Embed Size (px)
Citation preview

Chapter 4 Experimental
4. 1 Preparation of Polymer Supports and Functionalization
4. 1. a. Materials and methods
The polymeric supports under study were synthesized and
characterized in the laboratory. IR spectra were recorded on a Shimadzu
IR-470 spectrophotometer using KBr pellets. I3c CP/MAS NMR was
recorded at 75.47 MHz on a Bruker 300 MSL CP/MAS instrument at the
Sophisticated Instrumentation Facility, Indian Institute of Science,
Bangalore.
4. 1. b. Source of chemicals
Styrene, TTEGDA and Polyvinyl alcohol (mol. wt. 72,000) were
purchased from Sigma Chemical Company, USA. Benzoyl peroxide was
obtained from SISCO, Bombay and was recrystallised before use.

4. I . c. Polymer synthesis
(i) Preparation of 2% TTEGDA-crosslinked polystyrene by suspension polymerization
Styrene was destabilized by washing with 1% sodium hydroxide
solution (20 ml x3) and then washed with distilled water (20 ml x 3). It
was then dried over anhydrous calcium chloride. A 1% solution of
polyvinyl alcohol (mol. wt. 72,000; 3.48 g) in water (348 ml) was prepared
and kept mechanically stirred in a cylindrical polymerization vessel under
nitrogen atmosphere on a water bath at 80°C. A mixture of styrene (22.46
ml; 98 mmol), TTEGDA (1.089 ml; 2 mmol), toluene (20 ml) as inert
diluent and benzoyl peroxide (I g) was prepared and flushed with nitrogen
gas. This mixture was then added to the PVA solution kept at 80°C with
stirring. The polymerization was allowed to proceed for 6 h. The white
shiny beads obtained were collected by filtration and washed thoroughly
with hot water, acetone (30 ml; 3 x 3 min), melhanol(30 rnl; 3 x 3 min) and
drained. The product resin was soxhleted using acetone to remove all the
low molecular weight impurities and linear polymers and dried in the oven
at 80°C. Yield 80%. Beads were sieved into different sizes using standard
sieves. Polymers of different TTEGDA crosslinks were prepared by
adjusting the relative amounts of the monomers.
4. 1. d. Functionalization of TTEGDA-crosslinked polystyrene support to incorporate chloromcthyl groups 222: Gcncral procedure.
The resin beads (200-400 mesh size; 2 g) were allowed to swell in
dry dichloromethane (20 ml) in a round bottomed flask. Chloromethyl
methylether (12 ml) and a solution of anhydrous zinc chloride in THF (1M;
0.4 ml) was added to the swollen resin under anhydrous conditions slowly
with shaking. The mixture was refluxed at 50°C with calcium chloride
guard tube for 4h. It was then cooled and filtered through a sintered glass

funnel (G-2), washed with THF (30 ml; 3 x 3 min), THFl4N HCI (30 ml; 3
x 3 min), THFIwater (30 ml; 3 x 3 min), water (till the filtrate was free of
chloride ions), finally with methanol (30 ml; 3 x 3 min) and drained. It was
soxhleted with THF and dried in oven.
(i) Preparation of chloromethyl methyl ether 223
To a mixture of formaldehyde (66 ml) and methanol (33 ml), kept
at O°C, a constant stream of dry HCI gas was passed. The formation of
ether was indicated by the appearance of oily layer, after one hour.
Administration of HCI was continued for half an hour more till the ether
was separated clearly &om the aqueous phase. The oily layer was separated
and dried over calcium chloride. Yield: 40 ml. This was used without
further purification.
(ii) Preparation of IManhydrous ZnCl2 in THF
Anhydrous zinc chloride (1.5 g) was placed in a 25 ml Erlenmeyer
flask and concentrated HC1 (3 drops) and distilled water (5 drops) were
added and the contents stirred and heated until the solid dissolved
completely. Tcmpcrature was gradually raised to cvaporatc thc watcr and
to leave a crust of solid which was then melted by stronger heating. When
the zinc chloride became a clear mobile liquid with no further evolution of
bubbles, the flask was placed in a dessicator and allowed to cool. The
resulting mass was dissolved in freshly distilled THF (10 ml). The exact
concentration of the resulting solution was determined by pipetting a sample
in to water containing several drops of nitric acid and titrating against 0.1M
silver nitrate solution.

(iii) Estimation of chlorine capacity by pyridine fusion method:224 General procedure
The chloromethyl resin (200 mg) was fused with pyridine (5 ml) in
a boiling tube at 1 10°C for 5 h. The resin was quantitatively transferred
with acetic acidlwater (1:1), diluted with water (25 ml). To this, silver
nitrate solution (O.lN, 10 ml) was added after acidifying with concentrated
nitric acid (5 ml) and titrated against ammonium thiocyanate solution
(0.1M) using ferric alum as indicator (modified Volhard's method).225 A
blank was also performed.
4. 2. Peptide Synthesis
4. 2. a. Source of chemicals
All side chain protected L-amino acids, tertiary butyl carbazate,
dicy clohexylcarbodiimide, 2(t-butyloxycarbonyloximino 2-phenyl
acetonitrile) (Boc-ON), trifluoroacetic acid, thioanisole, 1,2-ethanedithiol
and cesium carbonatc were purchased from Sigma Chcmical Company,
USA. Boc-Gly, Boc-Ala, Boc-Leu, Boc-Ile and Boc-Val were prepared in
the laboratory following Schnabel's procedure and Boc-Phe by Boc-ON bo:, .,
I 11'
method. All solvents were of reagent grade and were obtained from E.
Merck, India. They were purified following literature procedures.
4. 2. b. Purification of reageGs and solvents
All the solvents used for peptide synthesis were purified before use.
Dichloromethane was dried by adding fused calcium chloride and kept
overnight.
Diethylether was dried over fused calcium chloride overnight.
Ethanol was double distilled and stored in airtight bottles.

Diisopropyl ethylamine was refluxed over ninhydrin for lh, distilled and
stored in amber coloured bottles.
Commercial ethyl acetate was distilled and used for extraction purposes.
Tetrahydrofuran (THF) was freed from peroxide by shaking with basic
alumina, dried using sodium till the water was removed, distilled and stored
in dark coloured bottles.
4.2. c. Physical measurements
An LKB BROMA high performance liquid chromatograph with
C18 reverse phase column (preparative) was used for the purification of
peptides. For checking the fractions, a Shimadzu C-R6A liquid
chromatograph with C18 reverse phase column (analytical) was used.
NMR studies were carried out on a Bxvker AMX-400 spectrometer. CD
spectra were recorded on a JASCO-J-500 spectropolarimeter attached with
a Jasco DP-501 N data processor using 1 mm path length cells. The mass
spectra were recorded on a Kratos PC-Kompact MALDI spectrometer and
Hcwlctt Packard Elcctrospray ionization spcctromctcr. Thc absorbing
matrix used was a-cyano-4-hydroxy cinnanlic acid.
4. 2. d. Detection
TLC was used to monitor the progress of the reaction and to check
the purity of the final product. TLC was performed on glass plates pre-
coated with silica gel containing calcium sulphate binder and activated by
heating for 6 h at 1 OO°C and cooled just before use.
4.2. e. Identification of the peptides on TLC
This is an inexpensive technique and is useful for detecting small
and medium range peptides. The methanol solution of the peptide is spotted

on the plate and developed in a suitable solvent of appropriate composition.
The solvent systems used include
Butanol-1 : acetic acid: water:pyridine (4: 1 : 1 : 1)
Butanol-1 : acetic acid: water (4:1:5)
Butanol-1: formic acid: water (10:3:8)
Chloroform: methanol: acetic acid (8.5: 1:0.5)
Methanol: chloroform (1 :9)
4. 2. f. Visualization
The developed chromatogram was visualized by the following
methods:
Ninhydrin spray:
Ninhydrin spray detects the presence of free amino groups. A 0.2%
pure ninhydrin in acetone was sprayed on the developed chromatogram and
heated in an air oven at 80-100°C for 10 min. Pink colour was developed
by free amino groups. N-terminal proline gives an yellow colour.
Iodine vapour.
The TLC plates were exposed to iodine vapours in a closed
chamber. Brown spots of amino acids and peptides were obtained.
4. 2. g. Preparation of reagents and amino acid derivatives
(4 Preparation of Boc-azidefrom t-butyl carbazate 226
Boc-azide was prepared from t-butyl carbazate following the
method of Carpino and coworkers. Tertiary butyl carbazate (20 g) was
dissolved in a mixture of glacial acetic acid (17.6 ml) and water (25 rnl).
Sodium nitrite (11.6 g) was added in small portions in about 15 min.

