Embed Size (px)
Citation preview

Vol. 160, No. 1JOURNAL OF BACTERIOLOGY, OCt. 1984, p. 153-1600021-9193/84/100153-08$02.00/0Copyright © 1984, American Society for Microbiology
Cloning and Characterization of recA Genes from Proteus vulgaris,Erwinia carotovora, Shigella flexneri, and Escherichia coli B/r
SHERRY L. KEENER, KEVIN P. McNAMEE, AND KEVIN McENTEE*
Department of Biological Chemistry, University of California at Los Angeles School of Medicine, Los Angeles, California90024
Received 2 April 1984/Accepted 3 July 1984
The recA genes of Proteus vulgaris, Erwinia carotovora, Shigellaflexneri and Escherichia coli B/r have beenisolated and introduced into Escherichia coli K-12. All the heterologous genes restore resistance to killing byUV irradiation and the mutagen 4-nitroquinoline-1-oxide in RecA- E. coli K-12 hosts. Recombinationproficiency is also restored as measured by formation ofLac' recombinants from duplicated mutant lacZ genesand the ability to propagate phage A derivatives requiring host recombination functions for growth (Fec-). Thecloned heterologous genes increase the spontaneous induction of prophage in lysogens of a recA strain.Addition of mitomycin C stimulates phage production in cells carrying the E. coli B/r and S. flexneri recAgenes, but little or no stimulation is seen in cells carrying the E. carotovora and P. vulgaris recA genes. Aftertreatment with nalidixic acid, the heterologous RecA proteins are synthesized at elevated levels, a resultconsistent with their regulation by the E. coli K-12 LexA repressor. Southern hybridization and preliminaryrestriction analysis indicate divergence among the coding sequences, but antibodies prepared against the E. coliK-12 RecA protein cross-react with the heterologous enzymes, indicating structural conservation among theseproteins.
Homologous recombination has been detected in a widevariety of organisms from simple bacteriophages to complexeucaryotic cells. Among the best studied of these systems isEscherichia coli K-12 where both genetic and biocherhicalinvestigations have defined roles for several enzymes need-ed in recombination. Of these, the RecA enzyme promotesfundamental and probably early steps in the processesleading to crossing over. Recent studies with purified RecAproteins and model DNA substrates support the conclusionthat RecA protein participates in the early steps of synapse,allowing alignment of DNA molecules before exchange, instrand transfer, where there is transfer of a single-strandedsegment to a recipient duplex to form a limited heteroduplexregion between the interacting DNAs and in the extension ofthis heteroduplex region by a reaction involving the concert-ed winding and unwinding of incoming and outgoing DNAchains, respectively. The hydrolysis of ATP by RecA pro-tein is required for these events in vitro (5, 21).The recA gene performs another equally important role in
cell metabolism by controlling expression of a group ofunlinked genes that aid in recovery of cells after exposure toDNA-damaging agents. This response, termed the SOSresponse, involves genes that pa-ticipate in repair of DNAdamage, mutagenesis, and coordination of cell divisionevehts (26). More than 11 genes are involved in this responseand are transcriptionally repressed by the LexA protein (11).In vivo, derepression of the SOS genes is thought to beaccomplished by the proteolytic removal of the LexA re-pressor (9). In vitro, in the presence of ATP and polynucleo-tide cofactors, RecA protein promotes proteolysis of theLexA repressor as well as the phage A repressor (3, 10, 11).Despite its multifunctional behavior, the RecA protein is amodestly sized protein of 37,842 daltons (23).Although the RecA protein is nonessential for viability (4,
15), mutations in this gene severely disable cells with respectto genetic recombination, DNA repair, and mutability by
* Corresponding author.
chemical agents. These and other observations suggest thatthe recA gene product might be conserved in a wide varietyof procaryotes and eucaryotes. Consistent with this view arethe reports of RecA-like activities isolated from Proteusmirabilis (25), Salmonella typhimurium (20), and the lowereucaryote Ustilago maydis (8). Although each of theseenzymes promotes DNA-exchange reactions coupled toATP hydrolysis, the details of the reactions differ.Our interest in RecA protein structure and function
prompted us to seek recA-like genes from other gram-negative bacteria. In this paper, we report the isolation andcharacterization of the recA genes and their products fromProteus vulgaris, Erwinia carotovora, Shigellaflexneri, andE. coli B/r. When introduced into E. coli K-12, each of thesegenes complemented repair and regulatory defects of recAmutations. Although we found evidence for considerablerecA nucleotide sequence divergence among these bacterialstrains, the RecA proteins were more highly conserved.These results argue that the RecA protein, together withother key components of the SOS regulon, has been struc-turally and functionally conserved through evolution.
MATERIALS AND METHODSBacterial strains and plasmids. The bacterial strains and
relevant genotypes are given in Table 1. Plasmids, insertDNAs, and vectors are given in Table 2.Growth media. Cells were grown in LB liquid medium or
LB medium plus 50 mg of ampicillin per ml (LB-amp) forplasmid-containing strains; Lac' papillae were detected onlactose MacConkey medium (Difco Laboratories) containing20 g of lactose per liter and 50 mg of ampicillin per liter.Selection for ampicillin-resistant (Amp9 transformants wasperformed on LB agar containing 50 mg of ampicillin perliter. RecA+ transformants were selected on LB agar con-taining 50 mg of ampicillin per liter and 10 mg of 4-nitroquinoline-1-oxide (NQO) (Sigma Chemical Co.) perliter.
