Embed Size (px)
Citation preview

1
UC J. León
Tema 27
Tema 27. Metabolismo de las cadenas hidrocarbonadas de los aminoácidos.
Panorámica de las rutas de degradación y destino de las cadenas de los aminoácidos . Co-enzimas transportadoras de restos monocarbonados: tetrahidrofolato, S-adenosil-metionina, cobalamina. Aminoácidos glucogénicos y cetogénicos. Destino metabólicos de los aminoácidos. El glutamato como precursor. Degradación de la fenilalanina. Fenilcetonuria, tirosinemias, alcaptonuria. Degradación de aminoácidos de cadenas ramificadas. Metilmalonil mutasa y 5'-dexosiadenosil cobalamina. Patología asociada. Esquema de la biosíntesis de los aminoácidos no esenciales. Aminoácidos como precursores de otras aminas biológicas. Síntesis y degradación del grupo hemo. Metabolismo del hierro. Porfirias.
BIOQUÍMICA-1º de Medicina Departamento de Biología Molecular
Javier León
2
UC J. León
Tema 27
Glucosa
AcidosGrasos
NADH+
CO2
+ H2O
Catabolismo de los esqueletos carbonados de los aminoácidos
Pentosas--fosfato
Glu
Arg, His, Gln, Pro
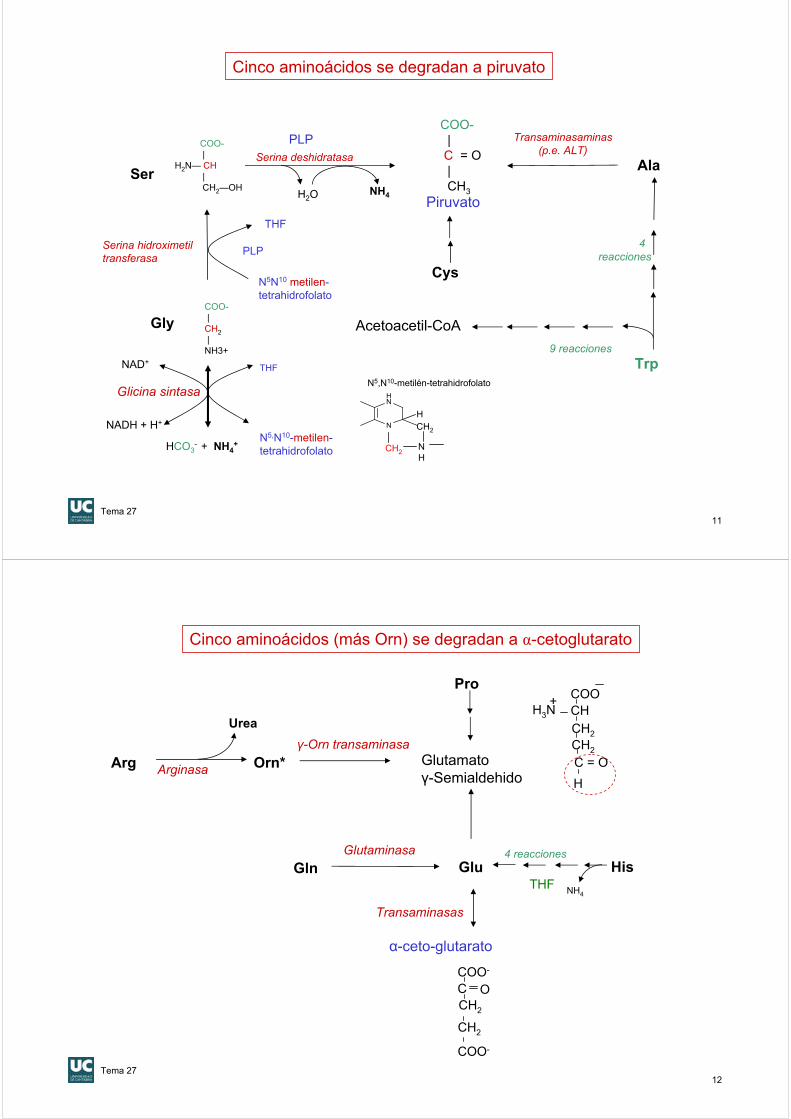
• Cetogénicos y glucogénicos:Ile, Phe, Tyr, Trp
Sólo glucogénicos:Gly, Ala, Val, Pro, Ser, ThrCys, Met,Asp, Asn, Glu, GlnHis, Arg
Sólo cetogénicos: Leu, Lys
Gly, Ala, SerCys, Trp
Lys, LeuIle, Phe,Tyr, Trp
Ciclo deKrebs
OAA
α-ceto-glutaratoSuccinil-CoA
Fumarato
Citrato
IsocitratoMalato
Succinato
Piruvato Acetil CoA
Phe, Tyr
Val, Ile, Thr, Met
Asn, Asp
Glucógeno
3
UC J. León
Tema 27
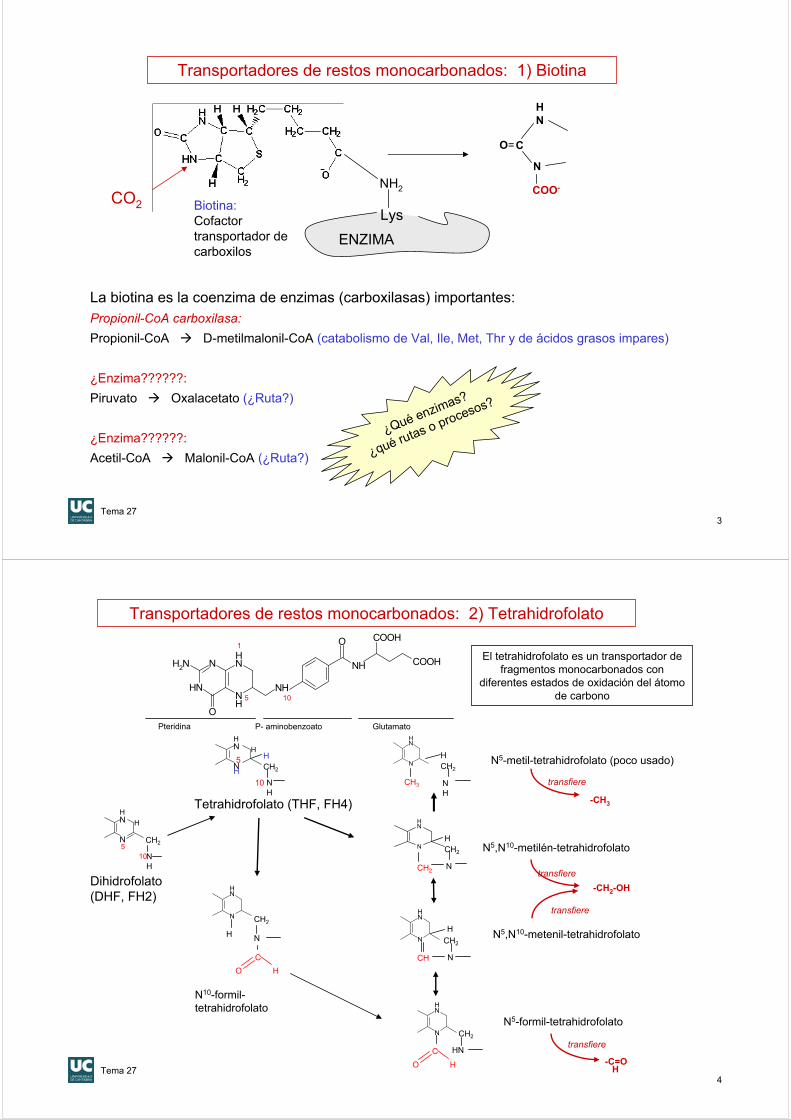
Biotina:Cofactor transportador de carboxilos
NH2
Lys
ENZIMA
CO2
Transportadores de restos monocarbonados: 1) Biotina
La biotina es la coenzima de enzimas (carboxilasas) importantes:Propionil-CoA carboxilasa:
Propionil-CoA D-metilmalonil-CoA (catabolismo de Val, Ile, Met, Thr y de ácidos grasos impares)
¿Enzima??????:
Piruvato Oxalacetato (¿Ruta?)
¿Enzima??????:
Acetil-CoA Malonil-CoA (¿Ruta?)
¿Qué enzimas?
¿qué rutas o procesos?
