Embed Size (px)
Citation preview

Case Scenario:
• 15 year old girl presented in ED with
aggressive behaviour and hallucinations.
• No associated fever, vomiting, seizures
or developmental concerns
• Previously well

Inborn Errors of Metabolism
Clinical Approach to Diagnosis
and Treatment
Joy Lee, M.D., FRACP

Acknowledgement
Asso. Prof. Avihu Boneh for the slides he
shared

Misconceptions on Inborn Errors of Metabolism
• IEMs are very RARE.
• These disorders are difficult to DIAGNOSE and to
TREAT.

STRESS TEST ...

Outline
Part I
Overview of IEM
When to suspect IEM?
What investigations to request?
What is expanded newborn screening?
Part II
What are the treatment principles?

Overview of IEM

Incidence - Prevalence
Individually very rare
Collectively ~1; 5000

Patterns of Inheritance
MENDELIAN
~60% autosomal
recessive
~20% autosomal
dominant
~15% X-linked
(Scriver, et al. 2001)
COMPLEX NON-
MENDELIAN
~5% mitochondrial

Classification
According to intracellular site
ex. mitochondrial disorders, peroxisomal disorders
lysosomal storage disorders
According to biochemical pathology
ex. disorders in intermediary metabolism
disorders of neurotransmitter metabolism
disorders of purines/pyrimidines
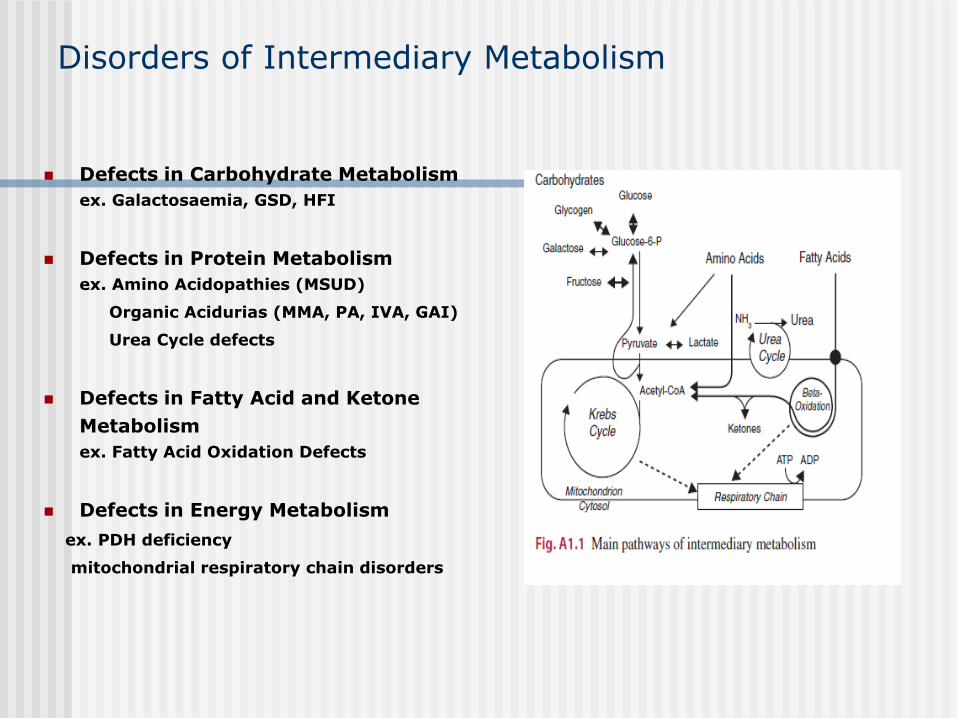
Disorders of Intermediary Metabolism
Defects in Carbohydrate Metabolism
ex. Galactosaemia, GSD, HFI
Defects in Protein Metabolism
ex. Amino Acidopathies (MSUD)
Organic Acidurias (MMA, PA, IVA, GAI)
Urea Cycle defects
Defects in Fatty Acid and Ketone
Metabolism
ex. Fatty Acid Oxidation Defects
Defects in Energy Metabolism
ex. PDH deficiency
mitochondrial respiratory chain disorders