Cloning recA gene sequences in E. coli K-12. High-molecu-
153

154 KEENER, McNAMEE, AND McENTEE
TABLE 1. Bacterial strains
Strain designation Relevant genotype Source (or
E. carotovora L. HeffernonEC100
S. flexneri BS12 L. Heffernon2A
E. coli B/r UCLAP. vulgaris UCLAE. coli K-12HB101 F- hsdS20 recAl3 (12)MC1061 F- hsr zAlacX74 recA+ (1)JC14604 F- lacMS286080II A. J. Clark (27)
lacBKI dl(srl-recA)hsr
C600 recA56 F- sri (TnlO) recA56 This labDM1187A21 F- lexA(Def) This lab
d121(srl-recA) sfiSK8710 C600 recA56(pMK710) This studySK9816 C600 recA56(pMK816) This studySK2184 C600 recA56(pMK184) This studySK8815 C600 recA56(pMK815) This studySK9100 C600 recA56(pBR322) This studySK9700 C600 recA56(Yrpl2) This studySK7710 JC14604(pMK710) This studySK8816 JC14604(pMK816) This studySK1184 JC14604(pMK184) This studySK7815 JC14604(pMK815) This studySK7840 JC14604(Yrpl2) This studySK7860 JC14604(pBR322) This studySK3500 C600 recA56(pBR-E. This study
coli recA+)
aL. Heffernon, Microbiology Department, University of California at LosAngeles (UCLA); A. J. Clark, Molecular Biology Department, University ofCalifornia at Berkeley.
lar-weight chromosomal DNA was prepared from S. flex-neri, E. carotovora, P. vulgaris, and E. coli B/r essentiallyby the procedure of Marmur (14).Chromosomal DNA and pBR322 plasmid DNA were
restricted with BamHI, using conditions recommended bythe manufacturer (Bethesda Research Laboratories). Thelinearized pBR322 DNA was treated with bacterial alkalinephosphatase for 60 min at 68°C, extracted with phenol (twotimes) and with chloroform (two times), and precipitatedwith 2 volumes of cold (-20°C) absolute ethanol. Therestricted chromosomal DNA was similarly treated withphenol and chloroform and precipitated by ethanol. Restrict-ed plasmid and chromosomal DNAs were mixed with 1 to 5U of T4 DNA ligase, 10 mM dithiothreitol, and 1 mM ATP.Ligations were performed for 12 to 18 h at 12°C. The ligationmix was used to transform competent HB101 cells to Ampr.Ampr colonies were tested for NQO resistance NQOr byreplica plating to drug plates. Colonies that grew in thepresence of 10 mg of NQO per liter were recloned on LB-amp plates and plasmid DNA was isolated from these strainsby a rapid boiling procedure (7). These crude plasmid DNA
preparations were used to transform strain C600 recA56 toAmpr, and the resulting transformants were tested forgrowth in the presence of NQO by replica plating. Clonesthat grew in the presence of the DNA-damaging agent were
examined for UV resistance (UVr) by a semiquantitativesurvival test (17). Strains that were NQOr and UV'r were
grown in LB-amp medium, and the plasmid DNA was
purified by equilibrium centrifugation in CsCI gradientscontaining ethidium bromide.A second selection for RecA' clones used recA strain
JC14604, which contains two defective copies of the lac
operon. The strain is Lac-, but Lac' cells can arise byrecombination upon introduction of a functional recA gene.
Strain JC14604 was transformed with a ligation mixture, andAmpr colonies were selected on lactose MacConkey platescontaining 50 mg of ampicillin per liter. Lac' papillae were
recloned on lactose MacConkey plates and tested for resist-ance to NQO or UV or both, as described earlier.
Southern hybridizations. Genomic DNA or purified cloneswere digested with a 2- to 10-fold excess of the indicatedrestriction enzyme. Digested DNA was electrophoresed inagarose gels (0.8%), denatured in situ, and transferred tonitrocellulose as described by Maniatis et al. (12). Nick-translated DNA probes were labeled with [ox-32P]dCTP to a
specific activity of ca. 107 cpm/p.g. Nitrocellulose filterswere hybridized from 22 to 42 h at 42°C in a solutioncontaining 50% formamide, 5x SSPE (0.9 M NaCl, 5 mMEDTA, 50 mM NaPO4 [pH 7.71), 500 jig of heat denaturedcalf thymus DNA per ml, and 0.3% sodium dodecyl sulfate(SDS). Filters were washed at 37°C in 2x SSPE-0.1% SDS(60 min) and 0.lx SSPE-0.1% SDS (60 min), and autoradi-ography was performed for 24 to 72 h at -70°C with DupontCronex film and intensifying screens.Western blotting. Antibodies were raised against purified
E. coli K-12 RecA protein (2) and against purified P. vulgarisRecA protein (a gift of J. Halbrook, Department of Biologi-cal Chemistry, UCLA) in male, New Zealand white rabbitsby administering into the backs of the rabbits two injections,1 month apart, of 0.5-mg of one of the proteins plus Freundcomplete adjuvant. Approximately 1 month after the secondinjection, the rabbits were bled (10 to 20 ml), and antiserawere prepared. The anti-RecA serum was diluted 100- to250-fold and used for the Western blotting procedure asdescribed (6). Mouse anti-rabbit immunoglobulin G-peroxi-dase conjugates were obtained from Miles Laboratories, Inc.UV survival measurements. Cells were grown in LB-amp
liquid medium to a cell density of 2 x 108 to 4 x 108 cells perml, collected by centrifugation, and suspended at 108 cellsper ml in M9 medium. The suspensions were irradiated forthe indicated period of time by using a masked, germicidal,low-pressure mercury lamp (GE G8T5). After irradiation,samples were placed on ice, serially diluted, and spread onLB-amp plates.
Induction of A prophage and other bacteriological tech-niques. Plasmid-containing strains were lysogenized with A
TABLE 2. Plasmids
Designation Insert DNA Vector Reference
pMK710 E. carotovora recA region pBR322 This studypMK816 S. flexneri recA region pBR322 This studypMK184 E. coli B/r recA region Yrpl2 This studypMK815 P. vulgaris recA region pBR322 This studyYrpl2-recA430 E. coli recA430 Yrpl2 This study, 24pBR322-recA+ E. coli recA+ pBR322 This study, 12