N
COO-
CO=
NH
4
UC J. León
Tema 27
Pteridina P- aminobenzoato Glutamato
CH2
H
NH
CH3
N
NH
CH2
H
NCH
N
NH
N
NH
NH
NH2
NH NH
O
O
NH
COOH
COOH
5 10
1
El tetrahidrofolato es un transportador de fragmentos monocarbonados con
diferentes estados de oxidación del átomo de carbono
CH2
H
NCH2
N
NH
N5,N10-metilén-tetrahidrofolato
N
NH
CH2
HNC
O H
N5-formil-tetrahidrofolato
N5,N10-metenil-tetrahidrofolato
N5-metil-tetrahidrofolato (poco usado)
N
NH
CH2
NH
N10-formil-tetrahidrofolato
CH2
H
NH
5
10
Tetrahidrofolato (THF, FH4)
NH
NH
H
Transportadores de restos monocarbonados: 2) Tetrahidrofolato
-CH3
transfiere
-C=OH
transfiere
-CH2-OH
transfiere
transfiere
C
O H
Dihidrofolato(DHF, FH2)
NH
CH25
10
N
NH
H

5
UC J. León
Tema 27
N
N
N
N
N H 2
O
OH OH
CH2P
P
P
COO
CH
CH2
CH2
H3N
S
+
CH3
Metionina
ATP
S-Adenosil metionina (AdoMet): transfiere metilos
COO
CH
CH2
CH2
H3N
S
+
CH3
N
N
N
N
N H 2
O
OH OH
CH2+
Metionina adenosiltransferasaPPi + Pi
COO
CH
CH2
CH2
H3N
S
+
N
N
N
N
N H 2
O
OH OH
CH2
S-Adenosil homocisteína
R R-CH3
Varias Metil-transferasas
+
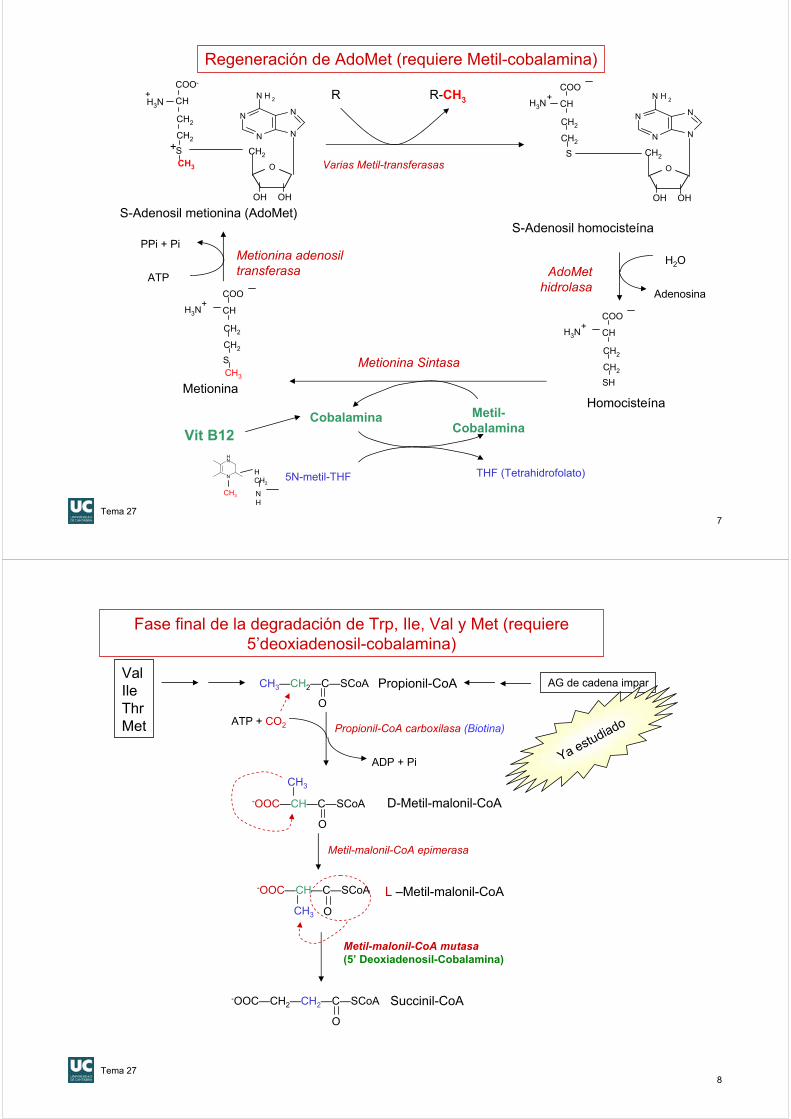
Transportadores de restos monocarbonados: 3) S-adenosilmetionina, AdoMet
6
UC J. León
Tema 27
Descubierta en estudios de anemia perniciosa en 1926Estructura de la ciano-B12 resuelta por Dorothy Crowfoot HodgkinSe presenta en dos formas que participan en dos rutas del metabolismo humano
Catabolismo AA y AG
Sintesis de Met y de AdoMet
Transportadores de restos monocarbonados: 4) Cobalamina
Dorothy Crowfoot Hodgkin (1910-1994). Premio Nobel de Química 1964 "for her determinations by X-ray techniques of the structures of important biochemical substances“ (sobre todoPenicilina y Vit. B12)
- “Biochemistry”. 5ª ed. Berg, J., Tymoczko, J. Stryer, L. Ed. W.H. Freeman. 2003.
7
UC J. León
Tema 27
COO-
CH
CH2
CH2
H3N
S
+
CH3
N
N
N
N
N H 2
O
OH OH
CH2
S-Adenosil metionina (AdoMet)
COO
CH
CH2
CH2
S
H3N+
N
N
N
N
N H 2
O
CH2
R R-CH3
+
Varias Metil-transferasas
Regeneración de AdoMet (requiere Metil-cobalamina)
OH OH
S-Adenosil homocisteína
COO
CH
CH2
CH2
H3N
SH
+
Homocisteína
AdoMethidrolasa
H2O
Adenosina
5N-metil-THF THF (Tetrahidrofolato)CH2
H
NH
CH3
N
NH
COO
CH
CH2
CH2
H3N
S
+
CH3
Metionina
Metil-Cobalamina
Cobalamina
Metionina Sintasa
Vit B12
Metionina adenosiltransferasa
ATP
PPi + Pi
8
UC J. León
Tema 27
CH3—CH2—C—SCoA
O
-OOC—CH—C—SCoA
CH3
-OOC—CH—C—SCoA
CH3
-OOC—CH2—CH2—C—SCoA
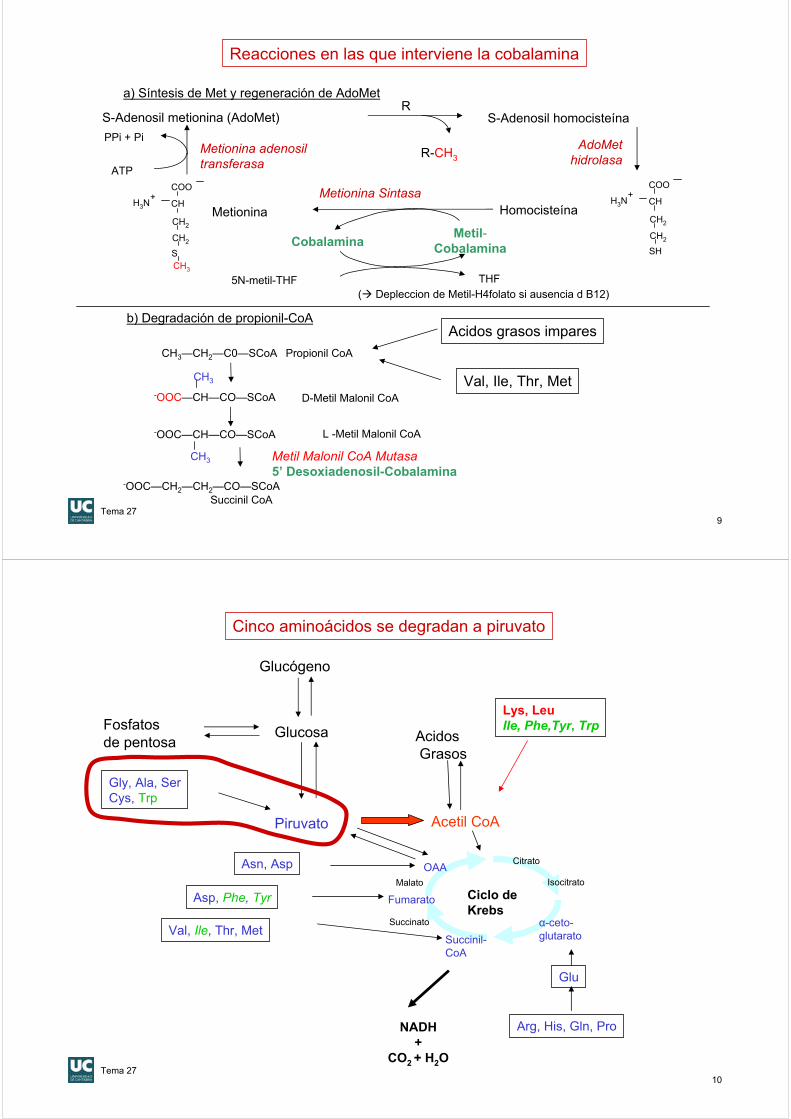
Propionil-CoA
Propionil-CoA carboxilasa (Biotina)ATP + CO2
D-Metil-malonil-CoA
Metil-malonil-CoA epimerasa
L –Metil-malonil-CoA
Metil-malonil-CoA mutasa(5’ Deoxiadenosil-Cobalamina)
Succinil-CoA
Fase final de la degradación de Trp, Ile, Val y Met (requiere 5’deoxiadenosil-cobalamina)
O
O
O
ValIleThrMet
ADP + Pi
AG de cadena impar
Ya estudiado
9
UC J. León
Tema 27
S-Adenosil metionina (AdoMet) S-Adenosil homocisteína
COO
CH
CH2
CH2
H3N
SH
+
Homocisteína
COO
CH
CH2
CH2
H3N
S
+
CH3
Metionina
Metil-CobalaminaCobalamina
Metionina Sintasa
Metionina adenosiltransferasa
AdoMethidrolasa
5N-metil-THF THF
ATP
PPi + Pi
a) Síntesis de Met y regeneración de AdoMet
Reacciones en las que interviene la cobalamina
-OOC—CH2—CH2—CO—SCoA
L -Metil Malonil CoA
Metil Malonil CoA Mutasa5’ Desoxiadenosil-Cobalamina
Succinil CoA
CH3—CH2—C0—SCoA Propionil CoA
-OOC—CH—CO—SCoA
CH3
D-Metil Malonil CoA
-OOC—CH—CO—SCoA
CH3
b) Degradación de propionil-CoAAcidos grasos impares
Val, Ile, Thr, Met
R-CH3
R
( Depleccion de Metil-H4folato si ausencia d B12)
10
UC J. León
Tema 27
Glucosa AcidosGrasos
Glucógeno
NADH+
CO2 + H2O
Fosfatos de pentosa
Glu
Arg, His, Gln, Pro
Gly, Ala, SerCys, Trp
Asp, Phe, Tyr
Val, Ile, Thr, Met
Asn, Asp
Ciclo deKrebs
OAA
α-ceto-glutaratoSuccinil-
CoA
Fumarato
Citrato
IsocitratoMalato
Succinato
Piruvato Acetil CoA
Cinco aminoácidos se degradan a piruvato
Lys, LeuIle, Phe,Tyr, Trp
11
UC J. León
Tema 27
HCO3- + NH4
+
THF
N5,N10-metilen-tetrahidrofolato
NADH + H+
NAD+
Glicina sintasa
COO-|CH2|NH3+
Gly
CH2
H
NH
CH2
N
NH
N5,N10-metilén-tetrahidrofolato
Cys
Ala
