Embed Size (px)
Citation preview

8/9/2019 CH2209 – Atomic and Molecular Spectroscopy, Quantum Chemistry
http://slidepdf.com/reader/full/ch2209-atomic-and-molecular-spectroscopy-quantum-chemistry 1/18
CH2209 – Atomic and Molecular Spectroscopy, Quantum Chemistry
Spectroscopy
Spectroscopy is the interpretation of interactions between matter and electromagnetic radiation.
There are two general types;
-absorptionCO+hν→CO*
-emission
H*+H*→H2
In both cases light, written as hν, is associated with a change in energy of an atom or molecule.
To understand spectra it is necessary to understand how atoms and molecules store and change
energy. This leads into quantum mechanics, the rules governing the behavior of electrons and nuclei.
Quantisation was first proposed by Planck, he proposed that energy comes in discrete packets
whereas before it had been believed that any energy was allowed.Planck stated that possible energies were defined by;
E=nhν
Where n is any integer, ν is the frequency (measured in Hz or s-1
) and h is Planck’s constant,
6.626x10-34 Js.
This quantisation led to the explanations of several phenomena previously inexplicable to classical
physics, for example the photoelectric effect. When firing a beam of light at a metal the frequency of
radiation and the type of metal determine the energy of the electrons emitted, at low frequencies
there is no emission no matter how intense the light beam is. Intensity is only important in
determining how many electrons are emitted. Since radiation consists of discrete packets of
radiation, photons, the kinetic energy of the emitted electrons can be defined as KE=hν-Φ where Φ
is the work function of the metal, equivalent to the ionization energy of an atom. Each photon can
be pictured as ‘knocking out’ an electron, hence more intense light means more photons and so
more electrons emitted.
Classical physics predicts that atomic spectra should be continuous, that there should be absorption
and emission at all wavelengths , however spectral absorption/emission lines are seen are very
distinct discrete wavelengths suggesting quantisation of energy levels in atoms, the lines are
transitions between these levels.
This led to a new description of the atom, Bohr proposed an alternative model in which electrons in
fixed spherical orbits surround a nucleus. Each of these orbits is denoted by its quantum number, n.
The radii of each orbit is proportional to n2, energy proportional to 1/n2 and transitions therefore

8/9/2019 CH2209 – Atomic and Molecular Spectroscopy, Quantum Chemistry
http://slidepdf.com/reader/full/ch2209-atomic-and-molecular-spectroscopy-quantum-chemistry 2/18
denoted as (1/n12-1/n2
2), these are the basis of the Rydberg formula.
Bohr’s model was able to explain all of the atomic transitions seen from atomic hydrogen however it
fails to explain the spectra of any more complex atom, e.g. He.
Davisson & Germer soon showed that electrons could be diffracted, a characteristic of a wave not a
particle. At the same time de Broglie was proposing that theoretically all particles have a wavelength
according to;
Since Davisson & Germer’s experiment agreed with this idea the notion of wave-particle duality was
born.
This was the final step in picturing the atom, electrons are not simply particles but instead have
wavelengths comparable to their orbit radii. Nuclei also have wavelengths, smaller than for electrons
as they are heavier and slower, this discovery led to the birth of quantum mechanics.
In the 1920’s, Schrodinger and Heisenberg discovered quantum mechanics. Instead of assigning a
definite position and momentum to particles, quantum mechanics takes a probabilistic view. The
average speed, position and momentum are all we can know about electrons and nuclei. The central
concept of quantum mechanics is that of wavefunction, particles act as waves and the wavefunction
describes where the waves are probabilistically likely to be found. The amplitude of the
wavefunction at a given point is related to the probability of finding the particle there, it is therefore
a probability distribution.
The Schrodinger equation is fundamental to quantum mechanics, it defines the relation between
wavefunction and the forces acting on the particle;
is the wavefunction of the particle, H is the Hamiltonian operator and E is the energy of the
particle.
The Hamiltonian describes the energy of the particle including all terms that contribute to it such as
kinetic energy and potential energy (H=T+V). Once H is known a wavefunction must be found that
obeys the Schrodinger equation. Since the Hamiltonian is an operator it performs a mathematical
operation on the wavefunction, Schrodinger adapted the classical Hamiltonian for the needs of
quantum mechanics.
In classical physics kinetic energy was defined as ½ mv2
(=p2
/2m)Schrodingers first postulate was to replace all momentum terms (mv) with ħ/i d/dx where ħ=h/2π
and i=√-1.
The second was to replace all position terms, x, with ‘multiply by x’ operators. For example for a 1d
quantum mechanical kinetic energy operator;
Or in 3d the kinetic energy operator is;