J. BACTERIOL.
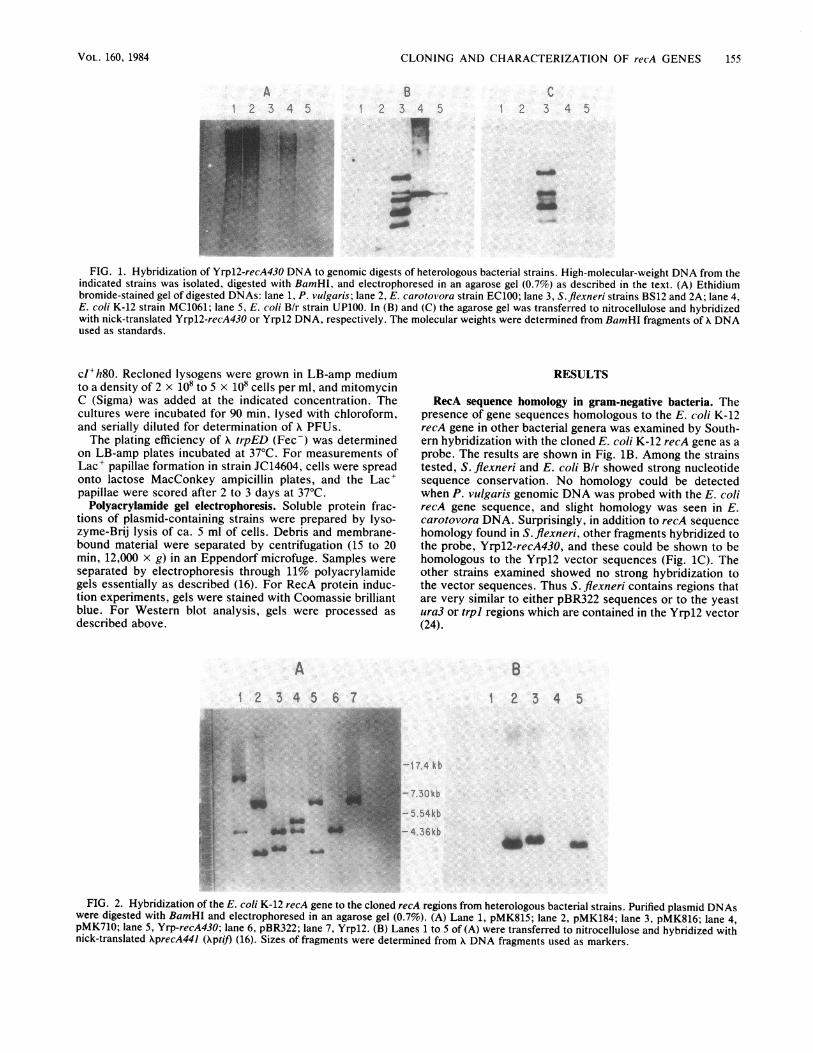
CLONING AND CHARACTERIZATION OF recA GENES 155
A1 2 3 4 5
Bl 2 3 4 5
.J0
a'o&
C1 2 3 4 5
U411
FIG. 1. Hybridization of Yrpl2-recA430 DNA to genomic digests of heterologous bacterial strains. High-molecular-weight DNA from theindicated strains was isolated, digested with BamHI, and electrophoresed in an agarose gel (0.7%) as described in the text. (A) Ethidiumbromide-stained gel of digested DNAs: lane 1, P. vulgaris; lane 2, E. carotovora strain EC100; lane 3, S. flexneri strains BS12 and 2A; lane 4,E. coli K-12 strain MC1061; lane 5, E. coli B/r strain UP100. In (B) and (C) the agarose gel was transferred to nitrocellulose and hybridizedwith nick-translated Yrpl2-recA430 or Yrpl2 DNA, respectively. The molecular weights were determined from BamHI fragments of A DNAused as standards.
cI+h80. Recloned lysogens were grown in LB-amp mediumto a density of 2 x 108 to 5 x 108 cells per ml, and mitomycinC (Sigma) was added at the indicated concentration. Thecultures were incubated for 90 min, lysed with chloroform,and serially diluted for determination of A PFUs.The plating efficiency of X trpED (Fec-) was determined
on LB-amp plates incubated at 37°C. For measurements ofLac' papillae formation in strain JC14604, cells were spreadonto lactose MacConkey ampicillin plates, and the Lac'papillae were scored after 2 to 3 days at 37°C.Polyacrylamide gel electrophoresis. Soluble protein frac-
tions of plasmid-containing strains were prepared by lyso-zyme-Brij lysis of ca. 5 ml of cells. Debris and membrane-bound material were separated by centrifugation (15 to 20min, 12,000 x g) in an Eppendorf microfuge. Samples wereseparated by electrophoresis through 11% polyacrylamidegels essentially as described (16). For RecA protein induc-tion experiments, gels were stained with Coomassie brilliantblue. For Western blot analysis, gels were processed asdescribed above.
A
I i,.~~~~~~~~~~~~~~~~~~~~~..,.
RESULTS
RecA sequence homology in gram-negative bacteria. Thepresence of gene sequences homologous to the E. coli K-12recA gene in other bacterial genera was examined by South-ern hybridization with the cloned E. coli K-12 recA gene as aprobe. The results are shown in Fig. 1B. Among the strainstested, S. flexneri and E. coli B/r showed strong nucleotidesequence conservation. No homology could be detectedwhen P. vulgaris genomic DNA was probed with the E. colirecA gene sequence, and slight homology was seen in E.carotovora DNA. Surprisingly, in addition to recA sequencehomology found in S. flexneri, other fragments hybridized tothe probe, Yrp12-recA430, and these could be shown to behomologous to the Yrp12 vector sequences (Fig. 1C). Theother strains examined showed no strong hybridization tothe vector sequences. Thus S. flexneri contains regions thatare very similar to either pBR322 sequences or to the yeastura3 or trpl regions which are contained in the Yrp12 vector(24).
