Embed Size (px)
Citation preview

CASE STUDIES FROM NEURONEXT
Christopher S. CoffeyDepartment of Biostatistics
University of Iowa
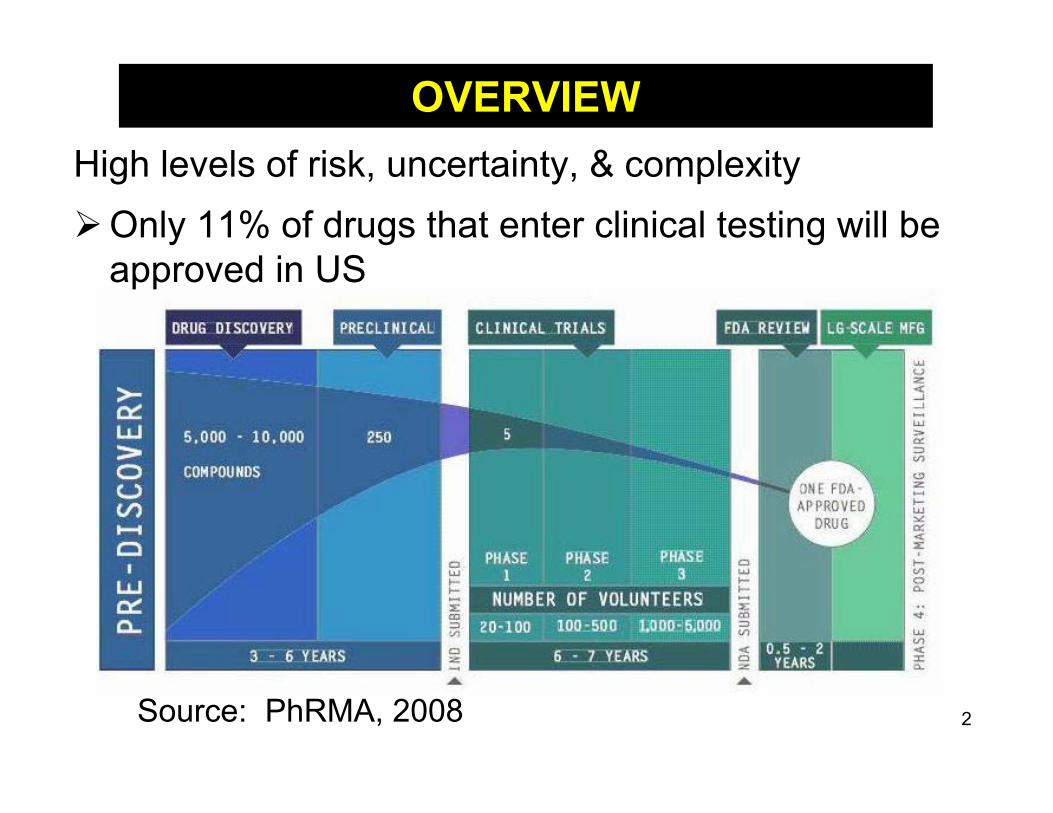
Source: PhRMA, 2008
High levels of risk, uncertainty, & complexityOnly 11% of drugs that enter clinical testing will be
approved in US
OVERVIEW
2

Three basic requirements for any clinical trial:
1) Trial should examine an important research question
2) Trial should use rigorous methodology to answer the question of interest
3) Trial must be based on ethical considerations and assure that risks to subjects are minimized
Due to size limitations, meeting these requirements in early phase clinical trials can be a challenge.As a consequence, the importance of study planning is magnified in these settings. 3
OBJECTIVES

Fundamental Point: Every clinical trial must have one or more primary
question(s). Should be of interest, and one that is capable of
being answered. Question on which sample size should be based Primary question(s) should be:
• Carefully selected• Clearly defined• Stated in advance
OBJECTIVES
4

Key Aspect - What is the Question? Phrase research question in concise, quantitative
terms Poor Question:
“Is drug A better than drug B” Better Question:
“In population W, is drug A at daily dose X more efficacious in reducing Z over a period of time T
than drug B at daily dose Y?”
OBJECTIVES
5

