Embed Size (px)
Citation preview

Comparison of mRNA Gene Expression Analysis between Quantitative-real time PCR Assay and DNA Microarray Based Assay
Su LJ1,a,*, Wu LC1,b and Tseng ALH1,c 1Institute of Systems Biology and Bioinformatics, National Central University, Jhongli City, Taoyuan
County, Taiwan [email protected], [email protected], [email protected]
*corresponding author
Keywords: Microarray/Q-RT-PCR comparisons, independent/pair-wised sample analysis, Q-RT-PCR internal control.
Abstract. The development of microarrays permits us to monitor transcriptomes on a genome-wide scale. To validate microarray measurements, quantitative-real time-reverse transcription PCR (Q-RT-PCR) is one of the most robust and common approaches. The new challenge in gene quantification analysis is how to explicitly incorporate statistical estimations into such studies. In the realm of statistical analysis, the various available methods of the probe level normalization for microarray analysis may result in distinctly different target selections as well as variation in the scores for any correlation between the microarray and Q-RT-PCR results. Here, we explore the differences in gene expression measurements between a short-oligonucleotide-based microarray based assay and Q-RT-PCR assays. The systematic analysis procedures used here focus on identifying genes with minimal variation in the microarray data, which facilitates essential internal control selection prior to Q-RT-PCR analysis. To obtain more reliable validation measurements, it is necessary, firstly, to identify the potential internal controls to be used for the Q-RT-PCR analyses and to explore the possibility that these internal controls are suitable for general use. To do this, it is necessary to examine the probe set characteristics and to rank genes with respect to variance and inter-quartile range. Secondly, correctly scoring the correlation between the microarray dataset and the Q-RT-PCR measurements is dependent on whether the raw dataset is normally distributed or not, which can be tested using the Shapiro-Wilk test. If the dataset is a normally distributed, the microarray and Q-RT-PCR data comparisons should use Pearson’s or Kendall’s tau correlations. If the dataset is not normally distributed, then Spearman’s rho test, which is the rank-based non-parametric equivalent of the more commonly used Pearson’s correlation calculation, should be used. Finally, systematic microarray and Q-RT-PCR analyses may then be carried out to reveal the appropriate biostatistical methods to use with different experimental designs and identify those that can adequately detect genes with minimal variation in a microarray dataset.
1. Introduction
In recent years, microarrays, by making use of the sequence resources created in genomic projects, have become a powerful technology capable of measuring the expression levels of thousands of genes simultaneously and have dramatically expedited a comprehensive understanding of gene expression profiles during disease development. For example, microarray technology has been used to compare gene expression profiles between normal and diseased cells and this has led to dramatic advances in our understanding of cellular processes at the molecular level. Oligonucleotide microarrays have
2011 International Conference on Agricultural and Natural Resources Engineering Advances in Biomedical Engineering, Vol.3-5
978-1-61275-996-8 /10/$25.00 ©2011 IERI ANRE2011
158

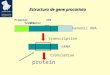
become a widely used alternative to cDNA microarray because of their superior specificity, reproducibility and ease of design. The short-oligonucleotide-based Affymetrix GeneChip® arrays, a commercial platforms, utilize multiple probes for each gene with automated control for the experimental process from hybridization to quantification and thus provide reliable and comparable data [1]. The multiple probe sets for each gene are typically widely scattered across the surface of the Affymetrix microarrays. Variations in intensity from probe to probe or chip to chip for samples need to be resolved to obtain a reliable level of expression. Various statistical algorithms are available for probe-cell level normalization and to summarize expression values. Presently, no standard definition of validation is available. Correlations of microarray and Q-RT-PCR data are seldom explicitly discussed in the literature and inconsistent results are rarely explained. It is well documented that both Q-RT-PCR and microarray analysis have inherent pitfalls [2, 3] that may significantly influence the data obtained by each method. Researchers are still confronted with challenging questions after completing the expression profiling of a cell type and one of these is how to validate and standardize the data processing using proper statistical analysis (Fig. 1). Q-RT-PCR is widely used and is a sensitive and robust technique for the detection and quantification of often rare mRNA targets [4]. By recording the amount of fluorescence emission after each cycle, it is possible to monitor the PCR reaction during the exponential phase where the first significant increase in copy number occurs and in these circumstances the amount of PCR product correlates with the initial amount of target template. More specifically, Q-RT-PCR has become one of the gold standards for gene expression studies and is the method of choice for corroborating microarray data [5, 6]. In this article, our aim was to discuss two unanswered questions associated with microarray target selection for Q-RT-PCR validation. Firstly, it is not certain what kind of method should be used to identify the gene or genes that will serve as the best internal controls for the Q-RT-PCR simply because there is no perfect internal control [7]. Secondly, a major challenge when scoring the correlation between microarray datasets and Q-RT-PCR measurements that remains unsettled is which of the many different probe level normalization methods should be used as the different methods may result in different gene correlations.