B1 2 3 4 5
-1 7.4 kb
-7,30kb
- 5.54kb
- 4.36kb*dod
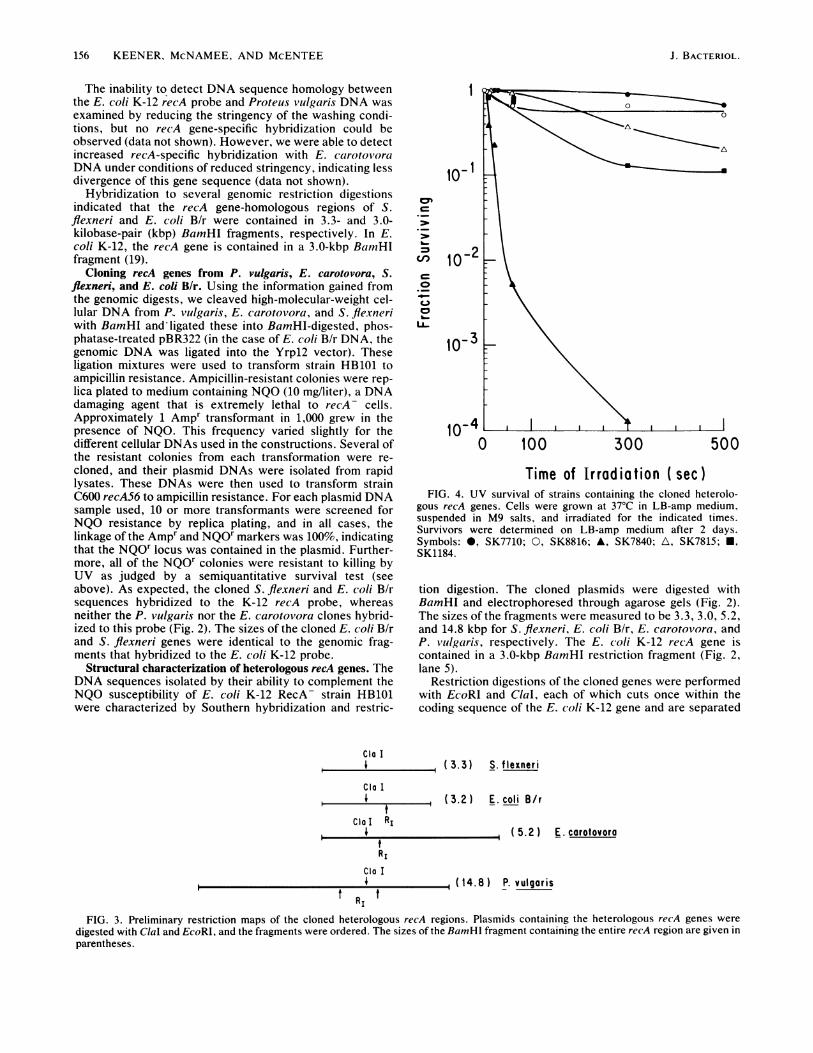
FIG. 2. Hybridization of the E. coli K-12 recA gene to the cloned recA regions from heterologous bacterial strains. Purified plasmid DNAswere digested with BamHI and electrophoresed in an agarose gel (0.7%). (A) Lane 1, pMK815; lane 2, pMK184; lane 3, pMK816; lane 4,pMK710; lane 5, Yrp-recA430; lane 6, pBR322; lane 7, Yrpl2. (B) Lanes 1 to 5 of (A) were transferred to nitrocellulose and hybridized withnick-translated XprecA441 (Aptij) (16). Sizes of fragments were determined from X DNA fragments used as markers.
1.ii...V
VOL. 160, 1984
156 KEENER, McNAMEE, AND McENTEE
The inability to detect DNA sequence homology betweenthe E. coli K-12 recA probe and Proteus vulgaris DNA wasexamined by reducing the stringency of the washing condi-tions, but no recA gene-specific hybridization could beobserved (data not shown). However, we were able to detectincreased recA-specific hybridization with E. carlotovoraDNA under conditions of reduced stringency, indicating lessdivergence of this gene sequence (data not shown).
Hybridization to several genomic restriction digestionsindicated that the recA gene-homologous regions of S.flexneri and E. coli B/r were contained in 3.3- and 3.0-kilobase-pair (kbp) BamHI fragments, respectively. In E.coli K-12, the recA gene is contained in a 3.0-kbp BamHIfragment (19).
Cloning recA genes from P. vulgaris, E. carotovora, S.flexneri, and E. coli B/r. Using the information gained fromthe genomic digests, we cleaved high-molecular-weight cel-lular DNA from P. vulgaris, E. carotovora, and S. flexneriwith BamHI and-ligated these into BamHI-digested, phos-phatase-treated pBR322 (in the case of E. coli B/r DNA, thegenomic DNA was ligated into the Yrp12 vector). Theseligation mixtures were used to transform strain HB101 toampicillin resistance. Ampicillin-resistant colonies were rep-lica plated to medium containing NQO (10 mg/liter), a DNAdamaging agent that is extremely lethal to recA- cells.Approximately 1 Ampr transformant in 1,000 grew in thepresence of NQO. This frequency varied slightly for thedifferent cellular DNAs used in the constructions. Several ofthe resistant colonies from each transformation were re-cloned, and their plasmid DNAs were isolated from rapidlysates. These DNAs were then used to transform strainC600 recA56 to ampicillin resistance. For each plasmid DNAsample used, 10 or more transformants were screened forNQO resistance by replica plating, and in all cases, thelinkage of the Ampr and NQOr markers was 100%, indicatingthat the NQOr locus was contained in the plasmid. Further-more, all of the NQOr colonies were resistant to killing byUV as judged by a semiquantitative survival test (seeabove). As expected, the cloned S. flexneri and E. coli B/rsequences hybridized to the K-12 recA probe, whereasneither the P. vulgaris nor the E. carotovora clones hybrid-ized to this probe (Fig. 2). The sizes of the cloned E. coli B/rand S. flexneri genes were identical to the genomic frag-ments that hybridized to the E. coli K-12 probe.
Structural characterization of heterologous recA genes. TheDNA sequences isolated by their ability to complement theNQO susceptibility of E. coli K-12 RecA- strain HB101were characterized by Southern hybridization and restric-
CIa II
CIa II
tClO IR
itRI
CIO Ii
t RIt
101
c
. _
10_
0
U-
0o-4 , I0 100 300 500
Time of Irradiation (sec)FIG. 4. UV survival of strains containing the cloned heterolo-
gous recA genes. Cells were grown at 37°C in LB-amp medium,suspended in M9 salts, and irradiated for the indicated times.Survivors were determined on LB-amp medium after 2 days.Symbols: 0, SK7710; 0, SK8816; A, SK7840; A, SK7815; *,SK1184.
tion digestion. The cloned plasmids were digested withBamHI and electrophoresed through agarose gels (Fig. 2).The sizes of the fragments were measured to be 3.3, 3.0, 5.2,and 14.8 kbp for S. flexneri, E. coli B/r, E. carotovora, andP. vulgaris, respectively. The E. coli K-12 recA gene iscontained in a 3.0-kbp BamHI restriction fragment (Fig. 2,lane 5).
Restriction digestions of the cloned genes were performedwith EcoRI and ClaI, each of which cuts once within thecoding sequence of the E. coli K-12 gene and are separated
( 3.3) S. flexneri
(3.2) E. coli B/r
-( 5.2 ) E. carotovora
.., (1(4.8) P. vulgoris
FIG. 3. Preliminary restriction maps of the cloned heterologous recA regions. Plasmids containing the heterologous recA genes weredigested with ClaI and EcoRl, and the fragments were ordered. The sizes of the BamHI fragment containing the entire recA region are given inparentheses.
