Embed Size (px)
Citation preview

Car-Parrinello Method and Car-Parrinello Method and ApplicationsApplications
Moumita SaharayJawaharlal Nehru Center for Advanced Scientific Research,
Chemistry and Physics of Materials Unit,Bangalore.

Outline
Difference between MD and ab initio MDWhy to use ab initio MD ?Born-Oppenheimer Molecular DynamicsCar-Parrinello Molecular DynamicsApplications of CPMDDisadvantages of CPMDOther methodsConclusions

DFT, MD, and CPMD
Properties of liquids/fluids depend a lot on configurational entropy
MD with improved empirical potentials
DFT calculation of a frozen liquid configuration
Configurational Entropy part of the free energy will be missing in that case.
Ab initio MD offers a path that mixes the goodness of both MD and
of DFT
AIMD is expensive.

Molecular simulationsClassical MDHardwired potential
No electronic degrees of freedom
No chemical reaction
Accessible length scale ~100 Å
Accessible time scale ~ 10 ns
Ab initio MDOn-the-fly potential
Electronic degrees of freedom
Formation and breaking of bonds
Accessible length scale ~ 20 Å
Accessible time scale ~ 10 ps

Livermore’s Nova Laser Sandia National Laboratories
Z accelerators
A short intense shock caused
the hydrogen to form a hot
plasma and become a
conducting metal
The experiments found different compressibilities which could affect the
equation of state of hydrogen and its isotope
Quantum simulations could give the proper reasons for different results
Conditions of the Nova and Z flyer were different : Time scales of the pulse were different

Why ab Initio MD ?Chemical processes
Poorly known inter atomic interactions e.g. at high Pressure and/or Temperature
Properties depending explicitly on electronic states ; IR spectra, Raman scattering, and NMR chemical shift
Bonding properties of complex systems

Born-Oppenheimer approximation
Electronic motion and nuclear motion can be separated due to huge difference in mass
Different time scale for electronic and ionic motion
Fast electrons have enough time to readjust and follow the slow ions

Born-Oppenheimer MD Electron quantum adiabatic evolution and classical ionic dynamicsEffective Hamiltonian :
HoI ? Ionic k.e. and ion-ion interaction
2nd term ? Free energy of an inhomogeneous electron gas in the presence of fixed ions at positions (RI)
Electronic ground state – electron density ?(r) – F({RI}) min
Born-Oppenheimer Potential Energy Surface

Minimization to BO potential surfaceE
{?(r
)}
?(r)? 0(r)

Born-Oppenheimer MDForces on the ions due to electrons in ground state
Ionic Potential Energy
? i (r) one particle electron wave function1st ? Electronic k.e. ; 2nd ? Electrostatic Hartree term
3rd ? integral of LDA exchange and correlation energy density ?xc
4th ? Electron-Ion pseudopotential interaction ; 5th ? Ion-Ion interaction

Born-Oppenheimer MD
Electronic density ; fi ? occupation number
EeI ? Electron-Ion coupling term includes local and nonlocal components
Kohn-Sham Hamiltonian operator
Time evolution of electronic variables
Time dependence of Hks ? slow ionic evolution given by Newton’s equations
Uks = minimum of Eks w.r.t. ? i-

Merits and Demerits of BOMDAdvantages Disadvantages
True electronic Adiabatic Evolution on the BO surface
Need to solve the self- consistent electronic-structure problem at each time step
Minimization algorithms require ~ 10 iterations to converge to the BO forces
Poorly converged electronic minimization ? damping of the ionic motion
Computationally demanding procedure

Car-Parrinello MDCP Lagrangian
? i ? classical fields ? ? mass like parameter [1 Hartree x 1 atu 2 ]
4th ? orthonormality of the wavefunctions
Constraints on the KS orbitals are holonomic No dissipation

Choice of ?Folkmar Bornemann and Christof Schutte demonstrate
If the gap between occupied and unoccupied states = 0
If the gap between occupied and unoccupied states = 0
(Insulators and semiconductors)
(Metals)
Fictitious kinetic energy of the electrons grow without control
Use electronic thermostat
? must be small ? small integration time step
? ~ 400 au , time step ~ 0.096x 10-15 s

CP Equations of motionEquations of motion from Lcp :
Ionic time evolution
Electronic time evolution
Constraint equation
Boundary conditions

Hellmann-Feynman TheoremIf ? is an exact eigenfunction of a Hamiltonian H, and E is the corresponding energy eigenvalue :
? is any parameter occurring in H
For an approximate wavefunction ?
For an exact ?
I
I
R
EF∂∂−=

Force on Ions
GI ? constraint
force+ GI
When, ?i is an eigenfunction
Force on the ions due to electronic configuration, when electronic wavefunction is an eigen function is zero

Constants of motion
Vibrational density of states of electronic degrees of freedom
Comparison with the highest frequency
phonon mode of nuclear subsystem

Constants of motion

Merits and Demerits of CPMDAdvantagesFast dynamics compared to BOMD
No need to perform the quenching of electronic wave function at each time step
DisadvantagesDynamics is different from the adiabatic evolution on BO surface
Forces on ions are different from the BO forces
Ground state
?i ? ?ksi ? good agreement
with the BOMD

Velocity Verlet algorithm for CPMD
.

