lwmm10 20 30 40
Fraction NUmber
FIGURE 7 Location of NAD(P)H oxidase activity in a linearsucrose gradient. The samples of this gradient were dialyzedovernight against PBS. The vertical axes represent totalactivities found in each fraction. NADPHconcentration was0.20 mM, NADH0.80 mM; incubation time was 60 min.
min/0. 10 mgprotein. If anything, this is slightly higherthan the average sp act of 24.4±1.6 nmol/30 min/0.10mg protein reported in Table I. (2) We obtained aleukocyte preparation from a patient with pronouncedeosinophilia. The specific activity of NADPHoxidasein a granule fraction isolated from phagocytizingcells containing 84% eosinophils was 3.2 times theactivity seen in a paired preparation containing 82%neutrophils and only 3% eosinophils.
Wenext attempted to localize the oxidase activitieson a linear sucrose gradient. Fig. 7 shows one suchexperiment. Lactoferrin and myeloperoxidase (MPO)indicate the locations of the specific and azurophilgranules, respectively, in the gradient. The lactoferrinpeak was at a density of approximately 1.18 g/cubiccentimeter, the MPOpeak at 1.22 g/cubic centimeter,while both oxidase activities appeared to migratefurther into the gradient, with the peak activity at adensity of approximately 1.24 g/cubic centimeter.
Fig. 8 shows another preparation in which the wholehomogenate was divided in two parts; one part wasinitially centrifuged at 126 g while the second wascentrifuged at 500g. The resulting low-speed super-nates were then loaded on separate sucrose gradients.This was done to determine if the oxidase activitieswere due to eosinophil granules. Since eosinophilgranules have a greater density than neutrophilgranules, they sediment lower in the gradient (12) andthis might have explained the very dense localizationof the oxidase activities. It was known that many of the
NADPHand NADHOxidase Activities in Human Polymorphonuclear Leukocytes 287

500 SUPERNATEg
Froction Number
FIGURE 8 Location of NAD(P)H oxidase activity in a linearsucrose gradient. The samples of the gradient were pooledand centrifuged. The vertical axes represent total activity ineach fraction. NADPHconcentration was 0.20 mM, NADH0.80 mM; incubation time was 60 min.
eosinophil granules sediment at 500 g (18); con-
sequently the whole homogenate in this experimentwas given the two low-speed centrifugations to see ifthere would be a difference in the oxidase activitiesof the two gradients. No such distinction was detected.In this experiment the fractions were pooled,centrifuged, and resuspended. The disadvantage of thisprocedure was the loss of resolution caused byaveraging relatively large groups of fractions. In spiteof this limitation, however, it was still apparent thatthe MPO and lactoferrin sedimented at differentdensities. Nonspecific peroxidase sedimented with theMPOas did part of the lysozyme, a marker for boththe specific and azurophil granules. The specificgranule peak for lysozyme did not resolve sharply,probably due to the pooling of the fractions, but theazurophil peak was well resolved and it coincidedwith the MPOpeak. Again the peak oxidase activitiessedimented to a slightly greater density. These twoexperiments are representative of six separate experi-ments; in each separate experiment the peak ofoxidase activities was found at a density greaterthan the MPOactivity.
The oxidase activities were purified by thisprocedure. The total oxidase activity recovered fromthe gradient exceeds 100%, suggesting removal of aninhibitor. In addition, the specific activity of the peakoxidase fraction in the gradients was 10-30 times thatof the crude granule fraction.
DISCUSSION
In the present work, we have adapted a fluorometricprocedure for the determination of pyridine nucleo-tides for use in measuring the reduced pyridinenucleotide oxidase(s) in human PMNL. This system issuperior to the oxygen electrode or spectrophotometricmethods presently in use in that it is at least 1,000-foldmore sensitive (9). It has several advantages over theisotopic system as well. It can measure both oxidaseactivities with as much sensitivity as the isotopicassay measures the NADPHoxidase. In addition, it issimpler and far less expensive to use than theisotopic procedure. We have applied this system tothe study of the basic characteristics of the oxidasesin granule fractions and have initiated purification ofthe enzyme(s) on sucrose density gradients.