During the addition, the solution was stirred vigorously and maintained at
0°C. After 90 min, the oily layer was separated kom the aqueous layer.
The aqueous layer was extracted with ether (10 ml x 3). The ether extracts
were mixed with the oily layer, washed with water, 1 M sodium bicarbonate
(NaHC03) and dried over sodium sulphate. On evaporating ether under
reduced pressure, Boc-azide was obtained as a golden yellow liquid. It was
stored under refrigeration and used directly without purification. Yield: 20
ml.
(ii) Synthesis of Boc-amino acids: Schnabel's General procedure
The amino acid was suspended in dioxanelwater (1: 1) mixture and
Boc-azide was added to it. The mixture was stirred at room temperature,
maintained the pI~I in the alkaline range using 4N sodium hydroxide. Water
(1 5 ml) was added to the solution after 24 h and extracted with ether. The
aqueous layer was cooled in an ice-bath, acidified with 2N HC1, saturated
with sodium chloride and extracted with ethylacetate. The organic layer
was dried over anhydrous sodium sulphate and rotary evaporated to get the
Boc-amino acid. In most cases, the Boc-amino acid was precipitated by
adding dry petroleum ether followed by trituration. In certain cases,
seeding by a trace of fresh Boc-amino acid followed by trituration
precipitated the Boc-amino acid as a white powder.
In the case of leucine, the acidified aqueous layer was extracted
with ether. Purity of Boc-amino acid was tested on precoated TLC plate.
Solvent for the development of the chromatogram includes a mixture of
chloroform, methanol and glacial acetic acid in the ratio 85:10:5 (vlv). The
chromatogram was visualized by ninhydrin spray which gave a pink spot.

(iii) Preparation of Boc-glycine
Glycine (1.5 g; 20 rnmol) was suspended in 1:l dioxane-water
mixture (20 ml) and Boc-azide (3.2 ml; 20 rnmol) was added to it. The
mixture was stirred at room temperature maintaining the pH in the alkaline
range with 4 N sodium hydroxide solution. After 24 h, water (I5 ml) was
added and the solution was extracted with ether (I0 mi). The aqueous layer
was cooled in an ice-bath, acidified with 2 N HCl, saturated with NaCl and
extracted with ethyl acetate (20 ml x 3). It was dried over anhydrous
sodium sulphate and ethyl acetate was removed by rotary evaporation.
Petroleum ether was added in excess, seeded with a trace of fresh and pure
Boe-Gly to induce precipitation and triturated. The white powder of Boc-
Gly precipitate was washed with fresh petroleum ether and vacuum dried.
Yield: 1.2 g (80%)
Boc-Ala, Boc-Leu, Boc-Ile, Boc-Val, Boc-Pro were prepared following
Schnabel's method.
Table 4. 1 Preparation of Boc-amino acids

(iv) Boc-ON method: 228 General procedure
Amino acid (10 mmol), 4-tertiary butyloxycarbonyl oximino 2-phenyl
acetonitrile (Boc-ON) (2.71 g; 11 mmol) and triethylarnine (2.1 ml; 15
rnmol) in 50% aqueous dioxane (12 ml) were stirred at room temperature
for 12 h. The reaction mixture was diluted with water (20 ml) and washed
with ethylacetate (15 ml x 3). The aqueous layer was cooled to O°C,
acidified with 1N HCI and extracted with ethyl acetate (15 ml x 3). The
organic layer was dried over anhydrous sodium sulphate and rotary
evaporated to remove ethyl acetate. On adding petroleum ether, the Boc-
amino acid was obtained. All Boc-amino acid preparations were obtained
with 80-90 %yield.
(v) Preparation of Boc-Phe
Phenylalanine (0.84 g; 5 rnmol), Boc-ON (1.35 g; 5.5 mrnol) and
TEA (1.39 ml; 5.5 mrnol) were dissolved in 1.1 dioxane-water (6 ml) and
the mixture was stirred at room temperature for 12 h. The reaction mixture
was diluted with water (10 rnl) and washed with ethyl acetate (2 x 12.5 rnl).
The aqueous layer was acidified with 2N HCI to pH 2 and it was extracted
with ethyl acetate (10 ml x 3). The organic layer was dried and rotary
evaporated to remove ethyl acetate. The white powder of Boc-Phe
precipitated was washed with petroleum ether and vacuum dried.
Yield: 0.71 g (85%); M. I? (obs.): (78°C); M. I! (lit.) 80°C.
4. 2. h. Preparation of 1-Hydroxybenzotriazole (HOBt) 229
HOBt was prepared following the procedure adopted by Konig and
Geiger. 0-chloronitrobenzene (32 g) was dissolved in ethanol (100 ml). EL I ' "
Hydrazine hydrate (30 g) was added and the solution refluxed for 5 h. After
distilling off the ethanol, the residue was diluted with water (100 ml) and

extracted with ether (20 ml x 3). The aqueous layer was acidified with
concentrated HC1,when HOBt got precipitated. It was recrystallised from
hot water. Yield: 20 g (80%); M. P: 154°C.
4.2. i. Gcncral procedure for solid phase peptide synthesis
Manual SPPS using Boc-strategy was done in a glass reaction
vessel. The first amino acid of the C-terminal portion of the peptide was
esterified to the resin via a benzyl ester linkage by the cesium salt of the
Boc-amino acid. Boc group was deprotected with 33% TFA in DCM and
neutralization was effected by 5% DIEA in DCM or 5% TEA in DCM.
Second Boc-amino acid was coupled to the aminoacyl resin by DCC
coupling method or by active ester procedure. Dichloromethane or N-
methyl-2-pyrrolidone (NMP) was used as the coupling medium and the
coupling time was usually lh. The same procedure was adopted for the
coupling of all amino acids. Progress of coupling was monitored at every
stage by semiquantitative ninhydrin test. In all couplings, a 2.5 fold molar
excess of the Boc-amino acid was used and double coupling was done to
ensure completion of the reaction. Final cleavage of peptide from the
support was obtained by TFA in the presence of acid scavengers.
4.2. j. Attachment of first amino acid onto the resin
(9 Gisin's cesium salt method: 230 General procedure
The Boc-amino acid (2.5 mmol) was dissolved in ethanol and
neutralized with saturated solution of cesium carbonate. Ethanol was
evaporated under vacuum and the residue was dried by co-evaporating with
benzene (20 ml x 6) as an azeotrope under vacuum until the white powdery
ccsium salt of thc Boc-amino acid was obtained. It was then dissolvcd in

minimum quantity of NMP and the resin (lg; 1 mmol) was suspended in it.
The suspension was stirred at 50-60°C in an oil bath. After 24 h, the resin
was filtered out, washed successively with NMP (20 ml; 3 x 3 rnin), water
(20 ml; 3 x 3 rnin), methanol (20 ml; 3 x 3 min) and DCM (20 ml; 3 x 3
min) and dried in vacuum.
4. 2. k. Estimation of first amino acid substitution: Picric acid test:23' General procedure
The Boc-aminoacyl resin (10 mg) was deprotected using 33% TFA
in DCM for 30 min. filtered, washed with DCM (6 times) to get rid of TFA
completely. It was then neutralized with 5% TEA in DCM for 5 min.and
again washed with DCM and dried. From the deprotected resin, exactly 5
mg was taken in a Gisin tube and treated with 0.1M picric acid (2 x 5 min).
The unbound picric acid was washed off with DCM (2 ml; 3 x 2 min). The
resin bound picrate was then carefully eluted with 5% TEA in DCM till the
washings were clear. It was then made up to a definite volume (15 ml)
using 95% ethanol. The optical density (OD) of this solution was measured
at 358 nm. From the extinction coefficient of picrate (~3~~=14,500), the OD
value and the weight of the resin taken, the substitution level of the first
amino acid was estimated.
4. 2. 1. Removal of t-butyloxycarbonyl group
'The t-butyloxy (t-Boc) protection of amino acids and peptides can
be deprotected using anhydrous TFA in DCM (33%). For deprotection, the
protected amino acid or peptide was treated with the above solution at room
temperature for 30 min. Excess TFA was removed by filtration followed by
washing with DCM and the salt thus obtained was neutralized with DIEA in
DCM (5%).

