Embed Size (px)
Citation preview

CENTER FOR DRUG EVALUATION AND RESEARCH
APPLICATION NUMBER: 022545Orig1s000
ADMINISTRATIVE and CORRESPONDENCE DOCUMENTS














DEPARTMENT OF HEALTH AND
HUMAN SERVICES PUBLIC HEALTH SERVICE
FOOD AND DRUG ADMINISTRATION
REQUEST FOR CONSULTATION
TO (Division/Office): Devi Kozeli, Regulatory Project Manager, Division of Cardiovascular and Renal Products (DCRP)
FROM(Division/Office): Emily Baker, Regulatory Review Officer, Division of Drug Marketing, Advertising, and Communications (DDMAC)
DATE: 10/26/10
IND NO.
NDA NO. 022545
TYPE OF DOCUMENT:
DATE OF DOCUMENTS:
NAME OF DRUG Tekamlo (aliskiren and amlodipine) tablets
PRIORITY CONSIDERATION YES
CLASSIFICATION OF DRUG:
DESIRED COMPLETION DATE: 11/17/10
NAME OF FIRM: Novartis Pharmaceuticals Corporation
REASON FOR REQUEST
I. GENERAL
NEW PROTOCOL PROGRESS REPORT NEW CORRESPONDENCE DRUG ADVERTISING ADVERSE REACTION
REPORT MANUFACTURING
CHANGE/ADDITION MEETING PLANNED BY
PRE--NDA MEETING END OF PHASE II MEETING RESUBMISSION SAFETY PAPER NDA CONTROL SUPPLEMENT
RESPONSE TO DEFICIENCY
LETTER FINAL PRINTED LABELING LABELING REVISION ORIGINAL NEW
CORRESPONDENCE FORMULATIVE REVIEW OTHER (SPECIFY BELOW):
COMMENTS/SPECIAL INSTRUCTIONS: DDMAC is currently reviewing promotional materials for Tekamlo (aliskiren and amlodipine) tablets. Please see questions below, and feel free to comment on any other concerns with the proposed pieces. This consult will be put into DARRTS and the promotional materials and references will be hand delivered to you. Please note that I will be out of the office from November 8-19, so please carbon copy Sheila Ryan ([email protected]) when you send your response. Thank you, Emily Baker 6-7524 SIGNATURE OF REQUESTER
METHOD OF DELIVERY (Check one)
DARRTS and hand deliver SIGNATURE OF RECEIVER
SIGNATURE OF DELIVERER

MEMORANDUM
DEPARTMENT OF HEALTH AND HUMAN SERVICES Food and Drug Administration
Center for Drug Evaluation and Research Division of Drug Marketing, Advertising, and Communications
Date: October 26, 2010 From: Emily Baker, Pharm.D. Regulatory Review Officer Division of Drug Advertising, Marketing, and Communications (DDMAC) To: Devi Kozeli Regulatory Project Manager Division of Cardiovascular and Renal Products (DCRP) Re: Consult request for Tekamlo (aliskiren and amlodipine) tablets NDA 022545 DDMAC is currently reviewing professional launch promotional materials for advisory comments for Tekamlo (aliskiren and amlodipine) tablets in the form of a proposed visual aid and journal advertisement. Please feel free to provide any additional comments on claims or presentations within the promotional materials. Please note that I will be out of the office from November 8-19, so please carbon copy Sheila Ryan ([email protected]) when you send your response. Thank you in advance for your comments.
(b) (4)
1 Page(s) has been Withheld in Full as b4 (CCI/TS) immediately following this page

Reference ID: 2855292
---------------------------------------------------------------------------------------------------------This is a representation of an electronic record that was signedelectronically and this page is the manifestation of the electronicsignature.---------------------------------------------------------------------------------------------------------/s/----------------------------------------------------
EMILY K BAKER10/26/2010

DEPARTMENT OF HEALTH & HUMAN SERVICES Public Health Service
Food and Drug Administration Rockville, MD 20857
NDA 22-545 Novartis Pharmaceuticals Corporation Attention: Todd Gruber, M.D., M.P.H. One Health Plaza East Hanover, NJ 07936-1080
Re: Request for a Waiver for Certain Post-Marketing Reporting Responsibilities Under 21 CFR 314.80
Dear Dr. Gruber: Thank you for your letter, dated August 31, 2010, in which you requested under 21 CFR 314.90(a), a waiver from the requirement under 21 CFR 314.80 to submit to the Food and Drug Administration (FDA), as part of your post-marketing periodic safety reporting responsibilities, FDA Form 3500A for each adverse experience that is determined to be both non-serious and labeled. This waiver applies to the specific approved new drug application (NDA) listed below. I note the written commitments in your letter: (1) to hold in your corporate drug product safety files the individual case reports of adverse experiences that are both non-serious and labeled; (2) to submit these individual case reports to FDA within five (5) calendar days after receipt of a request by FDA to do so; and (3) to continue to include the non-serious, labeled adverse experiences in each periodic safety report you submit to FDA for this NDA, in the section that includes a summary tabulation by body system of all adverse experience terms and counts of occurrences submitted during the reporting period. Provided you continue to abide by the commitments in paragraph two of this letter, your requested waiver is hereby granted, as per 21 CFR 314.90(b), for the following approved NDA:
NDA 22-545 Tekamlo (aliskiren and amlodipine) tablets The waiver outlined in this letter will be in effect until you are notified in writing that it has been discontinued. Also, please note that this waiver in no way affects your other reporting responsibilities under our regulations except as specifically outlined in this letter (e.g., this waiver does not affect your expedited reporting responsibilities for adverse experiences that are both serious and unlabeled).

NDA 22-545 Page 2
If you have any questions about this waiver, please contact Ms. Jean Chung, Regulatory Analyst, at (301) 796-2380. Sincerely, {See appended electronic signature page}
Gerald Dal Pan, M.D., M.H.S. Director
Office of Surveillance and Epidemiology Center for Drug Evaluation and Research

ApplicationType/Number
SubmissionType/Number Submitter Name Product Name
-------------------- -------------------- -------------------- ------------------------------------------NDA-22545 GI-1 NOVARTIS
PHARMACEUTICALS CORP
ALISKIREN/AMLODPINE(SPA100A)FIXED COMBO
---------------------------------------------------------------------------------------------------------This is a representation of an electronic record that was signedelectronically and this page is the manifestation of the electronicsignature.---------------------------------------------------------------------------------------------------------/s/----------------------------------------------------
GERALD J DALPAN09/13/2010

Version: 12/4/09
ACTION PACKAGE CHECKLIST
APPLICATION INFORMATION1 NDA # 022545 BLA #
NDA Supplement # BLA STN # If NDA, Efficacy Supplement Type:
Proprietary Name: Tekamlo Established/Proper Name: aliskiren/amlodipine Dosage Form: Tablets
Applicant: Novartis Agent for Applicant (if applicable):
RPM: Michael Monteleone Division: Cardiovascular and Renal Products
NDAs: NDA Application Type: 505(b)(1) 505(b)(2) Efficacy Supplement: 505(b)(1) 505(b)(2) (A supplement can be either a (b)(1) or a (b)(2) regardless of whether the original NDA was a (b)(1) or a (b)(2). Consult page 1 of the NDA Regulatory Filing Review for this application or Appendix A to this Action Package Checklist.)
505(b)(2) Original NDAs and 505(b)(2) NDA supplements: Listed drug(s) referred to in 505(b)(2) application (include NDA/ANDA #(s) and drug name(s)): NDA 019787 amlodipine besylate NDA 021985 aliskiren Provide a brief explanation of how this product is different from the listed drug. This is a combination of aliskiren and amlodipine.
If no listed drug, check here and explain: Prior to approval, review and confirm the information previously provided in Appendix B to the Regulatory Filing Review by re-checking the Orange Book for any new patents and pediatric exclusivity. If there are any changes in patents or exclusivity, notify the OND ADRA immediately and complete a new Appendix B of the Regulatory Filing Review. No changes Updated Date of check: If pediatric exclusivity has been granted or the pediatric information in the labeling of the listed drug changed, determine whether pediatric information needs to be added to or deleted from the labeling of this drug. On the day of approval, check the Orange Book again for any new patents or pediatric exclusivity.
Actions
• Proposed action • User Fee Goal Date is 08-29-2010 AP TA CR
• Previous actions (specify type and date for each action taken) None
1 The Application Information section is (only) a checklist. The Contents of Action Package section (beginning on page 5) lists the documents to be included in the Action Package.