8/9/2019 CH2209 – Atomic and Molecular Spectroscopy, Quantum Chemistry
http://slidepdf.com/reader/full/ch2209-atomic-and-molecular-spectroscopy-quantum-chemistry 3/18
The Schrodinger equation for one particle of mass, m, moving in one dimension is;
The potential energy operator, V, varies. For a free particle it is 0, for a harmonic oscillator it is ½kx2
and for a hydrogen atom it is 1/r.
One of the most famous consequences of quantum mechanics is the uncertainty principle. It is
impossible to simultaneously specify both the exact position and momentum of a particle. A
quantitative form of this is given by;
Δp is the uncertainty in momentum and Δq the uncertainty in position.
The uncertainty principle shows that as a particle’s position is more precisely located, its speed or
momentum becomes less well defined, the opposite also being try. In this context position and
momentum are known as complementary variables.
Solving Schrodingers Equation
To solve Schrodingers equation a wavefunction must be found that satisfies the eigenvalue form.
This is dependent on the nature of the Hamiltonian, in particular V. For a free particle in which V=0
and so;
In this scenario any function whose 2nd derivative equals itself will be a solution. Since the second
derivative of (sin x) and (cos x) are (-sin x) and (-cos x) respectively any sine or cosine wave will form
a solution of the equation for a free particle.
The wavefunction contains all information about the system, wavefunction squared is proportional
to the probability of finding the particle at any given distance, x. Ψ2 dx will give the probability of
finding a particle between x and dx, in 3d this becomes dτ – this is the Born interpretation.
As a result of this there are several requirements for acceptable wavefunctions. Ψ must be
normalized, finite, single-valued and continuous. To normalize Ψ it is multiplied by the normalizationconstant, N. This is to ensure the overall probability of finding the electron is 1, as it should be, while

8/9/2019 CH2209 – Atomic and Molecular Spectroscopy, Quantum Chemistry
http://slidepdf.com/reader/full/ch2209-atomic-and-molecular-spectroscopy-quantum-chemistry 4/18
also preventing Ψ from having an infinite value at any point. By single-valued it is meant that there
cannot be two values of Ψ2 at any point, x. The wavefunction must be continuous, as must its first
derivative, since it is impossible to differentiate a non-continuous curve and the second derivative is
required for Ψ. It is these restrictions that give rise to quantisation of energy in quantum mechanics,
since many solutions fail these tests the energies of such solutions can never be attained – i.e. a
particle may only possess certain energies as otherwise it would have an unacceptable
wavefunction.
The Particle In a Box
A classic problem in quantum physics, the particle in a box describes an environment in which a
molecule is held within a container in one dimension, at the walls of the container the potential is
infinite, within the container the potential is equal to 0.
As well as the previously defined restrictions on Ψ there is now a boundary condition too. A sine
wave is able to obey all of these restrictions and so the wavefunction has the form;
C is the normalization constant, as such;
∫
√ ⁄
k is chosen to ensure the wave fits into the box so that at L, Csin(kL)=0 which in radians means that
kL=nπ (where n is a positive integer, since sin(nπ) in radians will always give a 0 value). Therefore;
And so the full wavefunctions are;
√ ⁄
This is confirmed as an eigenfunction by applying the Hamiltonian operator. Since V=0 in the region
of interest, only T is important;

8/9/2019 CH2209 – Atomic and Molecular Spectroscopy, Quantum Chemistry
http://slidepdf.com/reader/full/ch2209-atomic-and-molecular-spectroscopy-quantum-chemistry 5/18
The result of this operation is a number times the original function and so this is an acceptable
eigenfunction solution for this problem.
By then including the –ħ2/2m constant from the kinetic energy, T, the energies of the solutions can
be calculated;
The first three solutions therefore look like this;
Standing waves, each with the same amplitude but progressively shorter wavelengths.
The quantum number, n, is alone enough to specify the wavefunction and calculate its energy. As n
increases, energy increases as n2 and the number of nodes increases since the number of nodes is
equal to (n-1). A node is a point at which the wavefunction is zero, a point at which there is no
probability of finding the particle at that distance, as a result, in general, more nodes correlates with
higher energy.
Since n cannot be zero the minimum possible energy is defined by;
This is the zero-point energy. It is also possible to work out the difference between adjacent energy
levels, in this case defined by;
As L increases, the separation between levels decreases. If the ‘box’ is instead a conjugated alkene
and the particle a delocalized electron then as this energy separation decreases the energy of
transition between HOMO and LUMO is decreasing, eventually reaching a wavelength at which
visible light occurs, longer chain conjugation therefore leads to coloured molecules.
Solving the Schrodingers Equation For a Hydrogen Atom
The hydrogen atom is one of the simplest ‘real’ systems for which the Schrodinger equation can be
exactly solved. The same principles would apply for any one electron ion, e.g. He+, U91+ etc.
The Hamiltonian for hydrogen is;