A B C
General Metabolic Concept
Enzyme + Co-factor
D

3 Sources of Diagnostic Confusion
Confusion with common acquired
conditions ex. sepsis
Confusion caused by association with
intercurrent illness
Confusion arising from genetic
heterogeneity ex. MPS
Clarke, A Clinical Guide to Inherited Metabolic diseases
3rd edition

MPS I - Spectrum

Disease progression: severe MPS I
10 months 12 months
22 months 34 months
39 months
Muenzer J. The mucopolysaccharidoses: A heterogeneous group of disorders with variable pediatric presentations. J Peds. 2004. S27 – S34.
Photos courtesy of US MPS Society

When to suspect IEM?

1) Good history taking
2) Physical examination including review
of systems

History
Pregnancy and Birth: HELLP syndrome, maternal diet, foetal
movements , prolonged jaundice
Family: Ethnic origin, consanguinity, unexplained
deaths/miscarriages (? sex), SIDS, unexplained MR in sibling/close
relative, other affected sibs
Present and Past: Recurrent unexplained symptoms ex. vomiting,
respiratory and ear infections, progressive CNS degeneration, MR in
absence of major congenital anomalies, recurrent surgeries ,
surgical procedures (recovery from Anaesthesia)

History
Dietary:
aversion to certain food such as protein
vegetarian diet
craving for certain food

Clinical Presentation of IEM
Variable age of onset and clinical symptoms
Neurologic
a) acute encephalopathy
b) chronic encephalopathy
psychomotor retardation, regression,
seizures, movement disorders, stroke,
ataxia, psychiatric, myopathy

Hoffman et al Inherited Metabolic Diseases A Clinical Approach

Clinical Presentation of IEM
Variable age of onset and clinical symptoms
Neurologic
a) acute encephalopathy
b) chronic encephalopathy
psychomotor retardation, regression,
seizures, movement disorders, stroke,
ataxia, psychiatric, myopathy

Clinical Symptoms of IEM
Acute encephalopathy
Vomiting (recurrent)
Feeding refusal
breathing problems
Lethargy / irritability
Floppiness /dystonia/weakness/ataxia
Seizures
…in relation to age, precipitating factors

Clarke, A Clinical Guide to Inherited Metabolic diseases
3rd edition

Disorders of Intermediary Metabolism
Defects in Carbohydrate Metabolism
ex. Galactosaemia, GSD, HFI
Defects in Protein Metabolism
ex. Amino Acidopathies (MSUD)
Organic Acidurias (MMA, PA, IVA, GAI)
Urea Cycle defects
Defects in Fatty Acid and Ketone
Metabolism
ex. Fatty Acid Oxidation Defects
Defects in Energy Metabolism
ex. PDH deficiency
mitochondrial respiratory chain disorders

Precipitating Factors
Feeding / fasting
Dietary overload (e.g. protein)
Dietary deficiency (e.g. vit. B6, B12, carnitine)
Intercurrent infection / fever
Surgery
Exercise
Specific toxins (e.g. valproic acid)

Clinical Presentation of IEM
Neurologic – chronic encephalopathy, movement disorder,
diurnal pattern, ataxia, muscle pain, abnormal tone
Hepatic – jaundice, liver failure, hypoglycaemia
Cardiac – cardiomyopathy, rhythm disturbances
Respiratory – sleep apnoea, recurrent chest infections
Renal – tubular dysfunction, renal stones
Eye – cataracts, optic atrophy, retinal abnormalities
Dermatological – alopecia, rash, “kinky hair”,
Ears – recurrent infections, hearing loss
Gastrointestinal – chronic diarrhoea
Growth - failure to thrive
Dysmorphism