Each objective should link to a primary endpoint(s)This response variable will be compared across interventions in order to answer the primary question of interest.Necessary properties:
• Reflects primary objective of trial• Able to measure in all study subjects• Specified before trial is started• As objective as possible
OBJECTIVES
6

Secondary Objectives: Need to be defined a priori (avoid post hoc “fishing
expedition”) Chosen parsimoniously (avoid false positives) Two broad types:
• Examine response variable different from primary endpoint
• Subgroup hypotheses
OBJECTIVES
7

Common Errors in Drug Development:Poorly designed and optimistically interpreted “proof
of concept” studies Insufficient exploration of dose, regimen, and route
of administrationProceeding to phase III without adequate early
phase data to guide study design• Population; Sample Size; Endpoints; Study Duration
Ignoring FDA advice regarding evidence necessary to support a marketing application
OBJECTIVES
8

Phase I Trials: First investigation of potential new drug in humans Conducted in small number of healthy volunteers Results guide dosing and monitoring of future trials Objectives:
• Safety – Assess most significant adverse events• Tolerability – Explore possible toxic effects &
determine a maximum tolerated dose• Pharmacokinetics – Study of how drug is absorbed,
metabolized, and eliminated from body• Pharmacodynamics – Study of biochemical &
physiological effects of drug on body
PHASE I STUDIES
9

PHASE I STUDIES
10
Early learning phase designs in areas with potentially toxic treatments (e.g. cancer/neurological diseases) seek to determine the maximum tolerated dose (MTD).MTD is highest dose of drug that can be given before some percent of subjects experience an unacceptable dose limiting toxicity (DLT).
If Y denotes binary response with Y=1 denoting occurrence of DLT, and q0 denotes toxicity limit, then we seek to find a dose XMTD where:
Pr( Y=1 | XMTD) = q0
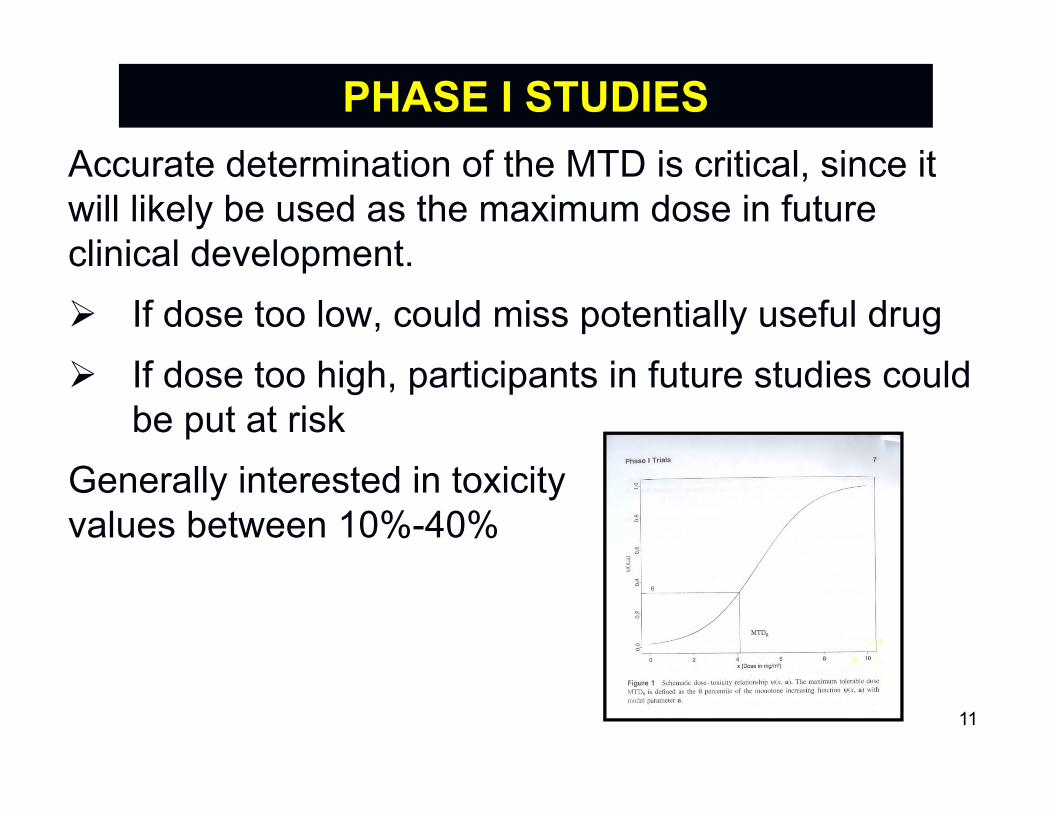
PHASE I STUDIES
11
Accurate determination of the MTD is critical, since it will likely be used as the maximum dose in future clinical development. If dose too low, could miss potentially useful drug If dose too high, participants in future studies could
be put at riskGenerally interested in toxicityvalues between 10%-40%