8/9/2019 CH2209 – Atomic and Molecular Spectroscopy, Quantum Chemistry
http://slidepdf.com/reader/full/ch2209-atomic-and-molecular-spectroscopy-quantum-chemistry 6/18
H=TNuc+TEl+V
H=Nuclear Kinetic Energy+Electron Kinetic Energy+Potential Energy (≠0)
Since the only thing of interest is the motion of the electron relatively to the nucleus and not the
motion of the entire atom this can simplified to give;
H=TInt+V
Since a proton is so much heavier than an electron, μ≈meso;
H=TEl+V
This is called the infinite-nucleus approximation, the motion of the electron is independent of themotion of the entire atom. This is a valid approximation for all but the most accurate of calculations.
The potential, V, is the Coulombic attraction between the electron and nucleus;
Where Z is the nuclear charge, e the charge on an electron, ε0 the vacuum permittivity and r the
electron-nucleus distance. V is dependent only on distance and not angle since the electron has a
spherically symmetrical potential. Due to this spherical symmetry the wavefunction can be split into
radial and angular parts using polar co-ordinates; r, ϕ and θ.
As such;
Ψ(r, ϕ, θ)=R(r) Y(θ, ϕ)
These can then be solved separately.
The radial Schrodinger equation is;
*
+
The first term being the Coulomb potential, the second term the
centrifugal potential.
Radial wavefunctions have the form;
R(r)=(polynomian in r) x e-βr
As with sine waves, e-βr is an eigenfunction of the kinetic energy operator since;
()
Solutions of the radial equation depend on the angular momentum, this is expressed in the Laguerre
polynomial. If l=0, Veff is attractive at all distances, if l≠0 the centrifugal potential repels the electron
from the nucleus and balances the Coulomb potential.
The energies of these radial solutions is given by the expression;

8/9/2019 CH2209 – Atomic and Molecular Spectroscopy, Quantum Chemistry
http://slidepdf.com/reader/full/ch2209-atomic-and-molecular-spectroscopy-quantum-chemistry 7/18
The collection of constants in the first term form the Rydberg constant. The presence of 1/n2 in this
equation also explains quantisation, arrived at entirely naturally.
The radial wavefunctions of a hydrogen atom are;
The angular Schrodinger equation is satisfied by the spherical harmonic functions. The shape
depends on the orbital angular momentum quantum number, l, and the magnetic quantum
number, ml. ml can take integer values from –l to +l. Spherical harmonics gives rise to the familiar
shapes of s, p and d orbitals, l gives shape and ml direction. An atomic orbital is a one-electron
wavefunction for an electron in an atom, for one electron atoms/ions it is specified by three
quantum numbers; n, l and ml – abbreviated to Ψn,l,ml. When an electron is described by this
wavefunction it is said to occupy that orbital – the orbital is the product of the radial and angularcontributions.
n determines the energy of the electron, l and m l specify the angular momentum around the
nucleus. An electron in an orbital with a quantum number l has a total angular momentum of
magnitude [l(l+1)]1/2 ħ where l=0,1,2..n-1. An electron in an orbital with a quantum number ml has a
z-component of angular momentum equal to ml ħ where ml=0, ±1, ±2.. ±l.
Orbitals with a given value of n are said to form a shell in which they are degenerate. Within each
shell different values of l form sub-shells, each denoted by a letter;

