Embed Size (px)
Citation preview

Utilização de Planilhas de Cálculo em QuímicaAnalítica
CAPÍTULO 3
A química é, essencialmente, uma ciência experimental. Este capítuloapresenta uma variedade de experimentos de laboratório, desde astitulações clássicas e a gravimetria até métodos instrumentais comocromatografia e espectroscopia. Instruções detalhadas são dadas paraa realização de cada experimento.
Métodos Selecionados de Análise
CAPÍTULO 37
© R
oyal
ty-fr
ee/C
orbi
s
Este capítulo contém instruções detalhadas para realizar uma variedade de análises químicas. Osmétodos foram selecionados para introduzi-lo a técnicas analíticas amplamente utilizadas pelos
químicos. Para muitas dessas análises, a composição das amostras é conhecida pelo professor. Assim,você será capaz de avaliar quão bem domina essas técnicas.
Suas chances de sucesso aumentarão muito se você reservar algum tempo antes de entrar no la-boratório para ler cuidadosamente e entender cada etapa do método e para planejar como e quandovocê vai realizar cada etapa.
A discussão nesta seção está voltada para ajudá-lo a desenvolver hábitos de trabalho eficientes no la-boratório e também para lhe oferecer algumas informações gerais sobre um laboratório de química analí-tica. Antes de iniciar uma análise, você deve entender a importância de cada etapa do procedimento paraevitar armadilhas e fontes potenciais de erros que são inerentes a todos os métodos analíticos. Informaçõessobre essas etapas podem ser normalmente encontradas (1) nas seções de discussão preliminares, (2) noscapítulos anteriores referenciados na seção de discussão, e (3) nas “Notas” que acompanham muitos dosprocedimentos. Se, depois de ler esses materiais, você ainda não entender a razão para realizar uma ou maisetapas do método, consulte seu instrutor antes de iniciar o trabalho no laboratório.
A Exatidão das Medições
Ao se deparar com um procedimento analítico, você deve decidir quais medições devem ser feitas com omáximo de precisão e, portanto, com o máximo de cuidado, e quais podem ser, ao contrário, realizadas ra-pidamente com pouca atenção à precisão. Geralmente, as medições que aparecem na equação usada paracalcular os resultados devem ser realizadas com o máximo de precisão. As medições restantes podem edevem ser feitas com menos cuidado para economizar tempo. As palavras cerca de e aproximadamente são

SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1001
com freqüência empregadas para indicar que uma medição não precisa ser feita cuidadosamente. Por exem-plo, você não deve desperdiçar tempo e esforço para medir um volume com precisão de �0,02 mL quan-do uma incerteza de � 0,5 mL ou até �5 mL não provocará efeitos marcantes no resultado.
Em alguns procedimentos, uma instrução como “pesar três amostras de 0,5 g com uma precisão de 0,1mg” pode ser encontrada. Nestes caso, as amostras de 0,4 a 0,6 g são aceitáveis, porém suas massas devemser conhecidas com uma precisão de 0,1 mg. O número de algarismos significativos na especificação deum volume ou massa também é um guia para o cuidado que se deve ter em uma medição. Por exemplo, ainstrução “adicionar 10,00 mL de uma solução em um béquer” indica que você deve medir, cuidadosa-mente, o volume com uma bureta ou uma pipeta, com uma incerteza de talvez �0,02 mL. Ao contrário, sea instrução for “adicione 10 mL”, a medição pode ser feita com uma proveta graduada.
Administração do Tempo
Você deve avaliar cuidadosamente o tempo requerido para todas as operações envolvidas em uma análiseantes de iniciar o trabalho. Essa avaliação deverá revelar aquelas operações que requerem um tempo deespera considerável, porém nenhum ou pouco tempo do operador. Exemplos dessas operações incluem asecagem de uma amostra em uma estufa, o resfriamento de uma amostra em um dessecador ou a evapo-ração de um líquido em uma chapa de aquecimento. Laboratoristas eficientes usam esses períodos pararealizar outras operações ou também para iniciar uma nova análise. Algumas pessoas consideram relevantepreparar por escrito uma programação para cada período de laboratório para evitar a perda de tempo.
O planejamento do tempo também é necessário para identificar períodos em que uma análise pode serinterrompida durante uma noite ou mais, assim como aquelas operações que devem ser finalizadas seminterrupções.