J. BACTERIOL.
lo-3

CLONING AND CHARACTERIZATION OF recA GENES 157
by 0.5 kbp. In Fig. 3, we have aligned the preliminaryrestriction maps with respect to the ClaI site. All but the S.flexneri clone contain an EcoRI site within 0.5 kbp of thisClaI site. A second EcoRI site is found in the P. vulgarisclone ca. 0.7 kbp on the other side of the ClaI site. Althoughwe have not located the coding sequence within theseclones, it is possible that the ClaI and EcoRI restriction siteslie within the structural sequence as they do in the E. coli K-12 gene.Complementation studies with heterologous recA genes. The
ability of the cloned heterologous genes to complement theDNA repair and recombination defects of strains JC14604and C600 recA56 was determined. Quantitative UV survivalexperiments (Fig. 4) indicated that cloned genes protectedthe JC14604 cells from killing by UV irradiation to ca. thesame extent at low doses. At higher doses the K-12 strainscarrying the P. vulgaris (SK7815) and E. coli B/r (SK1184)recA regions showed somewhat less survival. In a separateexperiment, we measured the survival of strain SK3500containing the E. coli K-12 recA region cloned into pBR322.At the highest UV dose examined (500 s), survival was 38%.The recombination proficiency of the strains carrying the
heterologous genes was determined in two ways. We mea-sured the plating efficiency of a phage X mutant that is unableto grow in a recA- strain (A Fec-) (13). The phage contains asubstitution of the trpE and trpD genes for the early region ofthe X chromosome replacing the red and -y genes of thephage. The plating efficiency of this phage on recA- strainC600 recA56 is 0.01, relative to that of recA+ strain MC1061(Table 3). All of the cloned heterologous genes were capableof restoring the plating efficiency of the A trpED phage tovalues approaching those measured on the Rec+ controlstrain. Introduction of plasmids pBR322 or Yrp12 into strainC600 recA56 had no effect on the A trpED efficiency ofplating.Recombination proficiency was also determined qualita-
tively in strain JC14604. This strain contains a duplication ofthe lacZ region, each copy containing a different lacZmissense mutation. Introduction of the heterologous recAplasmids into the JC14604 background resulted in the ap-pearance of Lac' papillae at high frequency (typically morethan 10% of the cells papillated after 48 h on lactoseMacConkey medium.) No Lac' colonies appeared in controlcells containing the pBR322 or Yrp12 plasmids alone. Weconclude that the cloned genes restore recombination profi-ciency to recA mutants.The inability to induce temperate prophages either sponta-
neously or after DNA damage is another defect in recAstrains. We prepared X lysogens of plasmid-containingstrains and measured the release of phage A from cells beforeand after exposure to mitomycin C (Table 4). In the absenceof mitomycin C, the level of spontaneous phage production
TABLE 3. Efficiency of plating of A trpED (Fec-) on recAplasmid-containing strains
Strain Platingefficiency'
MC1061........................................... 1.0SK8710 ........................................... 0.64SK9816 ............................................ 0.86SK2184 ............................................ 0.76SK8815 ........................................... 0.98SK9100 ............................................ 0.03SK9700 ........................................... 0.02
a Relative to the efficiency of plating on RecA+ strain MC1061.
TABLE 4. Spontaneous and mitomycin C-stimulated induction ofA prophage in plasmid-bearing strains
Prophage induction (PFU/ml) in strains treatedwith (per ml):
StrainNo mitomycin 0.4 p.g of 1.0 ,ug of
C mitomycin C mitomycin C
SK7710 1.7 x 106 ND" 2.6 x 106SK8816 2.0 x 105 8.6 x 106 3.0 x 107SK1184 2.0 x 106 5.0 x 107 2.4 x 107SK7815 1.6 x 105 ND 1.2 x 106SK7860 0 ND 10SK7840 40 ND 10MC1061 7 x 104 3.5 x 106 8.5 x 106
a ND, Not determined.
varied over a 10-fold range, from 1.6 x 105 PFU/ml for cellscontaining the P. vulgaris recA gene (SK7815) to 2 x 106PFU/ml for the E. coli B/r recA gene-containing strain. Nospontaneous phage release was detected in the recA- hostcontaining either pBR322 or Yrpl2. Addition of mitomycin Cto the cultures increased the production of free phage instrains SK1184 and SK8816. In the former strain, maximuminduction (25-fold) was obtained at 0.4 ,ug/ml, whereas in thelatter, prophage induction was higher at 1 ,ug/ml (150-fold).Strain SK7815 showed only an eightfold increase in freephage titer, and little or no increase was observed for strainSK7710 when it was exposed to 1 ,ug of mitomycin C per ml.Thus, these cloned recA genes behave differently withrespect to derepression of prophage A after DNA-damagingtreatment.