Both oxidase activities were increased with phago-cytosis, and both were insensitive to cyanide asevidenced by the fact that all assays were performedin 2.0 mMKCN. Although both activities increasedwith increasing substrate concentration, time ofincubation and protein, each may have had a slightlydifferent pH optimum. Oxidation of NADPH wasmaximal at pH 5.5 in agreement with previousreports (8), while oxidation of NADHappeared to beoptimal at pH 6.0. Phagocytosis enhanced neitheroxidase activity in granules isolated from CGDcells.Both activities sedimented to the same place in linearsucrose gradients.
A major question in the area of leukocyte metabolismis which enzyme (or more exactly which nucleotideoxidation) is responsible for the respiratory burstaccompanying phagocytosis in human PMNL. Thepresent work suggests that NADPHoxidation is likelyto have more physiological significance than NADHoxidation. As illustrated in Figs. 3 and 6, thedegree of NADPH oxidation was always greaterthan that of NADHwhen the substrate concentra-tions were equal (except at extremely high levels).In fact, in the range of 0.20-0.80 mMthe oxidationrate of NADPHwas usually four to five times thatof NADH. This was true every time the assay wasperformed, therefore, the procedure adopted for com-paring the activities was to assay at 0.20 mMNADPHand 0.80 mMNADH. Table I shows a compilationfrom several such experiments. In spite of the factthat the NADHconcentration was four times that ofNADPH, in the phagocytizing samples there was
288 D. Iverson, L. DeChatelet, J. Spitznagel, and P. Wang
126 SUPERNATEg

significantly more oxidation of NADPHthan NADH.This difference cannot be explained by a differencein protein because all assays were run at the sameprotein levels. Based on these data it seems reasonableto say that the oxidation of NADPHis responsiblefor the metabolic burst, since NADPH oxidationoccurs at a lower concentration of substrate thandoes oxidation of NADH. This is particularly significantsince the concentration of both reduced pyridinenucleotides within the human PMNLis estimated tobe 0.05 mM(19). In terms of the metabolic processesthat occur in the cell after phagocytosis, NADPHfitsinto the scheme much more easily than does NADH.Oxidation of NADPHcan directly explain the increasein oxygen consumption (as one of the reactantsin the oxidase reaction), the increase in H202production (as one of the products of the oxidasereaction) and the increase in the hexose mono-phosphate shunt activity (by providing NADP, the ratelimiting substrate in the hexose monophosphate shuntactivity).
Subcellular localization of the oxidase activity hasbeen of some concern to workers in the field.Some investigators feel the plasma membrane is thesite of this enzyme activity (20). Others have suggestedthat MPOwould be a logical candidate for oxidaseactivity (21) since the oxidase behaves very much likea peroxidase and purified peroxidases are known tocatalyze the oxidation of reduced pyridine nucleo-tides (22). Serious questions about the validity of thesetwo hypotheses are raised by the sucrose gradientwork presented here. Because there is no markeras yet for the membrane of human PMNL, it is im-possible to argue absolutely against membranelocalization, although the localization in the gradientwould make this relatively unlikely.
The other possibility, then, is that the oxidase(s) arelocalized in the azurophil granule. Patriarca et al. (23)reported NADPHoxidase activity in the azurophilgranule of rabbit PMNL, which seemed to support thehypothesis of Roberts and Quastel (21) that the oxidasewas really MPO. The data here case some doubt onthat possibility in human PMNL. Figs. 7 and 8 showthat the peak activities of MPOand pyridine nucleo-tide oxidase did not co-sediment in a linear sucrosegradient. Although there was some overlap of the twoactivities, their peaks were distinctly separate. In Fig.8, the nonspecific peroxidase coincided with theimmunologically determined MPO, and it too peakedbefore the oxidase. These data suggest that theoxidase is not MPO, nor does the oxidase seem tohave peroxidative activity based on the o-dianisidineassay. MPO is found primarily in the lighterazurophil granule as reported by Spitznagel et al. (12),so if the oxidase peak is not coincident with the MPOpeak, then it is not located in the light azurophil
granule. Lysozyme is found in the specific granulesand in the heavy azurophil granule (12). Fig. 8 showsthe distribution of lysozyme in those particulargradients and it can be seen that the oxidaseactivities are located deeper in the gradient thanlysozyme. Other gradients show this also. The dataindicate that the oxidase activities are associated withvery dense particulate material which sediments to50-51% sucrose in 2 h time. The nature of theparticle is presently unknown, but it does not seem tobe the normal azurophil granule as it is presentlyknown.