NDA/BLA # Page 2
Version: 12/4/09
If accelerated approval, were promotional materials received? Note: For accelerated approval (21 CFR 314.510/601.41), promotional materials to be used within 120 days after approval must have been submitted (for exceptions, see http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm069965.pdf). If not submitted, explain
Received
Application Characteristics 2
Review priority: Standard Priority Chemical classification (new NDAs only):
Fast Track Rx-to-OTC full switch Rolling Review Rx-to-OTC partial switch Orphan drug designation Direct-to-OTC
NDAs: Subpart H BLAs: Subpart E
Accelerated approval (21 CFR 314.510) Accelerated approval (21 CFR 601.41) Restricted distribution (21 CFR 314.520) Restricted distribution (21 CFR 601.42)
Subpart I Subpart H Approval based on animal studies Approval based on animal studies
Submitted in response to a PMR Submitted in response to a PMC Submitted in response to a Pediatric Written Request
Comments:
BLAs only: RMS-BLA Product Information Sheet for TBP has been completed and forwarded to OBPS/DRM (approvals only) Yes, date
BLAs only: is the product subject to official FDA lot release per 21 CFR 610.2 (approvals only) Yes No
Public communications (approvals only)
• Office of Executive Programs (OEP) liaison has been notified of action Yes No
• Press Office notified of action (by OEP) Yes No
• Indicate what types (if any) of information dissemination are anticipated
None HHS Press Release FDA Talk Paper CDER Q&As Other
2 Answer all questions in all sections in relation to the pending application, i.e., if the pending application is an NDA or BLA supplement, then the questions should be answered in relation to that supplement, not in relation to the original NDA or BLA. For example, if the application is a pending BLA supplement, then a new RMS-BLA Product Information Sheet for TBP must be completed.
NDA/BLA # Page 3
Version: 12/4/09
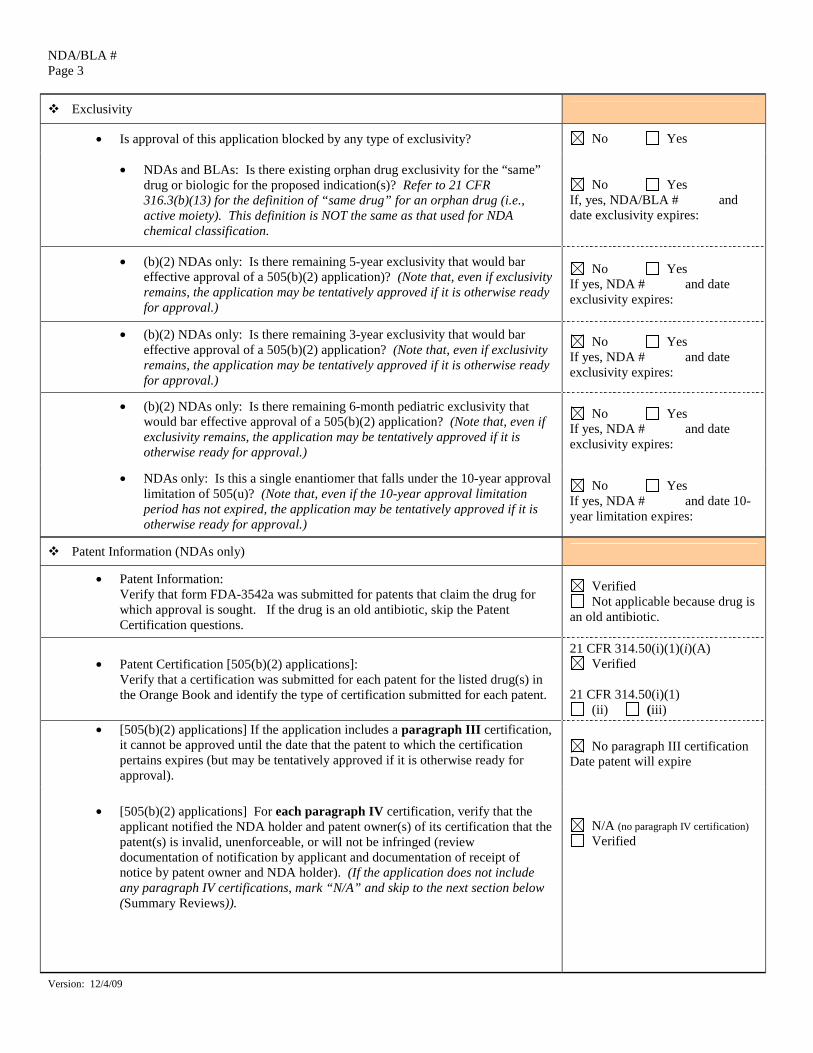
Exclusivity
• Is approval of this application blocked by any type of exclusivity? No Yes
• NDAs and BLAs: Is there existing orphan drug exclusivity for the “same” drug or biologic for the proposed indication(s)? Refer to 21 CFR 316.3(b)(13) for the definition of “same drug” for an orphan drug (i.e., active moiety). This definition is NOT the same as that used for NDA chemical classification.
No Yes If, yes, NDA/BLA # and date exclusivity expires:
• (b)(2) NDAs only: Is there remaining 5-year exclusivity that would bar effective approval of a 505(b)(2) application)? (Note that, even if exclusivity remains, the application may be tentatively approved if it is otherwise ready for approval.)
No Yes If yes, NDA # and date exclusivity expires:
• (b)(2) NDAs only: Is there remaining 3-year exclusivity that would bar effective approval of a 505(b)(2) application? (Note that, even if exclusivity remains, the application may be tentatively approved if it is otherwise ready for approval.)
No Yes If yes, NDA # and date exclusivity expires:
• (b)(2) NDAs only: Is there remaining 6-month pediatric exclusivity that would bar effective approval of a 505(b)(2) application? (Note that, even if exclusivity remains, the application may be tentatively approved if it is otherwise ready for approval.)
No Yes If yes, NDA # and date exclusivity expires:
• NDAs only: Is this a single enantiomer that falls under the 10-year approval limitation of 505(u)? (Note that, even if the 10-year approval limitation period has not expired, the application may be tentatively approved if it is otherwise ready for approval.)
No Yes If yes, NDA # and date 10-year limitation expires:
Patent Information (NDAs only)
• Patent Information: Verify that form FDA-3542a was submitted for patents that claim the drug for which approval is sought. If the drug is an old antibiotic, skip the Patent Certification questions.
Verified Not applicable because drug is
an old antibiotic.
• Patent Certification [505(b)(2) applications]: Verify that a certification was submitted for each patent for the listed drug(s) in the Orange Book and identify the type of certification submitted for each patent.
21 CFR 314.50(i)(1)(i)(A) Verified
21 CFR 314.50(i)(1)
(ii) (iii) • [505(b)(2) applications] If the application includes a paragraph III certification,
it cannot be approved until the date that the patent to which the certification pertains expires (but may be tentatively approved if it is otherwise ready for approval).
No paragraph III certification Date patent will expire
• [505(b)(2) applications] For each paragraph IV certification, verify that the
applicant notified the NDA holder and patent owner(s) of its certification that the patent(s) is invalid, unenforceable, or will not be infringed (review documentation of notification by applicant and documentation of receipt of notice by patent owner and NDA holder). (If the application does not include any paragraph IV certifications, mark “N/A” and skip to the next section below (Summary Reviews)).
N/A (no paragraph IV certification) Verified

NDA/BLA # Page 4
Version: 12/4/09
• [505(b)(2) applications] For each paragraph IV certification, based on the
questions below, determine whether a 30-month stay of approval is in effect due to patent infringement litigation.
Answer the following questions for each paragraph IV certification:
(1) Have 45 days passed since the patent owner’s receipt of the applicant’s
notice of certification?
(Note: The date that the patent owner received the applicant’s notice of certification can be determined by checking the application. The applicant is required to amend its 505(b)(2) application to include documentation of this date (e.g., copy of return receipt or letter from recipient acknowledging its receipt of the notice) (see 21 CFR 314.52(e))).
If “Yes,” skip to question (4) below. If “No,” continue with question (2).
(2) Has the patent owner (or NDA holder, if it is an exclusive patent licensee)
submitted a written waiver of its right to file a legal action for patent infringement after receiving the applicant’s notice of certification, as provided for by 21 CFR 314.107(f)(3)?
If “Yes,” there is no stay of approval based on this certification. Analyze the next paragraph IV certification in the application, if any. If there are no other paragraph IV certifications, skip the rest of the patent questions. If “No,” continue with question (3).
(3) Has the patent owner, its representative, or the exclusive patent licensee filed a lawsuit for patent infringement against the applicant?
(Note: This can be determined by confirming whether the Division has received a written notice from the (b)(2) applicant (or the patent owner or its representative) stating that a legal action was filed within 45 days of receipt of its notice of certification. The applicant is required to notify the Division in writing whenever an action has been filed within this 45-day period (see 21 CFR 314.107(f)(2))).
If “No,” the patent owner (or NDA holder, if it is an exclusive patent licensee) has until the expiration of the 45-day period described in question (1) to waive its right to bring a patent infringement action or to bring such an action. After the 45-day period expires, continue with question (4) below.
(4) Did the patent owner (or NDA holder, if it is an exclusive patent licensee)
submit a written waiver of its right to file a legal action for patent infringement within the 45-day period described in question (1), as provided for by 21 CFR 314.107(f)(3)?
If “Yes,” there is no stay of approval based on this certification. Analyze the next paragraph IV certification in the application, if any. If there are no other paragraph IV certifications, skip to the next section below (Summary Reviews). If “No,” continue with question (5).
Yes No
Yes No
Yes No
Yes No