8/9/2019 CH2209 – Atomic and Molecular Spectroscopy, Quantum Chemistry
http://slidepdf.com/reader/full/ch2209-atomic-and-molecular-spectroscopy-quantum-chemistry 8/18
l= 0 1 2 3 4 5 6
s p d f g h i
A shell with principal quantum number, n, has n sub-shells and contains a total of n2 degenerate
orbitals.
The ground-state (1s) wavefunction for hydrogen can be written as;
⁄
Where a0 is the radius of the first Bohr orbit (0.52918Å). The maximum value is found at r=0 and
decays exponentially from there, this gives a spherically symmetrical electron density distribution
with no nodes – a classical s orbital, spherical.
p-orbitals have l=1 and so a total angular momentum of √1(1+1)ħ, or √2ħ.
The direction of the angular momentum is given by ml which can take values of -1, 1 and 0.
When ml=0 there is no angular momentum about z
When ml=1 there is angular momentum about z equal to ħ (clockwise about z)
When ml=-1 there is angular momentum about z equal to –ħ (anti-clockwise about z)
po is identical to pz but p+1 and p-1 are located in the xy-plane, the familiar px and py orbitals are
combinations of p+1 and p-1;
All three contain a nodal plane, the wavefunction changes sign from one side to the other.
Spectroscopic Transitions
When an electron moves from one orbital to another, energy is lost (emission) or gained
(absorption) as a photon of energy. A photon has an angular momentum equivalent to l=1, there is a
law, the conservation of angular momentum, that means only certain transitions are allowed, givingrise to selection rules.
The selection rules for hydrogenic atoms are;
Δl=±1 (+1 for absorption, -1 for emission)
i.e p→s, d→p, s→p
Δml=0, ±1
i.e. p0→s, p1→s
As long as these rules are obeyed there is no restriction on Δn.
p0
= pz p
x p
+1 – p
-1 p
y p
+1+ p
-1

8/9/2019 CH2209 – Atomic and Molecular Spectroscopy, Quantum Chemistry
http://slidepdf.com/reader/full/ch2209-atomic-and-molecular-spectroscopy-quantum-chemistry 9/18
Allowed transitions are summarized by Grotrian diagrams;
Solving the Schrodinger Equation For Complex Atoms
The Schrodinger equation for heavier atoms is more complex than for hydrogenic atoms/ions. The
specific interactions between too electrons are too complex to solve exactly, this is the three body
problem;
H=TNuc+ΣTEl+VNuc-El+VEl
Approximations must be made. The full wavefunction for helium is a function of both electron co-
ordinates (Ψ(r1,r2)). The first approximation is that each electron occupies its own orbital, so this can
be split; Ψ(r1,r2)=Ψ(r1) Ψ(r2).
The orbitals are those from hydrogenic orbitals (1s, 2s, 2p etc.). The list of occupied orbitals in an
atom is known as its configuration, this is not necessarily the ground state. The ground state
configuration of helium is 1s2. Electrons, as fermions, have intrinsic spin – they rotate around their
own axes. Each electron has two spin states, spin-up and spin-down or α and β. The presence of two
spin states was confirmed by Stern and Gerlach. Upon firing a beam of silver atoms through an
inhomogeneous magnetic field only two spots of silver atoms were found on the detector, indicating
only two spin states. This spin is vital for multiple-electron atoms and is assigned a new spinquantum number, ms=±½ .
To determine how electrons fill up orbitals the Pauli exclusion principle states that ‘no two electrons
may have the same set of quantum numbers; n, l, m l and ms’. The two electrons in the 1s orbital of
helium are therefore separated only by their spin-state. This is key to atomic structure and helps to
explain several trends in the periodic table including why an atom of pn configuration will behave the
same as an atom ofp6-n configuration (the same for dn and d10-n) – it is this simplification that explains
why the Stern-Gerlach experiment worked and why silver atoms ([Kr]5s24d9)acted as single
electrons.
Unlike in hydrogen, s and p orbitals are not degenerate in many-electron atoms due to the screeningeffects of electrons. At some points both electrons in helium will feel full attraction to the nucleus, at