IEM and Dysmorphism
Disorders of cholesterol biosynthesis - SLO
Peroxisomal disorders
Mitochondrial disorders
Lysosomal disorders (ex. MPS)
CDG syndromes
Glutaric aciduria type II
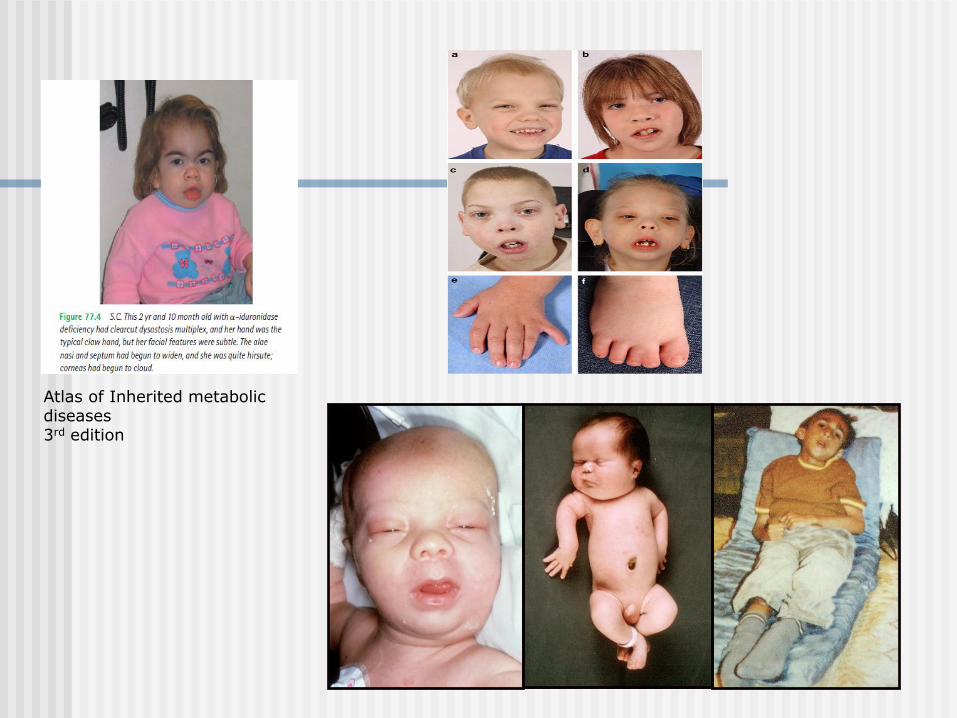
Atlas of Inherited metabolic diseases 3rd edition

Clinical Clues
Some characteristics of psychomotor retardation that should
“alert” clinicians.
1) It tends to be global affecting all spheres of development
to some extent.
2) Severe irritability, impulsitivity/aggressiveness, hyperactivity and
nocturnal restlessness are more common among patients with MR
caused by IEM than non-metabolic.
Clarke, A Clinical Guide to Inherited Metabolic diseases
3rd edition

Clinical Clues
Some characteristics of psychomotor retardation that
should “alert” clinicians.
3) Psychomotor retardation is usually progressive – gap wider
with time when compared with other children.
4) Psychomotor retardation is usually asso. with other objective
evidence of neurologic dysfunction – abnormal tone, impairment
of senses, seizures, pyramidal tract signs, cranial nerve deficits.
Clarke, A Clinical Guide to Inherited Metabolic diseases
3rd edition