It is not possible to determine by these datawhether there is one oxidase with different affinitiesfor each nucleotide or two separate enzymes. Thefact that the activities co-sediment in a linear sucrosegradient suggests, but does not prove, there is onlyone enzyme. In addition, the fact that in CGDbothactivities are defective further supports a one-enzymehypothesis. But a final answer to this question willcome only with the purification of the enzyme.
A very recent paper (24) described an NADHde-pendent nitro blue tetrazolium dye reductase with avery low Kmfor NADHwhich appeared to be absent incells from patients with CGD. The relationship of thisenzyme to the oxidase remains to be established; al-though we previously reported a low KmNADHdia-phorase in human PMNL(19), we have been unable todetect oxidase activity toward low concentrations ofNADHin either normal or CGDcells. Further, thelocation of the diaphorase in a sucrose densitygradient (24) was quite different from that of theoxidase activities reported here. These discrepanciesremain to be resolved.
Finally, in regard to purification of this enzyme, thework here shows the sucrose gradient centrifugationwill be an invaluable tool in accomplishing thisgoal. Not only was more than 100%o of the oxidaseactivity routinely recovered from the gradient, but thespecific activity of the peak fractions in the gradientwas 10-30 times that of a granule fraction preparedin parallel with the gradient. This will constitutethe first major step in the purification of the enzyme(s).Weare presently working toward that objective.
ACKNOWLEDGMENTSWe wish to thank Larry Martin for his excellent technicalassistance. This research was supported by a grant fromthe Forsyth Cancer Service, by grant ER-40-1-3628, andby U. S. Public Health Service grants AI-10732, AI-02430,and CA-12197.
REFERENCES1. Cagan, R. H., and M. L. Kamovsky. 1964. Enzymatic
basis of the respiratory stimulation during phagocytosis.Nature (Lond.). 204: 255-257.
NADPHand NADHOxidase Activities in Human Polymorphonuclear Leukocytes 289

2. Baehner, R. L., and M. L. Karnovsky. 1968. Deficiencyof reduced nicotinamide-adenine dinucleotide oxidase inchronic granulomatous disease. Science (Wash. D. C.).162: 1277-1279.
3. Wilkinson, R. W., D. R. Powars, and P. Hochstein.1975. New evidence for the role of NADHoxidase inphagocytosis by human granulocytes. Biochem. Med. 13:83-88.
4. Patriarca, P., R. Cramer, S. Moncalvo, F. Rossi, andD. Romeo. 1971. Enzymatic basis of metabolic stimulationin leukocytes during phagocytosis: the role of activatedNADPHoxidase. Arch. Biochem. Biophys. 145: 255-262.
5. Hohn, D. C., and R. I. Lehrer. 1975. NADPHoxidasedeficiency in X-linked chronic granulomatous disease.
J. Clin. Invest. 55: 707-713.6. Evans, A. E., and N. 0. Kaplan. 1966. Pyridine nucleo-
tide transhydrogenase in normal human and leukemicleukocytes.J. Clin. Invest. 45: 1268-1272.
7. Reed, P. W. 1969. Glutathione and the hexose monophos-phate shunt in phagocytizing and hydrogen peroxide-treated rat leukocytes. J. Biol. Chem. 244: 2459-2464.