8/9/2019 CH2209 – Atomic and Molecular Spectroscopy, Quantum Chemistry
http://slidepdf.com/reader/full/ch2209-atomic-and-molecular-spectroscopy-quantum-chemistry 10/18
others the repulsion between the electrons will balance the attraction. Screening affects s,p and d-
orbitals differently and so their degeneracy is now broken. Screening of electron-nucleus charge
gives rise to the idea of effective nuclear charge, Zeff . s-orbitals are less screened than p-orbitals
which in turn are less screened than d-orbitals and so forth, this is due to amount of time electrons
in each orbital spend at the nucleus, the closer to the nucleus the less shielding can occur. Outer
electrons are unable to feel the full attractive force of the nucleus, for helium the optimum value for
Zeff is found to be 1.688.
The second rule in filling orbitals is Hunds maximum multiplicity rule, ‘an atom’s ground state adopts
a configuration with the greatest number of unpaired electrons’ (arranged parallel to each other),
2pxpy will therefore be equivalent to 2pxpz however 2px2 will be an excited state.
The spectra of multiple-electron atoms show some similarities to hydrogen with distinct
absorption/emission lines, however due to electron repulsion these lines no longer correspond
exactly to orbital differences as the interactions change between states. The Rydberg formula is not
an exact fit for multiple-electron atoms however it does still work in some cases, in particular for the
alkali metals. The lines still correspond to changes in states of individual electrons and obey thesame selection rules as before however there is another to consider in a multiple-electron atom,
that of spin multiplicity, S. S=Σms, if S=0 it is a singlet state and if S=1 it is a triplet state. A singlet-
transition is forbidden and so the last selection rule is ΔS=0. Two separate spectra can then be
observed, one for singlet states and a second for triplet states.
This is shown on the Grotrian diagram for helium;
The spin of electrons has another effect on spectra – spin-orbit coupling. A moving charge generates
a magnetic field, the spin of electrons creates magnetism. Electrons with orbital angular momentum
(i.e. l>0) generate a magnetic field from this orbital motion. Spin and orbit magnetic fields interact
depending on the orientation of the magnetic moments. An anti-parallel spin has a lower energy
than a parallel spin, the size of the coupling depends on the size of the magnetic fields.
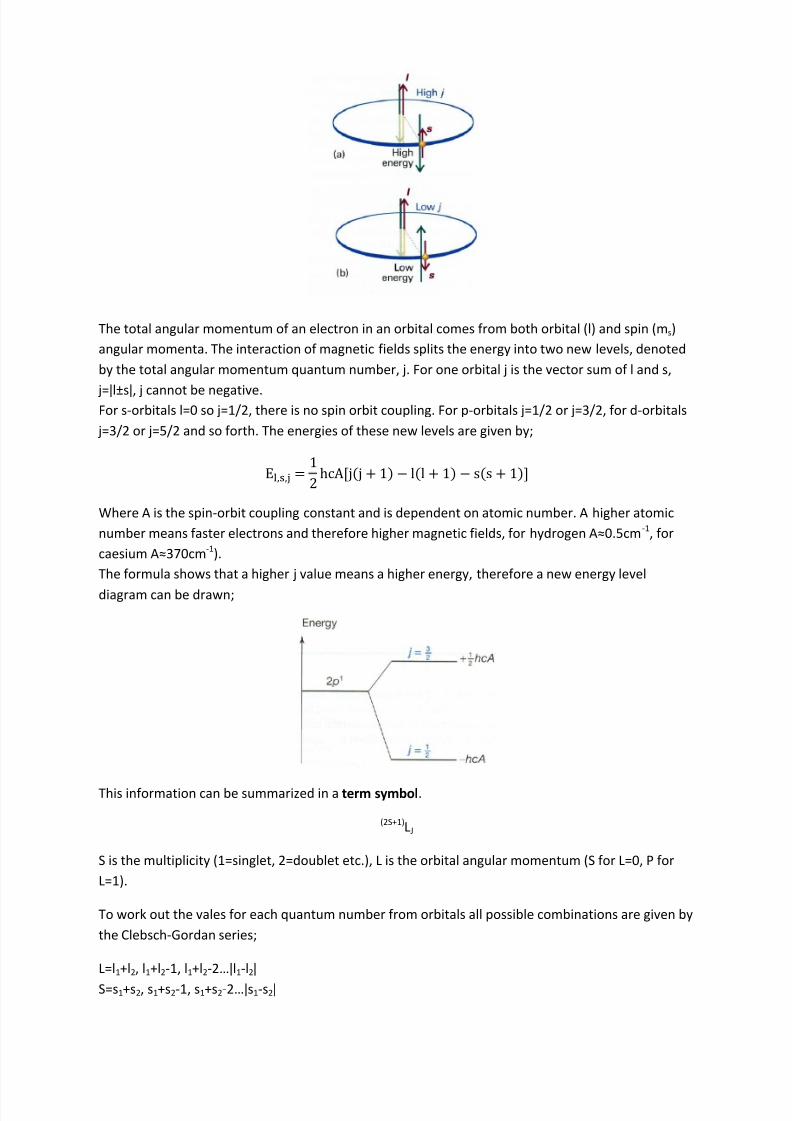
8/9/2019 CH2209 – Atomic and Molecular Spectroscopy, Quantum Chemistry
http://slidepdf.com/reader/full/ch2209-atomic-and-molecular-spectroscopy-quantum-chemistry 11/18
The total angular momentum of an electron in an orbital comes from both orbital (l) and spin (ms)
angular momenta. The interaction of magnetic fields splits the energy into two new levels, denoted
by the total angular momentum quantum number, j. For one orbital j is the vector sum of l and s,
j=ǀl±sǀ, j cannot be negative. For s-orbitals l=0 so j=1/2, there is no spin orbit coupling. For p-orbitals j=1/2 or j=3/2, for d-orbitals
j=3/2 or j=5/2 and so forth. The energies of these new levels are given by;
Where A is the spin-orbit coupling constant and is dependent on atomic number. A higher atomic
number means faster electrons and therefore higher magnetic fields, for hydrogen A≈0.5cm-1, for
caesium A≈370cm-1).
The formula shows that a higher j value means a higher energy, therefore a new energy level
diagram can be drawn;
This information can be summarized in a term symbol.
(2S+1)LJ
S is the multiplicity (1=singlet, 2=doublet etc.), L is the orbital angular momentum (S for L=0, P for
L=1).
To work out the vales for each quantum number from orbitals all possible combinations are given by
the Clebsch-Gordan series;
L=l1+l2, l1+l2-1, l1+l2-2…ǀl1-l2ǀ
S=s1+s2, s1+s2-1, s1+s2-2…ǀs1-s2ǀ