4. 2. m. Methods of activation and coupling
Mainly two methods were used for the activation of carboxyl group
by dicyclohexyl carbodiimide (DCC).
(i) Dicyclohexylcarbodiimide coupling "'
In the conventional DCC coupling method, the amino acid and
DCC were used in 1:1 ratio and they form 0-acylisourea as the active
intermediate which readily reacts with other N-protected amino acids to
give the symmetrical anhydride and dicyclohexyl urea (DCU). The
precipitated DCU was removed by washing with 33% methanol in DCM.
The extent of coupling was monitored by Kaiser reagent. If the test was
positive, a second coupling was done a3 described earlier. In some cases, a
third coupling was necessary to ensure complete reaction.
(ii) Active ester method 233
Certain difficult couplings were performed by the active ester
method. Active ester can be preformed or can be prepared in situ. In this
method, the Boc-amino acid, DCC and HOBt were used in 1 : 1 : 1 ratio to get
the active ester along with DCU. The precipitated DCU was removed by
washing with 33% methanol in DCM. This method was very usefbl for
activating Asn and Gln.
4. 2. n. Cleavage of the peptide from the resin: TFA/Thioanisole method234
The peptidyl resin (100 mg) was suspended in TFA (10 ml) and to this,
thioanisole (0.1 mi), m-cresol (0.1 rnl) and 1,2-ethanedithiol (0.1 ml) were
added. The reaction mixture was left for 20 h at room temperature. It was
filtered and the TFA solution was rotary evaporated to remove TFA. The

peptide was then precipitated by addition of ice cold ether and washed
thoroughly with ether, centrifuged ( I 0-1 5 times) and dried.
4. 2. o. Purification
(i) Thin layer chromatography (TLC)
TLC was used for estimating the purity both of starting materials
for SPPS and of synthesized short peptides. Aqueous or methanolic
solution of the peptide was spotted on the silica plate and developed in a
suitable solvent mixture of appropriate composition. The following solvent
system in indicated ratios were used.
Butanol-1 : acetic acid: water (4: 1 : 5 )
Butanol-1 : acetic acid: water: pyridine (4: 1 : 1 : 1)
Chloroform: methanol: acetic acid (8.5:1:0.5)
Methanol: chloroform (1 :9)
Butanol- 1 : formic acid: water (10:3:8)
Acetonitrile: water (3: 1)
Identification sprays
The following reagents were used to visualize the spots on the TLC
plate.
Ninhydrin spray:
The Boc group was removed by exposing the plate to vapours of
conc. IICl contained in a chamber. The spots were then developed by
spraying ninhydrin reagent and heating in an oven for 5-10 minutes. Violet
spots were obscwcd in thc case of free primary amino groups.
Chlorine gas/Starch/KI reagent (Rydon 's reagent):

This spray can be used for protected peptides not visible with
ninhydrin test. This test is given by almost all compounds containing -NH
groups. The plates were exposed to chlorine gas and sprayed with a
mixture containing equal volumes of 1% (wlv) aqueous starch and
potassium iodide solutions. Blue black spots over blue background were
observed.
Iodine:
The plates were exposed to iodine vapours in a closed chamber.
Brown spots were observed in the case of amino acids and peptides.
(ii) High performance liquid chromatography
Solid phase peptide synthesis became an established procedure only
after the introduction of efficient HPLC. It has extremely high resolving
power and is very useful in establishing the homogeneity of peptides.
HPLC analyses were done using a Shirnadzu two pump system equipped
with a controller unit. A Vydac C18 column 218 TP (particle size 5 pm)
and an injection loop of 20 p1, 0.8 mllmin flow rate and detection at 226 nm
were used.
Peptide purification was carried out on LKB HPLC system. Thc
column used was C18 (4 x 250 mm, particle size, 10 pm) and an injection
loop of 50 p1 and a flow rate of 1.5 rnllmin was used. Repetitive injections
of lmg per run were carried out and the desired fractions were collected by
the use of a LKB Superrac fraction collector. Details of gradients used
were re injected on the Shimadzu HPLC to confirm their homogeneity.
Detections were done at 226 nm.

(iii) Mass spectroscopy
Mass spectrometric analysis can be used for the accurate
determination of molecular weight of proteins, peptides and
oligonucleotides. Mass spectral interpretation helps determination of
identity of a molecule by deducing information from its mass spectrum.
ES is a soft ionization technique that allows the mass spectrornctric
analysis of large biomolecules such as polypeptides as well as non-volatile
and thermally unstable compounds. It allows deposition of multiple chargcs
on a protein without causing fragmentation of the protein. This allows
accurate mass determination down to a few daltons. In the electrospray
ionization process, a flow of sample solution is pumped through a narrow
bore metal capillary held at a potential of a few kilovolts relative to a
counter electrode. Charging of the liquid occurs and as a result, it sprays
(aerosol) fi-om the capillary orifice as a mist of very fine, charged droplets.
Solvent is striped away by a heated inert gas. This spraying process takes
place at atmospheric pressure in a specially designed chamber, outside the
vacuum region of the mass spectrometer. The whole ionization process is,
in fact a form of atmospheric pressure ionization (API).
The charged droplets upon solvent evaporation, decreases in size
until they become unstable and explode (coulomb explosion) to form a
number of small droplets. Finally, at a still smaller size, the field due to the
excess charge is large enough to cause the desorption of ionized sample
molecules from the droplet. These ions, which are field desorbed from the
droplets at atmospheric pressure, are then sampled through a series of
skimmers and lenses focus the ions into a beam. The nebulising and drying
gases are pumped away transferring the ions into the vacuum system for
mass analysis. A particular feature of the ES ionization spectra is that the

molecular ions recorded are multiple charged, (M+nH),"'in the positive-ion
mode, or (M-nH)"- in the negative-ion mode and also cover a range of
charge states. On average, one charge is added per 1000 Da in mass. ES
method offers methods for molecular weight determination of proteins and
other biopolymers with acceptable accuracy, convenient access, particularly
by insource dissociation, to some fragment ion information.
ESMS characteristics are:
Determination of mol. weights from 50 to morc than 100,000 Da with
0.02% accuracy.
Femtogram to picogram sensitivity.
Compatible with a wide range of compounds.
Compatible with a wide range of inlets such as HPLC.
MALDI is a soft ionization technique that produces (quasi)
molecular ions from large non-volatile molecules such as proteins,
oligonucleotides, polysaccharides and synthetic polymers with minimum
fragmentation. The first attempt to use organic matrices to facilitate laser
desorption and ionisation of non-volatile aminoacids and peptides was
reported by Karas, Bachmann and Hillenkamp in 1985 using nicotinic acid
as the organic matrix. Hillenkamps group further developed this approach
and produced molecular ion mass spectra of proteins with masses in excess
of 10,000 Da .
The use of organic matrices has become routine for MALDI, which
is one of the most powerhl tools for mass analysis of high molecular
weight compounds. MALDI generates high mass ions by irradiating a solid
mixture of an analyte dissolved in a suitable matrix compound with a pulsed
laser beam. The laser pulse desorbs and indirectly ionises the analyte

molecules. A short-pulse UV laser is typically used for the desorption.
Different wavelengths such as IR have been investigated recently as
alternatives. MALDI consists of two steps, sample preparation and mass
spectral analysis. The key to a successful MALDI analysis depends
primarily on uniformly mixing the matrix and the analyte. The samples are
typically prepared in a suitable solvent such as water, acetone or THF. A
few microlitres of this mixture is deposited onto a substrate and dried, and
the solid matrix is then placed into the mass spectrometer.
4. 2. p. Synthesis of model peptides
0) Synthesis of Ala-Phe-Gly
Attachment of Boc-Gly to the chloromethyl resin
Boc-Gly (83.lmg; 0.475 mmol) was dissolved in ethanol and
converted to cesium salt by adding a saturated solution of cesium carbonate
till the solution is neutral. EtOH was evaporated under pressure and water
removed by azeotropic distillation with benzene. The cesium salt was
dissolved in minimum volume of NMP and chloromethyl resin (100 mg;
0.19 mmol) was added and kept at 50°C for 48 h. The resin was washed
with NMP (20 ml x 3), 1:l NMP-water (20 ml x 3), water, methanol (20 ml
x 3), DCM (20 ml x 3) and dried in vacuum.
Synthesis of Ala-Phe-GIy
The Boc-protection on the resin was removed with 33% TFA in
DCM. The resin was washed with DCM (20 ml x 3) and then neutralized
with 5% DIEA in DCM (20 ml x 6) . The resin was washed with DCM (20