NDA/BLA # Page 5
Version: 12/4/09
(5) Did the patent owner, its representative, or the exclusive patent licensee
bring suit against the (b)(2) applicant for patent infringement within 45 days of the patent owner’s receipt of the applicant’s notice of certification?
(Note: This can be determined by confirming whether the Division has received a written notice from the (b)(2) applicant (or the patent owner or its representative) stating that a legal action was filed within 45 days of receipt of its notice of certification. The applicant is required to notify the Division in writing whenever an action has been filed within this 45-day period (see 21 CFR 314.107(f)(2)). If no written notice appears in the NDA file, confirm with the applicant whether a lawsuit was commenced within the 45-day period).
If “No,” there is no stay of approval based on this certification. Analyze the next paragraph IV certification in the application, if any. If there are no other paragraph IV certifications, skip to the next section below (Summary Reviews). If “Yes,” a stay of approval may be in effect. To determine if a 30-month stay is in effect, consult with the OND ADRA and attach a summary of the response.
Yes No
CONTENTS OF ACTION PACKAGE Copy of this Action Package Checklist3
Officer/Employee List List of officers/employees who participated in the decision to approve this application and
consented to be identified on this list (approvals only) Included
Documentation of consent/non-consent by officers/employees Included
Action Letters
Copies of all action letters (including approval letter with final labeling) Action(s) and date(s) Approval 08-26-2010
Labeling
Package Insert (write submission/communication date at upper right of first page of PI)
• Most recent draft labeling. If it is division-proposed labeling, it should be in track-changes format. 08-19-2010
• Original applicant-proposed labeling 10-29-2009
• Example of class labeling, if applicable
3 Fill in blanks with dates of reviews, letters, etc.

NDA/BLA # Page 6
Version: 12/4/09
Medication Guide/Patient Package Insert/Instructions for Use (write submission/communication date at upper right of first page of each piece)
Medication Guide Patient Package Insert Instructions for Use None
• Most-recent draft labeling. If it is division-proposed labeling, it should be in ttrack-changes format.
• Original applicant-proposed labeling 10-29-2009
• Example of class labeling, if applicable
Labels (full color carton and immediate-container labels) (write submission/communication date on upper right of first page of each submission)
• Most-recent draft labeling 08-20-2010
Proprietary Name • Acceptability/non-acceptability letter(s) (indicate date(s)) • Review(s) (indicate date(s))
Acceptable 02/02/2010 Review 02/02/2010; 08-12-2010
Labeling reviews (indicate dates of reviews and meetings)
RPM DMEDP 08/25/2010 DRISK 07/22/2010 DDMAC 06/11/2010 CSS Other reviews
SEALD 7-16-2010
Administrative / Regulatory Documents Administrative Reviews (e.g., RPM Filing Review4/Memo of Filing Meeting) (indicate
date of each review) 12/23/2009
NDAs only: Exclusivity Summary (signed by Division Director) Included
Application Integrity Policy (AIP) Status and Related Documents http://www.fda.gov/ICECI/EnforcementActions/ApplicationIntegrityPolicy/default.htm
• Applicant in on the AIP Yes No
• This application is on the AIP
o If yes, Center Director’s Exception for Review memo (indicate date)
o If yes, OC clearance for approval (indicate date of clearance communication)
Yes No
Not an AP action
Pediatrics (approvals only) • Date reviewed by PeRC 06/30/2010
If PeRC review not necessary, explain: • Pediatric Page (approvals only, must be reviewed by PERC before finalized)
Included
Debarment certification (original applications only): verified that qualifying language was not used in certification and that certifications from foreign applicants are cosigned by U.S. agent (include certification)
Verified, statement is acceptable
Outgoing communications (letters (except action letters), emails, faxes, telecons)
Internal memoranda, telecons, etc.
4 Filing reviews for scientific disciplines should be filed behind the respective discipline tab.

NDA/BLA # Page 7
Version: 12/4/09
Minutes of Meetings
• Pre-Approval Safety Conference (indicate date of mtg; approvals only) Not applicable
• Regulatory Briefing (indicate date of mtg) No mtg
• If not the first review cycle, any end-of-review meeting (indicate date of mtg) N/A or no mtg
• Pre-NDA/BLA meeting (indicate date of mtg) No mtg
• EOP2 meeting (indicate date of mtg) No mtg
• Other milestone meetings (e.g., EOP2a, CMC pilot programs) (indicates dates)
Advisory Committee Meeting(s) No AC meeting
• Date(s) of Meeting(s)
• 48-hour alert or minutes, if available (do not include transcript)
Decisional and Summary Memos
Office Director Decisional Memo (indicate date for each review) None
Division Director Summary Review (indicate date for each review) None 08/16/2010
Cross-Discipline Team Leader Review (indicate date for each review) None 08/09/2010
PMR/PMC Development Templates (indicate total number) None
Clinical Information5 Clinical Reviews
• Clinical Team Leader Review(s) (indicate date for each review)
• Clinical review(s) (indicate date for each review) 07/07/2010
• Social scientist review(s) (if OTC drug) (indicate date for each review) None Financial Disclosure reviews(s) or location/date if addressed in another review
OR If no financial disclosure information was required, check here and include a review/memo explaining why not (indicate date of review/memo)
Clinical Review 07/07/2010, pg 16
Clinical reviews from immunology and other clinical areas/divisions/Centers (indicate date of each review) None
Controlled Substance Staff review(s) and Scheduling Recommendation (indicate date of each review) Not applicable
Risk Management • REMS Document and Supporting Statement (indicate date(s) of submission(s)) • REMS Memo (indicate date) • Risk management review(s) and recommendations (including those by OSE and
CSS) (indicate date of each review and indicate location/date if incorporated into another review)
None
DSI Clinical Inspection Review Summary(ies) (include copies of DSI letters to investigators) None requested
5 Filing reviews should be filed with the discipline reviews.

NDA/BLA # Page 8
Version: 12/4/09
Clinical Microbiology None
Clinical Microbiology Team Leader Review(s) (indicate date for each review) None
Clinical Microbiology Review(s) (indicate date for each review) None
Biostatistics None
Statistical Division Director Review(s) (indicate date for each review) None
Statistical Team Leader Review(s) (indicate date for each review) None 06/02/2010
Statistical Review(s) (indicate date for each review) None 06/02/2010
Clinical Pharmacology None
Clinical Pharmacology Division Director Review(s) (indicate date for each review) None
Clinical Pharmacology Team Leader Review(s) (indicate date for each review) None 06/16/2010; 07/21/2010
Clinical Pharmacology review(s) (indicate date for each review) None 06/16/2010; 07/21/2010
DSI Clinical Pharmacology Inspection Review Summary (include copies of DSI letters) None 07/09/2010
Nonclinical None Pharmacology/Toxicology Discipline Reviews
• ADP/T Review(s) (indicate date for each review) None
• Supervisory Review(s) (indicate date for each review) None 06/03/2010 • Pharm/tox review(s), including referenced IND reviews (indicate date for each
review) None 06/03/2010
Review(s) by other disciplines/divisions/Centers requested by P/T reviewer (indicate date for each review) None
Statistical review(s) of carcinogenicity studies (indicate date for each review) No carc
ECAC/CAC report/memo of meeting None Included in P/T review, page
DSI Nonclinical Inspection Review Summary (include copies of DSI letters) None requested
Product Quality None Product Quality Discipline Reviews
• ONDQA/OBP Division Director Review(s) (indicate date for each review) None
• Branch Chief/Team Leader Review(s) (indicate date for each review) None 06/09/2010 08/04/2010
• Product quality review(s) including ONDQA biopharmaceutics reviews (indicate date for each review)
None 06/09/2010 08/04/2010
Microbiology Reviews NDAs: Microbiology reviews (sterility & pyrogenicity) (OPS/NDMS) (indicate
date of each review) BLAs: Sterility assurance, microbiology, facilities reviews
(DMPQ/MAPCB/BMT) (indicate date of each review)
Not needed
Reviews by other disciplines/divisions/Centers requested by CMC/quality reviewer (indicate date of each review)
None Biopharm 06/11/2010

NDA/BLA # Page 9
Version: 12/4/09
Environmental Assessment (check one) (original and supplemental applications)
Categorical Exclusion (indicate review date)(all original applications and all efficacy supplements that could increase the patient population)
Review & FONSI (indicate date of review) 06/28/2010
Review & Environmental Impact Statement (indicate date of each review)
Facilities Review/Inspection
NDAs: Facilities inspections (include EER printout) (date completed must be within 2 years of action date)
Date completed: 04/27/2010 Acceptable Withhold recommendation
BLAs: TB-EER (date of most recent TB-EER must be within 30 days of action date)
Date completed: Acceptable Withhold recommendation
NDAs: Methods Validation (check box only, do not include documents)
Completed Requested Not yet requested Not needed