Fig. 1. Illustration of major challenges in data comparison between microarray and Q-RT-PCR measurements.
2. Problems associated with using microarrays for gene expression studies
DNA microarrays provide an unprecedented capacity for whole genome profiling. However, the major complaint about this technology has been that it is unreliable and microarray experiments can demonstrate any outcome the investigator wishes to prove [8, 9]. The quality of gene expression data obtained from microarrays can vary greatly with the platform and procedures used. Compared to other molecular biology techniques, microarrays are relatively new. As such, their users are
159

challenged by a number of issues (Fig. 1). In the following sections, we briefly discuss noise and normalization issues with respect to microarrays.
2.1 Noise
Due to their nature, microarrays tend to be noisy. There are two principal sources of noise in a microarray experiments: biological noise and technical noise. Biological noise consists of the variation among the experimental samples or animals and involves the makeup of sample groups together with the heterogeneity of the genetic material within each sample due to cell to cell variation. Biological noise cannot be corrected for but it can be accounted for by the use of replicates of the treatments or conditions and statistics [10]. However, the noise derived from experimental techniques is reproducible and its boundaries can be modeled. Technical noise consists of differences in sample preparation and experiment variables, which include nonspecific cross hybridization, differences in the efficiency of the labeling reactions and production differences between the microarrays [11]. This is true even with the advantages afforded by the Affymetrix chip design, which is most popular microarray platform worldwide, namely, that each high-density array provides multiple, independent measurements for each transcript. Multiple probes mean that you can obtain a complete dataset with high reliability and reproducibility from every experiment. This is because about one third of the expression signals (half of the transcripts) undergo cross hybridization contamination when an Affymetrix Genechip® is used (Table 1). Several studies have tried to reduce noise signal by removing the contamination probes [12-14]; however, additional experimental evidences are required to support their methodologies. Up to now, even through the noise is known to exist and is not totally solved, microarrays are still a commonly used high throughput method for detecting differentially gene expression patterns.
Table 1. The summary of probe set characteristics on HG_U133A chip.
2.2 Normalization
The aim of the normalization is to account for the systematic differences across different array chips. The selection of different image analysis algorithms, the first step in the normalization processes, usually produces different analysis results. For example, several popular image analysis algorithms are available that are used with the Affymetrix array system [10]. For example, Microarray Suite software 5.0 (MAS5), developed by Affymetrix [15] (http://www.affymetrix.com /support/technical/byproduct.affx?product=mas), is often used to carry out the probe-pairs adjustment and to transformed the images into text files as intensity information. Common alternatives include Robust Multichip Average via quantile adjustment (RMA) [16], GC_RMA, which is similar to RMA, but pools the probes with comparable numbers of G-C bonds to achieve a stable mismatch adjustment [17] and dChip (DNA Chip analyzer) [18]. In this report, we used three of the above array processing methods, namely MAS5, RMA and GC-RMA, to normalize the Affymetrix expression signals for the transcripts based on corresponding probe pairs of oligonucleotides. In this study, we recalculated the lung cancer datasets published by Su and colleagues [19] and showed that there is a 70% overlap between these three image analysis algorithms (Fig. 2). If we compared the results for pairs of two algorithms, there is over 80% overlap. This suggests that there may be smaller analysis variance in such a case or that the more reliable results help to create a stable high throughput platform.