ml x 4) and NMI' (20 ml x 2). Then, Boc-Phe (100.8 mg; 0.38 mmol) atid
Boc-Ala (71.82 mg; 0.38 mmol) were coupled successively to the resin as
their HOBt active ester. Active ester was prepared by shaking the
respective Boc-amino acid with HOBt (51.3 mg; 0.38 mmol) and DCC
(78.28 mg; 0.38 mrnol) in NMP. DCU formed was filtered off and the
active ester was added to the resin. After the synthesis, the peptide resin
was washed with NMP (20 ml x 3), MeOH (20 ml x 3), DCM (20 ml x 3)
and dried in vacuum. The procedure for one synthetic cycle is given below.
1. Wash the resin with DCM (1 x 5 min)
2. Deblocking using 33% TFA in DCM (1 x 30)
3. Wash with DCM (6 x 1.5 min)
4. Prewash using 5% DIEA in DCM
5. Neutralization with 5% DIEA in DCM (I x 5 min)
6. DCM wash (4 x 1.5 min)
7. NMP wash (2 x 1.5 min)
8. Coupling of Boc-amino acid in presence of DCC and HOBt (60 min) (1:l:l)
9. DCU removal by washing with 33% methanol in DCM (3 x 2 min)
10. DCM wash (2 x 1.5 min)
11. Repetition of steps 7-10 to ensure maximum coupling, tested by Kaiser reagent.
Cleavage of the IJe~tide from the resin
50 mg of the peptidyl resin was treated with TFA (5 ml),
thioanisole (0.05 ml) and 1,2-ethanedithiol (0.05 ml) at room temperature
for 18 h. The resin was removed by filtration and TFA removed by rotary
evaporation. The peptide was precipitated by addition of ice-cold ether.

The precipitated peptide was washed several times with ether to get rid of
TFA and scavengers , dried and weight noted (1 8 mg).
(ii) Synthesis of Gly-Gly-Gly
Attachment of Boc-Gly to the chloromethvl resin
Boc-Gly (83.1 mg; 0.475 mmol) was dissolved in minimum
quantity of ethanol. Then, saturated solution of cesium carbonate is added
till the solution becomes neutral. It was kept for sometime and rotary
evaporated to remove ethanol. Water was removed by azeotropic
distillation with benzene. This was continued till a white powder of cesium
salt of Boc-Gly was obtained. It was dried under vacuum. Cesium salt of
Boc-Gly was dissolved in NMP, chloromethyl resin (100 mg; 0.19 mmol)
was added and heated at 50°C for 48 h. The resin was filtered, washed with
NMP (20 ml x 3), NMP-water (I: I; 20 ml x 3), water, methanol (20 ml x
3), DCM (20 ml x 3), drained and dried under vacuum.
Svnthesis of Gly-Gly-Glv
The deprotection of Boc-Gly on the resin was done with 33% TFA
in DCM for 30 minutes and the resin was washed with DCM (20 ml x 6)
and neutralized with 5% DIEA in DCM. The resin was washed with DCM
(20 ml x 4) and NMP (20 ml x 2). The Boc-glycines were then
successively coupled by HOBt active ester method. Boc-Gly (66.5 mg;
0.38 rnmol) was dissolved in NMP, HOBt (5 1.3 mg; 0.38 mmol) was added,
and the solution was shaken with DCC (78.28 mg; 0.38 mmol) for lh, DCU
formed was filtered off and the filtrate added to the resin. The mixture was
shaken for 60 minutes. After synthesis, peptidyl resin was washed with

NMP (20 ml x 3), MeOH (20 ml x 3), DCM (20 ml x 3) and dried in
vacuum. The procedure for one synthetic cycle is given below.
1. Wash with DCM (1 x 5 min)
2. Deblocking using 33% TFA in DCM (1 x 30)
3. Wash with DCM (6 x 1.5 min)
4. Prewash with 5% DIEA in DCM
5. Neutralization with 5% DIEA in DCM (1 x 5 min)
6. Wash with DCM (4 x 1.5 min)
7. Wash with NMP (2 x 1.5 min)
8. Coupling of Boc-amino acid in presence of DCC and 11OBt (60 min)
9. Washing off DCU with 33% methanol in DCM (3 x 2 min)
10. Wash with DCM (2 x 1.5 min)
I I . Repetition of steps 7-10 for maximum coupling, tested by Kaiser reagent.
Cleavage . of the ue~tide from the resin
50 mg of the peptidyl resin was treated with TFA (5 ml),
thioanisole (0.05 ml) and 1,2-ethanedithiol (0.05 ml) at room temperature
for 20 h. The resin was removed by filtration and TFA evaporated by
rotary evaporation. The peptide was precipitated by ice-cold ether. The
precipitated peptide was washed with ether several times to remove TFA
and scavengers, dried and weight noted (18 mg). A small amount of the
peptide was dissolved in methanol and spotted onto a TLC plate to check
the purity.

(iii) Synthesis of Gly-Ala-Ala
Attachment of Boc-Ala to the chloromethvl resin
A 2% TTEGDA-PS resin was used for this synthesis. Boc-Ala
(52.5 mg; 0.475 mmol) was dissolved in minimum volume of ethanol.
Then, saturated solution of cesium carbonate is added to make the solution
neutral. Ethanol was rotary evaporate$ water was removed by azeotropic
distillation with benzene. This was repeated to get a white powder of
cesium salt and was dried under vacuum. Cesium salt of Boc-Ala was
dissolved in NMP, chloromethyl resin (100 mg; 0.19 mmol) was added and
heated at 50°C for 48h. The resin was filtered, washed with NMP (20 ml x
3); NMP-water (20 mi x 3); water; methanol (20 ml x 3), DCM (20 ml x 3),
drained and dried under vacuum. The substitution level of Boc-Ala was
determined.
Svnthesis of Glv-Ala-Ala
Deprotection of Boc-Ala resin was done using 33% TFA in DCM
for 30 minutes. After washing with DCM (20 ml x 6), the resin was
neutralized with 5% DIEA in DCM. The rest of the amino acids were
coupled in NMP by HOBt active ester method. Active ester was prepared
by shaking the respective amino acids with HOBt (5 1.3 mg; 0.38 mmol) and
DCC (78.28; 0.38 mmol) in NMP. DCU formed was filtered off and the
active ester was added to the resin. After the synthesis, the peptide resin
was washed with NMP (20 ml x 3), MeOH (20 mi x 3), DCM (20 rnl x 3)
and dried under vacuum. The protocol for the synthesis is given below.
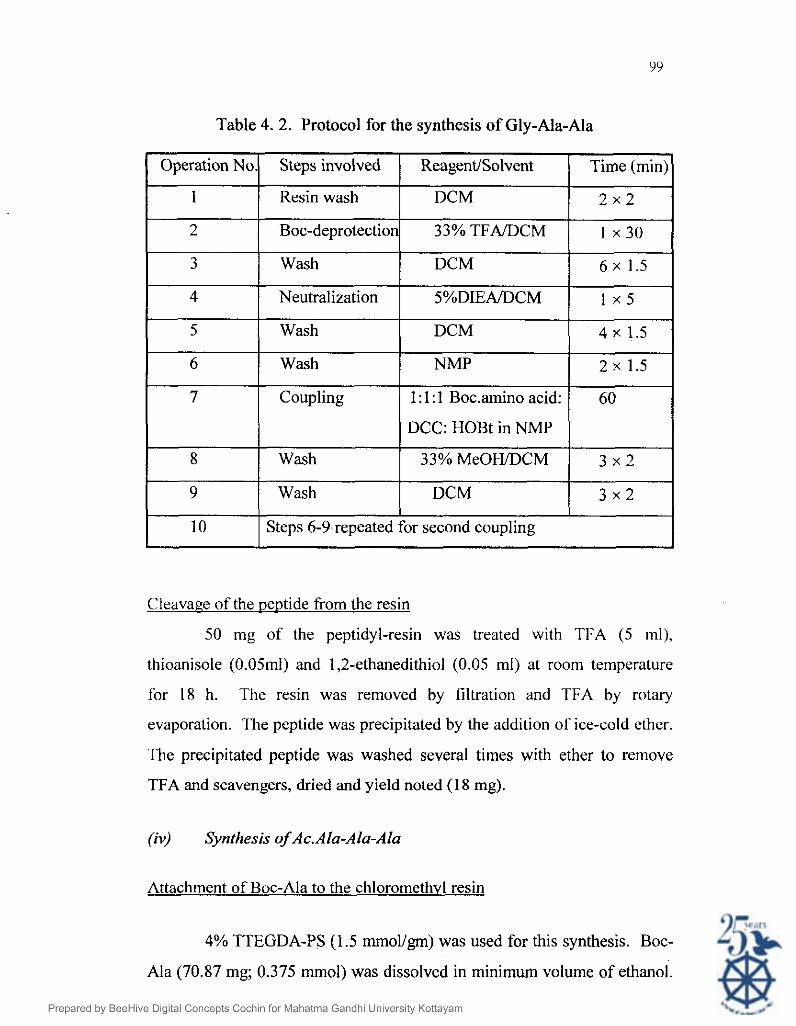
Table 4. 2. Protocol for the synthesis of Gly-Ala-Ala
Cleavage of the DeDtide from the resin
50 mg of the peptidyl-resin was treated with TFA (5 ml),
thioanisole (0.05ml) and 1,2-ethanedithiol (0.05 ml) at room temperature
for 18 h. The resin was removed by filtration and TFA by rotary
evaporation. ?'he peptide was precipitated by the addition of ice-cold ether.
'The precipitated peptide was washed several times with ether to remove
TFA and scavengers, dried and yield noted (18 mg).
DCC: HOBt in NMP
(iv) Synthesis of Ac.Ala-Ala-Ala
9
10
Attachment of Boc-Ala to the chloromethyl resin
4% TTEGDA-PS (1.5 mmollgm) was used for this synthesis. Boc-
Ala (70.87 mg; 0.375 mmol) was dissolved in minimum volume of ethanol.
Wash
Steps 6-9 repeated for second coupling
DCM 3 x 2