Generally desired to proceed very cautiously: Begin with small doses
• Well below dose expected to cause toxicity• But, also likely well below effective dose
Enroll in small cohorts (1-3 subjects) Any given dose escalation cannot increase by
more than one level• Ethical conflict is to increase dose slowly to avoid
unacceptable toxicity, but quickly enough to provide a therapeutic benefit
PHASE I STUDIES
12

PHASE I STUDIES
13
Conventional 3+3 methods, developed for oncology settings, employ an ad-hoc approach to identify MTD.Subjects are treated in groups of three, starting at initial low doseAlgorithm iterates moving dose up or down depending on number of toxicities observedThe MTD is identified from the data – highest dose studied with less than 2 toxicities out of a maximum of six treated patients.

PHASE I STUDIES
14
Bayesian paradigm allows incorporating ethical and statistical concerns from pre-clinical studies and sources outside of trial. Quantify prior information and uncertainty into a
probability distribution Update information and easily implement a
sequential design strategy Use all data to model dose-toxicity curve Most common approach is Continual
Reassessment Method [CRM – See Garrett-Meyer (Stat Med, 2005) for excellent tutorial]

Desired Study Parameters: Placebo group included for secondary comparison
of intracranial bleeding rates Enroll approximately 100 subjects
• 88 subjects in groups of four (3 treated / 1 placebo)• Default to placebo during safety review pauses
Dose limiting toxicities (DLTs) assessed from first dose to 48 hours following last dose of study treatment
NEURONEXT NN104 STUDY
15

Dose Levels: Four dose levels under consideration (dose level
can’t be increased by > one level at a time)• 120 µg/kg• 360 µg/kg
Target Toxicity Rate: MTD defined as highest dose with an estimated
dose limiting toxicity (DLT) rate of 10% or lessDesign: Continual Reassessment Method
• 240 µg/kg• 540 µg/kg
NEURONEXT NN104 STUDY
16