160

Fig. 2. A Venn diagram illustrating the overlap among genes prioritized by three different image analysis algorithms.
The gene expression patterns of lung adenocarcinoma were clustered by a non-parametric test using three different image analysis algorithms, namely MAS5, RMA and GC-RMA, which were applied to oligo-array data (p<10-6). There was 70% overlap between these three image analysis algorithms using the same analysis strategy under different significant level conditions (the p value is 10-6). If the results are compared between each two algorithms, there was over 80% overlap.
3. Q-RT-PCR validation system
Integration of the PCR-based assay with laser scanning technology to excite fluorescent dyes is currently being used in the specially designed TaqMan® probe system. It is a fully integrated system for Q-RT-PCR detection. The system includes a built-in thermal cycler, a laser to induce fluorescence, a CCD (charge-coupled device) detector, real-time sequence detection software and the TaqMan® reagents that are necessary for the fluorogenic 5'-Nuclease assay. An appropriate internal control for Q-RT-PCR should be expressed stably across all data samples and if this is true, the measurement of genes relative to the internal control will reflect their real gene expression. This implies that a reference gene should have a small variance together with sufficient intensity when applied as an appropriate internal control. Most published studies have focused on the identification of reference genes that can be used to normalize expression of a gene across patient samples or tissue types rather than within one specific type of tissue or cell line [20, 21]. Generally speaking, housekeeping genes, such as ACTB (actin), GAPDH (glyceraldehyde-3-phosphate dehydrogenase) and 18S ribosomal RNA, are commonly employed in Q-RT-PCR analysis [22-24]. Although 18S ribosomal RNA has been shown to be a reliable control in many studies [20, 21, 25], it does not undergo reverse transcription with oligo (dT) primers and is inappropriate for use when such primers are used. However, several studies have also demonstrated that the gene expression patterns of many commonly used internal controls may vary between tissue types, experimental conditions or pathological state [7, 26-28]. The “perfect” control gene for all Q-RT-PCR does not exist because variability in Q-RT-PCR data can also stem from differences in the expression of the reference gene, for example GAPDH and ATCB and it is on this that the measured expression of all the other genes is based [29]. In fact, aberrational results may be created from a good dataset when a variable internal control is used [30].
Up to the present, it is clear that there is no single housekeeping gene is suitable for all experimental conditions and a number of papers have been published looking at the viability of using specific genes as the housekeeping genes in different systems using various methodologies [23, 24, 28, 31-34]. Szabo et al. developed statistical models to assist in identifying appropriate housekeeping genes that can be used as Q-RT-PCR normalization controls in one or multiple types of tissue samples [35]. However, their rigorous approach heavily relies on an assumption that there is a multivariate normal distribution of microarray expression levels across the dataset and this may not fit most practical situations, especially without a large number of arrays. In addition, their models are
161

only applicable to the analysis of random samples, not paired samples that have been collected from individual patients as is the data used here.
4. Identification of a novel internal control for Q-RT-PCR through variation in gene expression levels.
4.1 Independent samples in experimental design
We employed acute lymphoblastic leukemia (ALL) datasets [36] to predict the suitable internal controls for Q-RT-PCR, for example. In total, 132 cases of pediatric ALL were selected from the original 327 diagnostic bone marrow aspirates for reanalysis of the Affymetrix HG_U133A microarrays (Table 3). Differentially expressed genes, identified by microarray through global normalization, require validation of their expression patterns by Q-RT-PCR, which generally employs one internal control for normalization. This distinct normalization method calls for an accurate internal control for the microarray and Q-RT-PCR data comparison.