8/9/2019 CH2209 – Atomic and Molecular Spectroscopy, Quantum Chemistry
http://slidepdf.com/reader/full/ch2209-atomic-and-molecular-spectroscopy-quantum-chemistry 12/18

8/9/2019 CH2209 – Atomic and Molecular Spectroscopy, Quantum Chemistry
http://slidepdf.com/reader/full/ch2209-atomic-and-molecular-spectroscopy-quantum-chemistry 13/18
Molecular Spectroscopy
The spectra of molecules is complicated by the inclusion of rotations and vibrations in their
absorption/emission patterns. Since these occur at different energies they are considered separately
however since they do have an effect on each other some corrections are required. In general a
change in dipole moment must be seen as part of the rotation or vibration for an absorption of
radiation to occur. This is due to the dipole interacting with the oscillating electric field of the
radiation. The transition dipole, μfi, is a key quantity defining the strength of the interaction;
∫
Where Ψf and Ψi are the wavefunctions of the initial and final states and μ is the dipole moment
operator.
Vibrations of molecules are higher in energy than rotations and so occur at a higher frequency/lower
wavelength. They occur in the infra-red region, with λ between 750nm and 1mm, most chemical
applications being found between 4000-650cm-1. The vibration of a diatomic molecule has a
potential energy curve of an anharmonic oscillator however close to equilibrium a harmonic
oscillator can be assumed. The harmonic approximation can be written as V(x)=½kx2 where x=(r-re)
and k is the force constant;
A large value of k indicates a stiffer bond and hence a steeper potential curve. The Schrodinger
equation for the relative motion of the two atoms is therefore;
Where μ is the reduced mass.
This Schrodinger equation can be solved exactly, the energy in Joules that result are;
⁄
This can be expressed directly in wavenumbers as;
v v
⁄
The energy levels depend inversely on the reduced mass, a heavier molecule will have more
compressed energy levels. Perhaps a more important point of interest is that even for v=0, the
lowest possible state, there is still vibrational energy equal to ½ħω J, ultimately this stems from the
uncertainty principle, the molecule can never be at rest.
The vibrational wavefunctions are similar to those for a particle in a box but decay to Ψ=0 slower
than sine waves. Functions of the form
are used as solutions, the value of a depending
on the vibrational state. The first two vibrational wavefunctions look like;