CRM model proceeds as follows:1) Enroll 4 subjects (3 treatment / 1 placebo) into a
cohort2) Determine # of evaluable subjects in cohort3) Observe # of evaluable subjects (out of 3 treated)
that have a DLT per study definition4) Refit dose-response curve using observed
information from all evaluable subjects to date5) Continue until pre-defined stopping criteria met
ADAPTIVE DOSE FINDING
17
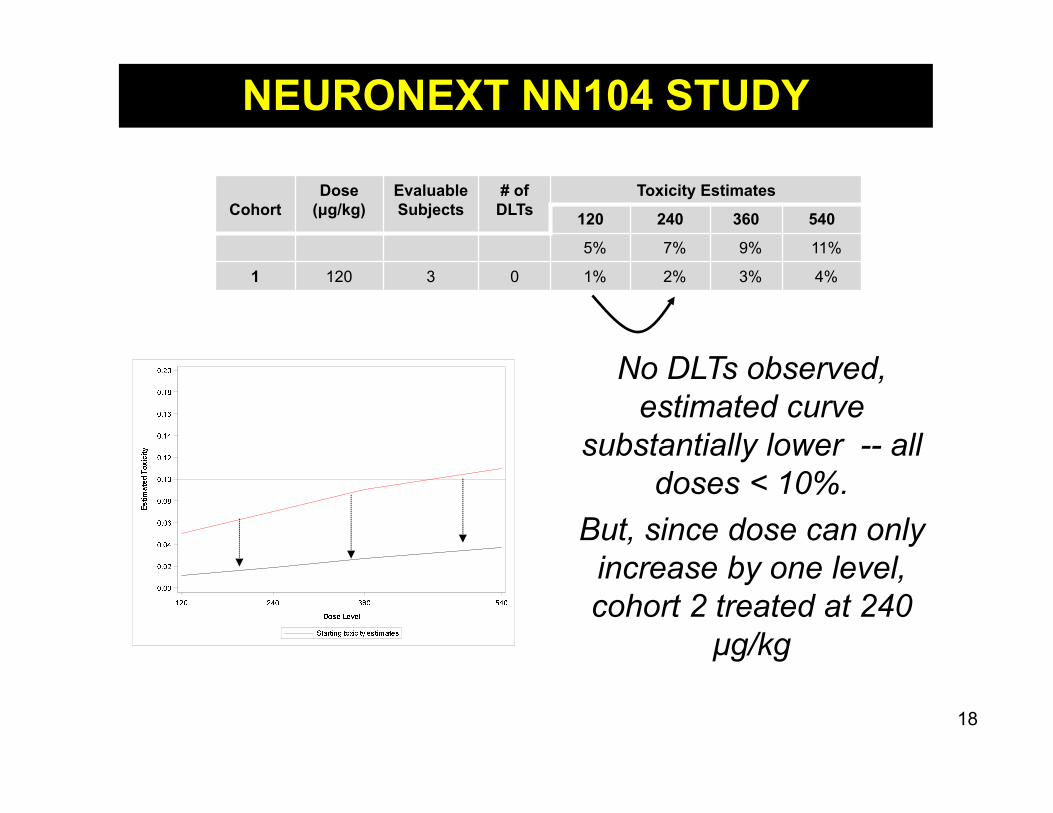
No DLTs observed, estimated curve
substantially lower -- all doses < 10%.
But, since dose can only increase by one level, cohort 2 treated at 240
μg/kg
CohortDose
(μg/kg)Evaluable Subjects
# of DLTs
Toxicity Estimates
120 240 360 540
5% 7% 9% 11%
1 120 3 0 1% 2% 3% 4%
NEURONEXT NN104 STUDY
18

DLT (Cerebral Hemorrhage) observed in subject treated
with 240 μg/kgAll DLT estimates now above 10% threshold.
Cohort 3 treated at 120 μg/kg
CohortDose
(μg/kg)Evaluable Subjects
# of DLTs
Toxicity Estimates
120 240 360 540
5% 7% 9% 11%
1 120 3 0 1% 2% 3% 4%
2 240 3 1 11% 14% 17% 20%
NEURONEXT NN104 STUDY
19

540 μg/kg is the maximum tolerated dose, with an estimated DLT rate around 7%
NEURONEXT NN104 STUDY
20

Phase II Trials: Performed in patients with disease of interest In general, phase II trials may be smaller than they
should be Results guide design of subsequent confirmatory
(phase III) trial
PHASE II STUDIES
21

Confirmatory Designs:• Large sample sizes
• Many standard designs
Learning Stage Designs:• Efficiency is critical
• Carefully evaluate alternative designs
22
PHASE II STUDIES

PHASE II STUDIES
23
Main Goals: Provide assessment of preliminary efficacy
• Screen out ineffective treatments• Determine if new treatment is sufficiently promising to
justify inclusion in large-scale randomized trials
Further characterize the safety profile Avoid early phase designs that look to simply
be an underpowered phase III study• “Treatment Effects” estimated in pilot studies often
tend to be over-estimates of true effect

PHASE II STUDIES
24
Due to concerns about missing an important effect, researchers may be tempted to include a large number of endpoints. If study design often calls for “going forward” if any
endpoint shows hint of an effect, then this type of design will almost always “go forward” – even if treatment does not work
Due to business implications, early phase studies with positive findings are more likely to be highlighted – could be real effect or ‘chance’ finding