To prioritize the potential internal controls for Q-RT-PCR analyses and to study the possibility of the general utilization of these potential internal controls, we first checked the probe set characteristics (Table 4) and ranked genes in terms of variance and inter-quartile range (IQR) (Fig. 3). This approach was used to evaluate the gene expression patterns of the various internal controls across the ALL microarray datasets with the goal of providing insight into which internal controls might be the best choice in this exercise. As an illustration of this, we first used RMA for the probe-cell level normalization and expression-value summary of the 132 microarrays in the ALL datasets. Associated with each gene, the appropriate averages and variances were used to evaluate five computed statistics in the form of a box plot that disclosed variation in gene expression. As a result, many potential internal controls were revealed. To prioritize the targets, we removed those with significantly lower signal intensities and the multiple probe sets on Affymetrix chip were also screened for potential cross hybridization contamination between genes using the NetAffx™ Analysis Center (https://www.affymetrix.com/site/login/login.affx). The copy number of the individual housekeeping gene chosen for relative quantification should be in a similar range to that of the target gene in order to make comparative quantification possible [37].
Table 3. Subgroup distribution of ALL case.
Eleven potential internal controls have the characteristics of small variance across different microarray intensity intervals and these consists of C1orf160 and NIBP (intensity range: 5 to 10), OTUB1 and GGA1 (intensity range: 10 to 20), SFRS10 and SKP1A (intensity range: 20 to 30), ACTR3 and YWHAQ (intensity range: 30 to 40), NONO and DDX17 (intensity range: 40 to 60) and CALM2 (intensity range: large than 60) (Fig. 3). These potential internal controls are presented with their different intensity ranges in order to identify appropriate normalization candidates for different target genes.
162

Table 4. Summary of probe set characteristics of 11 novel and 10 well-known internal controls for Q-RT-PCR in
Affymetrix HG-U133A chip.
In addition, the box plots for 10 well-known internal controls (http://medgen.ugent.be
/~jvdesomp/genorm) [7] (containing 23 probe sets) are also shown in Fig 3. However, 16 out of 23 probe sets were found to have various levels of cross hybridization as identified by the NetAffx™ Analysis Center (Table 4). The unique characteristics of these well-known internal controls for Q-RT-PCR are their high expression levels, but these genes also show huge variances with the exception oft YWHAZ. This finding further supports the view that the intensities of normalized microarray data and the copy number for Q-RT-PCR detection when measuring gene expression patterns should be in a similar range. The approach used here allows the identification of potential internal controls that have a similar range of expression as the selected target genes. We do not deny the limitations of the present analysis and therefore we needed to confirm our statistical analysis by using these novel internal controls on other ALL samples to produce absolute quantization by Q-RT-PCR.
4.2 Pair-wise sample experimental design
In contrast to the experimental design of the ALL dataset [36], pair-wise sample experimental design should be analyzed by suitable statistical methods; this will assess the variation in the microarray expression level for a specified gene in an experiment of moderate sample size that includes paired samples. Considering Su’s study was a paired design with a moderate sample size, the block bootstrapping technique was applied and the gene expression patterns of the various internal controls evaluated using the lung cancer dataset (as described in Su’s study); this was followed by
163

experimental Q-RT-PCR analysis for validation. A total of 39 blocks, resulting from 27 adjacent normal/tumor matched samples and 12 un-paired samples of microarrays, were used in the block bootstrap. Bootstrapping is a breakthrough statistical approach using a computationally intensive re-sampling technique and it allows complex problems to be solved in which the accuracy of a devised statistical procedure can not normally be analytically evaluated [38, 39]. Block bootstrapping was originally used for re-sampling methods in dependent cases, especially time series data [40]. The basic bootstrap generates artificial samples that allow the making of an inference of interest through re-sampling the original data with replacement in which all observations are assumed to be mutually independent and from the same distribution. To guarantee the structure of independence in bootstrap re-sampling, the concept of blocking for the paired data by treating each individual patient, mixture tissue or cell line as a block was used. By selecting an observation within the block with equal probability when combined with all the other un-paired samples, we can obtain an independently re-sampled dataset. Su and his colleagues created a bootstrap sample by randomly sampling the blocks in the dataset and computed the bootstrap replicates of the relevant summary statistics of the expression levels. After repeating this bootstrap re-sampling scheme sufficient times, we then used the averages of these bootstrap replicates to reveal the variation in expression summaries corresponding to a specific gene across the microarrays. Appropriate internal controls, such as DEAD (Asp-Glu-Ala-Asp) box polypeptide 5 (DDX5), were selected by ranking the variation in gene expression.