Reagentes
Instruções para a preparação de reagentes acompanham muitos procedimentos. Antes de preparar essesreagentes, assegure-se de conferir se já existe algum preparado e disponível para uso geral.
Se um reagente é reconhecidamente perigoso, você deve planejar antes do período no laboratório asetapas que deve seguir para evitar danos ou acidentes. Além disso, você deve estar familiarizado comas regras que se aplicam em seu laboratório para a disposição de resíduos líquidos e sólidos. Essas regrasvariam de uma instituição para outra e até mesmo entre laboratórios de um mesmo local.
Água
Alguns laboratórios empregam desionizadores para purificar água, outros empregam destiladores para essepropósito. Os termos “água destilada” e “água desionizada” são utilizados nas instruções a seguir. Ambosos tipos são aceitáveis para os procedimentos descritos neste capítulo.
Você deve utilizar água de torneira apenas para a limpeza preliminar das vidrarias. A vidraria lavada éentão enxaguada com pelo menos três pequenas porções de água destilada ou desionizada.
37A UM EXPERIMENTO INTRODUTÓRIO
O objetivo desse experimento é introduzir algumas das ferramentas, técnicas e habilidades necessárias parase trabalhar em um laboratório de química analítica. As técnicas são consideradas, uma a uma, como opera-ções unitárias. É importante dominar essas técnicas e adquirir habilidades individuais antes de se depararcom os demais experimentos de laboratório.
37A-1 O Emprego da Balança Analítica
Discussão
Nesse experimento, você obterá a massa de 5 moedas de cinco centavos, primeiro, determinando a massade cada uma individualmente. Então, você vai determinar a massa de todas as cinco moedas juntas, retirar

1002 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON
uma moeda de cada vez, e calcular a massa individual de cada uma encontrando a diferença. As duas mas-sas determinadas para cada moeda pelos dois métodos devem ser similares com uma diferença de apenasalguns miligramas. A partir desses dados, você vai determinar os valores médios e medianos, o desviopadrão e o desvio padrão relativo para as massas das moedas.
Você vai então pesar um cilindro de alumínio de massa desconhecida e obter a massa desse objeto.
PROCEDIMENTO
1. Depois de ter sido instruído no uso da balança e estar familiarizado com o seu emprego, obtenha umaporção de moedas, um cilindro de alumínio e um par de pinças com o professor.
2. Não pegue as moedas ou o cilindro com os dedos; sempre utilize as pinças. Se você está usando umabalança mecânica, assegure-se de que ela se encontre na posição “desligada” ou em “trava completa”toda vez que for remover ou colocar algo no prato da balança.
3. Antes de iniciar a determinação das massas, zere cuidadosamente sua balança analítica. Selecione cincomoedas aleatoriamente e pese cada uma na sua balança. Anote os valores em seu caderno de laboratório.Identifique as moedas colocando cada uma em uma folha de papel rotulada.
4. Zere sua balança. Coloque as cinco moedas no prato da balança, determine a massa total e anote o valor.5. Retire uma das moedas da balança, obtenha a massa das quatro restantes e anote a massa.6. Repita esse processo, removendo uma moeda de cada vez. Obtenha a massa de cada moeda pela dife-
rença. Esse processo é conhecido como pesagem por diferença, que é a maneira como muitas determi-nações de massa são feitas em um laboratório analítico.
7. Finalmente, verifique o zero em sua balança e encontre a massa do cilindro de alumínio.
37A-2 Fazendo Transferências Quantitativas
Discussão
O experimento a seguir foi elaborado para torná-lo hábil no emprego correto do balão volumétrico.
PROCEDIMENTO
1. Pese um béquer de 50 mL em uma balança mecânica ou em uma balança eletrônica apropriada.2. Ajuste a balança para uma massa adicional de 0,4 g e adicione KMnO4 no béquer até que o braço da ba-
lança esteja novamente balanceado. Se você estiver utilizando uma balança eletrônica com a função detara, pressione o botão para ajustar o zero da balança. Depois, adicione KMnO4 até que a balançaindique aproximadamente 0,4 g. Observe que os produtos químicos nunca devem ser devolvidos aofrasco original, para evitar contaminações.
3. Dissolva o permanganato de potássio do béquer com cerca de 20 mL de água destilada. Agite cuida-dosamente para evitar perdas. Essa solução é quase saturada e atenção deve ser dada para assegurar adissolução completa dos cristais.