8. DeChatelet, L. R., L. C. McPhail, D. Mullikin, andC. E. McCall. 1975. An isotopic assay for NADPHoxidaseactivity and some characteristics of the enzyme fromhuman polymorphonuclear leukocytes. J. Clin. Invest.55: 714-721.
9. Lowry, 0. H., N. R. Roberts, and J. I. Kapphahn. 1957.The fluorometric measurement of pyridine nucleotides.
J. Biol. Chem. 224: 1047-1064.10. Kaplan, N. O., S. P. Colowick, and C. C. Barnes.
1951. Effect of alkali on diphosphopyridine nucleotide.J. Biol. Chem. 191: 461-472.
11. Lowry, 0. H., N. J. Rosebrough, A. L. Farr, and R. J.Randall. 1951. Protein measurement with the Folinphenol reagent.J. Biol. Chem. 193: 265-275.
12. Spitznagel, J. K., F. G. Dalldorf, M. S. Leffel, J. D. Folds,I. R. H. Welch, M. H. Cooney, and L. E. Martin. 1974.Character of azurophil and specific granules purified fromhuman polymorphonuclear leukocytes. Lab. Invest. 30:774-785.
13. Ubhlen, P., S. Stein, W. Dairman, and S. Udenfriend. 1973.
Fluorometric assay of proteins in the nanogram range.Arch. Biochem. Biophys. 155: 213-220.
14. Osserman, E. F., and D. P. Lawlor. 1966. Serum andurinary lysozyme (muramidase) in monocytic and mono-myelocytic leukemia. J. Exp. Med. 124: 921-952 (andplates 91, 92).
15. Worthington Biochemical Corporation. 1972. Worthing-ton enzyme manual: peroxidase. Worthington Bio-chemical Corp., Freehold, N. J. 43.
16. Holmes, B., A. R. Page, and R. A. Good. 1967. Studiesof the metabolic activity of leukocytes from patientswith a genetic abnormality of phagocytic function.
J. Clin. Invest. 46: 1422-1432.17. McPhail, L. C., L. R. DeChatelet, and P. S. Shirley. 1975.
NADPHoxidase activity of human neutrophils.J. Reticu-loendothel. Soc. 18: 9b. (Abstr.)
18. Archer, G. T., and J. G. Hirsch. 1963. Isolation of granulesfrom eosinophil leukocytes and study of their enzymecontent. J. Exp. Med. 118: 277-286 (and plates 17-19).
19. DeChatelet, L. R., L. C. McPhail, D. Mullikin, andC. E. McCall. 1974. Reduced nicotinamide adeninedinucleotide and reduced nicotinamide adenine dinu-cleotide phosphate diaphorase activity in human poly-morphonuclear leukocytes. Infect. Immun. 10: 528-534.
20. Briggs, R. T., D. B. Drath, M. L. Karnovsky, and M. J.Karnovsky. 1975. Localization of NADHoxidase on thesurface of human polymorphonuclear leukocytes by a newcytochemical method. J. Cell. Biol. 67: 566-586.
21. Roberts, J., and J. H. Quastel. 1964. Oxidation of reducedtriphosphopyridine nucleotide by guinea pig poly-morphonuclear leukocytes. Nature (Lond.). 202: 85-86.
22. Akazawa, T., and E. E. Conn. 1958. The oxidation ofreduced pyridine nucleotides by peroxidase. J. Biol.Chem. 232: 403-415.
23. Patriarca, P., R. Cramer, P. Dri, L. Fant, R. E. Basford,and F. Rossi. 1973. NADPHoxidizing activity in rabbitpolymorphonuclear leukocytes: localization in azuro-philic granules. Biochem. Biophys. Res. Commun.53: 830-837.
24. Segal, A. W., and T. J. Peters. 1976. Characterization ofthe enzyme defect in chronic granulomatous disease.Lancet. I: 1363-1365.
290 D. Iverson, L. DeChatelet, J. Spitznagel, and P. Wang





