Clinical Clues:
Physical Examination
Multisystem involvement:
review of systems is important!
Pattern of abnormalities – typical of certain disorders
ex. Kinky hair, laxity of joints/skin, developmental delay
Pattern and degree of involvement of other organs and
tissues
ex. Coarse features, hepatosplenomegaly, joint contractures,
umbilical hernias, obstructive sleep apnoea
Unusual smell – ex. fishy odour, sweaty feet
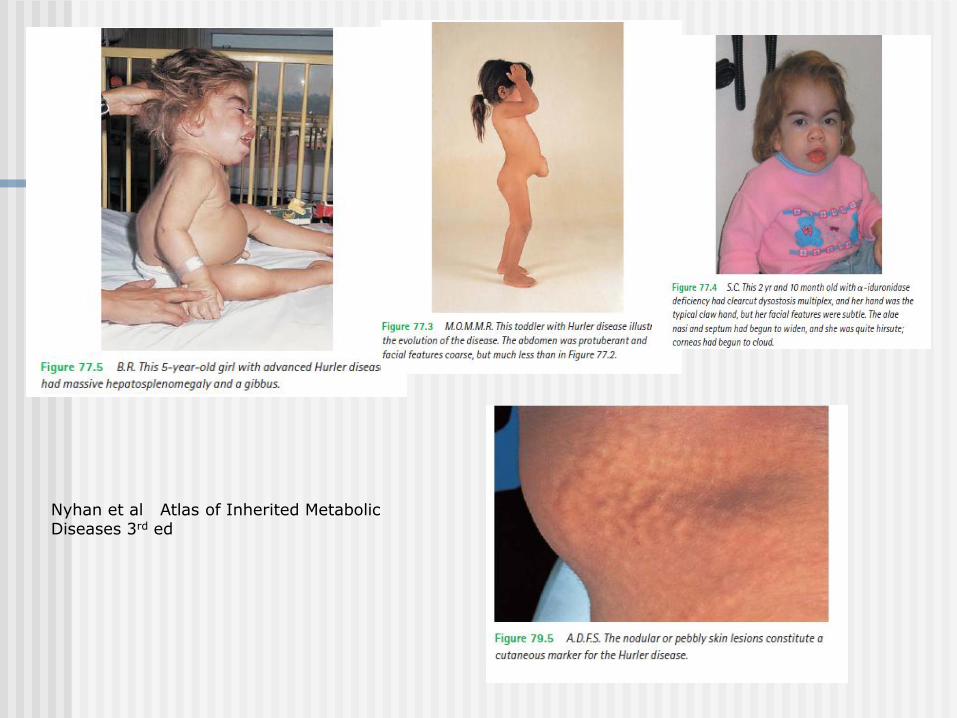
Nyhan et al Atlas of Inherited Metabolic Diseases 3rd ed

Nyhan et al Atlas of Inherited Metabolic Diseases 3rd ed

Nyhan et al Atlas of Inherited Metabolic Diseases 3rd ed

Nyhan et al Atlas of Inherited Metabolic Diseases 3rd ed

Nyhan et al Atlas of Inherited Metabolic Diseases 3rd ed

Nyhan et al Atlas of Inherited Metabolic Diseases 3rd ed

Nyhan et al Atlas of Inherited Metabolic Diseases 3rd ed

What investigations to request?

Laboratory Tests (1)
Acid-Base / Electrolytes
Glucose
Lactate
Ammonia
Urine ketones
Calculate for Anion Gap – normal < 16

Results in 30 minutes
or less!

Questions you should ask yourself
Is there Metabolic Acidosis?
Is there Hypoglycaemia?
Is there Ketosis?
Is there Hyperammonaemia?