Then, saturated solution of cesium carbonate was added till solution became
neutral. It was stirred for sometime and rotary evaporated to remove
ethanol. Benzene was added and the azeotropic mixture was evaporated till
a white powder of cesium salt of Boc-Ala was obtained. It was dried under
vacuum. The cesium salt was dissolved in NMP, chloromethyl resin (100
mg; 0.15 mmol) was added and heated at 50°C for 48h. The resin was
filtered, washed with NMP (20 ml x 3), NMP-water (1:l; 20 ml x 3), water,
methanol (20 ml x 3) and DCM (20 ml x 3), drained and dried under
vacuum.
Synthesis of Ala-Ala-Ala
The deprotection of Boc-Ala resin was done with 33% TFA in
DCM for 30 minutes. The resin washed with DCM (20 ml x 6) and
neutralized with 5% DIEA in DCM. The resin was washed with DCM (20
ml x 4) and NMP (20 ml x 2). The remaining residues were attached by the
HOBt active ester method. Boc-Ala (56.7 mg; 0.30 mmol) was dissolved in
NMP, HOBt (40.5 mg; 0.30 mmol) was added and the solution was shaken
with DCC (61.8 mg; 0.30 mmol) for 45 minutes. DCU formed was filtered
off and the filtrate added to the resin. The mixture was shaken for 60
minutes. After synthesis, the peptidyl-resin was washed with NMP (20 ml
x 2), MeOH-DCM mixture (20 ml x 6), DCM (20 ml x 6) and dried in
vacuum. The steps involved in the synthesis are given below.
1. Wash with DCM (1 x 5 min)
2. Boc-deprotection using 33% TFA in DCM (1 x 30)
3. DCM wash (6 x 1.5 rnin)
4. Prewash with 5% DIEA in DCM
5. Neutralization with 5% DIEA in DCM (1 x 5 min)

- - 6. Wash with DCM (4 x 1.5 min)
7. NMP wash (2 x 1.5 rnin)
8. Coupling with 1 : 1 : 1 Boc-amino acid, DCC and HOBt (60 ~~ .. ~~ ~ ...
9. Washing off DCU with 33% methanol in DCM (3 x 2 min) r . .
10. DCM wash (2 x 1.5 min)
I I . Rcpcat steps 7-10 to cnsurc maximum coupling, tcstcd by Kaiscr reagent.
Acetylation of Ala-Ala-Ala resin (Acetic anhydride method)235
Suspend the peptidyl resin in DMF. Prepare an acetylating mixture
by adding 5 mmol (0.47 ml) acetic anhydride and 5 mmol (0.70 ml) TEA to
15 ml of DMF. Add the acetylating mixture to the resin suspension and
shake for 30 minutes until the Kaiser test is negative.236
Cleavarzc of the veutide from the resin
50 mg of the peptidyl-resin is treated with TFA (5 ml), thioanisole
(0.05 ml), and 1,2-ethanedithiol (0.05 ml) at room temperature for 20 h.
The resin was removed by filtration, TFA evaporated by rotary evaporation.
The peptide was precipitated by the addition of ice-cold ether. The
precipitated peptide was washed several times with ether to remove TFA
and scavengers, dried and weight noted (18 mg).The purity of the
synthesized peptide was checked by TLC.

(v) Synthesis of Asn-Ala-Gly-Ala
Attachment of Boc-Ala to the chloromethvl resin
Boc-Ala (89.75 mg; 0.475 mmol) was dissolved in ethanol and
converted to cesium salt by adding a saturated solution of cesium carbonate
till the solution becomes neutral. Ethanol was evaporated under pressure
and water removed by azeotropic distillation with benzene. The cesium salt
was dissolved in minimum volume of NMP and chloromethyl resin (100
mg; 0.19 mmol) was added and kept at 50°C for 48 h. The resin was
washed with NMP (20 ml x 3), 1:l NMP-water (20 ml x 3), water,
methanol (20 ml x 3), DCM (20 ml x 3) and dried under vacuum.
Synthesis of Asn-Ala-Gly-Ala
The Boc-Ala protection was removed by 33% TFA in DCM. The
resin was washed with DCM (20 ml x 6) and then neutralized with 5%
DIEA in DCM (20 ml x 6). The resin was washed with DCM (20 ml x 4)
and NMP (20 ml x 2). Then, Boc-Gly (66.5 mg; 0.38 rnmol), Boc-Ala
(71.82 mg; 0.38 mmol) and Boc-Asn (88.25 mg; 0.38 mmol) were
successively coupled to the resin as their HOBt active esters. Active ester
was prepared by shaking the respective Boc-amino acid with HOBt (51.3
mg; 0.38 mmol) and DCC (78.28 mg; 0.38 mmol) in NMP. DCU formed
was filtered off and the active ester was added to the resin. After the
synthesis, the peptide resin was washed with NMP (20 ml x 3), MeOH (20
ml x 3), DCM (20 ml x 3) and dried in vacuum. The steps involved in the
synthesis of Asn-Ala-Gly-Ala are shown in Table 4.3.

Table 4. 3. Synthesis of Asn-Ala-Gly-Ala
:DCC:HOBt in NMP
Cleavage of the ueutide from the resin
50 mg of the peptidyl resin was treated with TFA (5 ml),
thioanisole (0.05 ml) and 1,2-ethanedithiol (0.05 ml) at room temperature
for 18 h. The resin was removed by filtration and TFA removed by rotary
evaporation. The peptide was precipitated by addition of ice-cold ether.
The precipitated peptide was washed several times with ether to get rid of
TFA and scavengers, dried and weight noted (16 mg).