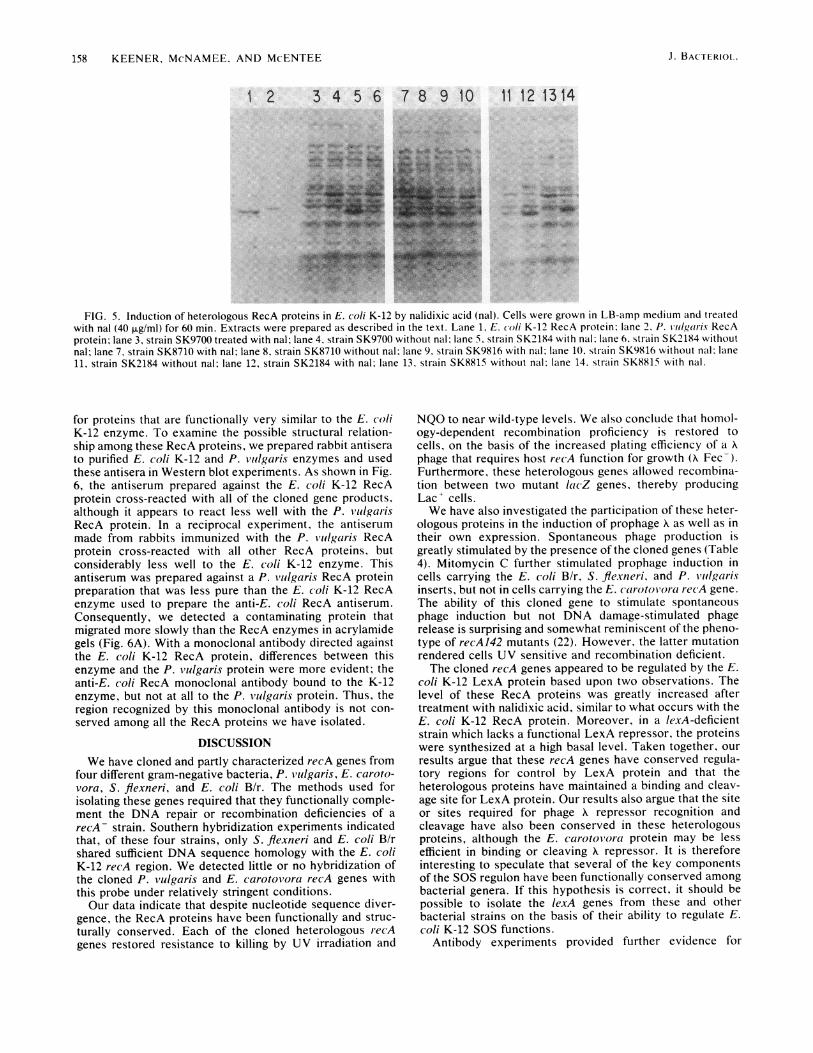
Protein production in strains carrying heterologous recAgenes. The failure to detect induction of X prophage in strainSK7710 after exposure to mitomycin C suggested that the E.carotovora RecA protein might be incapable of efficientlyrecognizing the K repressor and possibly the LexA repressorwhich regulates recA gene expression in E. coli K-12. To testthis possibility, we investigated RecA protein production inthis and other plasmid-containing strains after they weretreated with nalidixic acid. Soluble protein extracts wereprepared from control and nalidixate-treated cells, electro-phoresed in polyacrylamide gels, and stained with Coomas-sie brilliant blue (Fig. 5). For each of the strains, includingstrain SK8710, treatment with nalidixic acid greatly in-creased the amount of a protein which migrated at or nearthe position of the E. coli K-12 RecA protein marker. Theprotein produced in strain SK8815 migrated somewhat moreslowly than the E. coli K-12 enzyme, having an apparentmolecular weight of ca. 39,000. These proteins were pro-duced to the same extent in strains deleted for the chromo-somal recA gene, indicating that the induction shown in Fig.5 was due to plasmid recA gene expression (data not shown).Moreover, when these plasmids are introduced into a lexA-deficient host, lacking a functional LexA repressor (18), theheterologous proteins are made at high, constitutive levels inthe absence of damaging treatment (data not shown). Weconclude that the E. coli LexA protein functions to repressthese heterologous genes in vivo. Treatments that blockDNA replication increase expression of these heterologousgenes presumably by a mechanism similar to that of the E.coli K-12 RecA enzyme.
Structural similarities among the heterologous RecA pro-teins. Although hybridization analysis of the cloned genesreveals nucleotide sequence divergence for the P. vulgarisand E. carotovora recA regions, these genes appear to code
VOL. 160, 1984
158 KEENER, McNAMEE, AND McENTEE
1 2 3 4 5 6 7 8 9 10 112 13 14
4
FIG. 5. Induction of heterologous RecA proteins in E. coli K-12 by nalidixic acid (nal). Cells were grown in LB-amp medium and treatedwith nal (40 ,ug/ml) for 60 min. Extracts were prepared as described in the text. Lane 1, E. coli K-12 RecA protein; lane 2. P. 1'id1g0(Iris RecAprotein; lane 3, strain SK9700 treated with nal; lane 4, strain SK9700 without nal: lane 5, strain SK2184 with nal: lane 6. strain SK2184 withoutnal; lane 7, strain SK8710 with nal; lane 8, strain SK8710 without nal; lane 9. strain SK9816 with nal; lane 10. strain SK9816 without nal: lane11, strain SK2184 without nal; lane 12, strain SK2184 with nal; lane 13, strain SK8815 without nal: lane 14. strain SK8815 with nal.
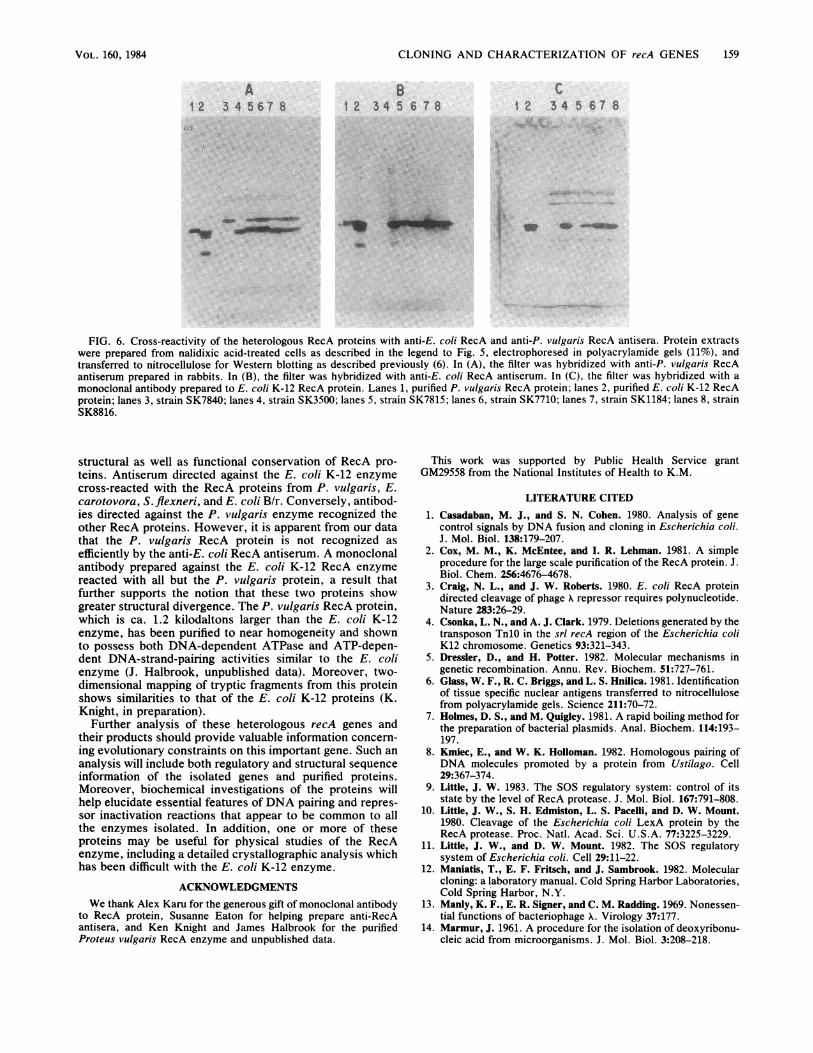
for proteins that are functionally very similar to the E. coliK-12 enzyme. To examine the possible structural relation-ship among these RecA proteins, we prepared rabbit antiserato purified E. coli K-12 and P. vl'ugaris enzymes and usedthese antisera in Western blot experiments. As shown in Fig.6, the antiserum prepared against the E. coli K-12 RecAprotein cross-reacted with all of the cloned gene products,although it appears to react less well with the P. vl'ugarisRecA protein. In a reciprocal experiment, the antiserummade from rabbits immunized with the P. 'ul/garis RecAprotein cross-reacted with all other RecA proteins, butconsiderably less well to the E. coli K-12 enzyme. Thisantiserum was prepared against a P. vl'ugaris RecA proteinpreparation that was less pure than the E. coli K-12 RecAenzyme used to prepare the anti-E. coli RecA antiserum.Consequently, we detected a contaminating protein thatmigrated more slowly than the RecA enzymes in acrylamidegels (Fig. 6A). With a monoclonal antibody directed againstthe E. coli K-12 RecA protein, differences between thisenzyme and the P. vlilgaris protein were more evident; theanti-E. coli RecA monoclonal antibody bound to the K-12enzyme, but not at all to the P. vulgaris protein. Thus, theregion recognized by this monoclonal antibody is not con-served among all the RecA proteins we have isolated.