Transaminasaminas(p.e. ALT)
Acetoacetil-CoA
Trp
4 reacciones
9 reacciones
Cinco aminoácidos se degradan a piruvato
COO-|
H2N— CH |CH2—OH
Ser
Serina hidroximetiltransferasa
THF
N5N10 metilen-tetrahidrofolato
PLP
COO-|
C = O|CH3
Piruvato
Serina deshidratasa
PLP
H2O NH4
12
UC J. León
Tema 27
Cinco aminoácidos (más Orn) se degradan a α-cetoglutarato
Glutamatoγ-Semialdehido
ProCOOCH
CH2
CH2
H3N
H
C = O
+
GlnGlutaminasa
α-ceto-glutarato
Transaminasas
COO-
CCH2
COO-
O
CH2
His
NH4
4 reacciones
THFGlu
Arg Orn*Arginasa
γ-Orn transaminasa
Urea

13
UC J. León
Tema 27
Asn Asp Oxalacetato
COO-
CCH2
COO-
O
COO
CH
CH2
H3N
C
+
OH2N
NH4+H2O
Asparaginasa Transaminasa
α-KG Glu
Asn y Asp se degradan a oxalacetato
14
UC J. León
Tema 27
Cuatro aminoácidos se degradan a succinil-CoA
Propionil-CoACH3-CH2-CO-S-CoA
α-cetobutiratoCH3-CH2-CO-COO-
Thr
NH4
H2O
Thrdeshidratasa
Val
7 reacciones
Met
5 reacciones
Ile
6 reacciones
Acetil-CoA
Metil-malonil-CoA
-OOC—CH —C—SCoA
O
CH3
4. Desaminación directa (Ser-dehidratasa y Thr dehidratasa)
Ser Piruvato + NH4+
Thr α-cetobutirato + NH4+
Recordatorio
Tema 26
CO2
Succinil-CoA
-OOC—CH2—CH2—C—SCoA
O
(PLP)
Biotina

15
UC J. León
Tema 27
CH3—CH2—C—SCoA
O
-OOC—CH—C—SCoA
CH3
-OOC—CH—C—SCoA
CH3
-OOC—CH2—CH2—C—SCoA
Propionil-CoA
Propionil-CoA-carboxilasa(Biotina)
ATP + CO2
D-Metil-malonil-CoA
Metil-malonil-CoA epimerasa
L –Metil-malonil-CoA
Metil Malonil-CoA Mutasa(5’ Desoxiadenosil-Cobalamina)
Succinil-CoA
Fase final de la degradación de Met, Trp, Ile, Val (pero no Leu)
O
O
O
Met, Thr, Ile, Val AG impares
Tema 23
16
UC J. León
Tema 27
Glucosa AcidosGrasos
Glucógeno
NADH+
CO2 + H2O
Fosfatos de pentosa
Glu
Arg, His, Gln, Pro
Gly, Ala, SerCys, Trp
Asp, Phe, Tyr
Val, Ile, Thr, Met
Asn, Asp
Acetoacetil-CoA
Lys, LeuPhe,Tyr, Trp
IleTrpLeu
Ciclo deKrebs
OAA
α-ceto-glutaratoSuccinil-
CoA
Fumarato
Citrato
IsocitratoMalato
Succinato
Piruvato Acetil CoA
Seis aminoácidos se degradan a acetil-CoA
Tiolasa
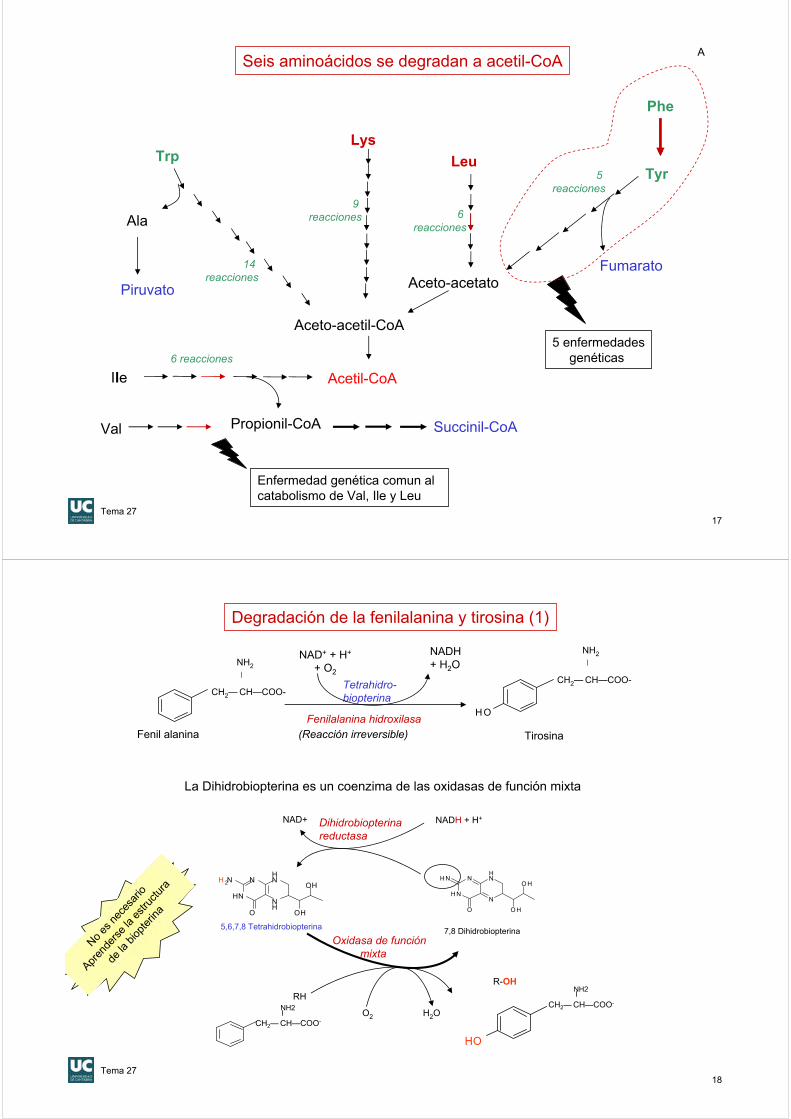
17
UC J. León
Tema 27
Seis aminoácidos se degradan a acetil-CoA
Aceto-acetil-CoA
Acetil-CoA
5 enfermedadesgenéticas
Aceto-acetato
Phe
Tyr5 reacciones
Fumarato
Lys
9 reacciones
Ile6 reacciones
Leu
6 reacciones
Trp
Ala
14 reacciones
Piruvato
Propionil-CoA Succinil-CoAVal
Enfermedad genética comun al catabolismo de Val, Ile y Leu
A
18
UC J. León
Tema 27
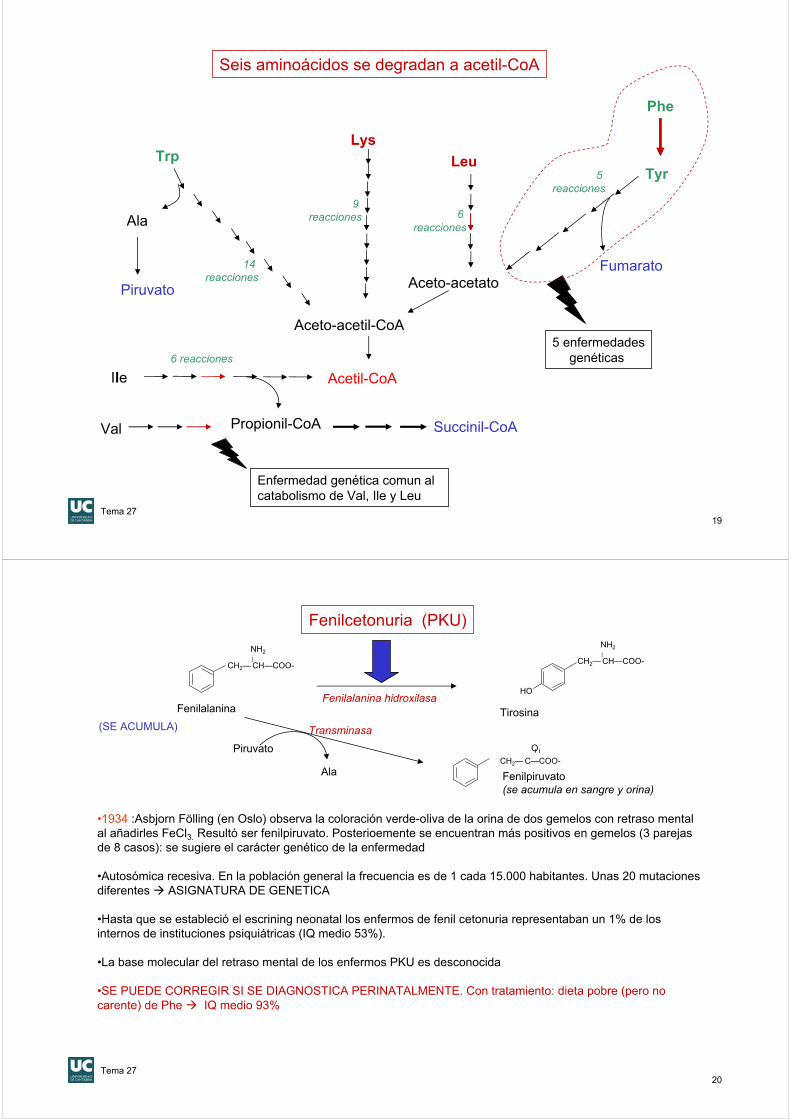
La Dihidrobiopterina es un coenzima de las oxidasas de función mixta
Degradación de la fenilalanina y tirosina (1)
N
NH
NH
O O H
O HNNH
5,6,7,8 Tetrahidrobiopterina 7,8 Dihidrobiopterina
NADH + H+NAD+ Dihidrobiopterinareductasa
RH
R-OH
O2 H2O
Oxidasa de función mixta
CH2— CH—COO-
OH
NH2
CH2— CH—COO-
NH2
N
NH
NH
NH 2
NH
O OH
OH
OH
NH2|
CH2— CH—COO-
Tirosina
NH2|
CH2— CH—COO-
Fenil alaninaFenilalanina hidroxilasa
(Reacción irreversible)
Tetrahidro-biopterina
NAD+ + H+
+ O2
NADH + H2O
No es
nec
esar
io
Apren
ders
e la
estru
ctur
a
de la
biop
terin
a
19
UC J. León
Tema 27
Seis aminoácidos se degradan a acetil-CoA
Aceto-acetil-CoA
Acetil-CoA
5 enfermedadesgenéticas
Aceto-acetato
Phe
Tyr5 reacciones
Fumarato
Lys
9 reacciones
Ile6 reacciones
Leu
6 reacciones
Trp
Ala
14 reacciones
Piruvato
Propionil-CoA Succinil-CoAVal
Enfermedad genética comun al catabolismo de Val, Ile y Leu
20
UC J. León
Tema 27
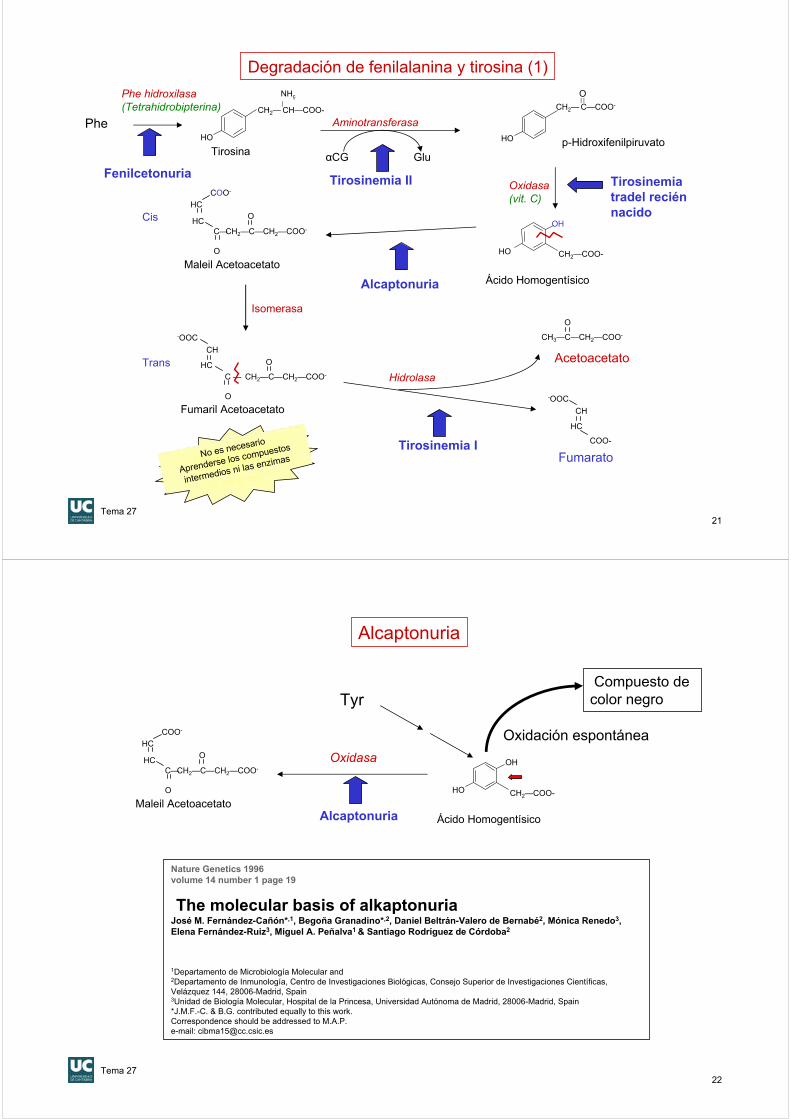
Fenilcetonuria (PKU)
•1934 :Asbjorn Fölling (en Oslo) observa la coloración verde-oliva de la orina de dos gemelos con retraso mental al añadirles FeCl3. Resultó ser fenilpiruvato. Posterioemente se encuentran más positivos en gemelos (3 parejas de 8 casos): se sugiere el carácter genético de la enfermedad
•Autosómica recesiva. En la población general la frecuencia es de 1 cada 15.000 habitantes. Unas 20 mutaciones diferentes ASIGNATURA DE GENETICA
•Hasta que se estableció el escrining neonatal los enfermos de fenil cetonuria representaban un 1% de los internos de instituciones psiquiátricas (IQ medio 53%).
•La base molecular del retraso mental de los enfermos PKU es desconocida