NDA/BLA # Page 10
Version: 12/4/09
Appendix A to Action Package Checklist An NDA or NDA supplemental application is likely to be a 505(b)(2) application if:
(1) It relies on published literature to meet any of the approval requirements, and the applicant does not have a written right of reference to the underlying data. If published literature is cited in the NDA but is not necessary for approval, the inclusion of such literature will not, in itself, make the application a 505(b)(2) application.
(2) Or it relies for approval on the Agency's previous findings of safety and efficacy for a listed drug product and the applicant does not own or have right to reference the data supporting that approval.
(3) Or it relies on what is "generally known" or "scientifically accepted" about a class of products to support the safety or effectiveness of the particular drug for which the applicant is seeking approval. (Note, however, that this does not mean any reference to general information or knowledge (e.g., about disease etiology, support for particular endpoints, methods of analysis) causes the application to be a 505(b)(2) application.)
Types of products for which 505(b)(2) applications are likely to be submitted include: fixed-dose combination drug products (e.g., heart drug and diuretic (hydrochlorothiazide) combinations); OTC monograph deviations(see 21 CFR 330.11); new dosage forms; new indications; and, new salts. An efficacy supplement can be either a (b)(1) or a (b)(2) regardless of whether the original NDA was a (b)(1) or a (b)(2). An efficacy supplement is a 505(b)(1) supplement if the supplement contains all of the information needed to support the approval of the change proposed in the supplement. For example, if the supplemental application is for a new indication, the supplement is a 505(b)(1) if:
(1) The applicant has conducted its own studies to support the new indication (or otherwise owns or has right of reference to the data/studies).
(2) And no additional information beyond what is included in the supplement or was embodied in the finding of safety and effectiveness for the original application or previously approved supplements is needed to support the change. For example, this would likely be the case with respect to safety considerations if the dose(s) was/were the same as (or lower than) the original application.
(3) And all other “criteria” are met (e.g., the applicant owns or has right of reference to the data relied upon for approval of the supplement, the application does not rely for approval on published literature based on data to which the applicant does not have a right of reference).
An efficacy supplement is a 505(b)(2) supplement if:
(1) Approval of the change proposed in the supplemental application would require data beyond that needed to support our previous finding of safety and efficacy in the approval of the original application (or earlier supplement), and the applicant has not conducted all of its own studies for approval of the change, or obtained a right to reference studies it does not own. For example, if the change were for a new indication AND a higher dose, we would likely require clinical efficacy data and preclinical safety data to approve the higher dose. If the applicant provided the effectiveness data, but had to rely on a different listed drug, or a new aspect of a previously cited listed drug, to support the safety of the new dose, the supplement would be a 505(b)(2).
(2) Or the applicant relies for approval of the supplement on published literature that is based on data that the applicant does not own or have a right to reference. If published literature is cited in the supplement but is not necessary for approval, the inclusion of such literature will not, in itself, make the supplement a 505(b)(2) supplement.
(3) Or the applicant is relying upon any data they do not own or to which they do not have right of reference. If you have questions about whether an application is a 505(b)(1) or 505(b)(2) application, consult with your ODE’s ADRA.

ApplicationType/Number
SubmissionType/Number Submitter Name Product Name
-------------------- -------------------- -------------------- ------------------------------------------NDA-22545 ORIG-1 NOVARTIS
PHARMACEUTICALS CORP
ALISKIREN/AMLODPINE(SPA100A)FIXED COMBO
---------------------------------------------------------------------------------------------------------This is a representation of an electronic record that was signedelectronically and this page is the manifestation of the electronicsignature.---------------------------------------------------------------------------------------------------------/s/----------------------------------------------------
MICHAEL V MONTELEONE08/26/2010

DEPARTMENT OF HEALTH AND HUMAN SERVICES
Food and Drug Administration Silver Spring MD 20993
NDA 022545 INFORMATION REQUEST
Novartis Pharmaceuticals Corporation Attention: Lori Kneafsey Associate Director, Drug Regulatory Affairs One Health Plaza East Hanover, NJ 07936 Dear Ms. Kneafsey: Please refer to your new drug application (NDA) dated October 28, 2009, received October 29, 2009, submitted pursuant to section 505(b)(2) of the Federal Food, Drug, and Cosmetic Act, for Tekamlo, (aliskiren/amlodipine) 150/5mg, 150/10mg, 300/5mg and 300/10mg Tablets. We are reviewing the carton and container labeling of your submission and have the following comments and information requests. We request a prompt written response in order to continue our evaluation of your NDA. Please note that these comments are in addition to the CMC comments conveyed in letters dated April 8 and May 28, 2010.
1. General Comments a. We note the color purple is used for the tradename across the Aliskerin product
line (i.e. Tekturna, Tekturna HCT, and Tekamlo). Additionally, the same circle graphic is used across the product line. In order to differentiate Tekamlo from the remaining Aliskerin products we recommend a different color be used for the tradename and the circle graphic be revised so that it is not the same across the product line.
b. Present the entire proprietary name in a single font color. As currently presented, portions of the
proprietary name make the name difficult to read. 2. Container Labels (30 count and 90 count)
a. See comments 1-A and 1-B. 3. Blister Labels
a. Differentiate the product strengths through the use of color, boxing, reverse-blocking, or some other means.
b. Include an asterisk at the beginning of the qualifying statement “each tablet contains… XX mg of amlodipine besylate” so that it is clear to what the asterisk at the end of the product strength is referring.
4. Unit-Dose Carton Labeling a. See comments 1-A and 1-B.
(b) (4)

NDA 022545 Page 2
b. We note different colors are used to differentiate the product strengths (e.g. yellow for 150 mg/5 mg strength, orange for 150 mg/10 mg strength), however, the color blue is still used predominantly in the labels and labeling thereby diminishing any differentiation offered by the differing colors. We recommend using the same colors used to differentiate the product strengths in place of where the color blue is used (e.g. the color band across the top of the carton labeling that contains the NDC number) as is used on the 30-count and 90-count trade container labels.
c. Remove the statement. The statement is a more accurate reflection of the contents of the carton.
If you have any questions, please call Michael Monteleone, Regulatory Project Manager, at (301) 796.1952.
Sincerely, {See appended electronic signature page} Norman Stockbridge, MD, PhD Director Division of Cardiovascular and Renal Products Office of Drug Evaluation I Center for Drug Evaluation and Research
(b) (4) (b) (4)
(b) (4)

ApplicationType/Number
SubmissionType/Number Submitter Name Product Name
-------------------- -------------------- -------------------- ------------------------------------------NDA-22545 ORIG-1 NOVARTIS
PHARMACEUTICALS CORP
ALISKIREN/AMLODPINE(SPA100A)FIXED COMBO
---------------------------------------------------------------------------------------------------------This is a representation of an electronic record that was signedelectronically and this page is the manifestation of the electronicsignature.---------------------------------------------------------------------------------------------------------/s/----------------------------------------------------
NORMAN L STOCKBRIDGE08/16/2010

Pediatric Research Equity Act (PREA) Waiver Request, Deferral Request/Pediatric Plan and Assessment Template(s)
BACKGROUND Please check all that apply: Full Waiver Partial Waiver Pediatric Assessment Deferral/Pediatric Plan BLA/NDA#: NDA 022545 PRODUCT PROPRIETARY NAME: TEKAMLO ESTABLISHED/GENERIC NAME:aliskiren/amlodipine APPLICANT/SPONSOR: Novartis Pharmaceuticals Corporation PREVIOUSLY APPROVED INDICATION/S: (1) N/A__________________________________ (2) ______________________________________ (3) ______________________________________ (4) ______________________________________ PROPOSED INDICATION/S: (1) Treatment of hypertension: As initial therapy in patients likely to need multiple drugs
to achieve their blood pressure goals; In patients not adequately controlled with monotherapy; May be substituted for titrated components.
(2) ______________________________________ (3) ______________________________________ (4) ______________________________________ BLA/NDA STAMP DATE: 10/29/2009 SUPPLEMENT TYPE: NA SUPPLEMENT NUMBER: NA

Does this application provide for (If yes, please check all categories that apply and proceed to the next question): NEW active ingredient(s) (includes new combination); indication(s); dosage form; dosing regimen; or route of administration? Has the sponsor submitted a Proposed Pediatric Study Request (PPSR) or does the Division believe there is an additional public health benefit to issuing a Written Request for this product, even if the plan is to grant a waiver for this indication? (Please note, Written Requests may include approved and unapproved indications and may apply to the entire moiety, not just this product.)
Yes No Is this application in response to a PREA (Postmarketing Requirement) PMR? Yes No If Yes, PMR # __________ NDA # __________ Does the division agree that this is a complete response to the PMR? Yes No If Yes, to either question Please complete the Pediatric Assessment Template. If No, complete all appropriate portions of the template, including the assessment template if the division
believes this application constitutions an assessment for any particular age group.