PHASE II STUDIES
25
Due to budgetary constraints, researchers may insist on sample size limitations, regardless of whether there is scientific justification. Study may not be able to adequately answer
question of interest Results become somewhat subjective
• A researcher who views treatment favorably will likely see positive trends
• A researcher who views treatment unfavorably will likely see negative trends

Types of Phase II Studies: Using Different Endpoints Than Phase III Endpoint
• Safety/Tolerability Designs• Surrogate Endpoint Designs
Using Phase III Endpoints – Not Powered for Efficacy (in Phase III sense)
• Predictive Probability Designs• N-of-1 Designs• Selection Designs• Futility Designs
PHASE II STUDIES
26

SAFETY/TOLERABILITY DESIGNPossible to design a trial to primarily address safety & tolerability.Trial powered to detect some specified level of
safety concerns (usually higher than level of minimal concern)
Trial also power to detect important differences in tolerability (completing study on assigned dose)
Efficacy data typically collected as an exploratory outcome
27

NN-105 STAIR Study: Primary Objective:
To assess the tolerability of SRX246 in irritable subjects with early symptomatic HD over a period of 12 weeks compared to placebo
NEURONEXT NN105 STUDY
Rand
omize
ScreeningPhase
Initial Screening
120 mg(n = 36)
Placebo(n = 36)
160 mg(n = 36)
(Up to 30 days)
28

Primary Tolerability Hypothesis:Assess whether either 120 mg or 160 mg daily
dosing of SRX246 is sufficiently tolerable, compared to placebo.• Assessed by comparing percentage of participants
enrolled in each dosage group who are able to complete study (12 weeks) on assigned dose Any participant removed from study drug or who fails to complete
the study (for any reason) will be deemed not to have tolerated their assigned medication
29
NEURONEXT NN105 STUDY

SURROGATE ENDPOINT DESIGNAs previously discussed, a surrogate endpoint (biomarker) can be measured earlier, easier, and more frequently than many standard clinical endpoints.
A surrogate endpoint design allows using surrogate for ‘go’ / ‘no go’ decision making.
This often leads to reduced sample size or length of study – allowing screening process to move more efficiently.
Subsequent confirmatory studies can still be powered on the gold standard clinical endpoint.
30

NN-102 SPRINT-MS Study: A Randomized, Double-Blind, Placebo-Controlled
Study to Evaluate Safety, Tolerability, and Activity of Ibudilast (MN-166) in Subjects with Progressive Multiple Sclerosis• PI: Robert Fox (Cleveland Clinic)
• Primary Objective:To evaluate the activity, safety, and tolerability of ibudilast(MN-166 – 100 mg/day) versus placebo administeredorally in subjects with primary progressive andsecondary progressive multiple sclerosis
NEURONEXT NN102 STUDY
31