•SE PUEDE CORREGIR SI SE DIAGNOSTICA PERINATALMENTE. Con tratamiento: dieta pobre (pero no carente) de Phe IQ medio 93%
OH
NH2|
CH2— CH—COO-
Tirosina
NH2|
CH2— CH—COO-
FenilalaninaFenilalanina hidroxilasa
CH2— C—COO-
O
Fenilpiruvato(se acumula en sangre y orina)
Ala
Piruvato
Transminasa(SE ACUMULA)
21
UC J. León
Tema 27
OH
NHç|
CH2— CH—COO-
OH
CH2— C—COO-
CH2—COO-
COO-
HC
HCC—
O
CH2—C—CH2—COO-
O
Maleil Acetoacetato
Ácido Homogentísico
p-HidroxifenilpiruvatoTirosina
Aminotransferasa
Oxidasa(vit. C)
Phe hidroxilasa(Tetrahidrobipterina)
Isomerasa
Hidrolasa
CH3—C—CH2—COO-
O
Acetoacetato
CH
HC
COO-
-OOC
Fumarato
Degradación de fenilalanina y tirosina (1)
No es necesario
Aprenderse los compuestos
intermedios ni las enzimas
Phe
CH
HCC —
O
CH2—C—CH2—COO-
O
-OOC
Fumaril Acetoacetato
αCG Glu
O| |
OH
OHCis
Trans
Fenilcetonuria Tirosinemia II Tirosinemiatradel recién nacido
Alcaptonuria
Tirosinemia I
22
UC J. León
Tema 27
Alcaptonuria
OH
OH
CH2—COO-
Ácido Homogentísico
Oxidasa
Alcaptonuria
COO-
HC
HCC—
O
CH2—C—CH2—COO-
O
Maleil Acetoacetato
Compuesto de color negro
Oxidación espontánea
Tyr
Nature Genetics 1996volume 14 number 1 page 19
The molecular basis of alkaptonuriaJosé M. Fernández-Cañón*,1, Begoña Granadino*,2, Daniel Beltrán-Valero de Bernabé2, Mónica Renedo3, Elena Fernández-Ruiz3, Miguel A. Peñalva1 & Santiago Rodríguez de Córdoba2
1Departamento de Microbiología Molecular and 2Departamento de Inmunología, Centro de Investigaciones Biológicas, Consejo Superior de Investigaciones Científicas, Velázquez 144, 28006-Madrid, Spain3Unidad de Biología Molecular, Hospital de la Princesa, Universidad Autónoma de Madrid, 28006-Madrid, Spain *J.M.F.-C. & B.G. contributed equally to this work. Correspondence should be addressed to M.A.P.e-mail: [email protected]

23
UC J. León
Tema 27
Fenilalanina
Tirosina
p-hidroxifenilpiruvato
Ac. Homogentísico
Maleil acetoacetato
Fumarato Acetoacetato
Fenilcetonuria (PKU)
Tirosinemia II
Alcaptonuria
Melanina
Albinismo
Fumaril acetoacetato
No es necesario
aprender la estructura ni los
nombres de los compuestos
intermedios
Resumen de enfermedades del catabolismo de la Phe
Phe
Cromatografía en capa fina de muestras de sangre de recién nacido
Leu –Phe –
Glu –
His –
F enilcetonúricos
Tyr –
Val -
Tyr -
Tirosinemia I
24
UC J. León
Tema 27
Aceto-acetil-CoA
Acetil-CoA
Aceto-acetato
Phe
Tyr5 reacciones
Fumarato
Lys
9 reacciones
Ile6 reacciones
Leu
6 reacciones
Trp
Ala
14 reacciones
Piruvato
Propionil-CoA Succinil-CoAVal
El fallo en esta reacción supone una enfermedad genéticacomun al catabolismo de Val, Ile y Leu
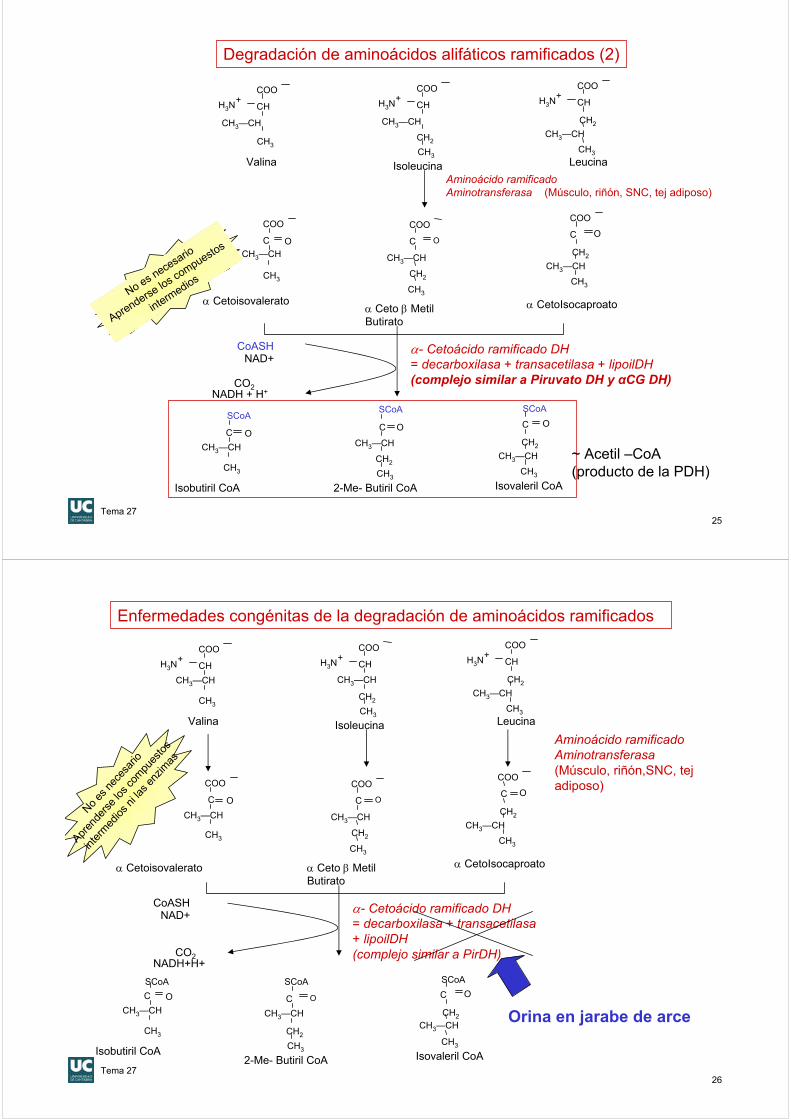
Degradación de aminoácidos alifáticos ramificados (1)
25
UC J. León
Tema 27
No es necesario
Aprenderse lo
s compuestos
intermedios
COO
CH
CH3—CH
H3N+
CH3
COO
CH
CH3—CH
H3N+
CH2
CH3
COO
CH
CH3—CH
H3N+
CH2
CH3
Aminoácido ramificadoAminotransferasa (Músculo, riñón, SNC, tej adiposo)
COO
C
CH3—CH
CH3
COO
C
CH3—CH
CH2
CH3
COO
C
CH3—CH
CH2
CH3
O OO
Valina Isoleucina Leucina
C
CH3—CH
CH3
CH3—CH
CH2
CH3
C
CH3—CH
CH2
CH3
OO
SCoASCoA SCoA
α- Cetoácido ramificado DH= decarboxilasa + transacetilasa + lipoilDH(complejo similar a Piruvato DH y αCG DH)
CoASH
CO2
NAD+
NADH + H+
Isobutiril CoA 2-Me- Butiril CoA Isovaleril CoA
α Cetoisovaleratoα Ceto β MetilButirato
α CetoIsocaproato
Degradación de aminoácidos alifáticos ramificados (2)
C O
~ Acetil –CoA(producto de la PDH)
26
UC J. León
Tema 27
COO
CH
CH3—CH
H3N+
CH3
COO
CH
CH3—CH
H3N+
CH2
CH3
COO
CH
CH3—CH
H3N+
CH2
CH3
Aminoácido ramificadoAminotransferasa(Músculo, riñón,SNC, tejadiposo)COO
C
CH3—CH
CH3
COO
C
CH3—CH
CH2
CH3
COO
C
CH3—CH
CH2
CH3
O OO
Valina Isoleucina Leucina
C
CH3—CH
CH3
C
CH3—CH
CH2
CH3
C
CH3—CHCH2
CH3
O O OSCoA SCoA SCoA
α- Cetoácido ramificado DH= decarboxilasa + transacetilasa+ lipoilDH(complejo similar a PirDH)
CoASH
CO2
NAD+
NADH+H+
Isobutiril CoA2-Me- Butiril CoA Isovaleril CoA
α Cetoisovalerato α Ceto β MetilButirato
α CetoIsocaproato
Enfermedades congénitas de la degradación de aminoácidos ramificados
Orina en jarabe de arce