DISCUSSIONWe have cloned and partly characterized recA genes from
four different gram-negative bacteria, P. vulgaris, E. caroto-vora, S. flexneri, and E. coli B/r. The methods used forisolating these genes required that they functionally comple-ment the DNA repair or recombination deficiencies of arecA- strain. Southern hybridization experiments indicatedthat, of these four strains, only S. flexneri and E. coli B/rshared sufficient DNA sequence homology with the E. coliK-12 recA region. We detected little or no hybridization ofthe cloned P. vulgaris and E. carotovora recA genes withthis probe under relatively stringent conditions.Our data indicate that despite nucleotide sequence diver-
gence, the RecA proteins have been functionally and struc-turally conserved. Each of the cloned heterologous recAgenes restored resistance to killing by UV irradiation and
NQO to near wild-type levels. We also conclude that homol-ogy-dependent recombination proficiency is restored tocells, on the basis of the increased plating efficiency of a Xphage that requires host recA function for growth (A Fec-).Furthermore, these heterologous genes allowed recombina-tion between two mutant 1acZ genes, thereby producingLae' cells.We have also investigated the participation of these heter-
ologous proteins in the induction of prophage X as well as intheir own expression. Spontaneous phage production isgreatly stimulated by the presence of the cloned genes (Table4). Mitomycin C further stimulated prophage induction incells carrying the E. coli B/r, S. flexneri, and P. vuli/garisinserts, but not in cells carrying the E. carotoi'ora recA gene.The ability of this cloned gene to stimulate spontaneousphage induction but not DNA damage-stimulated phagerelease is surprising and somewhat reminiscent of the pheno-type of recA142 mutants (22). However, the latter mutationrendered cells UV sensitive and recombination deficient.The cloned recA genes appeared to be regulated by the E.
coli K-12 LexA protein based upon two observations. Thelevel of these RecA proteins was greatly increased aftertreatment with nalidixic acid, similar to what occurs with theE. coli K-12 RecA protein. Moreover, in a lexA-deficientstrain which lacks a functional LexA repressor, the proteinswere synthesized at a high basal level. Taken together, ourresults argue that these recA genes have conserved regula-tory regions for control by LexA protein and that theheterologous proteins have maintained a binding and cleav-age site for LexA protein. Our results also argue that the siteor sites required for phage X repressor recognition andcleavage have also been conserved in these heterologousproteins, although the E. carotov'ora protein may be lessefficient in binding or cleaving X repressor. It is thereforeinteresting to speculate that several of the key componentsof the SOS regulon have been functionally conserved amongbacterial genera. If this hypothesis is correct, it should bepossible to isolate the lexA genes from these and otherbacterial strains on the basis of their ability to regulate E.coli K-12 SOS functions.Antibody experiments provided further evidence for
J BAC1TERIOIL.
CLONING AND CHARACTERIZATION OF recA GENES 159
A12 345678
*_-
B2 34 5 6 78
*:
C1 2 34 5 67 8
U
~~~~~~~~~~~~~-.~~~~~~~~~~~~~~~~~~~~~~
FIG. 6. Cross-reactivity of the heterologous RecA proteins with anti-E. coli RecA and anti-P. vulgaris RecA antisera. Protein extractswere prepared from nalidixic acid-treated cells as described in the legend to Fig. 5, electrophoresed in polyacrylamide gels (11%), andtransferred to nitrocellulose for Western blotting as described previously (6). In (A), the filter was hybridized with anti-P. vulgaris RecAantiserum prepared in rabbits. In (B), the filter was hybridized with anti-E. coli RecA antiserum. In (C), the filter was hybridized with amonoclonal antibody prepared to E. coli K-12 RecA protein. Lanes 1, purified P. vulgaris RecA protein; lanes 2, purified E. coli K-12 RecAprotein; lanes 3, strain SK7840; lanes 4, strain SK3500; lanes 5, strain SK7815; lanes 6, strain SK7710; lanes 7, strain SK1184; lanes 8, strainSK8816.
structural as well as functional conservation of RecA pro-teins. Antiserum directed against the E. coli K-12 enzymecross-reacted with the RecA proteins from P. vulgaris, E.carotovora, S. flexneri, and E. coli B/r. Conversely, antibod-ies directed against the P. vulgaris enzyme recognized theother RecA proteins. However, it is apparent from our datathat the P. vulgaris RecA protein is not recognized asefficiently by the anti-E. coli RecA antiserum. A monoclonalantibody prepared against the E. coli K-12 RecA enzymereacted with all but the P. vulgaris protein, a result thatfurther supports the notion that these two proteins showgreater structural divergence. The P. vulgaris RecA protein,which is ca. 1.2 kilodaltons larger than the E. coli K-12enzyme, has been purified to near homogeneity and shownto possess both DNA-dependent ATPase and ATP-depen-dent DNA-strand-pairing activities similar to the E. colienzyme (J. Halbrook, unpublished data). Moreover, two-dimensional mapping of tryptic fragments from this proteinshows similarities to that of the E. coli K-12 proteins (K.Knight, in preparation).Further analysis of these heterologous recA genes and
their products should provide valuable information concern-ing evolutionary constraints on this important gene. Such ananalysis will include both regulatory and structural sequenceinformation of the isolated genes and purified proteins.Moreover, biochemical investigations of the proteins willhelp elucidate essential features of DNA pairing and repres-sor inactivation reactions that appear to be common to allthe enzymes isolated. In addition, one or more of theseproteins may be useful for physical studies of the RecAenzyme, including a detailed crystallographic analysis whichhas been difficult with the E. coli K-12 enzyme.