Clarke, A Clinical Guide to Inherited Metabolic diseases, 3rd edition
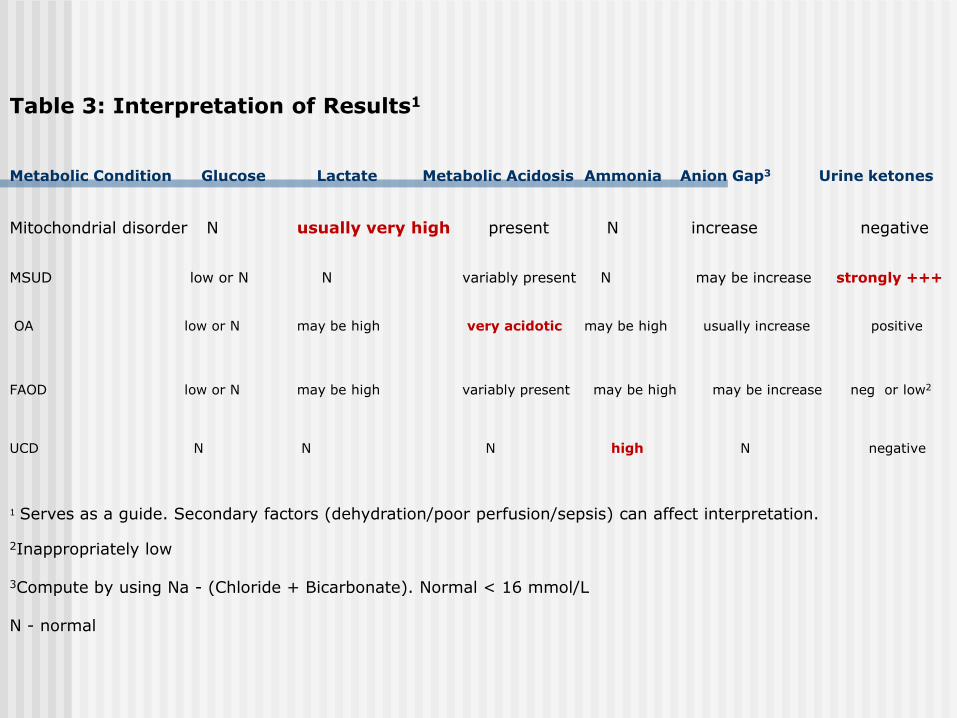
Table 3: Interpretation of Results1
Metabolic Condition Glucose Lactate Metabolic Acidosis Ammonia Anion Gap3 Urine ketones
Mitochondrial disorder N usually very high present N increase negative
MSUD low or N N variably present N may be increase strongly +++ OA low or N may be high very acidotic may be high usually increase positive FAOD low or N may be high variably present may be high may be increase neg or low2
UCD N N N high N negative 1 Serves as a guide. Secondary factors (dehydration/poor perfusion/sepsis) can affect interpretation. 2Inappropriately low 3Compute by using Na - (Chloride + Bicarbonate). Normal < 16 mmol/L N - normal

Laboratory Tests – Blood (2)
Biochemical
Plasma amino acids
Guthrie card amino acids/acylcarnitine profile
Plasma carnitine
Ketones, free fatty acids
Pyruvate – needs special tube
Ancillary:
Full blood count – ex. Neutropenia, pancytopenia
LFT, coagulation screen, alpha feto protein
Lipid profile, uric acid
Creatine kinase

Blood Spots on Guthrie Card
for a rapid analysis of : acylcarnitines
amino acids
DNA extraction for mutation analysis
Dry in room air; store/send in a paper envelope

Laboratory Tests: Urine
Smell
pH
Glucose
Ketones
Protein
Reducing Substances (Clinitest, Clinistix)

Laboratory Tests: Urine
Amino Acids
Organic Acids
GAGs
Others: Sulphocysteine, Purines and
Pyrimidines, Bile acids, P6C
Store and freeze extra urine for further
analysis

Laboratory Tests - CSF
Glucose
Protein
Lactate
Pyruvate
Amino Acids
Neurotransmitters – special tubes
*Paired: Blood/CSF – glucose, lactate,
pyruvate and amino acids*
Store and freeze extra sample for further analysis

Sample Handling/Collection
Blood
ammonia – free flowing, in ice, send to lab ASAP
lactate – free flowing, no tourniquet
Do not freeze whole blood.
Separate plasma/serum and freeze at -20 C or -70 C
Urine: Freeze at -20 C or -70 C
CSF:
A bloody sample is not informative
If only a few RBC: spin and freeze at -20 C or -70 C.

Post-Mortem Samples
Collect and freeze plasma, urine and CSF
Blood spots on Guthrie Card
Skin biopsy: put in culture medium or sterile
Normal Saline; store at 4 C
Biopsies: Liver, heart and heart
Guidelines on PM samples on RCH website
PM kit available in core lab with instructions

How NOT to order metabolic testing

Expanded Newborn Screening

Current Australian newborn screening panels
• Congenital hypothyroidism
enzyme linked immunosorbent assay (commercial kit)
• Cystic fibrosis
ELISA or FIA (commercial kit) followed by mutation testing
• Phenylketonuria + 22 other inborn errors of metabolism
Tandem mass spectrometry
(single test --- multiple disorders)
• Galactosaemia (not in Vic)

Collection of Guthrie card at 48 to 72 hours of age
Testing is voluntary with consent >99.5% of babies have the test
Program is funded by the Victorian government ~ 74,000 Vic babies tested per year (2011)
~ 77,400 babies in 2012