No es
nec
esar
io
Apren
ders
e lo
s co
mpu
esto
s
inter
med
ios n
i las
enzim
as

27
UC J. León
Tema 27
C
CH3—CH
CH3
C
CH3—CH
CH2
CH3
C
CH3—CH
CH2
CH3
O O OSCoA SCoA SCoA
Isobutiril CoA 2-Metil-Butiril CoA Isovaleril CoA
Complejo Acil-CoA DH (FAD)
C
CH3—C
CH2CH
CH3
O
S-CoA
Metilacrilil CoA Tiglil CoA β- Metil crotonill CoA
Propionil CoA Acetil CoA Acetoacetato
Succinil CoA
Degradación de aminoácidos alifáticos ramificados (3)
Val Ile Leu
-OOC—CH2—CH2—C—SCoA
O
FADH2
C
CH3—C
O
S-CoA
C
CH3—C
O
S-CoA
CH3
FADH2FADH2
28
UC J. León
Tema 27
CH3—CH2—C—SCoA
O
-OOC—CH—C—SCoA
CH3
-OOC—CH—C—SCoA
CH3
-OOC—CH2—CH2—C—SCoA
Propionil-CoA
Propionil-CoA-carboxilasa(Biotina)
ATP + CO2
D-Metil-malonil-CoA
Metil-malonil-CoA epimerasa
L –Metil-malonil-CoA
Metil Malonil-CoA Mutasa(5’ Desoxiadenosil-Cobalamina)
Succinil-CoA
Fase final de la degradación de Met, Trp, Ile, Val (pero no Leu)
O
O
O
Met, Thr, Ile, Val AG impares
Acidmia metilmalónica
29
UC J. León
Tema 27
Aminoácidos esenciales
Aromáticos: Fenilalanina, Triptófano
Alifáticos ramific: Valina, Leucina, Isoleucina
Con azufre: Metionina
Alcohol: Treonina
Básicos: Histidina, Lisina
“Esenciales condicionales”:
Arginina, Glutamina, Cisteina, Glicina, Prolina, Tirosina
Todos los aminoácidos
Aromáticos: Fenilalanina, Triptófano, Tirosina
Alifáticos: Glicina, Alanina, Prolina
Alifáticos ramific.: Valina, Leucina, Isoleucina
Con azufre: Metionina, Cisteína
Alcohol: Serina, Treonina
Básicos: Arginina, Histidina, Lisina
Acidos: Aspartato, Glutamato
Amidas: Asparagina, Glutamina
AA no esenciales = BIOSINTETIZADOS
30
UC J. León
Tema 27
Biosíntesis de aminoácidos no esenciales
Piruvato transaminasa Alaα-Cetoglutarato transaminasa Glu Glu Gln sintetasa GlnGlu (4 pasos) ProOAA transaminasa Asp Asp Asn sintetasa AsnPhe Phe hidroxilasa Tyr3-fosfoglicerato (3 pasos) SerMet + Ser (3 pasos) Cys Ser Serhidroximetiltransferasa GlyHCO3
- + NH4+ Gly sintasa Gly
(Asp + Orn Arg-succinato liasa Arg)
PRECURSOR RUTA AMINOACIDO

31
UC J. León
Tema 27
Biosíntesis de aminoácidos no esenciales
Fenilalanina Tirosina
Glucosa
3-Fosfoglicerato
Piruvato
Alanina
Citrato
Isocitrato
Alfa- cetoglutarato Glutamato
Glutamina
Glutamato semialdehido
Prolina
CicloKrebs
OAA AcetilCoA
Serina
Glicina
CisteínaMetionina
HCO3- + NH4
+
Aspartato
Asparragina
Glutamina
Glu
32
UC J. León
Tema 27
COO-|
CH2
|NH2
COO-|
H2N— CH |CH2—OH
Serina
Piruvato
Serina deshidratasa
Glicina
Serina hidroximetil transferasa
THF
N5N10- metilen-tetrahidrofolato
Sintesis de serina y glicina
COO-|
C = O|CH3
COO-
|CH-OH|CH2-O-
3-Fosfoglicerato
H2O + NH4+
HCO3- + NH4
+
THF
N5,N10-metilen-tetrahidrofolatoNADH + H+
NAD+
Glicina sintasa
CH2
H
NH
CH2
N
NH
Glicina
Proteínas, especialmente colágeno
Glutation, creatina, porfirina, purinas
Recordatorio
P
NADH+H+ αKG Pi

33
UC J. León
Tema 27
COO+
COO
CH
CH2
CH2
H3N
S
+
CH3
N
N
N
N
N H 2
O
OH OH
CH2
S-Adenosil metionina (AdoMet)
+
Síntesis de cisteína: Adomet como donadora de átomos de azufre
CH
CH2
CH2
H3N
S
N
N
N
N
N H 2
O
OH OH
CH2
S-Adenosil homocisteína
COO
CH
CH2
CH2
H3N
SH
+
Homocisteína
Adenosilhomocisteínahidrolasa
Met
ATP PP+Pi
Homocistinuria
COO
CH
CH2OH
H3N+
COO
CH
CH2
CH2
H3N
S
+
COO
CH
CH2
H3N+
Serina
β-Cistationina
COO-
CH
CH2
H3N+
SH
NH4+
Cisteína
COO-
C
CH2
CH3
O
Alfa cetobutirato
Cistationina βsintasa(PLP)
Cistationina liasa(PLP)
H2O
34
UC J. León
Tema 27
Fotosensibilidad, ataxia, seudo-pelagra
Transportador de Val, Leu, Ile, Tyr, Trp, Phe
7Hartnup
Letargia, convulsiones, muerte prematura
Carbamilfosfato sintetasa I (ciclo de la urea)
0.5Hiperamonemia tipo I
Retraso mentalPhe hidroxilasa8Fenilcetonuria
Retraso mental, convulsiones, muerte temprana
Complejo deshidrogenasa de α-cetoácidos ramificados
0.4Orina en jarabe de arce
Retraso mental, convulsiones, muerte temprana
Metilmalonil-CoA mutasa0.5Acidemia metilmalónica
Retraso mentalCistationina sintasa1Homocistinuria
Orina oscuraHomogentísico oxigenasa0,4Alcaptonuria
No pigmentaciónTirosinasa3Albinismo
EfectosEnzima defectivaIncidencia
(por 105 nac.)