5. Q-RT-PCR validation and comparison with microarray.
As Morey suggests [41], subsequent to microarray data analysis a set of genes was chosen for validation using Q-RT-PCR based on their expression change levels’ p-value. Correlation between the microarray and Q-RT-PCR results for this gene set was then performed for each experiment and the statistical significance of the correlations determined. For the microarray, the data input into the correlation analysis was the relative expression value of the normalized average for each gene on the array representing all samples. However, we usually used the mean relative expression value of the internal control or housekeeping gene as reported by Q-RT-PCR from all experimental samples (Fig. 1). We also agree with Morey’s viewpoints that prior to performing correlation analysis, the dataset should be tested for normality using the Shapiro-Wilk test. In this case the data points were not normally distributed and therefore Spearman’s rho test was used. Spearman’s ρ test is the rank-based non-parametric equivalent of the more commonly used Pearson’s correlation calculation [41]. In a pair-wise experimental design, we also employed microarray datasets to elucidate the gene expression profiles of the adjacent-normal/tumor matched samples. Student’s t-test is inappropriate for such a design and a signed-rank test is much better because it is distribution-free and adjusts for the paired design. Based on the paired adjacent normal and tumor samples, Wilcoxon signed-rank test should be applied to identify any differentially expressed genes [42]. Specific results that needed confirmation of a correlation between the normalized microarray data and the Q-RT-PCR results were analyzed by Pearson’s and Kendall’s tau correlations [19].
164

Fig. 3. A series of potential internal controls with relatively small variance with different microarray intensity intervals.
The box plot results show the best eleven internal control candidates, all of which exhibit consistent expression intensity in the ALL microarray dataset. The following potential internal controls have the characteristics of small variance at different microarray intensity intervals and no cross-hybridization: C1orf160 and NIBP (intensity range: 5 to 10), OTUB1 and GGA1 (intensity range: 10 to 20), SFRS10 and SKP1A (intensity range: 20 to 30), ACTR3 and YWHAQ (intensity range: 30 to 40), NONO and DDX17 (intensity range: 40 to 60) and CALM2 (intensity range: large than 60). The relative expression patterns of ten well-known Q-RT-PCR internal controls, which are contained in a 23 probe sets on the HG-U133A chip, are also shown on the right for comparison. These panels of different intensity genes may be considered as alterative internal candidates for Q-RT-PCR. These are shown as #1-23 in x-axis. The detailed probe set characteristics are shown in Table 4.
6. Conclusions
The microarray experiments should conform to the MIAME (minimal information about a microarray experiment) standards of the Microarray Gene Expression Data Society (MGED; http://www.www.mged.org) no matter what kind of platform is used. There should be good agreement for the mRNA gene expression measurement between the Q-RT-PCR and DNA microarray results for genes with moderate levels of expression that have PCR primers located close to the microarray probes [43]. For more reliable validation measurements, it is necessary, firstly, to prioritize the potential internal controls for the Q-RT-PCR analyses and to study the possibility of general utilization of suitable internal controls; these should be analyzed in terms of the probe set characteristics and the genes ranked with respect to variance and IQR. Secondly, it needs to be noted that correct scoring of the correlation between microarray and Q-RT-PCR measurements remains dependent on the experimental design and the sample characteristics.
Acknowledgement
This research was supported by grants from National Science Council (NSC99-2112-M-008-012 and NSC99-2811-M-008-053 to Su LJ).
165

References
[1] R. J. Lipshutz et al.: Nat Genet Vol.21 (1999), p. 20.
[2] Y. H. Yang et al.: Nucleic Acids Res Vol. 30 (2002), e15.
[3] E. Wurmbach, T. Yuen, and S. C. Sealfon: Methods Vol. 31 (2003), p. 306.
[4] S. A. Bustin: J Mol Endocrinol Vol. 25 (2000), p. 169.
[5] J. J. Yun et al.: Nucleic Acids Res Vol. 34 (2006), e85.