(vi) Synthesis of Val-Gly-Val-Ala-Pro-Gly
Attachment of Boc-Gly to the chloromethvl resin
A 2% TTEGDA-PS resin was used for this synthesis. Boc-Gly
(83.125 mg; 0.475 mmol) was dissolved in ethanol and converted to cesium
salt. Ethanol was evaporated under pressure and water removed by
azeotropic distillation with benzene. The cesium salt was dissolved in
minimum volume of NMP and chloromethyl resin (100 mg; 0.19 mmol)
was added and kept at 50°C for 48 h. The resin was washed with NMP (20
ml x 3), 1 : 1 NMP-water (20 ml x 3), water, MeOH (20 ml x 3), DCM (20
ml x 3) and dried under vacuum.
Synthesis of Val-Glv-Val-Ala-Pro-Gly
Boc-protection was removed with 33% TFA in DCM. The resin
was washed with DCM (20 ml x 6) and then neutralized with 5% DIEA in
DCM. The resin was washed with DCM (20 ml x 4) and NMP (20 ml x 2).
Then, Boc-Pro (81.7 mg; 0.38 mmol), Boc-Ala (71.82 mg; 0.38 mmol) ,
Boc-Val (82.56 mg; 0.38 mmol), Boc-Gly (66.5 mg; 0.38 mmol) and Boc-
Val (82.56 mg; 0.38 mmol) were coupled successively to the resin as their
HOBt active esters. Active ester was prepared by shaking the respective
Boc-amino acid with HOBt (51.3 mg; 0.38 mmol) and DCC (78.24 mg;
0.38 mmol) in NMP. DCU formed was filtered off and the active ester was
added to the resin. Atter the synthesis, the peptide resin was washed with
NMP (20 ml x 3), MeOH (20 ml x 3), DCM (20 ml x 3) and dried in
vacuum. The protocol followed for the synthesis is shown below.

105
Table 4.4. Protocol for the synthesis of Val-Gly-Val-Ala-Pro-Gly
Cleavage of the ueutide from the resin
50 mg of the peptidyl resin was treated with TFA (5 ml),
thioanisole (0.05 ml), and 1,2-ethanedithiol (0.05 ml) at room temperature
for 20 h. The resin was removed by filtration and TFA by rotary
evaporation. Then, the peptide was precipitated by the addition of ice-cold
ether . The precipitated peptide was washed with ether several times to get
rid of TFA and scavengers, dried and weight noted (19 mg).
DCC: HOBt in NMP
4. 2. q. Synthesis of hydrophobic peptides
9
10
(i, Synthesis of Ala-Cys-Leu-Phe- Val-Dpro-Gly-Leu- Val- Val-Cys-Ala
Attachment of Boc-Ala to the chloromethyl resin
Wash
Steps 6-9 repeated for second coupling
DCM 3x2

Boc-Ala (1 13.4 mg; 0.6 mmol) was dissolved in ethanol and the
cesium salt was prepared by adding a saturated solution of cesium
carbonate. Ethanol was evaporated under vacuum and water removed by
azeotropic distillation with benzene. The cesium salt was dissolved in
minimum volume of NMP and chloromethyl resin (400 mg; 0.24 mmol)
was added and kept at 50°C for 48 h. The resin was washed with NMP (40
ml x 2), 1:l NMP-water (40 ml x 3), water, methanol (40 ml x 3), DCM
(40 ml x 3) and dried under vacuum.
Synthesis of Ala-Cvs-Leu-Phe-Val-Dvro-Glv-Leu-Val-Val-Cys-Ala
The Boc-protection on the resin was removed by 33% TFA in
DCM. After neutralization with 5% DIEA in DCM, the resin was washed
with DCM (40 ml x 4) and NMP (40 ml x 2). Then, Boc-Cys.Acm (140
mg; 0.48 mmol), Boc-Val (104 mg; 0.48 mmol), Boc-Val (104 mg; 0.48
mmol), Boc-Leu (11 1.0 mg; 0.48 mmol), Boc-Gly (84 mg; 0.48 mmol),
Boc-Dpro (103.2 mg; 0.48 mmol), Boc-Val (104 mg; 0.48 mmol), Boc-Phe
(127.3 mg; 0.48 mmol), Boc-Leu (1 11.0 mg; 0.48 mmol), Boc-Cys.Acm
(140 mg; 0.48 mmol) and Boc-Ala (90.7 mg; 0.48 mmol) were successively
coupled to the resin as their HOBt active esters. Active ester was prepared
by shaking the respective Boc-amino acid with HOBt (64.8 mg; 0.48 mmol)
and DCC (98.8 mg; 0.48 mmol) in NMP. DCU formed was filtered off and
the active ester was added to the resin. After the synthesis, the peptide resin
was washed with NMP (40 ml x 3), MeOH-DCM mixture (40 ml x 3),
DCM (40 ml x 3) and dried in vacuum. The steps involved in the synthesis
are shown below.

Table 4. 5. Protocol for the synthesis of 12 residue peptide
Cleavage and purification
200 mg of the peptidyl resin was treated with TFA (20 ml),
thioanisole (0.2 ml) and 1,2-ethanedithiol (0.2 ml) at room temperature for
22 h. The resin was removed by filtration and TFA removed by rotary
evaporation. The peptide was precipitated by the addition of ice-cold ether.
The precipitated peptide was washed several times with ether to get rid of
TFA and scavengers, dried and yield noted (80 mg). A small aliquot of the
peptide dissolved in methanol was injected to a Shimadzu C-R6A liquid
chromatograph with C18 rpc and eluted using 0.1% TFA in 100% water (A)
and 0.1% TFA in 80% acetonitrile: 20% water (B) to check the purity of the
peptide. The peptide was then purified to homogeneity by C18 reversed

phase preparative HPLC. The purified peptide was analyzed by mass
spectrometry, CD and NMR. [Section 3. 2 (ii) (a)]
(ii) Synthesis of Val-Leu-Gly-Phe-Leu-Gly-Phe-Leu-Ala-Thr-Ala-Gly- Ser-Ala-Met-Gly-Ala-Ala-Ser-Leu
Attachment of Boc-Leu to the chloromethvl resin
Boc-Leu (103.95 mg; 0.45 mmol) was dissolved in ethanol and
converted to the ccsium salt by adding a saturated solution of cesium
carbonate till the solution is neutral. Ethanol was evaporated under vacuum
and water removed by azeotropic distillation with benzene. The cesium salt
was dissolved in minimum volume of NMP and chloromethyl resin (100
mg; 0.45 mmol) was added and kept at 50°C for 48 h. The resin was
washed with NMP (20 ml x 2), 1:l NMP-water (20 ml x 3), water,
methanol (20 ml x 3), DCM (20 ml x 3) and dried. The substitution level
of Boc-Leu was determined.
Synthesis of Val-Leu-Gly-Phe-I~eu-Gl~he-Leu-Ala-Thr-Ala-Glv-Ser-Ala- Met-Glv-Ala-Ala-Ser-Leu
The deblocking of Boc-Leu resin was carried with 33% TFA in
DCM. The resin was washed with DCM (20 ml x 6) and neutralized with
5% DIEA in DCM. The resin was washed with DCM (20 ml x 4) and with
NMP (20 ml x 2). Then, Boc-Ser.OBzl (106.3 mg; 0.36 mmol), Boc-Ala
(68.04 mg; 0.36 mmol), Boc-Ala (68.04 mg; 0.36 mmol), Boc-Gly (63.0
mg; 0.36 mmol), Boc-Met (89.74 mg; 0.36 mmol), Boc-Ala (68.04 mg; 0.36
mmol), Boc-Ser.OBzl (106.3 mg; 0.36 mmol), Boc-Gly (63.0 mg;0.36
mrnol), Boc-Ala (68.04 mg; 0.36 mmol), Boc-Thr.OBzl (1 11.38 mg; 0.36
mmol), Boc-Ala (68.04 mg; 0.36 mmol), Boc-Leu (83.16 mg; 0.36 mmol),
Boc-Phe (95.5 mg; 0.36 mmol), Boc-Gly (63.0 mg; 0.36 mrnol), Boc-Leu

(83.16 mg; 0.36 mmol), Boc-Phe (95.5 mg; 0.36 mmol), Boc-Gly'(63.0 mg;
0.36 mmol), Boc-Leu (83.16 mg; 0.36 mmol), and Boc-Val (78.2 mg; 0.36
mmol) were successively coupled to the resin as their HOBt active esters.
Active ester was prepared by shaking the respective Boc-amino acid with
HOBt (48.6 mg; 0.36 mmol) and DCC (74.16 mg; 0.36 mmol) in NMP.
DCU formed was filtered off and the active ester was added to the resin.
After the synthesis, the peptide resin was washed with NMP (30 mi x 3),
MeOH-DCM mixture (30 ml x 3), DCM (30 ml x 3) and dried in vacuum.
The protocol for the synthesis is given below.
Table 4. 6. Protocol for the synthesis of 20 residue peptide
mino acid in NMP
Cleavage and purification of the pe~tide
300 mg of the peptidyl resin was treated with TFA (30 ml),
thioanisole (0.3 ml) and 1,2-ethanedithiol (0.3 ml) at room temperature for