Enfermedad
Enfermedades del metabolismo de aminoácidos
No es necesario
aprenderse las incidencias

35
UC J. León
Tema 27
Sintesis de prolina
Arg Orn Glutamatoγ-Semialdehido
Prolina
Arginasa
*seis con la Orn
Ciclo deKrebs
α-ceto-glutaratoSuccinil-
CoA
Glu
Arg, His, Gln, Pro
Citrato
Isocitrato
GluGlutamina
α-ceto-glutarato
Transaminasas
COO
CH
CH2
CH2
H3N
HC = O
+
His
NH4
4 reaccionesGlutaminasa
Recordatorio
Purinas
Pirimidinas
36
UC J. León
Tema 27
Biosíntesis de aminoácidos no esenciales: Tyr a partir de Phe
OH
TirosinaFenilalanina
Fenilalanina hidroxilasa= Fenilalanina monoxigenasa
(Reacción irreversible)
Tetrahidro-biopterina
NAD+ + H+
+ O2
NADH + H2O
NH3+
CH2— CH—COO-NH3+
CH2— CH—COO-
Recordatorio

37
UC J. León
Tema 27
Los aminoácidos son precursores de otras moléculas importantes en la fisología
Glutamato GABA, poliaminas (putrescina, espermidina)
Triptofano Niacina, Serotonina
Histidina Histamina
Tirosina Dopamina, adrenalina, melanina
Lisina Carnitina
Serina Etanolamina, colina
Arginina Creatina, creatinina, oxido nítrico
Glicina Porfirinas, creatina, glutation
38
UC J. León
Tema 27
GlutamatoAlfa-cetoglutarato
Glutamina
GABA
Glutamatosemialdehido
Prolina
Ornitina
El glutamato como precursor de otras moléculas
Dador de NH2 a otros cetoácidos
Poliaminas
CH2
CH2
H2N CH2
COOH
Gamma-aminobutirato(GABA)
CO2
PLP
CH2
CH2
H2N - CH - COO-
COO-
Glutamato
39
UC J. León
Tema 27
H3N+
CH2
CH2
CH2
H2N – CH - COO-
Ornitina
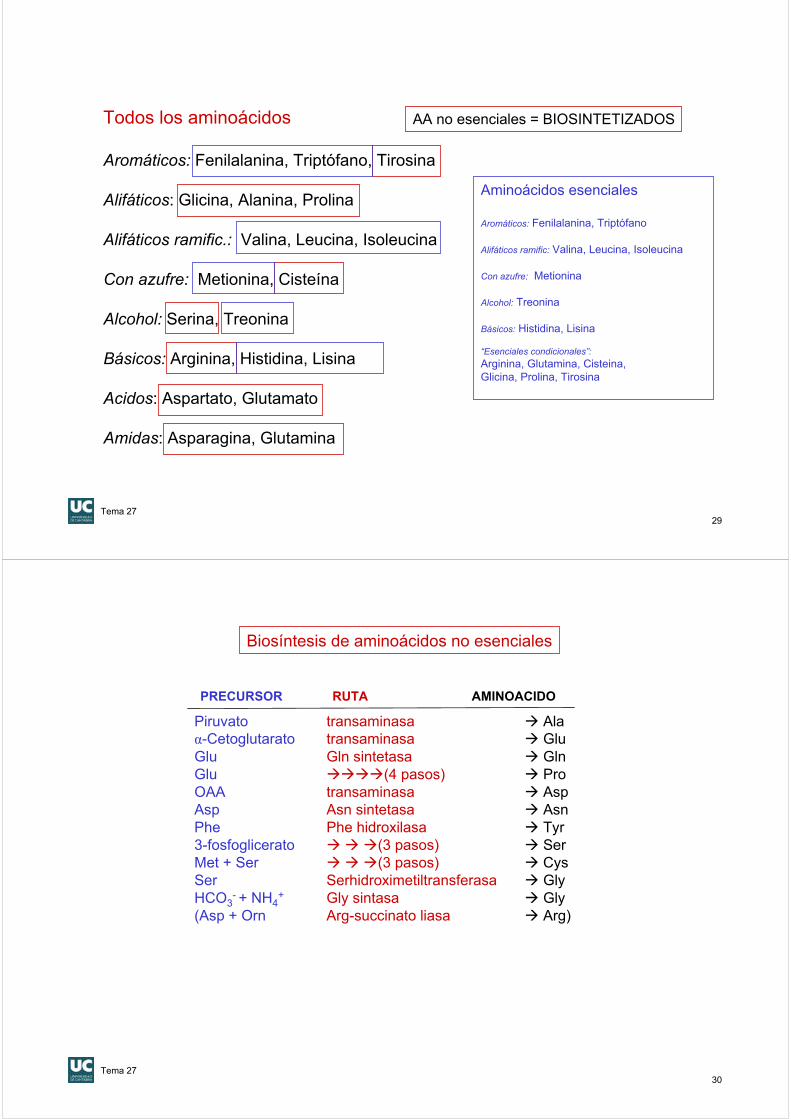
Ornitina descarboxilasa
CO2
H3N+
CH2
CH2
CH2
H2N CH2
Putrescina
Síntesis de poliaminas
H3N+
CH2
CH2
CH2
CH2
HN
CH2
CH2
CH2
H3N+
COO-
CH
CH2
CH2
H3N
S
+
CH3
N
N
N
N
N H2
O
OH OH
CH2
S-Adenosil metionina (AdoMet)
Espermidina
S
CH3
N
N
N
N
N H2
O
OH OH
CH2
Metil-tioadenosina
+CO2
40
UC J. León
Tema 27
DOPA Descarboxilasa = Decarboxilasa de AA aromáticos
(PLP)
H
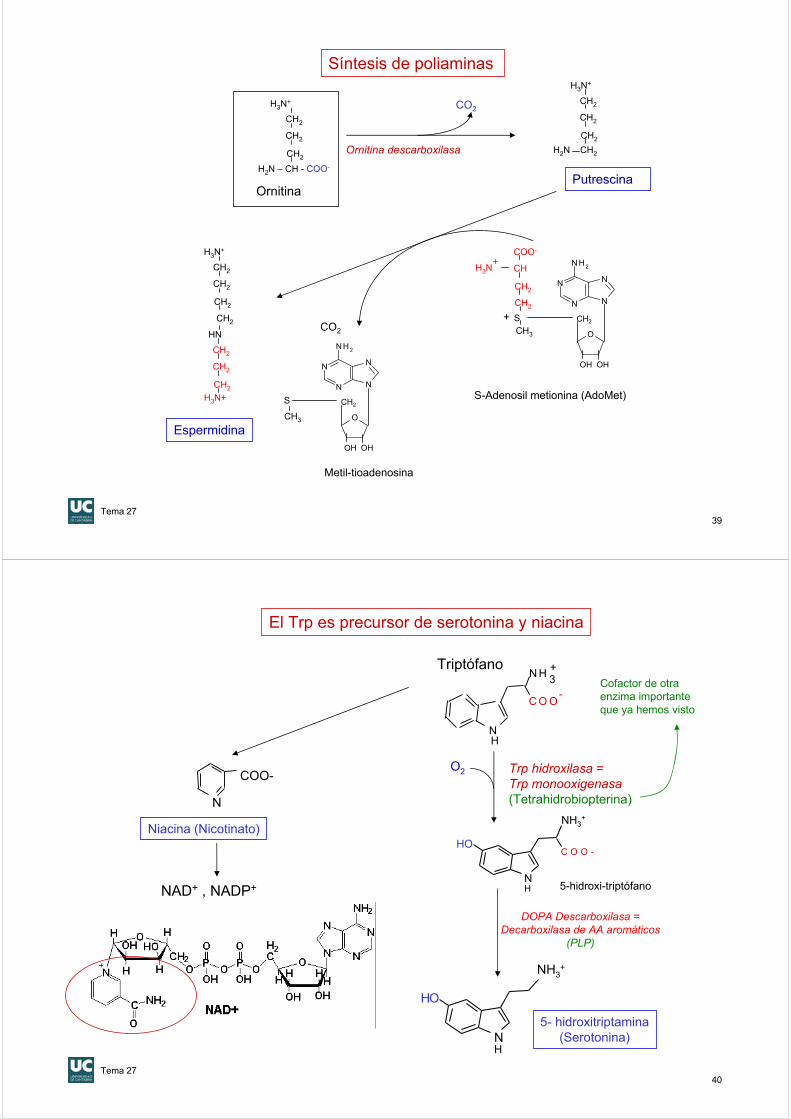
Triptófano
N
C O O-
N H 3+
Trp hidroxilasa = Trp monooxigenasa(Tetrahidrobiopterina)
O2
5- hidroxitriptamina(Serotonina)N
H
NH3+
OH
NH
NH3+
OHC O O -
5-hidroxi-triptófano
COO-
N
Niacina (Nicotinato)
NAD+ , NADP+
El Trp es precursor de serotonina y niacina
Cofactor de otraenzima importanteque ya hemos visto
41
UC J. León
Tema 27
OH
CH2
CH
O
NH3+
O
OH
CH2
CH2
NH3+
OH
OH
CH2
CH
NH3+
OH
O
O
Tirosina
L-DOPA(L-3,4-dihidroxi-
Fenilalanina)
Dopamina
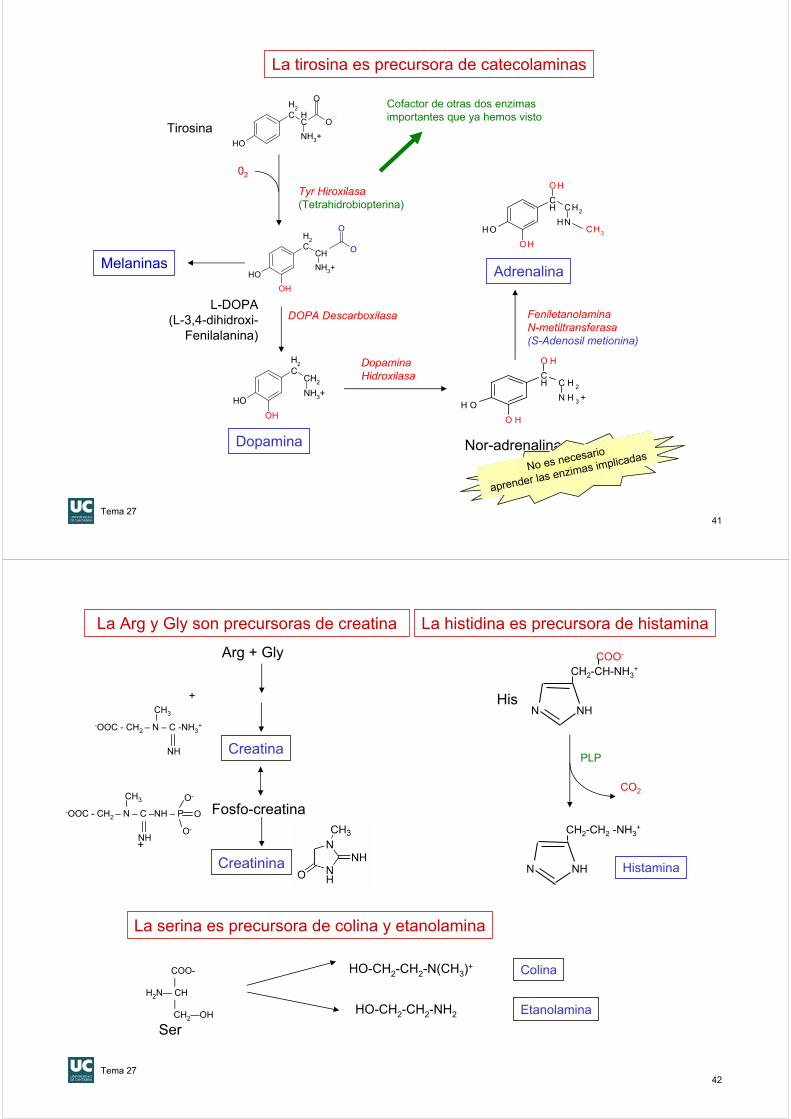
Tyr Hiroxilasa(Tetrahidrobiopterina)
DOPA Descarboxilasa
La tirosina es precursora de catecolaminas
OH
CH C H 2
N H 3 +
O H
O H
OH
CH CH2
NH
OH
OH
CH3
Nor-adrenalina
Adrenalina
DopaminaHidroxilasa
FeniletanolaminaN-metiltransferasa(S-Adenosil metionina)
No es necesario
aprender las enzimas implicadas
Melaninas
02
Cofactor de otras dos enzimasimportantes que ya hemos visto
42
UC J. León
Tema 27