WAIVER REQUEST Please attach: Draft Labeling (If Waiving for Safety and/or Efficacy) from the sponsor unless the Division plans to change.
If changing the sponsor’s proposed language, include the appropriate language under Question 4 in this form. Pediatric Record
1. Pediatric age group(s) to be waived. FULL WAIVER
2. Reason(s) for waiving pediatric assessment requirements (Choose one. If there are different reasons for different age groups or
indications, please choose the appropriate reason for each age group or indication. This section should reflect the Division’s thinking.)
Studies are impossible or highly impractical (e.g. the number of pediatric patients is so small or is geographically
dispersed). (Please note that in the DARRTS record, this reason is captured as “Not Feasible.”) If applicable, chose from adult- related conditions on the next page
The product would be ineffective and/or unsafe in one or more of the pediatric group(s) for which a waiver is being
requested. Note: If this is the reason the studies are being waived, this information MUST be included in the pediatric use section of labeling. Please provide the draft language you intend to include in the label. The language must
be included in section 8.4 and describe the safety or efficacy concerns in detail.
The product fails to represent a meaningful therapeutic benefit over existing therapies for pediatric patients and is unlikely to be used in a substantial number of all pediatric age groups or the pediatric age group(s) for which a waiver is being requested.
Reasonable attempts to produce a pediatric formulation for one or more of the pediatric age group(s) for which the
waiver is being requested have failed. (Provide documentation from Sponsor) Note: Sponsor must provide data to support this claim for review by the Division, and this data will be publicly posted. (This reason is for Partial Waivers Only)

3. Provide justification for Waiver: Tekamlo is a combination antihypertensive agent. There are single agent products studied and labeled for use in pediatrics, and most pediatric patients are not treated with combination antihypertensives (supported by The Fourth Report on the Diagnosis, Evaluation, and Treatment of High Blood Pressure in Children and Adolescents, Pediatrics 2004;114;555-576). 4. Provide language Review Division is proposing for Section 8.4 of the label if different from sponsor’s proposed language:
(b) (4)

24 Page(s) of Draft Labeling have been Withheld in full as b4 (CCI/TS) immediately following this page

ApplicationType/Number
SubmissionType/Number Submitter Name Product Name
-------------------- -------------------- -------------------- ------------------------------------------NDA-22545 ORIG-1 NOVARTIS
PHARMACEUTICALS CORP
ALISKIREN/AMLODPINE(SPA100A)FIXED COMBO
---------------------------------------------------------------------------------------------------------This is a representation of an electronic record that was signedelectronically and this page is the manifestation of the electronicsignature.---------------------------------------------------------------------------------------------------------/s/----------------------------------------------------
MICHAEL V MONTELEONE08/11/2010

DEPARTMENT OF HEALTH AND HUMAN SERVICES
PUBLIC HEALTH SERVICE FOOD AND DRUG ADMINISTRATION
REQUEST FOR CONSULTATION
TO (Office/Division): PMHS
FROM (Name, Office/Division, and Phone Number of Requestor): Michael Monteleone, Division of Cardiorenal, x61952
DATE
7-9-10
IND NO.
NDA NO. 22545
TYPE OF DOCUMENT Labeling
DATE OF DOCUMENT 7-9-10
NAME OF DRUG
Tekamlo
PRIORITY CONSIDERATION
Standard
CLASSIFICATION OF DRUG
combination
DESIRED COMPLETION DATE
July 19, 2010 NAME OF FIRM: Novartis
REASON FOR REQUEST
I. GENERAL
NEW PROTOCOL PROGRESS REPORT NEW CORRESPONDENCE DRUG ADVERTISING ADVERSE REACTION REPORT MANUFACTURING CHANGE / ADDITION MEETING PLANNED BY
PRE-NDA MEETING END-OF-PHASE 2a MEETING END-OF-PHASE 2 MEETING RESUBMISSION SAFETY / EFFICACY PAPER NDA CONTROL SUPPLEMENT
RESPONSE TO DEFICIENCY LETTER FINAL PRINTED LABELING LABELING REVISION ORIGINAL NEW CORRESPONDENCE FORMULATIVE REVIEW OTHER (SPECIFY BELOW):
II. BIOMETRICS
PRIORITY P NDA REVIEW END-OF-PHASE 2 MEETING CONTROLLED STUDIES PROTOCOL REVIEW OTHER (SPECIFY BELOW):
CHEMISTRY REVIEW PHARMACOLOGY BIOPHARMACEUTICS OTHER (SPECIFY BELOW):
III. BIOPHARMACEUTICS
DISSOLUTION BIOAVAILABILTY STUDIES PHASE 4 STUDIES
DEFICIENCY LETTER RESPONSE PROTOCOL - BIOPHARMACEUTICS IN-VIVO WAIVER REQUEST
IV. DRUG SAFETY
PHASE 4 SURVEILLANCE/EPIDEMIOLOGY PROTOCOL DRUG USE, e.g., POPULATION EXPOSURE, ASSOCIATED DIAGNOSES CASE REPORTS OF SPECIFIC REACTIONS (List below) COMPARATIVE RISK ASSESSMENT ON GENERIC DRUG GROUP
REVIEW OF MARKETING EXPERIENCE, DRUG USE AND SAFETY SUMMARY OF ADVERSE EXPERIENCE POISON RISK ANALYSIS
V. SCIENTIFIC INVESTIGATIONS
CLINICAL
NONCLINICAL
COMMENTS / SPECIAL INSTRUCTIONS: Please review substantially complete PI for new NDA 022545, Tekamlo (aliskiren/amlodipine) combination. All primary reviews are in DARRTS, word labeling will be sent via email to Tammy Brent Howard. SIGNATURE OF REQUESTOR
Mike Monteleone
METHOD OF DELIVERY (Check one)
DFS EMAIL MAIL HAND
PRINTED NAME AND SIGNATURE OF RECEIVER
PRINTED NAME AND SIGNATURE OF DELIVERER

ApplicationType/Number
SubmissionType/Number Submitter Name Product Name
-------------------- -------------------- -------------------- ------------------------------------------NDA-22545 ORIG-1 NOVARTIS
PHARMACEUTICALS CORP
ALISKIREN/AMLODPINE(SPA100A)FIXED COMBO
---------------------------------------------------------------------------------------------------------This is a representation of an electronic record that was signedelectronically and this page is the manifestation of the electronicsignature.---------------------------------------------------------------------------------------------------------/s/----------------------------------------------------
MICHAEL V MONTELEONE07/09/2010

DEPARTMENT OF HEALTH AND HUMAN SERVICES
Food and Drug Administration Silver Spring MD 20993
NDA 22-545 INFORMATION REQUEST
Novartis Pharmaceuticals Corporation Attention: Lori Ann Kneafsey
Associate Director, Drug Regulatory Affairs One Health Plaza East Hanover, NJ 07936-1080
Dear Ms. Kneafsey: Please refer to your New Drug Application (NDA) submitted under section 505(b) of the Federal Food, Drug, and Cosmetic Act for SPA100 (aliskiren and amlodipine) film-coated tablets. We are reviewing the Chemistry, Manufacturing, and Controls sections of your submission and have the following comments and information requests. We request a prompt written response in order to continue our evaluation of your NDA.
1. Provide an updated amlodipine besylate drug substance specification that will include a limit for total of
not to exceed ppm. 2. Based on the review of the overall dissolution information provided in Amendment dated
April 26, 2010, we consider that your proposal for the dissolution acceptance criteria of your drug product is acceptable, as follows: • Amlodipine: Q-value of in 30 minutes for SPA100 tablets of all strengths. • Aliskiren: Q-value of in 30 minutes for SPA100 tablets of all strengths.
3. Provide updated drug product specification that includes all revisions made according to FDA comments (i.e. revised Description and Dissolution of SPA 100 tablets).
4. . Clarify why
carton labels for 30 count and 90 count trade HDPE bottles are not provided. 5. Include a reference in the text of the labels for 150 mg aliskiren as follows
, and respective reference for 300 mg aliskiren:
6. Revise containers labels to include the following statement:
(b) (4)
(b) (4)
(b) (4)
(b) (4)
(b) ( )
(b) (4)
(b) (4)
(b) (4)
(b) (4)

NDA 22-545 Page 2 If you have any questions, call Don Henry, Regulatory Project Manager, at (301) 796-4227.
Sincerely, {See appended electronic signature page} Ramesh Sood, Ph.D. Branch Chief Division of New Drug Quality Assessment I Office of New Drug Quality Assessment Center for Drug Evaluation and Research

ApplicationType/Number
SubmissionType/Number Submitter Name Product Name
-------------------- -------------------- -------------------- ------------------------------------------NDA-22545 ORIG-1 NOVARTIS
PHARMACEUTICALS CORP
ALISKIREN/AMLODPINE(SPA100A)FIXED COMBO
---------------------------------------------------------------------------------------------------------This is a representation of an electronic record that was signedelectronically and this page is the manifestation of the electronicsignature.---------------------------------------------------------------------------------------------------------/s/----------------------------------------------------
RAMESH K SOOD05/28/2010