4. Transfira quantitativamente a solução para um balão volumétrico de 100 mL com o auxílio de umpequeno funil. Para evitar que a solução derrame pela parte externa do béquer, utilize um bastão de vidro para escoar a solução e depois toque levemente o bastão na boca do béquer para remover aúltima gota. Adicione mais água no béquer, agite e repita o procedimento.
5. Repita o procedimento até que não reste nenhum indicativo da cor do permanganato no béquer. Anote onúmero de lavagens necessárias para transferir quantitativamente o permanganato do béquer para obalão volumétrico.
6. Transfira para o balão qualquer resquício da solução presente no bastão de vidro com um pouco deágua. Enxágüe também o funil e remova-o. Dilua a solução do balão até que a parte inferior do meniscoatinja a marca graduada de 100 mL. Tampe o balão volumétrico, vire-o para baixo e agite. Retorne obalão à posição original e deixe que as bolhas de ar voltem para a sua parte superior.

7. Repita esse procedimento cerca de dez vezes até que a solução fique completamente homogênea.Guarde a solução para a etapa escrita na Parte 37A-3.
37A-3 Transferência de uma Alíquota
Discussão
Toda vez que uma bureta ou uma pipeta é usada para retirar uma alíquota de uma solução, o líquido con-tido antes da medida deve ter a mesma composição da solução a ser transferida. As operações a seguir sãodestinadas a ilustrar como lavar e preencher uma pipeta e como transferir uma alíquota de uma solução.
PROCEDIMENTO
1. Preencha uma pipeta com a solução de permanganato de potássio e deixe escoar.2. Aspire com a pipeta alguns mililitros de água destilada contida em um béquer de 50 mL, enxágüe toda
a superfície interna da pipeta e descarte a solução de lavagem. Não preencha a pipeta completamente;esse procedimento consome tempo, causa desperdício e é ineficiente. Aspire apenas uma pequena quan-tidade, vire a pipeta na horizontal e gire-a para lavar a superfície interna.
3. Determine o número mínimo de vezes necessário para que o procedimento de lavagem remova comple-tamente a cor do permanganato da pipeta. Se a sua técnica for eficiente, três lavagens serão suficientes.
4. Preencha novamente a pipeta com a solução de permanganato e proceda à lavagem, como descrito ante-riormente. Dessa vez, colete a água de lavagem em uma proveta graduada e determine o volume mínimoda água necessário para remover a cor do permanganato. Menos de 5 mL são suficientes com uma téc-nica eficiente. Durante a operação de lavagem a água contida no béquer de 50 mL foi contaminada pelopermanganato? Se uma coloração rosada aparecer no béquer repita todo o procedimento com maiscuidado.
5. Como um teste para a sua habilidade técnica, peça ao professor para observar e comentar a seguinteoperação: enxágüe uma pipeta de 10 mL várias vezes com a solução de permanganato de potássio quevocê preparou.
6. Pipete 10 mL da solução de permanganato em um balão volumétrico de 250 mL.7. Dilua cuidadosamente a solução até completar o volume, tentando misturar o menos possível os com-
ponentes.8. Misture a solução virando o balão para baixo e agitando repetidas vezes. Observe o esforço necessário
para dispersar a cor do permanganato uniformemente na solução.9. Enxágüe a pipeta com a solução contida no balão volumétrico. Pipete uma alíquota de 10 mL em um
frasco de erlenmeyer.
37A-4 Calibração de uma Pipeta
Discussão
A técnica apropriada para a calibração de uma pipeta analítica de transferência é facilmente aprendida coma prática, cuidado e atenção aos detalhes. Com exceção de determinações de massas, esse experimentoconstitui potencialmente o conjunto de medidas mais exato e preciso que você realizará.
PROCEDIMENTO
1. Limpe uma pipeta de 10 mL. Quando uma pipeta, uma bureta ou outra vidraria volumétrica são limpasapropriadamente, nenhuma gota do reagente permanece na superfície interna quando essas vidrarias sãoescoadas. Isso é muito importante para a obtenção de resultados exatos e reprodutíveis. Se um reagenteadere na parte interna de uma pipeta, você não pode transferir o volume nominal da pipeta. Se vocêlimpar uma pipeta ou qualquer outra vidraria com uma solução alcoólica de KOH, utilize o recipiente
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1003

contendo a solução de lavagem somente dentro da pia e lave-o corretamente antes de guardá-lo. Não ocoloque diretamente na bancada; isso pode danificar a sua superfície. A solução é muito corrosiva. Sevocê sentir seus dedos escorregadios após o uso, ou se alguma parte de seu corpo desenvolver uma irri-tação, lave a área copiosamente com água.