NN102: SPRINT-MS Study In a phase 2 trial involving patients
with progressive MS, ibudilast wasassociated with slower progressionof brain atrophy than placebo
Ibudilast was associated with higherrates of gastrointestinal side effects,headache, and depression
NEURONEXT NN102 STUDY
32
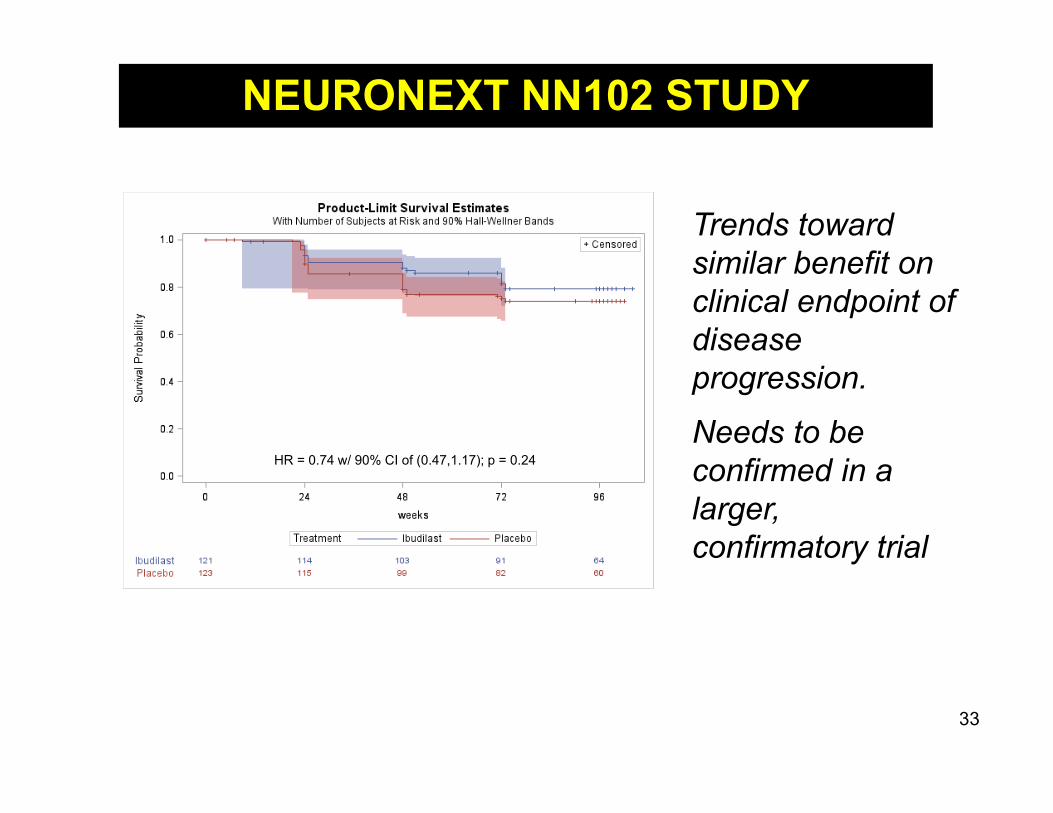
Trends toward similar benefit on clinical endpoint of disease progression.
Needs to be confirmed in a larger, confirmatory trial
NEURONEXT NN102 STUDY
HR = 0.74 w/ 90% CI of (0.47,1.17); p = 0.24
33

FUTILITY DESIGNA futility (non-superiority) design is a screening tool to identify whether agents should be candidates for phase III trials while minimizing costs/sample size. If “futility” is declared, results would imply not cost
effective to conduct a future phase III trial If “futility” is not declared, suggests that there could
be a clinically meaningful effect which should be explored in a larger, phase III trial
34
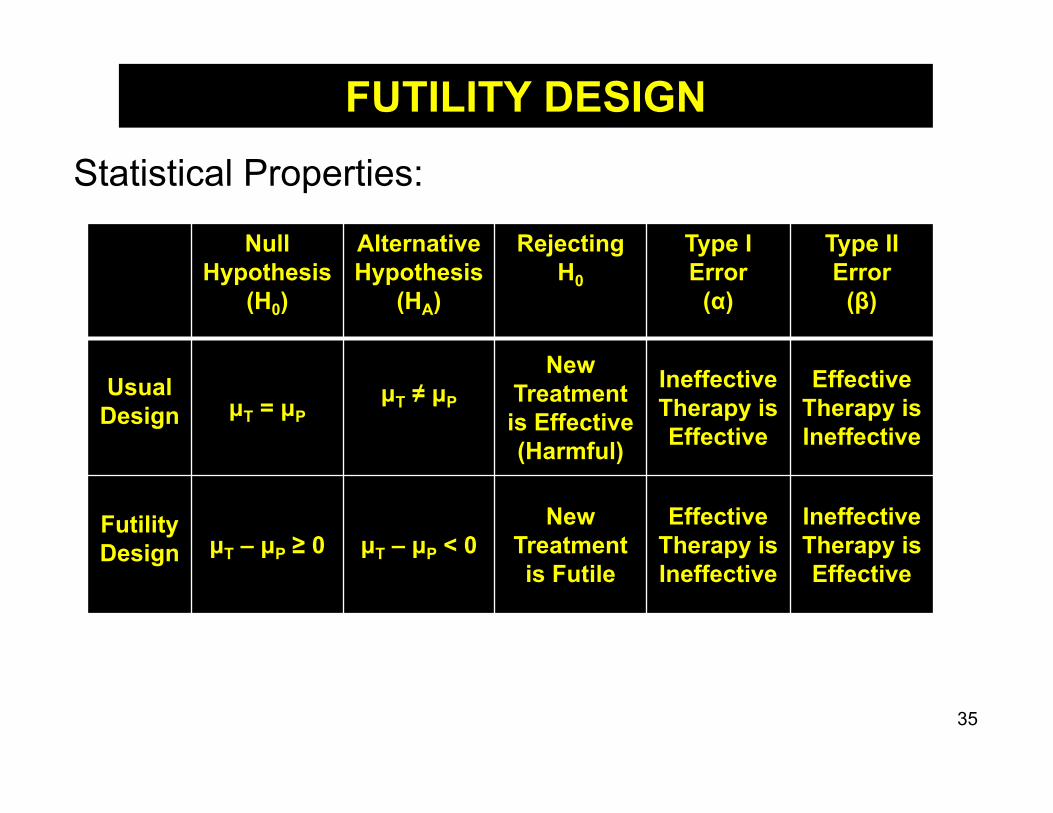
Statistical Properties:
NullHypothesis
(H0)
AlternativeHypothesis
(HA)
Rejecting H0
Type I Error
(α)
Type II Error
(β)
UsualDesign μT = μP
μT ≠ μP
New Treatment is Effective(Harmful)
Ineffective Therapy is Effective
Effective Therapy is Ineffective
FutilityDesign μT – μP ≥ 0 μT – μP < 0
New Treatment is Futile
EffectiveTherapy is Ineffective
Ineffective Therapy is Effective
FUTILITY DESIGN
35