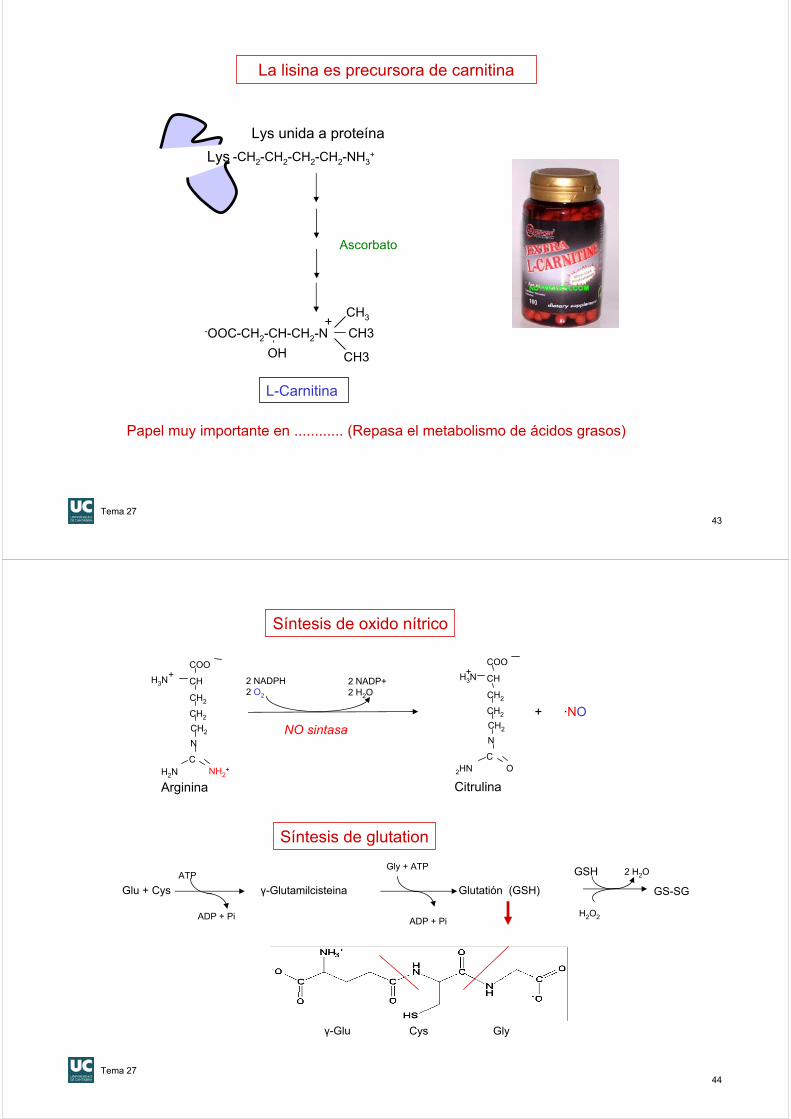
La serina es precursora de colina y etanolamina
COO-|
H2N— CH |CH2—OH
HO-CH2-CH2-N(CH3)+
HO-CH2-CH2-NH2
Ser
Etanolamina
Colina
La Arg y Gly son precursoras de creatina
Arg + Gly
Creatina
Fosfo-creatina
Creatinina
-OOC - CH2 – N – C -NH3+
NH
CH3
-OOC - CH2 – N – C –NH – P
NH
CH3
+
+
O
O-
O-
La histidina es precursora de histamina
Histamina
CO2
PLP
CH2-CH-NH3+
N NH
COO-
His
N NH
CH2-CH2 -NH3+
43
UC J. León
Tema 27
La lisina es precursora de carnitina
Lys -CH2-CH2-CH2-CH2-NH3+
-OOC-CH2-CH-CH2-N
OH
L-Carnitina
Lys unida a proteína
CH3
CH3
+CH3
Ascorbato
Papel muy importante en ............ (Repasa el metabolismo de ácidos grasos)
44
UC J. León
Tema 27
Síntesis de oxido nítrico
COO
CH
CH2
H3N+
CH2
CH2
N
NH2+H2N
C
COO
CH
CH2
H3N+
CH2
CH2
N
O2HNC
2 NADPH2 O2
+ ·NO
2 NADP+2 H2O
NO sintasa
GSH
H2O2
2 H2O
GS-SGGlu + Cys γ-Glutamilcisteina Glutatión (GSH)
ATPGly + ATP
Síntesis de glutation
ADP + Pi ADP + Pi
γ-Glu Cys Gly
CitrulinaArginina

45
UC J. León
Tema 27
CH2
CH2
C
COO
SCoA
O
CH2— NH2|COO-
Succinil CoA Glicina
CH2
CH2
C
COO-
O
CH2|NH2
δ- Aminolevulinato( δ-ALA)
Biosíntesis del hemo (1)
CO2CoA-SH
(MITOCONDRIA)δ-ALA Sintasa
-
• Inhibición alostérica• Represión de la expresión génica
Ferroquelatasa(MITOCONDRIA)
Hemo
N H
N
NH
N
Fe2+
CH3CH3
CH=CH2CH3
CH=CH2
CH3
COO-
CH2
CH2
COO-
CH2
CH2
Fe2+
+ PLP
δ-Ala dehidratasa=Porfobilinógeno sintasa
(Zn)(CITOSOL)H2O
CH2
CH2
C
COO
CH2|NH2
+
δ-Ala
CH2|NH2
CH2
COO CH2
CH2
COO
NH
Porfobilinógeno
O
CH=CH2
Protoporfirina IX
N H
N
NH
N
CH3CH3
CH=CH2CH3
CH3
COO-
CH2
CH2
COO-
CH2
CH2
5 pasos (4 en CITOSOL)
4 Porfobilinógenos
4 NH4+
No hay que aprenderse las estructuras a partir del δ-ALA
46
UC J. León
Tema 27
Bioíntesis de hemo (2)
Porfobilinógeno
Succinil CoA+Glicina
δ- Aminolevulinato( δ-Ala)
δ-Ala
δ-Ala
Uroporfirinógeno I
Uroporfirinógeno III
UroporfirinógenoCosintasa Decarboxilasa
4NH4+
4CO2
Protoporfirina IX
Oxidasa
Ferroquelatasa
Fe++
2CO2
Coproporfirinógeno III
Protoporfirinogeno IX
δ-Ala sintasa
δ-Ala dehidratasaH2O
HEMO
3PBG
Coproporfirinógeno III
Oxidasa
CITOSOL
Sitios de síntesis
1) Hemglobina en eritroblastos de médula ósea ( 80% del hemo)
2) Mioglobina en músculo
3) Citocromos a, b, c en todos los tejidos
4) P450 en hígado
5) Otras enzimnas en todos los tejidos (peroxidasa, catalasa, etc)
δ-Ala deaminasa
PLP -
Pb
Pb
Pb

47
UC J. León
Tema 27
Defectos en la ruta de biosíntesis de Porfirinas = Porfirias
Porfobilinógeno
Coproporfirinógeno III
Uroporfirinógeno III
Protoporfirinogeno IX
Protoporfirina IX
Hemo
δ-ALA dehidratasa
δ-Ala deaminasa
UroporfirinógenoCosintasa
Decarboxilasa
Oxidasa
Oxidasa
FerroquelatasaFe++
Porfiria aguda intermitente
Porfiria cutánea tardía
Coproprofiria hereditaria
Protoporfiria eritropoyética
Porfiria variegada
Porfiria congénita eritropoyética
δ-Ala
NH
N
NH