[6] L. Shi et al.: Nat Biotechnol Vol. 24 (2006), p. 1151.
[7] J. Vandesompele et al.: Genome Biol Vol. 3 (2002), RESEARCH0034.
[8] S. Michiels, S. Koscielny, and C. Hill: Lancet Vol. 365 (2005), p. 488.
[9] R. Shields: Trends Genet Vol. 22 (2006), p. 65.
[10] S. O. Zakharkin et al.: BMC Bioinformatics Vol. 6 (2005), p. 214.
[11] V. M. Aris et al.: BMC Bioinformatics Vol. 5 (2004), p. 185.
[12] H. Liu et al.: Bioinformatics Vol. 23 (2007), p. 2385.
[13] J. Lu et al.: BMC Bioinformatics Vol. 8 (2007), p. 108.
[14] P. Du, W. A. Kibbe, and S. M. Lin: Biol Direct Vol. 2 (2007), p. 16.
[15] E. Hubbell, W. M. Liu, and R. Mei: Bioinformatics Vol. 18 (2002), p. 1585.
[16] R. A. Irizarry et al.: Biostatistics Vol. 4 (2003), p. 249.
[17] Z. Wu, and R. A. Irizarry: J Comput Biol Vol. 12 (2005), p. 882.
[18] C. Li, and W. H. Wong: Proc Natl Acad Sci U S A Vol. 98 (2001), p. 31.
[19] L. J. Su et al.: BMC Genomics Vol. 8 (2007), p. 140.
[20] J. L. Aerts, M. I. Gonzales, and S. L. Topalian: Biotechniques Vol. 36 (2004), p. 84.
[21] B. R. Kim et al.: Biotechnol Lett Vol. 25 (2003), p. 1869.
[22] Y. S. Lin et al.: Gene Expr Vol. 13 (2006), p. 15.
[23] T. Suzuki, P. J. Higgins, and D. R. Crawford: Biotechniques Vol. 29 (2000), p. 332.
[24] O. Thellin et al.: J Biotechnol Vol. 75 (1999), p. 291.
[25] D. G. Ginzinger: Exp Hematol Vol. 30 (2002), p. 503.
[26] J. Bereta, and M. Bereta: Biochem Biophys Res Commun Vol. 217 (1995), p. 363.
[27] P. J. Gibbs et al.: Transpl Immunol Vol. 12 (2003), p. 89.
[28] H. K. Hamalainen et al.: Anal Biochem Vol. 299 (2001), p. 63.
[29] D. J. Moore et al.: Biochim Biophys Acta Vol. 1521 (2001), p. 107.
[30] R. M. Murphy et al.: Physiol Genomics Vol. 12 (2003), p. 163.
[31] D. L. Foss, M. J. Baarsch, and M. P. Murtaugh: Anim Biotechnol Vol. 9 (1998), p. 67.
[32] Y. Gong, L. Cui, and G. Y. Minuk: Hepatology Vol. 23 (1996), p. 734.
[33] H. Yamada et al.: Biochem Biophys Res Commun Vol. 231 (1997), p. 835.
[34] H. Zhong, and J. W. Simons: Biochem Biophys Res Commun Vol. 259 (1999), p. 523.
166

[35] A. Szabo et al.: Genome Biol Vol. 5 (2004), R59.
[36] M. E. Ross et al.: Blood Vol. 102 (2003), p. 2951.
[37] M. Jung et al.: Biochemica Vol. 4 (2002), p. 9.
[38] A. C. Davison, and D. V. Hinkley: Bootstrap Methods and Their Application. (Cambridge University Press., 1997).
[39] B. Efron: Ann Stat, Vol. 7(1979), p. 1.
[40] S. N. Lahiri: Springer-Verlag, New York (2003).
[41] J. S. Morey, J. C. Ryan, and F. M. Van Dolah: Biol Proced Online Vol. 8 (2006), p. 175.
[42] E. Lehmann: Nonparametrics: Statistical Methods Based on Ranks (Holden-Day, Inc., San Francisco, 1975).
[43] W. Etienne et al.: Biotechniques Vol. 36 (2004), p. 618.
167