DEPARTMENT OF HEALTH AND HUMAN SERVICES
PUBLIC HEALTH SERVICE FOOD AND DRUG ADM NISTRATION
REQUEST FOR DDMAC LABELING REVIEW CONSULTATION
**Please send immediately following the Filing/Planning meeting** TO: CDER-DDMAC-RPM
FROM: (Name/Title, Office/Division/Phone number of requestor) Mike Monteleone, RPM DCRP x61952
REQUEST DATE 4-19-2010
IND NO.
NDA/BLA NO. 022545
TYPE OF DOCUMENTS (PLEASE CHECK OFF BELOW)
NAME OF DRUG Tekamlo (aliskiren/amlodipine)
PRIORITY CONSIDERATION Standard
CLASSIFICATION OF DRUG
DESIRED COMPLETION DATE (Generally 1 week before the wrap-up meeting) July 01, 2010
NAME OF FIRM:
Novartis
PDUFA Date: August 29, 2010
TYPE OF LABEL TO REVIEW
TYPE OF LABELING: (Check all that apply) ⌧ PACKAGE INSERT (PI) ⌧ PATIENT PACKAGE INSERT (PPI)
CARTON/CONTAINER LABELING MEDICATION GUIDE INSTRUCTIONS FOR USE(IFU)
TYPE OF APPLICATION/SUBMISSION ⌧ ORIGINAL NDA/BLA
IND EFFICACY SUPPLEMENT SAFETY SUPPLEMENT LABELING SUPPLEMENT PLR CONVERSION
REASON FOR LABELING CONSULT ⌧ INITIAL PROPOSED LABELING
LABELING REVISION
EDR link to submission: In DARRTS
Please Note: There is no need to send labeling at this time. DDMAC reviews substantially complete labeling, which has already been marked up by the CDER Review Team. The DDMAC reviewer will contact you at a later date to obtain the substantially complete labeling for review. COMMENTS/SPECIAL INSTRUCTIONS: Mid-Cycle Meeting: [April 7, 2010] Wrap-Up Meeting: [July 7, 2010]
SIGNATURE OF REQUESTER Michael Monteleone SIGNATURE OF RECEIVER
METHOD OF DELIVERY (Check one)
eMAIL ⌧DARRTS HAND

ApplicationType/Number
SubmissionType/Number Submitter Name Product Name
-------------------- -------------------- -------------------- ------------------------------------------NDA-22545 ORIG-1 NOVARTIS
PHARMACEUTICALS CORP
ALISKIREN/AMLODPINE(SPA100A)FIXED COMBO
---------------------------------------------------------------------------------------------------------This is a representation of an electronic record that was signedelectronically and this page is the manifestation of the electronicsignature.---------------------------------------------------------------------------------------------------------/s/----------------------------------------------------
MICHAEL V MONTELEONE04/19/2010

DEPARTMENT OF HEALTH AND HUMAN SERVICES
Food and Drug Administration Silver Spring MD 20993
NDA 22-545 INFORMATION REQUEST
Novartis Pharmaceuticals Corporation Attention: Lori Ann Kneafsey
Associate Director, Drug Regulatory Affairs One Health Plaza East Hanover, NJ 07936-1080
Dear Ms. Kneafsey: Please refer to your New Drug Application (NDA) submitted under section 505(b) of the Federal Food, Drug, and Cosmetic Act for SPA100 (aliskiren and amlodipine) film-coated tablets. We are reviewing the Chemistry, Manufacturing, and Controls sections of your submission and have the following comments and information requests. We request a prompt written response in order to continue our evaluation of your NDA.
Drug Substance
1. The DMF , which you are referencing for drug substance Amlodipine Besylate, is currently inadequate. A deficiency letter was sent to the DMF holder. Please be advised that satisfactory resolution of these deficiencies will be necessary before your NDA may be approved.
2. Besides the individual limits for in the amlodipine besylate drug substance specification, include a limit for total of
not to exceed ppm [refer to the Test Specification (Test 30001.01) “Impurities by HPLC” of the Novartis’s Test Specification for Amlodipine Besylate from
3. Provide validation of the HPLC Test 53001.01 (Impurities by HPLC) included in the Test Specification for Amlodipine Besylate from since it has been modified to include additional specified impurities as compared to HPLC Test 54001.01.
4. Provide exact numerical values for the total combined yeasts /moulds count in the Microbial Enumeration test for three batches of amlodipine besylate manufactured by
and for three drug substance batches manufactured by . using ). We note that your batch analysis
data show values whereas the specification limit is Drug Product
1. Include “non-scored” in the Description of SPA100 tablets in the drug product
specification for each dosage strength.
(b) (4) (b) (4)
(b) (4)
(b) (4)
(b) (4)
(b) (4)
(b) (4) (b) (4)
(b) (4) (b) (4)
(b) ( )
(b) (4)
(b) (4)
(b) (4) (b) (4)

NDA 22-545 Page 2
2. Provide confirmation that all excipients of the drug product comply with the USP<467> for residual solvents.
3. The aliskiren degradation impurity was qualified at the level of in the previous NDA 22-217 (Valturna). Provide toxicology data that qualifies impurity at the limit of in the drug product specification.
4. Provide toxicology studies supporting qualification of the aliskiren impurity at level of in the drug product specification.
5. Since the 12-month stability data at long-term and intermediate conditions do not justify the proposed limits, tighten the shelf-life limit of for impurity and limit of
for sum of the degradation products in the drug product specification. 6. Clarify your conclusion in the validation report for test method AM38011C(AS6220) that
this method is judged to be suitable for determination only of even though the method is intended to control the sum of
at the limit of ppm. Provide a rationale for developing a limit test for these impurities instead of a regular numerical test.
7. Provide structural characterization of the reference standard for specified degradation product originating from amlodipine besylate, and for aliskiren specified impurity
. Provide a source and additional purification steps, if applicable, for these reference standards.
8. To support the bracketing design for stability studies, provide data on head space volume for each bottle/count configuration and results of the USP Container Test <671> for
for each combination of bottle and cap intended for commercial and sample use.
9. Based on the results of the comparative dissolution testing, we recommend the following revision to the drug product dissolution specification. Provide updated drug product specifications according to this revision:
a. the Q-value for amlodipine should be release of amlodipine at 20 minutes for SPA100 tablets of all strengths.
b. the Q-value for aliskiren should be release of aliskiren at 20 minutes for SPA100 tablets of all strengths.
Provide Q-value data at 20 minutes at release and on stability for 150/5 mg, 150/10 mg, 300/5 mg and 300/10 mg strengths tablets.
Labeling & Package Insert
Container/Carton Labels 1. Provide full representation in color of the updated container/carton labels (not the drafts)
since the trade name is approved. 2. Include a reference on the text of the labels for 5 mg amlodipine as follows: “each tablet
contains of amlodipine besylate”, and the corresponding reference for amlodipine.
3. Explain why carton and bottle labels for HDPE bottles of 100 counts and of 14 counts are not provided.
4. Provide carton labels for HDPE bottles if applicable. 5. Delete the USP designation for amlodipine from the text of labels since the USP
monograph is for amlodipine besylate not amlodipine .
(b) (4)
(b) (4)
(b) (4)
(b) (4)
(b) (4)
(b) (4)
(b) (4)
(b) (4)
(b) (4)
(b) (4)
(b) (4)
(b) (4)
(b) (4)
(b) (4)
(b) (4)
(b) (4)
(b) (4)
(b) (4) (b) (4)

NDA 22-545 Page 3
Package Insert 6. Include the full description of the individual dosage strengths with the NDC codes in the
HOW SUPPLIED/ STORAGE AND HANDLING section of Package Insert. 7. Explain why the HOW SUPPLIED/ STORAGE AND HANDLING section of the
Package Insert lists only HDPE bottles of 30 counts and 90 counts tablets, and not HDPE bottles of 100 counts.
If you have any questions, call Don Henry, Regulatory Project Manager, at (301) 796-4227.
Sincerely, {See appended electronic signature page} Ramesh Sood, Ph.D. Branch Chief Division of Pre-Marketing Assessment I Office of New Drug Quality Assessment Center for Drug Evaluation and Research

ApplicationType/Number
SubmissionType/Number Submitter Name Product Name
-------------------- -------------------- -------------------- ------------------------------------------NDA-22545 ORIG-1 NOVARTIS
PHARMACEUTICALS CORP
ALISKIREN/AMLODPINE(SPA100A)FIXED COMBO
---------------------------------------------------------------------------------------------------------This is a representation of an electronic record that was signedelectronically and this page is the manifestation of the electronicsignature.---------------------------------------------------------------------------------------------------------/s/----------------------------------------------------
KASTURI SRINIVASACHAR04/08/2010