ReferencesR. Car and M. Parrinello; Phys. Rev. Lett. 55 (22), 2471 (1985)
D. Marx, J. Hutter; http://www.fz-juelich.de/nic-series/
F. Buda et. al; Phys. Rev. A 44 (10), 6334 (1991)
D.K. Remler, P.A. Madden; Mol. Phys. 70 (6), 921 (1990)
B.M. Deb; Rev. Mod. Phys. 45 (1), 22 (1973)
M. Parrinello; Comp. Chemistry 22, (2000)
M.C. Payne et. al; Rev. Mod. Phys. 64 (4), 1045 (1992)

CPMDCPMD code is available at http://www.cpmd.org
Code developers : Michele Parrinello, Jurg Hutter, D. Marx, P. Focher, M.
Tuckerman, W. Andreoni, A. Curioni, E. Fois, U. Roethlisberger,
P. Giannozzi, T. Deutsch, A. Alavi, D. Sebastiani, A. Laio, J.
VandeVondele, A. Seitsonen, S. Billeter and others
PWscf (Plane Wave Self Consistent field) http://www.pwscf.org
PINY-MD http://homepages.nyu.edu/~mt33/PINY_MD/PINY.html

Applications

Autoionization in Liquid Water
Chandler, Parrinello et. al Science 2001, 291, 2121
pH determination of water by CPMDIntact water molecules dissociate ? OH- + H3O+ Rare event ~ 10 hours >>>> fs
Transition state separation between the charges ~ 6Å
Proposed theory ? Autoionization occurs due to specific solvent
structure and hydrogen bond pattern at transition state
Diffusion of ions from this transition state
Role of solvent
structure in
autoionization
Diffusion of ions
Microsecond motion of a system as it crosses
transition state can not be resolved experimentally
pH = - log [H+]

Nature of proton transfer in waterGrotthuss’s idea : Proton has very high mobility in liquid water which is due to the rearrangement of bonds through a long chain of water molecule; effective motion of proton than the real movement
+ +

Charge separation
Chandler, Parrinello et. al Science 2001, 291, 2121
1
Dissociation:
Fluctuation in
solvent electric field ;
cleavage of OH bond
2
H3O+ moves by
proton transfer
within 30 fs
3
4
Conduction of
proton through
H-bond network
60 fs5
Crucial
fluctuations carries
system to
transition state ;
breaking of H-
bond : 30 fs 6
NO fast ion
recombination

Order parameter for autoionizationFluctuations that control routes for proton :
No. of hydrogen bond connecting the ions : ?
? = 2 ; recombination occurs within 100 fs
reactant ? = 0 ; product ? ? 3
Critical ion separation is 6 Å
At ? = 2 , sometimes reactant basin ; Thus ? is not the only order
parameterPotential of proton in H-bonded wire ? fluctuation
q ? configuration description ; q = 1 neutral ; q = 0 charge
separated? E = E[r(1) – r(0)] ? solvent preference for separated ions over neutral
molecules

Potential of protons in hydrogen bonded wires connecting H3O+ and OH- ions
Chandler, Parrinello et. al Science 2001, 291, 2121
Neutral state, bond destabilizing
electric field has not appeared
Electric field starts to
appear ; metastable
state w.r.t. proton
motion ; 2kcal/mol
higher than neutral
state
Field fluctuations
increase ; stable charge
separated state ;
20kcal/mol more stable

Nature of the hydrated excess proton in liquid water
Two proposed theories : 1. Formation of H9O4+ (by Eigen)
2. Formation of H5O2+ (by Zundel)
Charge migration happens in a few
picoseconds
Tuckermann, Parrinello et. al J. Chem. Phys. 1997, 275, 817
+ +
H9O4+
H5O2+
+
Hydrogen bonds in solvation shells of the ions break and reform and the local
environment reorders
Ab initio calculations show
that transport of H+ and OH-
are significantly different