Thus, futility designs are good at identifying ineffective treatments, but not very good at identifying effective treatments.
However, improvement over running underpowered efficacy trials in phase II or conducting phase III trials as first rigorous test of efficacy for a new treatment.
FUTILITY DESIGN
36
High negative predictive values:If “futility” declared, treatment likely not effective
Low positive predictive values:Lack of “futility” does not imply treatment is effective

NN-103 - BeatMG• PI: Richard Nowak (Yale)• A previous study at Yale demonstrated that ~80% of subjects
treated with rituximab achieved at least a 75% reduction in their prednisone dose at 52 weeks
• Primary Objective: Determine whether large benefit observed in prior Yale study can be refuted in a multi-site trial, or looks promising enough to justify a future phase III trial (“go”)
NEURONEXT NN103 STUDY
37

Suppose a 30% increase in favorable response rates (achieving a 75% reduction in prednisone dose) with rituximab over placebo is clinically meaningful.This futility design tests the following hypothesis:
H0: Rituximab improves outcome by at least 30% compared to placebo(pR – pP ≥ 0.30 – not futile)
versus
HA: Rituximab does not improve outcome by at least 30% copmared to placebo
(pR – pP < 0.30 – futile)
NEURONEXT NN103 STUDY
38

Power to declare “futility” for range of true response rates for rituximab (assumes placebo rate of 40%):
Table demonstrates benefits of using futility design, if true rituximab success rate: Near/below placebo rate, declare “futility” with high probability
Well above placebo rate, low probability of declaring “futility”
In between, uncertainty to address in subsequent trial
RituximabRate (pR) 30% 35% 40% 45% 50% 60% 70% 80% 90%
Pr (Futility) 92% 84% 74% 63% 50% 25% 10% 2% <1%
NEURONEXT NN103 STUDY
39

40
Reduction in Mean Daily Prednisone
Dose ≥ 75%
1-Sided Odds Ratios (90% CI) Rituximab vs.
PlaceboGroup Rituximab Placebo
Primary 60% 56% 1.14 (0 , 2.41)Observed 65% 63% 1.11 (0 , 2.43)
Primary results show “futility” for showing pre-defined clinically meaningful difference with rituximab (p = 0.03).
NEURONEXT NN103 STUDY






