8/9/2019 CH2209 – Atomic and Molecular Spectroscopy, Quantum Chemistry
http://slidepdf.com/reader/full/ch2209-atomic-and-molecular-spectroscopy-quantum-chemistry 14/18
The gross selection rule for infra-red absorption is that a change in dipole must be induced by the
vibration, there is no need for a permanent dipole moment since an asymmetrical vibration can
induce a temporary dipole. This does mean that homo-nuclear diatomic molecules such as H2 are
inactive in infra-red spectroscopy since at any bond length there is no dipole. The specific selection
rule for a harmonic oscillator is Δν=±1. Most molecules at room temperature are in the vibrational
ground state as the thermal energy at this temperature is approximately 200cm-1. The most intense
absorption seen is therefore from ν=0 to ν=1. This is the fundamental transition.As the higher excitations are achieved the behavior of the molecules breaks away from harmonic
oscillation and becomes anharmonic. The potential in these regions is no longer proportional to x2.
The Morse potential energy function is;
De is the depth of the potential minimum.
D0 defines the dissociation energy and so De=D0+Zero-point energy.
The Schrodinger equation can again be solved for the Morse potential giving energy levels equal to;
v
v v
χ e is the anharmonicity constant.
Unlike the harmonic case the energy levels are not evenly spaced, according to the anharmonic
approximation towards vmax the energy levels converge, above this value the molecule dissociates.
A plot of ΔG against ν is known as a Birge-Sponer extrapolation, by extrapolating to zero νmax can be
found. In the anharmonic approximation the Δν=±1 selection rule can be broken, if only by weaktransitions known as overtone bands. The intensity of these overtones depends on χ e – the more
anharmonic the vibration the more intense the overtone band. At higher temperatures hot bands
are also seen, these are transitions from ν=1→ν=2 and they increase in intensity as temperature
raises – this is simply due to the Boltzmann distribution again, as more the temperature rises more
molecules can occupy an excited state and so transitions from the excited state increase in
probability.
Rotational-Vibrational Spectroscopy
The separation between vibration and rotation is only an approximation – in practise each
vibrational level has many superimposed rotational levels. When a mole vibrates it can also changerotational state, both changes occur simultaneously and the energies are coupled. At high resolution

8/9/2019 CH2209 – Atomic and Molecular Spectroscopy, Quantum Chemistry
http://slidepdf.com/reader/full/ch2209-atomic-and-molecular-spectroscopy-quantum-chemistry 15/18
the vibrational spectra consists of many closely spaced bands consisting of two branches – a P-
branch (transitions going up a vibrational level and down a rotational level) and an R-branch
(transitions going up a vibrational level and up a rotational level) – this is due to the selection rule
ΔJ=±1, however molecules with unpaired spins can have ΔJ=0 in which a Q -branch is sometimes
seen. The energy (cm-1 of a ro-vibrational band is given by (ignoring anharmonicity);
S(ν, J)=(ν+½)+BJ(J+1)
Polyatomic molecules have several vibrations, in general 3N-6 for non-linear molecules or 3N-5 for
linear molecules. These are best described as normal modes, synchronous movement of atoms that
leave the centre of mass unchanged. Since they are independent of each other the excitation of each
is separate. Normal modes are usually treated as a set of independent harmonic oscillators where;
v v
⁄
Where v q is the wavenumber of the individual mode, q. This depends on the force constant, kq and
the mass, mq. mq is not the reduced mass of the molecule but the amount of mass moving in a
particular mode.
As with diatomics the gross selection rule means each normal mode must give rise to a change in
dipole to be observed in infrared.
Rotational Spectroscopy
The rotation of molecules is a low energy process and so occurs are low frequency/long wavelength,
typically in the microwave region. The gross selection rule is that a molecule must have a permanent
dipole moment to show rotational absorption/emission. The size of the dipole moment determinesthe intensity of the absorption, while the geometry and mass of the molecule determine the energy
levels. They key parameter in determining the energy levels is the moment of inertia, I. The moment
of inertia can be thought of as angular mass, the less concentrated the mass is around the axis the
harder it is to rotate and in turn the larger the moment of inertia.
∑
ri is the distance of the atom from the axis of rotation, mi is the atomic mass.
In general there are 3 moments of inertia about a perpendicular axes, Ia, Ib and Ic. By convention
these axes are chosen such that Ic≥Ib≥Ia with all passing through the centre of mass.
For linear molecules ri=0 about the internuclear axis for all atoms so Ia=0. Molecular symmetry can
be used to classify molecules by their moments of inertia;
-Spherical rotors have Ic=Ib=Ia, e.g. CH4, SF6
For a tetrahedral and octahedral molecule the moment of inertia is equal to;