Proton transport
Tuckermann, Parrinello et. al Nature 1997, 275, 817
Proton diffusion does not occur via
hydrodynamic Stokes diffusion of a rigid
complex
Continual interconversion between the
covalent and hydrogen bonds
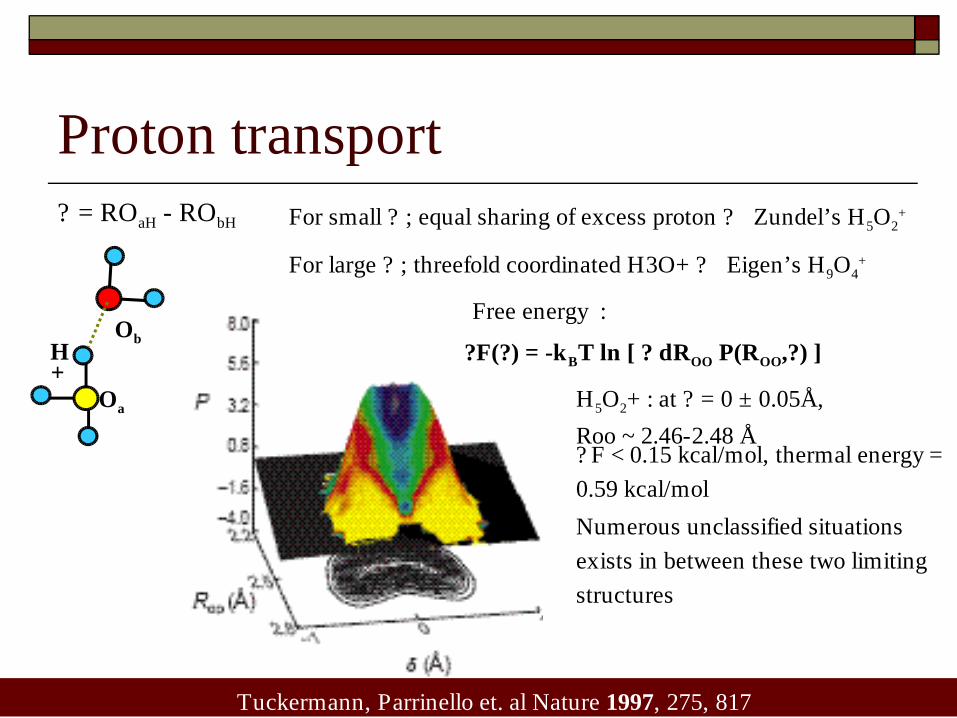
Proton transport? = ROaH - RObH
+Oa
ObH
For small ? ; equal sharing of excess proton ? Zundel’s H5O2+
For large ? ; threefold coordinated H3O+ ? Eigen’s H9O4+
Tuckermann, Parrinello et. al Nature 1997, 275, 817
?F(?) = -kBT ln [ ? dROO P(ROO,?) ]
Free energy :
H5O2+ : at ? = 0 ± 0.05Å,
Roo ~ 2.46-2.48 Å? F < 0.15 kcal/mol, thermal energy =
0.59 kcal/mol
Numerous unclassified situations
exists in between these two limiting
structures

Breaking bonds by mechanical stress
Frank et. al J. Am. Chem. Soc. 2002, 124, 3402
Reactions induced by mechanical stress in PEG1. Formation of ions corresponds to heterolytic bond cleavage 2. Motion of electrons during the reaction Polymer is
expanded with
AFM tip
Unconstrained reactions can not be observed by classical MD
Quantum chemical approaches are more powerful in
describing the general chemical reactivity of complex
systems

H
H
C2
C C
C
O1
O2 H
H H H
H H H H
O
H
H
Solvent
Small piece of PEG in water
Breaking bonds by mechanical stress
Method ?E (C-O) kcal/mol ?E (C-C) kcal/mol
BLYP
Exp
83.979.1
85.0 83.0
Radicaloid bond breaking
After equilibration, distance between O1 and O2 was
increased continuously by 0.0001 au/time
Reaction started at 250 K ; C2O1 ~ 3.2 Å

Snapshots of the reaction mechanisms
O
O
H
O
H
H O
H
H OH
+
-
OH
OH
O
H
O H
H
OH
O
O
H
O
H HO
HH
O
H
O
O
H
O
H HO
H HO
H
OO
HO
H
HO
H
H OH
+
-
O
OH
O
H
O H
H
OH
H
250 K
320 K
Frank et. al J. Am. Chem. Soc. 2002, 124, 3402

Hydrogen bond driven chemical reaction
Parrinello et. al J. Am. Chem. Soc. 2004, 126, 6280
Beckmann rearrangement of Cyclohexanone Oxime into ?-Caprolactam in
SCWSCW accelerates and make selective synthetic organic
reactionsSystem
description : CPMD simulation , BLYP exchange correlation
MT norm conserving pseudo potential
Plane wave cut-off 70 Ry, Nose-Hoover thermostat
T = 673K, 300K
64 H2O + 1 solute, 18 ps analysis + 11 ps equil.
Disrupted hydrogen bond network of SCW alters the solvation of O and N

Proton attack on the Cyclohexanone Oxime
Parrinello et. al J. Am. Chem. Soc. 2004, 126, 6280

Problems
Computationally costly
Can not simulate slow chemical processes that take place beyond time scales of 10 ps
Inaccuracy in the assumption of exchange and correlation potential
Limitation in the number of atoms and time scale of simulation
Inaccurate van der Waals forces, height of the transition energy barrierBOMD not applicable for photochemistry; transition between different electronic energy levels

Other methodsQM/MM – quantum mechanics / molecular mechanics
Classical MD AIMD e.g. catalytic part in enzyme
Path-sampling approach combined with ab-initio MD for slow chemical processes
Metadynamics, for slow processes

Conclusions
CPMD : nuclear and electronic degrees of freedomInteraction potential is evaluated on-the-flyBond formation and breaking is accessible in CPMD : direct access to the chemistry of materialsTransferability over different phases of matterCPMD is computationally expensive

Acknowledgement
Prof. S. Balasubramanian
Dr. M. Krishnan, Bhargava, Sheeba, Saswati

THANK YOU





