N
porfirinógeno
CH2|NH2
CH2
COO CH2
CH2
COO
NH
4NH4+
4CO2
2CO2
MITO-CONDRIA
(CITOSOL)
δ-ALA sintasa (MITOCONDRIA)
Gly + Succinil-CoA
δ-ALA
PLP
Uroporfirinógeno I
No hay que aprenderse las enzimas ni loscompuestos intremedios
48
UC J. León
Tema 27
Hemooxigenasa
BiliverdinaBiliverdínReductasa
Bilirrubina
N O HNH
NH
NOHMACROFAGOS,Células del SRE, BAZO
N H
N
NH
N
Fe2+
CH3CH3
CH=CH2CH3
CH=CH2
CH3COO-
CH2
CH2
CH=CH2 CH=CH2CH3CH3CH3
COO-
CH2
CH2
COO-
CH2
CH2CH3
Fe3+ + CO + NADP+
NADPH+ O2
ERITROCITOS
Bilirrubina transportada con albúmina
SANGRE
BILIS
Bilirrrubin-diglucurónido
Si está en exceso se acumula en sangre= ICTERICIA (prehepática)
Deficiencia transitoria de la transferasa en recién nacidos
Bilirrrubin-diglucurónido (B. conjugada)
HIGADO 2 UDP-glucuronato
2 UDPTransferasa
Ferritin Hb
ICTERICIASi no drena bienla vesícloa
Catabolismo del hemo
Expresión baja congénita de la transferasa = síndorme de Gilbert
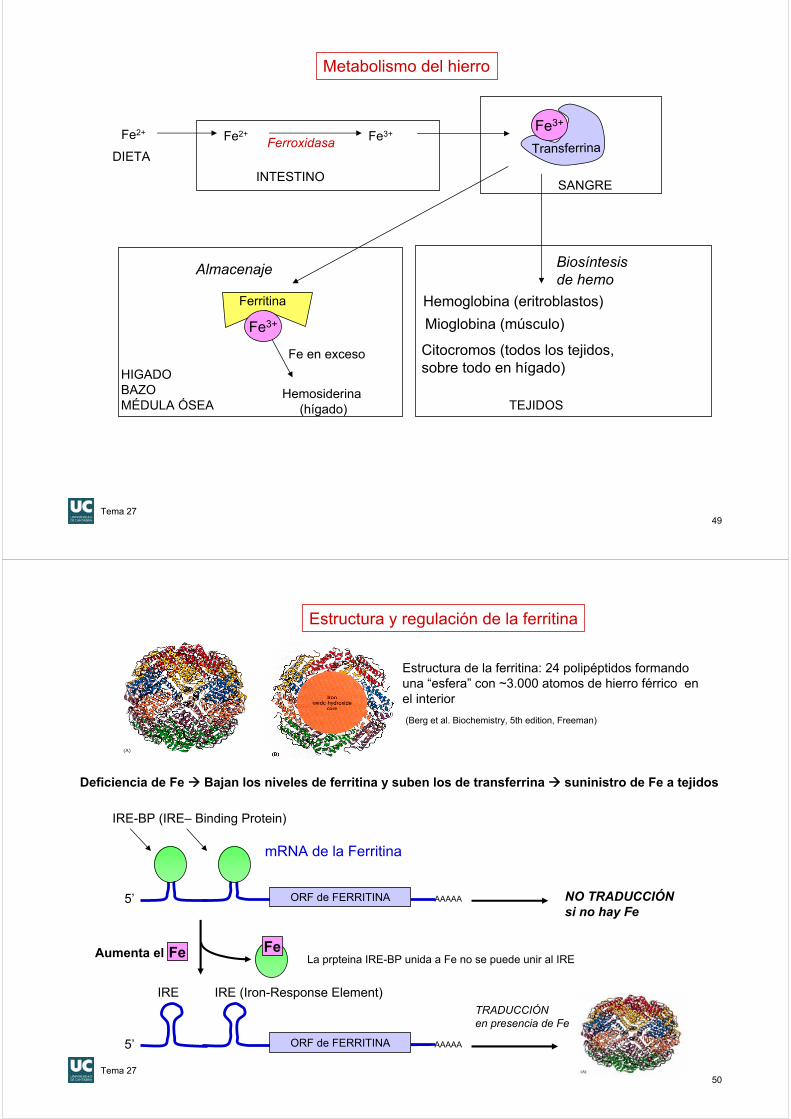
49
UC J. León
Tema 27
Hemoglobina (eritroblastos)
Mioglobina (músculo)
Citocromos (todos los tejidos,sobre todo en hígado)
Biosíntesis de hemo
TEJIDOS
Ferritina
Fe en exceso
Hemosiderina(hígado)
Almacenaje
HIGADOBAZOMÉDULA ÓSEA
Metabolismo del hierro
Fe2+ Fe2+ Fe3+
INTESTINOSANGRE
FerroxidasaDIETA
Fe3+
Transferrina
Fe3+
50
UC J. León
Tema 27
Estructura y regulación de la ferritina
Estructura de la ferritina: 24 polipéptidos formando una “esfera” con ~3.000 atomos de hierro férrico en el interior
(Berg et al. Biochemistry, 5th edition, Freeman)
AAAAAORF de FERRITINA5’ NO TRADUCCIÓN si no hay Fe
IRE-BP (IRE– Binding Protein)
mRNA de la Ferritina
Deficiencia de Fe Bajan los niveles de ferritina y suben los de transferrina suninistro de Fe a tejidos
AAAAAORF de FERRITINA5’
IRE IRE (Iron-Response Element)TRADUCCIÓN en presencia de Fe
La prpteina IRE-BP unida a Fe no se puede unir al IREAumenta el FeFe Fe

51
UC J. León
Tema 27
Hemocromatosis
Dolor abdominal fatiga, artritis, piel bronceada, en algunos casos cáncer de hígadoAutosómico recesivo. La enfermedad metabolica más frecuente: 1/200-400 en USA. Entre la poblacion de origen celta es portador ¼
Mutacion en el gen de una proteína (HFE1) que se une al receptor de transferrina y funciona comolimitador del transporte de Fe. Si está mutado, se transporta excesivo hiero
HFE1 normal: Unión normal a transferrina
HFE1
Receptor de transferrina
Fe3+
Transferrina
Receptor de transferrina
HFE1 mutado: Unión de alta afinidad
HFE1
Excesiva entrada de hierro
Fe3+
Transferrina
52
UC J. León
Tema 27
Dieta ANIMAL
B12 Factorintrínseco
ESTÖMAGO
Células parietales
Células del epitelio intestinal
ILEON
Factorintrínseco
Factorintrínseco
B12
B12
HÏGADO
Absorción y transporte de vitamina B12
B12
Transcobalamina
B12
SANGRE
B12Transcobalamina





