8/9/2019 CH2209 – Atomic and Molecular Spectroscopy, Quantum Chemistry
http://slidepdf.com/reader/full/ch2209-atomic-and-molecular-spectroscopy-quantum-chemistry 16/18
-Linear rotors have Ic=Ib and Ia=0, e.g. HCl, HC=CH
For a diatomic linear rotor;
For a symmetric linear rotor (i.e. A-B-A);
For an asymmetric linear rotor (i.e. A-B-C);
-Symmetric rotors have two equal moments, either Ic=Ib or Ib=Ia, e.g. NH3, CH3Cl
-Anything else is an asymmetric rotor
The expressions for the moment of inertia of symmetric and asymmetric rotors are much more
complex.
Like any other form of energy, molecular rotation is quantised and restricted to certain, definite
energy levels. Angular momentum is again key – the rotational energy is given by;
Where Ja is the angular momentum about a, and Ia the moment of inertia about a.
For linear rotors this reduces to E=J2/2I, this is then modified by quantum mechanics to give;
The energy levels of a spherical rotor are therefore given by;
In wavenumbers this is written as;
The difference between energy levels for a linear rotor is defined by ΔEJ=EJ-EJ+1=2B(J+1)cm-1.
Since B∝1/I a heavier molecular will have more closely spaced energy levels. I is dependent on mass
and geometry so measurement of B can give detailed information on molecular structure. For
diatomics this is straight forwards as there is only one variable (bond length), for more complex
tetrahedral I = 8/3 mAR
2
octahedral I = 4mAR
2

8/9/2019 CH2209 – Atomic and Molecular Spectroscopy, Quantum Chemistry
http://slidepdf.com/reader/full/ch2209-atomic-and-molecular-spectroscopy-quantum-chemistry 17/18

8/9/2019 CH2209 – Atomic and Molecular Spectroscopy, Quantum Chemistry
http://slidepdf.com/reader/full/ch2209-atomic-and-molecular-spectroscopy-quantum-chemistry 18/18
ensure the differences can be observed.
The scattered light of the same frequency comes from elastic collisions, termed Rayleigh scattering.
Changes in frequency are from inelastic collisions, where energy is exchanged between the photon
and molecule, this is Raman scattering. The amount of energy transferred to or from the molecule
follows the same rules as absorption/emission spectra. If energy is transferred to the molecule the
scattered light will have a lower frequency that the incident beam, this is Stokes radiation. If the
energy is transferred from the molecule to the photon the scattered light will have a higher
frequency than the incident beam, this is anti-Stokes radiation. For anti-Stokes radiation the
molecule must already have some rotational or vibrational energy, as such anti-Stokes peaks are
usually less intense than Stokes radiation peaks. The key quantity in Raman spectroscopy is the
polarisability of the molecule, the measure of how dipole moment changes in an applied electric
field;
Where α is the polarisability and μ0 is the dipole in no field.
To be Raman active a rotation or vibration must change the polarisability. Most molecules have
anisotropic polarisabilities; they are distorted differently according to the direction of the applied
electric field. The polarisability of a diatomic molecule is greater along the bond axis than
perpendicular to it, rotation of a diatomic molecule therefore gives rise to a change in polarisability.
This anisotropy means that Raman spectra can be observed for rotation of non-polar molecules such
as H2. Spherical molecules do not show polarisability anisotropy and so are Raman inactive.
The specific selection rule for Raman rotational spectroscopy is ΔJ=0, ±2 only. The ΔJ=0 transition
corresponds to Rayleigh scattering, +2 to Stokes scattering and -2 to anti-Stokes scattering. A
rotational Raman spectrum has lines at B(4J+6)cm-1 either side of the incident radiation frequency.As with rotational spectra infrared absorption is useless for homo-nuclear diatomics, Raman spectra
can be used as vibrations can induce a change in polarisability. In H2 for example a longer bond
length means electrons are less tightly held than at equilibrium and so polarisability is increased.
Again there is Rayleigh scattering (Δν=0) and Stokes scattering (Δν is positive) however since most
molecules are in their ground state at room temperature anti-Stokes scattering (Δν is negative) are
much less intense. The energy levels and selection rules are essentially identical to infra-red
absorption.
v
v
For a diatomic molecule, since the energies involved are quite small, only a single Stokes line is likely
to be seen (the transition from ν0 to ν1) as well as a weaker corresponding anti-Stokes line. In very
accurate work it is possible to resolve the rotational fine structure around the vibrational Raman
lines.
For more complex molecules not all normal mode vibrations are Raman active, detailed analysis
requires symmetry and group theory arguments but there are some simple rules. Symmetric
vibrations will give intense Raman lines, asymmetric vibrations are usually weak or totally inactive.
Also a molecule with a centre of symmetry can have on vibration that is both Raman and infrared
active.




