ACKNOWLEDGMENTSWe thank Alex Karu for the generous gift of monoclonal antibody
to RecA protein, Susanne Eaton for helping prepare anti-RecAantisera, and Ken Knight and James Halbrook for the purifiedProteus vulgaris RecA enzyme and unpublished data.
This work was supported by Public Health Service grantGM29558 from the National Institutes of Health to K.M.
LITERATURE CITED1. Casadaban, M. J., and S. N. Cohen. 1980. Analysis of gene
control signals by DNA fusion and cloning in Escherichia coli.J. Mol. Biol. 138:179-207.
2. Cox, M. M., K. McEntee, and I. R. Lehman. 1981. A simpleprocedure for the large scale purification of the RecA protein. J.Biol. Chem. 256:4676-4678.
3. Craig, N. L., and J. W. Roberts. 1980. E. coli RecA proteindirected cleavage of phage A repressor requires polynucleotide.Nature 283:26-29.
4. Csonka, L. N., and A. J. Clark. 1979. Deletions generated by thetransposon TnlO in the srl recA region of the Escherichia coliK12 chromosome. Genetics 93:321-343.
5. Dressier, D., and H. Potter. 1982. Molecular mechanisms ingenetic recombination. Annu. Rev. Biochem. 51:727-761.
6. Glass, W. F., R. C. Briggs, and L. S. Hnilica. 1981. Identificationof tissue specific nuclear antigens transferred to nitrocellulosefrom polyacrylamide gels. Science 211:70-72.
7. Holmes, D. S., and M. Quigley. 1981. A rapid boiling method forthe preparation of bacterial plasmids. Anal. Biochem. 114:193-197.
8. Kmiec, E., and W. K. Holloman. 1982. Homologous pairing ofDNA molecules promoted by a protein from Ustilago. Cell29:367-374.
9. Little, J. W. 1983. The SOS regulatory system: control of itsstate by the level of RecA protease. J. Mol. Biol. 167:791-808.
10. Little, J. W., S. H. Edmiston, L. S. Pacelli, and D. W. Mount.1980. Cleavage of the Escherichia coli LexA protein by theRecA protease. Proc. Natl. Acad. Sci. U.S.A. 77:3225-3229.
11. Little, J. W., and D. W. Mount. 1982. The SOS regulatorysystem of Escherichia coli. Cell 29:11-22.
12. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecularcloning: a laboratory manual. Cold Spring Harbor Laboratories,Cold Spring Harbor, N.Y.
13. Manly, K. F., E. R. Signer, and C. M. Radding. 1969. Nonessen-tial functions of bacteriophage X. Virology 37:177.
14. Marmur, J. 1961. A procedure for the isolation of deoxyribonu-cleic acid from microorganisms. J. Mol. Biol. 3:208-218.
VOL. 160, 1984

160 KEENER, McNAMEE, AND McENTEE
15. McEntee, K. 1977. Genetic analysis of the Escherichia coli K-12srl region. J. Bacteriol. 132:904-911.
16. McEntee, K. 1977. Protein X is the product of the recA gene ofEscherichia coli. Proc. Natl. Acad. Sci. U.S.A. 74:5275-5279.
17. McEntee, K. 1983. Cloning and expression of the E. coli recAgene, p. 285-293. In E. C. Friedberg and P. C. Hanawalt (ed.),DNA repair: a laboratory manual of research procedures, vol.II. Marcel Dekker, New York.
18. Mount, D. W. 1977. A mutant of Escherichia coli showingconstitutive expression of the lysogenic induction and error-
prone DNA repair pathways. Proc. Nat]. Acad. Sci. U.S.A.74:300-304.
19. Ogawa, T., H. Wabiko, T. Tsurimoto, T. Herii, H. Masukatu,and H. Ogawa. 1979. Characteristics of purified RecA proteinand the regulation of its synthesis in vivo. Cold Spring HarborSymp. Quant. Biol. 43:909-915.
20. Pierre, A., and C. Paoletti. 1983. Purification and characteriza-tion of RecA protein from Salmonella typhimurium. J. Biol.Chem. 258:2870-2874.
21. Radding, C. M. 1982. Homologous pairing and strand exchange
in genetic recombination. Annu. Rev. Genet. 16:405-437.22. Roberts, J. W., and C. W. Roberts. 1975. Proteolytic cleavage of
bacteriophage lambda repressor in induction. Proc. Natl. Acad.Sci. U.S.A. 72:147-151.
23. Sancar, A., C. Stachelek, W. Koningsberg, and W. D. Rupp.1980. Sequences of the recA gene and protein. Proc. Natl. Acad.Sci. U.S.A. 77:2611-2615.
24. Struhl, K., D. T. Stinchcomb, S. Scherer, and R. W. Davis. 1979.High frequency transformation of yeast: autonomous replica-tion of hybrid DNA molecules. Proc. Nat]. Acad. Sci. U.S.A.76:1035-1039.
25. West, S. C., J. K. Countryman, and P. Howard-Flanders. 1983.Purification and properties of the RecA protein from Proteusmirabilis. J. Biol. Chem. 258:4648-4654.
26. Witkin, E. M. 1976. Ultraviolet mutagenesis and inducible DNArepair in Escherichia coli. Bacteriol. Rev. 40:869-907.
27. Zieg, J., V. F. Maples, and S. R. Kushner. 1978. Recombinationlevels of Escherichia coli K-12 mutants deficient in variousreplication, recombination, or repair genes. J. Bacteriol.134:958-966.
J. BACTERIOL.





