Sorting the cards
Punching out Microtitre plate
Tandem mass spectrometry test Hypothyroidism ELISA test
Adding reagents


Amino acids
Amino acid Metabolic Possibilities
Phe PKU, hyperphe, BH4 metabolism defect
Valine, xleu Classic MSUD
Glycine NKH
Methionine Homocystinuria, MAT deficiency
ASA Argininosuccinic aciduria
Citrulline Citrillunemia I or II
Tyrosine Tyrosinemia I , Tyrosinemia II or III

Acylcarnitines
Acylcarnitine Metabolic Possibilities
Free carnitine(C0) Carnitine uptake defect
Propionylcarnitine(C3) MMA, PA, cobalamin defect, B12 def.
Octanoylcarnitine(C8) MCAD deficiency
Glutarylcarnitine(C5DC) GAI
Tetra-decanoylcarnitine(C14) VLCAD deficiency
3-hydroxy-isovalerylcarnitine (0HC5) holocarboxylase def., MCCC def.

Normal newborn profile
-5
-4
-3
-2
-1
0
1
2
3
4
5
GU
AN
IDIN
OA
CE
TIC
OH
PR
OL
INE
D5 P
HE
(IN
T S
TD
)
GL
Y
AL
A
VA
L
ISO
LE
U_L
EU
_O
HP
RO
OR
N
LY
S_G
LN
ME
T
PH
E
GL
YC
YL
-PR
OL
INE
AR
G
CIT
RU
LL
INE
TY
R
HO
MO
CIT
RU
LL
INE
AR
GIN
INO
SU
CC
INIC
C8 C
AR
N (
INT
ST
D)
D9F
C_U
ND
ER
IV
FR
EE
CA
RN
C2 C
AR
N
C3 C
AR
N
C4 C
AR
N
C5:1
CA
RN
C5 C
AR
N
OH
C4 C
AR
N
C6 C
AR
N
OH
C5 C
AR
N
C8 C
AR
N
C3 D
C C
AR
N
C10 C
AR
N
C5 D
C C
AR
N
C12 C
AR
N_C
6:1
DC
C6 D
C C
AR
N
OH
C5 D
C C
AR
N
C14:1
CA
RN
C14 C
AR
N
C16 C
AR
N
OH
C16 C
AR
N
C18 C
AR
N
OH
C18:1
CA
RN
C0/(
C16+C
18)
-10 = LOW (undetected with lower cutoff>0)
<-1 = LOW (divisors of lower cutoff)
-1 to 0 = NORMAL (between lower cutoff and median)
0 to 1 = NORMAL (between median and cutoff)
>1 = HIGH (multiples of cutoff)D:\Newborn\Mar2004\02.PRO\Data\A27247-AA-1Select sample:

Newborn screening MSMS panel example (a)
C8 and C6 carnitine, supported by C8/C10 ratio
• Almost diagnostic for medium chain acyl CoA dehydrogenase deficiency
Amino acids acyl carnitines ratios

What conditions are detected by tandem mass spectrometry?
Group A: potentially or often serious, well detected PKU, homocystinuria, tyrosinemia II Citrullinemia type I, argininosuccinic aciduria, classic MSUD methylmalonic acidaemias, isovaleric acidaemia, propionic acidemia GAI, multiple carboxylase def. medium-chain acyl CoA dehydrogenase (MCADD), LCHAD, CPT I or II Carnitine transport defect
Group B: less serious
3-methylcrotonyl CoA carboxylase
2-methylbutyryl CoA dehydrogenase Short-chain acyl CoA dehydrogenase

Group C: potentially or often serious, poorly detected Tyrosinaemia type 1, Citrullinemia II
OTC deficiency, argininaemia Glycine encephalopathy, cobalamin C def.
What conditions are detected by tandem mass spectrometry?
Group D: maternal disorders vitamin B12 deficiency
3-methylcrotonyl CoA carboxylase deficiency Carnitine uptake defect

Finally…
‘Think Metabolic’
Do ‘the basics’
Call for assistance






