22 h. The resin was removed by filtration and TFA removed by rotary
evaporation. The peptide was precipitated by the addition of ice-cold ether.
The precipitated peptide was washed several times with ether to get rid of
TFA and scavengers, dried and yield noted (240 mg).
A small aliquot of the peptide was dissolved in acetonitrile-water
mixture and injected to a Shimadzu C-R6A liquid chromatograph with C 18
rpc and eluted using 0.1% TFA in 100% water (A) and 0.1% TFA in 80%
acetonitrile : 20% water (B) to check the purity of'the peptide. The peptide
was then purified to homogeneity by C18 reversed phase preparative HPLC.
The purified peptide was analyzed by MALDI mass spectroscopy [Section
3. 2 (ii) (b)] .
(iii) Synthesis of Glu-Thr-Thr-Ala-Leu- Val-Ala-Asp-Asn-Gly
Attachment of Boc-Gly to the chlorometh~l resin
Boc-Gly (78.75 mg; 0.45 rnmol) was dissolvcd in ethanol and
converted to the cesium salt by adding a saturated solution of cesium
carbonate till the solution is neutral. Ethanol was evaporated under vacuum
and water removed by azeotropic distillation with benzene. The cesium salt
was dissolved in minimum volume of NMP and chloromethyl resin (100
mg; 0.18 rnmol) was added and kept at 50°C for 48 h. The resin was
washed with NMP (20 ml x 2), 1:l NMP-water (20 ml x 3), water,
methanol (20 ml x 3), DCM (20 ml x 3) and dried under vacuum.
Synthesis of Glu-Thr-Thr-Ala-Leu-Val-Ala-Asv-Asn-Gly
The Boc-protection on the resin was removed by 33% TFA in
DCM. The resin was washed with DCM (20 ml x 6) and then neutralized

with 5% DIEA in DCM. The resin was washed with DCM (20 ml x 4) and
NMP (20 ml x 2). Then, Boc-Asn (104.49 mg; 0.45 mmol), Boc-Asp.OBzl
(145.48 mg; 0.45 mmol), Boc-Ala (85.05 mg; 0.45 mmol), Boc-Val (97.78
mg; 0.45 mmol), Boc-Leu (93.6 mg; 0.45 mmol), Boc-Ala (85.05 mg; 0.45
mmol), Boc-Thr.OBz1 (139.23 mg; 0.45 mmol), Boc-Thr.OBzl(139.23 mg;
0.45 mmol) and Boc-Glu.OBz1 (151.83 mg; 0.45 mmol) were successively
coupled to the resin as their HOBt active esters. Active ester was prepared
by shaking the respective Boc-amino acid with HOBt (60.75 mg; 0.45
mmol) and DCC (92.70 mg; 0.45 mmol) in NMP. DCU formed was filtered
off and the active ester was added to the resin. After the synthesis, the
peptide resin was washed with NMP (30 ml x 3), MeOH-DCM mixture (30
ml x 3), DCM (30 ml x 3) and dried in vacuum. The steps involved in the
synthesis are shown below.
Table 4. 7 . Protocol for the synthesis of 10 residue peptide

Cleavage and ~urification
200 mg of the peptidyl resin was treated with TFA (20 ml),
thioanisole (0.2 ml) and 1,2-ethanedithiol (0.2 ml) at room temperature for
22 h. The resin was removed by filtration and TFA removed by rotary
evaporation. The peptide was precipitated by the addition of ice-cold ether.
The precipitated peptide was washed several times with ether to get rid of
TFA and scavengers, dried and yield noted (130 mg). A small aliquot of the
peptide was dissolved in acetonitrile-water mixture and injected tn a
Shimadzu C-R6A liquid chromatograph with C18 rpc and eluted using 0.1%
TFA in 100% water (A) and 0.1% TFA in 80% acetonitrile: 20% water (B)
to check the purity of the peptide. The peptide was then purified to
homogeneity by C18 reversed phase preparative WLC. The purified
peptide was analyzed by MALDI mass spectra [Section 3. 2 (ii) (c)].
(iv) Synthesis of Tyr-Ala-Gly-AIa-Val-Val-Asn-Asp-Leu-Tyr-Gly-Ala- Val- Val-Asn-Asp-Leu
Attachment of Boc-Leu to the chloromethvl resin
Boc-Leu (219.45 mg; 0.95 rnmol) was dissolved in ethanol and
converted to the cesium salt by adding a saturated solution of cesium
carbonate till the solution is neutral. Ethanol was evaporated under vacuum
and water removed by azeotropic distillation with benzene. The cesium salt
was dissolved in minimum volume of NMP and chloromethyl resin (200
mg; 0.38 rnmol) was added and kept at 50°C for 48 h. The resin was
washed with NMP (30 ml x 2), 1:l NMP-water (30 ml x 3), water,
methanol (30 ml x 3), DCM (30 ml x 3) and dried under vacuum.
Synthesis of Tyr-Ala-Glv-Ala-Val-Val-Asn-~~-Leu-Tvr-Gly-Ala-Val-Val- Asn-Asv-Leu

The Boc-protection was removed by 33% TFA in DCM.
The resin was washed with DCM (30 ml x 6) and then neutralized with 5%
DIEA in DCM. The resin was washed with DCM (30 ml x 4) and NMP (30
ml x 2). Then, Boc-Asp.OBzl (310 mg; 0.95 mmol), Boc-Asn (220 mg;
0.95 mmol), Boc-Val (206 mg; 0.95 mmol), Boc-Val (206 mg; 0.95 mmol),
Boc-Ala (180 mg; 0.95 mmol), Boc-Gly (170 mg; 0.95 mmol), Boc-
Tyr.OBzl (355 mg; 0.95 mmol) Boc-Leu (219.45 mg; 0.95 mmol), Boc-
Asp.OBz1 (310 mg; 0.95 mmol), Boc-Asn (220 mg; 0.95 mmol), Boc-Val
(206 mg; 0.95 mmol), Boc-Val (206 mg; 0.95 mmol), Boc-Ala (180 mg;
0.95 rnmol), Boc-Gly (170 mg; 0.95 mmol), Boc-Ala (180 mg; 0.95 mmol)
and Boc-Tyr.OBz1 (355 mg; 0.95 mmol) were successively coupled to the
resin as their HOBt active esters. Active ester was prepared by shaking the
respective Boc-amino acid with HOBt (130 mg; 0.95mmol) and DCC (195
mg; 0.95 mmol) in NMP. DCU formed was filtered off and the active ester
was added to UIG resin. Aner the synthesis, the peplide resin was washed
with NMP (30 ml x 3), MeOH-DCM mixture (30 ml x 3), DCM (30 ml x
3) and dried in vacuum. The protocol for the synthesis is given below.
1. DCM wash (1 x 5 min)
2. Deprotection of Boc groups using 33 % TFA in DCM (1 x 30)
3. DCM wash (6 x 1.5 min)
4. Prewash with 5% DIEA in DCM
5. Neutralization with 5% DIEA in DCM ( I x 5 min)
6. Wash with DCM (4 x 1.5 min)
7. NMP wash (2 x 1.5 min)
8. Coupling of Boc-amino acid with DCC and HOBt in 1 : 1 : 1 ratio (60 min)
9. Washing off DCU with 33% methanol in DCM (3 x 2 min)
1 0. Wash with DCM (2 x 1.5 min)
I I. Repeating steps 7-1 0 to ensure maximum coupling.