DEPARTMENT OF HEALTH AND HUMAN SERVICES
PUBLIC HEALTH SERVICE FOOD AND DRUG ADM NISTRATION
REQUEST FOR CONSULTATION
TO (Division/Office): Mail: OSE
FROM: Michael Monteleone (x61952), OND / ODE1 / DCRP
DATE 4-8-10
IND NO.
NDA NO.
022545
TYPE OF DOCUMENT
Risk Management Plan
DATE OF DOCUMENT
10-29-09 NAME OF DRUG Tekamlo (aliskiren/amlodipine)
PRIORITY CONSIDERATION Standard
CLASSIFICATION OF DRUG Combination (aliskiren/amlodipine)
DESIRED COMPLETION DATE
5-8-10 NAME OF FIRM: Novartis
REASON FOR REQUEST
I. GENERAL
NEW PROTOCOL PROGRESS REPORT NEW CORRESPONDENCE DRUG ADVERTISING ADVERSE REACTION REPORT MANUFACTURING CHANGE/ADDITION MEETING PLANNED BY
PRE--NDA MEETING END OF PHASE II MEETING RESUBMISSION SAFETY/EFFICACY PAPER NDA CONTROL SUPPLEMENT
RESPONSE TO DEFICIENCY LETTER FINAL PRINTED LABELING LABELING REVISION ORIGINAL NEW CORRESPONDENCE FORMULATIVE REVIEW
⌧ OTHER (SPECIFY BELOW):
II. BIOMETRICS STATISTICAL EVALUATION BRANCH
STATISTICAL APPLICATION BRANCH
TYPE A OR B NDA REVIEW END OF PHASE II MEETING CONTROLLED STUDIES PROTOCOL REVIEW OTHER (SPECIFY BELOW):
CHEMISTRY REVIEW PHARMACOLOGY BIOPHARMACEUTICS OTHER (SPECIFY BELOW):
III. BIOPHARMACEUTICS
DISSOLUTION BIOAVAILABILTY STUDIES PHASE IV STUDIES
DEFICIENCY LETTER RESPONSE PROTOCOL-BIOPHARMACEUTICS IN-VIVO WAIVER REQUEST
IV. DRUG EXPERIENCE
PHASE IV SURVEILLANCE/EPIDEMIOLOGY PROTOCOL DRUG USE e.g. POPULATION EXPOSURE, ASSOCIATED DIAGNOSES CASE REPORTS OF SPECIFIC REACTIONS (List below) COMPARATIVE RISK ASSESSMENT ON GENERIC DRUG GROUP
REVIEW OF MARKETING EXPERIENCE, DRUG USE AND SAFETY SUMMARY OF ADVERSE EXPERIENCE POISON RISK ANALYSIS
V. SCIENTIFIC INVESTIGATIONS
CLINICAL
PRECLINICAL
COMMENTS/SPECIAL INSTRUCTIONS: DRISK, please review the sponsor’s Risk Management Plan, they propose routine pharmacovigilance and post marketing studies. Application is in DARRTS. SIGNATURE OF REQUESTER Michael Monteleone
METHOD OF DELIVERY (Check one)
MAIL ⌧DARRTS HAND SIGNATURE OF RECEIVER
SIGNATURE OF DELIVERER

ApplicationType/Number
SubmissionType/Number Submitter Name Product Name
-------------------- -------------------- -------------------- ------------------------------------------NDA-22545 ORIG-1 NOVARTIS
PHARMACEUTICALS CORP
ALISKIREN/AMLODPINE(SPA100A)FIXED COMBO
---------------------------------------------------------------------------------------------------------This is a representation of an electronic record that was signedelectronically and this page is the manifestation of the electronicsignature.---------------------------------------------------------------------------------------------------------/s/----------------------------------------------------
MICHAEL V MONTELEONE04/08/2010

DEPARTMENT OF HEALTH AND HUMAN SERVICES
Food and Drug Administration Silver Spring MD 20993
NDA 22-545 INFORMATION REQUEST
Novartis Pharmaceuticals Corporation Attention: Lori Ann Kneafsey
Associate Director, Drug Regulatory Affairs One Health Plaza East Hanover, NJ 07936-1080
Dear Ms. Kneafsey: Please refer to your New Drug Application (NDA) submitted under section 505(b) of the Federal Food, Drug, and Cosmetic Act for aliskiren-amlodipine tablets. We are reviewing the Chemistry, Manufacturing, and Controls sections of your submission and have the following comments and information requests. We request a prompt written response in order to continue our evaluation of your NDA.
Your dissolution data on the 150/10 mg strength could not be located in the submission. Before the dissolution data and the proposed dissolution methodology and specifications can be reviewed thoroughly, you need to submit the mean and individual dissolution data
on the 150/10 mg strength (biobatch No. AEUS/2008/0183) using the above proposed methodology. If you already submitted the data, please provide the location (i.e., Module, Section, Volume, and Page Nos.).
If you have any questions, call Don Henry, Regulatory Project Manager, at (301) 796-4227.
Sincerely, {See appended electronic signature page} Ramesh Sood, Ph.D. Branch Chief Division of Pre-Marketing Assessment I Office of New Drug Quality Assessment Center for Drug Evaluation and Research
(b) (4)

ApplicationType/Number
SubmissionType/Number Submitter Name Product Name
-------------------- -------------------- -------------------- ------------------------------------------NDA-22545 ORIG-1 NOVARTIS
PHARMACEUTICALS CORP
ALISKIREN/AMLODPINE(SPA100A)FIXED COMBO
---------------------------------------------------------------------------------------------------------This is a representation of an electronic record that was signedelectronically and this page is the manifestation of the electronicsignature.---------------------------------------------------------------------------------------------------------/s/----------------------------------------------------
RAMESH K SOOD02/26/2010

DEPARTMENT OF HEALTH & HUMAN SERVICES
Public Health Service
Food and Drug Administration
Silver Spring, MD 20993
NDA 022545 PROPRIETARY NAME REQUEST CONDITIONALLY ACCEPTABLE
Novartis Pharmaceuticals Corporation One Health Plaza East Hanover, New Jersey 07936 Attention: Lori Ann Kneafsey Associate Director, Drug Regulatory Affairs Dear Ms. Kneafsey: Please refer to your New Drug Application (NDA) dated October 28, 2009, received October 29, 2009, submitted under section 505(b) of the Federal Food, Drug, and Cosmetic Act for Aliskiren and Amlodipine Tablets, 150 mg/5 mg, 150 mg/10 mg, 300 mg/5 mg, and 300 mg/10 mg. We also refer to your October 30, 2009, correspondence, received November 4, 2009, requesting review of your proposed proprietary name, Tekamlo. We have completed our review of the proposed proprietary name, Tekamlo and have concluded that it is acceptable. The proposed proprietary name, Tekamlo, will be re-reviewed 90 days prior to the approval of the NDA. If we find the name unacceptable following the re-review, we will notify you. If any of the proposed product characteristics as stated in your October 30, 2009 submission are altered prior to approval of the marketing application, the proprietary name should be resubmitted for review. If you have any questions regarding the contents of this letter or any other aspects of the proprietary name review process, contact Nina Ton, Safety Regulatory Project Manager in the Office of Surveillance and Epidemiology, at 301-796-1648. For any other information regarding this application contact the Office of New Drugs (OND) Regulatory Project Manager, Michael Monteleone at 301-796-1952.
Sincerely, {See appended electronic signature page}
Denise Toyer, Pharm.D. Deputy Director Division of Medication Error Prevention and Analysis Office of Surveillance and Epidemiology Center for Drug Evaluation and Research

ApplicationType/Number
SubmissionType/Number Submitter Name Product Name
-------------------- -------------------- -------------------- ------------------------------------------NDA-22545 ORIG-1 NOVARTIS
PHARMACEUTICALS CORP
ALISKIREN/AMLODPINE(SPA100A)FIXED COMBO
---------------------------------------------------------------------------------------------------------This is a representation of an electronic record that was signedelectronically and this page is the manifestation of the electronicsignature.---------------------------------------------------------------------------------------------------------/s/----------------------------------------------------
DENISE P TOYER02/02/2010

DEPARTMENT OF HEALTH AND HUMAN SERVICES
Food and Drug Administration Silver Spring MD 20993
NDA 022545 FILING COMMUNICATION Novartis Pharmaceuticals Corporation Attention: Lori Kneafsey Associate Director, Drug Regulatory Affairs One Health Plaza East Hanover, NJ 07936 Dear Ms. Kneafsey: Please refer to your new drug application (NDA) dated October 28, 2009, received October 29, 2009, submitted pursuant to section 505(b)(2) of the Federal Food, Drug, and Cosmetic Act, for Tekamlo, (aliskiren/amlodipine) 150/5mg, 150/10mg, 300/5mg and 300/10mg Tablets. We also refer to your submissions dated October 4, 16 and December 7, 2009. We have completed our filing review and have determined that your application is sufficiently complete to permit a substantive review. Therefore, this application is considered filed 60 days after the date we received your application in accordance with 21 CFR 314.101(a). The review classification for this application is Standard. Therefore, the user fee goal date is August 29, 2010. We are reviewing your application according to the processes described in the Guidance for Review Staff and Industry: Good Review Management Principles and Practices for PDUFA Products. Therefore, we have established internal review timelines as described in the guidance, which includes the timeframes for FDA internal milestone meetings (e.g., filing, planning, mid-cycle, team and wrap-up meetings). Please be aware that the timelines described in the guidance are flexible and subject to change based on workload and other potential review issues (e.g., submission of amendments). We will inform you of any necessary information requests or status updates following the milestone meetings or at other times, as needed, during the process. If major deficiencies are not identified during the review, we plan to communicate proposed labeling and, if necessary, any postmarketing commitment requests by June 29, 2010. At this time, we are notifying you that, we have not identified any potential review issues. Please note that our filing review is only a preliminary evaluation of the application and is not indicative of deficiencies that may be identified during our review. Under the Pediatric Research Equity Act (PREA) (21 U.S.C. 355c), all applications for new active ingredients, new indications, new dosage forms, new dosing regimens, or new routes of administration are required to contain an assessment of the safety and effectiveness of the

NDA 020545 Page 2
product for the claimed indication(s) in pediatric patients unless this requirement is waived, deferred, or inapplicable. We acknowledge receipt of your request for a full waiver of pediatric studies for this application. Once we have reviewed your request, we will notify you if the full waiver request is denied and a pediatric drug development plan is required. If you have any questions, please call Michael Monteleone, Regulatory Project Manager, at (301) 796-1952.
Sincerely, {See appended electronic signature page} Norman Stockbridge, M.D., Ph.D Director Division of Cardiovascular and Renal Products Office of Drug Evaluation I Center for Drug Evaluation and Research

ApplicationType/Number
SubmissionType/Number Submitter Name Product Name
-------------------- -------------------- -------------------- ------------------------------------------NDA-22545 ORIG-1 NOVARTIS
PHARMACEUTICALS CORP
ALISKIREN/AMLODPINE(SPA100A)FIXED COMBO
---------------------------------------------------------------------------------------------------------This is a representation of an electronic record that was signedelectronically and this page is the manifestation of the electronicsignature.---------------------------------------------------------------------------------------------------------/s/----------------------------------------------------
NORMAN L STOCKBRIDGE12/23/2009

DEPARTMENT OF HEALTH AND HUMAN SERVICES
PUBLIC HEALTH SERVICE FOOD AND DRUG ADMINISTRATION
REQUEST FOR CONSULTATION
TO (Office/Division): Raanan Bloom, OPS/PARS, (301)796- 2185
FROM (Name, Office/Division, and Phone Number of Requestor): Don Henry Project Manager, ONDQA, 301-796-4227 on behalf of L. Soldatova/K. Srinivasachar
DATE
12/8/2009
IND NO.
NDA NO. 22-545
TYPE OF DOCUMENT original submission
DATE OF DOCUMENT 10/29/2009
NAME OF DRUG
Tekamlo(aliskiren-amlodipine)
PRIORITY CONSIDERATION
standard
CLASSIFICATION OF DRUG
cardio-renal
DESIRED COMPLETION DATE
3/30/2010
NAME OF FIRM: Novartis Pharmaceuticals
REASON FOR REQUEST
I. GENERAL
NEW PROTOCOL PROGRESS REPORT NEW CORRESPONDENCE DRUG ADVERTISING ADVERSE REACTION REPORT MANUFACTURING CHANGE / ADDITION MEETING PLANNED BY
PRE-NDA MEETING END-OF-PHASE 2a MEETING END-OF-PHASE 2 MEETING RESUBMISSION SAFETY / EFFICACY PAPER NDA CONTROL SUPPLEMENT
RESPONSE TO DEFICIENCY LETTER FINAL PRINTED LABELING LABELING REVISION ORIGINAL NEW CORRESPONDENCE FORMULATIVE REVIEW OTHER (SPECIFY BELOW):
II. BIOMETRICS
PRIORITY P NDA REVIEW END-OF-PHASE 2 MEETING CONTROLLED STUDIES PROTOCOL REVIEW OTHER (SPECIFY BELOW):
CHEMISTRY REVIEW PHARMACOLOGY BIOPHARMACEUTICS OTHER (SPECIFY BELOW):
III. BIOPHARMACEUTICS
DISSOLUTION BIOAVAILABILTY STUDIES PHASE 4 STUDIES
DEFICIENCY LETTER RESPONSE PROTOCOL - BIOPHARMACEUTICS IN-VIVO WAIVER REQUEST
IV. DRUG SAFETY
PHASE 4 SURVEILLANCE/EPIDEMIOLOGY PROTOCOL DRUG USE, e.g., POPULATION EXPOSURE, ASSOCIATED DIAGNOSES CASE REPORTS OF SPECIFIC REACTIONS (List below) COMPARATIVE RISK ASSESSMENT ON GENERIC DRUG GROUP
REVIEW OF MARKETING EXPERIENCE, DRUG USE AND SAFETY SUMMARY OF ADVERSE EXPERIENCE POISON RISK ANALYSIS
V. SCIENTIFIC INVESTIGATIONS
CLINICAL
NONCLINICAL
COMMENTS / SPECIAL INSTRUCTIONS: The expected introduction concentration for one of the active moieties, amlodipine besylate, is less that 1 ppb. The expected introduction concentration for the second active moiety, aliskiren hemifumarate, exceeds the acceptable limit of 1 ppb and therefore, the Environmental Assessment requires evaluation. This is an electronic submission. SIGNATURE OF REQUESTOR
{See appended electronic signature page}
METHOD OF DELIVERY (Check one)
DFS EMAIL MAIL HAND
PRINTED NAME AND SIGNATURE OF RECEIVER
PRINTED NAME AND SIGNATURE OF DELIVERER
(b) (4)

ApplicationType/Number
SubmissionType/Number Submitter Name Product Name
-------------------- -------------------- -------------------- ------------------------------------------NDA-22545 ORIG-1 NOVARTIS
PHARMACEUTICALS CORP
ALISKIREN/AMLODPINE(SPA100A)FIXED COMBO
---------------------------------------------------------------------------------------------------------This is a representation of an electronic record that was signedelectronically and this page is the manifestation of the electronicsignature.---------------------------------------------------------------------------------------------------------/s/----------------------------------------------------
DON L HENRY12/08/2009
RAMESH K SOOD12/08/2009

DEPARTMENT OF HEALTH AND HUMAN SERVICES
Food and Drug Administration Silver Spring MD 20993
NDA 022545 NDA ACKNOWLEDGMENT Novartis Pharmaceuticals Corporation Attention: Lori Ann Kneafsey Associate Director Drug Regulatory Affairs One Health Plaza East Hanover, NJ 07936 Dear Ms. Kneafsey: We have received your new drug application (NDA) submitted section 505(b)(2) of the Federal Food, Drug, and Cosmetic Act (FDCA) for the following: Name of Drug Product: Tekamlo™ (aliskiren-amlodipine) Tablets Date of Application: October 28, 2009 Date of Receipt: October 29, 2009 Our Reference Number: NDA 022545 Unless we notify you within 60 days of the receipt date that the application is not sufficiently complete to permit a substantive review, we will file the application on December 28, 2009 in accordance with 21 CFR 314.101(a). If you have not already done so, promptly submit the content of labeling [21 CFR 314.50(l)(1)(i)] in structured product labeling (SPL) format as described at http://www.fda.gov/oc/datacouncil/spl.html. Failure to submit the content of labeling in SPL format may result in a refusal-to-file action under 21 CFR 314.101(d)(3). The content of labeling must conform to the content and format requirements of revised 21 CFR 201.56-57. The NDA number provided above should be cited at the top of the first page of all submissions to this application. Send all submissions, electronic or paper, including those sent by overnight mail or courier, to the following address:
Food and Drug Administration Center for Drug Evaluation and Research Division of Cardiovascular and Renal Products 5901-B Ammendale Road Beltsville, MD 20705-1266

NDA 022545 Page 2
All regulatory documents submitted in paper should be three-hole punched on the left side of the page and bound. The left margin should be at least three-fourths of an inch to assure text is not obscured in the fastened area. Standard paper size (8-1/2 by 11 inches) should be used; however, it may occasionally be necessary to use individual pages larger than standard paper size. Non-standard, large pages should be folded and mounted to allow the page to be opened for review without disassembling the jacket and refolded without damage when the volume is shelved. Shipping unbound documents may result in the loss of portions of the submission or an unnecessary delay in processing which could have an adverse impact on the review of the submission. For additional information, please see http://www.fda.gov/cder/ddms/binders.htm. If you have any questions, please contact:
Mr. Michael Monteleone, M.S. Regulatory Health Project Manager (301) 796-1952
Sincerely, {See appended electronic signature page} Edward Fromm, R.Ph., RAC Chief, Project Management Staff Division of Cardiovascular and Renal Products Office of Drug Evaluation I Center for Drug Evaluation and Research

ApplicationType/Number
SubmissionType/Number Submitter Name Product Name
-------------------- -------------------- -------------------- ------------------------------------------NDA-22545 ORIG-1 NOVARTIS
GRIMSBY LTDALISKIREN/AMLODPINE(SPA100A)FIXED COMBO
---------------------------------------------------------------------------------------------------------This is a representation of an electronic record that was signedelectronically and this page is the manifestation of the electronicsignature.---------------------------------------------------------------------------------------------------------/s/----------------------------------------------------
EDWARD J FROMM11/03/2009





