Cleavage of the vevtide from the resin
250 mg of the peptidyl resin was treated with TFA (25 mi),
thioanisole (0.25 ml) and 1,2-ethanedithiol (0.25 ml) at room temperature
for 22 h. The resin was removed by filtration and TFA removed by rotary
evaporation. The peptide was precipitated by the addition of ice-cold ether.
The precipitated peptide was washed several times with ether to get rid of
TFA and scavengers, dried and yield noted (170 mg).
Transfer hydrogenation with formic acid
The protected peptide was dissolved in 98% formic acid in a 50 ml
round bottomed flask containing 10% palladium charcoal. The mixture was
continuously stirred while hydrogen gas was bubbled slowly through the
solution for 6 h. The reaction can be followed by TLC. After completion,
the catalyst is filtered off and washed with formic acid. The washings were
collected and evaporated in vacuo at room temperature. The residue
obtained was purified by IIPLC. 'l'he purified peptide was analyzed by
mass spectroscopy [Section 3. 2 (ii) (d)].
4.2.r. Synthesis of cystine peptides
(i) Synthesis of Cys-Pro-Leu-Cys-Gly-Ala
Attachment of Boc-Ala to the chloromethvlated resin
Boc-Ala (340 mg; 1.8 mmol) was attached to the chloromethylated
resin (400 mg; 0.72 mmol) by cesium salt method. The resin was washed
with NMP (40 rnl x 2); 1 : 1 NMP-water (40 rnl x 3); water, methanol (40 ml
x 3), DCM (40 rnl x 3) and dried under vacuum.

Svnthesis of Cys-Pro-Leu-Cvs-Glv-Ala
The Boc-protection on the resin was removed by 33% TFA in
DCM. The resin was washed with DCM (40 ml x 6) and then neutralized
with 5% DIEA in DCM. The resin was washed with DCM (40 ml x 4) and
NMP (40 ml x 2). Then, Boc-Gly (315 mg; 1.8 mmol), Boc-Cys.Acm (526
mg; 1.8 mmol), Boc-Leu (416 mg; 1.8 mmol), Boc-Pro (387 mg; 1.8 mmol),
Boc-Cys.Acm (526 mg; 1.8 mmol) were successively coupled to the resin as
their HOBt active esters. Active ester was prepared by shaking the
respective Boc-amino acid with HOBt (243 mg; 1.8 mmol) and DCC (370
mg; 1.8 rnrnol) in NMP. DCU formed was filtered off and the active ester
was added to the resin. After the synthesis, the peptide resin was washed
with NMP (40 ml x 3), MeOH-DCM mixture (40 ml x 3); DCM (40 ml x
3) and dried in vacuum. The protocol used for the synthesis is given below
in Table 4. 8.
Table 4. 8. Protocol for the synthesis of Cys-Pro-Leu-Cys-Gly-Ala

Cleavage and ~urification
200 mg of the peptidyl resin was treated with TFA (20 ml),
thioanisole (0.2 ml) and 1,2-ethanedithiol (0.2 ml) at room temperature for
22 h. TFA was removed by rotary evaporation. The peptide was
precipitated by the addition of ice-cold ether. The precipitated peptide was
washed several times with ether to get rid of TFA and scavengers, dried and
yield noted (90 mg). The crude peptide was purified by HPLC. A
preparative C18 rpc column was used. The solvent system used was
acctonitrilc-water containing 0.1% TFA and water containing 0.1% TFA.
The fraction corresponding to the major peak was collected and solvent
evaporated and lyophilized to get the pure peptide. Purity was further
confirmed by TLC. The purified peptide was subjected to NMR, CD and
mass spectral analysis [Section 3. 2 (iii) (a)].
Oxidation and disulfide bond formation
A mixturc of acctic acid and water in a 4: 1 ratio was prcparcd. 10
pmol (7.04 mg) of the purified bis (Acm) protected peptide was dissolved in
15 ml of the aforementioned mixture. Solid iodine (mol. wt. 253.8; 5 mg)
corresponding to 2 equiv./S-Acm function was dissolved in minimum
amount of 80% acetic acid and added to the peptide solution with vigorous
mixing at 25'C. M e r 2 h, the reaction was quenched by dilution with
water. The iodine was then removed with a 10% solution of sodium
thiosulphate and the mixture was concentrated by evaporation. It was then
purified by HPLC. The fraction corresponding to the oxidized peptide was
collected and the purity of the peptide was checked in three different
solvent systems.

(ii) Synthesis of Cys-Tyr-Zle-Gln-Asn-Cys-Pro-Leu-Gly
Attachment of Boc-Glv to the chloromethvlated resin
Boc-Gly (207 mg; 1.18 mmol) was converted to cesium salt. The
cesium salt was dissolved in minimum amount of NMP and chloromethyl
resin (250 mg; 0.475 mmol) was added and kept at 50°C for 48 h. The resin
was washed with NMP (30 ml x 3), 1:l NMP-water (30 ml x 3), water,
methanol (30 ml x 3), DCM (30 ml x 3) and dried under vacuum.
Synthesis of Cvs-Tyr-Ile-Gin-Asn-Cys-Pro-Leu-Gly
Thc Boc-group on thc rcsin was rcmovcd with 33% 'I'l:A in DCM.
The resin was washed with DCM (30 ml x 6) and then neutralized with 5%
DIEA in DCM. The resin was washed with DCM (30 ml x 4) and NMP (30
ml x 2). Then, Boc-Leu (220 mg; 0.95 rnmol), Boc-Pro (220 mg; 0.95
mmol), Boc-Cys.Acm (280 mg; 0.95 mmol), Boc-Asn (220 mg; 0.95
mmol), Boc-Gln (235 mg; 0.95 mmol), Boc-Ile (220 mg; 0.95 mmol), Boc-
'l'yr.0-Hz1 (307 mg; 0.95 mmol), Boc-Cys.Acm (280 mg; 0.95 mmol) were
successively coupled to the resin as their HOBt active esters. The active
ester was prepared by shaking the respective Boc-amino acid with HOBt
(128 mg; 0.95 rnmol) and DCC (196 mg; 0.95 mmol) in NMP. DCU
formed was filtered off and the active ester was added to the resin. After
the synthesis, the peptide-resin was washed with NMP (30 ml x 2), MeOM
(30 ml x 6), DCM (30 ml x 3) and dried under vacuum. The steps involved
in the synthesis are given below.
1. Wash with DCM (I x 5 min)
2. Deblocking using 33% TFA in DCM (1 x 30)
3. Wash with DCM (6 x 1.5 min)

4. Prewash with 5% DIEA in DCM
5. Neutralization with 5% DIEA in DCM (1 x 5 min)
6. Wash with DCM (4 x 1.5 min)
7. Wash with NMP (2 x 1.5 min)
8. Coupling of Boc-amino acid in presence of DCC and HOBt (60 min)
9. Washing off DCU with 33% methanol in DCM (3 x 2 min)
10. Wash with DCM (2 x 1.5 min)
11. Repeat steps 7-10 to for maximum coupling, tested by Kaiser reagent.
Cleavage ofthe ue~tide from the resin
300 mg of the peptidyl-resin was treated with 1'FA (30 ml),
thioanisole (0.3 ml) and l,2-ethanedithiol (0.3 ml) at room temperature for
20 h. The resin was removed by filtration and TFA by rotary evaporation.
The peptide was precipitated by adding ice-cold ether and the peptide was
washed several times with ether to remove TFA and scavengers, dried, yield
noted (145 mg).
Transfer hydrogenation with formic acid
The cleaved peptide was subjected to hydrogenation to remove the
benzyl group. A solution of the peptide in 98% formic acid was treated
with 10% palladium charcoal (5 times). The mixture was stirred
continuously while hydrogen gas was slowly bubbled through the solution
for 6 h. The mixture was filtered and the filtrate evaporated in vacuo. The
residue obtained was purified by HPLC. The purified peptide was analysed
by NMR, CD and mass spectroscopy [Section 3. 2 (iii) (b)].

Oxidation and simultaneous devrotection of Cvs-Tvr-Ile-Gln-Asn-Cvs-Pro- Leu-Gly
A mixture of acetic acid and water in a 4: 1 ratio was prepared. 10
~pmol (5.5 mg) of the purified S-Acm protected peptide was dissolved in 15
ml of the aforementioned mixture. Solid iodine (mol.wt.253.8; 2.5 mg)
corresponding to 2 equiv./S-Acm function was dissolved in minimum
volume of 80% acetic acid and added to the peptide solution with vigorous
mixing at 25OC. After 2 h, the reaction was quenched by dilution with
water. The iodine was removed by treatment with 10% solution of sodium
thiosulphate. The mixture was concentrated by evaporation. It was then
purified by HPLC and the fraction corresponding to oxidised pept~de was
collected, solvent evaporated and lyophilized. The purity was checked by
'I'LC in three different solvent systems.





