2. Obtenha uma pêra de sucção, um erlenmeyer de 50 mL com uma tampa seca, um béquer de 400 mLcontendo água destilada à temperatura ambiente e um termômetro.
3. Determine a massa do frasco com tampa e anote o valor com uma precisão de 0,1 mg. Não toque ofrasco com seus dedos após a pesagem. Utilize pinças ou uma tira dobrada de papel manteiga paramanipulá-lo.
4. Meça e anote a temperatura da água.5. Pipete 10,00 mL de água destilada no frasco usando a técnica descrita na página 4L. Tampe o frasco,
determine a massa do frasco contendo a água destilada e anote.6. Da mesma maneira, transfira uma segunda alíquota de 10,00 mL de água destilada com a pipeta para o
frasco; remova a tampa apenas antes da adição. Recoloque a tampa e, mais uma vez, determine e anotea massa do frasco e da água. Em cada etapa do experimento, determine a massa de água adicionada nofrasco com a pipeta.
7. Repita esse procedimento até que você tenha determinado, consecutivamente, quatro massas de águacom uma precisão de 0,02 g. Se as determinações das massas de água dispensadas pela pipeta não apre-sentarem uma precisão nessa faixa, sua técnica de pipetagem pode estar incorreta. Consulte seu profes-sor para ajudá-lo a encontrar a fonte de erro e, então, repita o experimento até ser capaz de dispensarquatro volumes de água consecutivos com a precisão necessária.
8. Corrija a massa em função do empuxo como descrito na página 27 e calcule o volume da pipeta emmililitros.
9. Informe a média, o desvio padrão e o desvio padrão relativo do volume de sua pipeta. Calcule e informeos resultados com um intervalo de 95% de confiança.
37A-5 Leitura de Buretas
Discussão
O exercício a seguir visa à prática da leitura de uma bureta e confirmar a exatidão de suas leituras.
PROCEDIMENTO
1. Obtenha um conjunto de cinco secções seladas de buretas com seu professor.2. Vire cada secção de bureta para baixo e bata levemente para remover qualquer solvente que possa ter
ficado na extremidade selada.3. Anote o número e as leituras de cada secção de bureta no formulário fornecido. Utilize um cartão mar-
cado para leituras de bureta para realizar as leituras com uma precisão de 0,01 mL.4. Compare suas leituras com os valores fornecidos pelo seu professor.
37A-6 Leitura de uma Bureta
Discussão
O exercício a seguir demonstra a maneira apropriada de se usar uma bureta.
PROCEDIMENTO
1. Monte uma bureta em um suporte e preencha-a com água destilada.2. Espere pelo menos 30 segundos antes de realizar a primeira leitura. Utilize um cartão de leitura para
bureta para realizar as leituras. Um cartão de leitura para bureta pode ser facilmente construído colando
1004 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON

um pedaço de fita adesiva preta em um cartão de 7,5 � 12 cm. Nuncaajuste o volume em uma bureta exatamente em 0,00 mL. A tentativa derealizar esse procedimento provocará distorções no processo de me-dição e perda de tempo.3. Agora, deixe escoar cerca de 5 mL em um erlenmeyer de 250 mL.
Espere cerca de 30 segundos e obtenha a “leitura final”. A quantidadede solução no frasco é igual à diferença entre a leitura final e a inicial.Anote a sua leitura final em seu caderno de laboratório e, então, peçapara seu professor obter a leitura final. Compare os dois resultados.Eles devem concordar dentro de uma precisão de 0,01 mL. Note queo dígito final da leitura da bureta é sua estimativa da distância entreduas marcas consecutivas de 0,1 mL da bureta.
4. Preencha novamente a bureta e tome um novo valor inicial. Agora,adicione 30 gotas no erlenmeyer e anote a leitura final. Calcule ovolume médio de uma gota; repita empregando 40 gotas e calculenovamente o volume médio de uma gota. Anote esses resultados ecompare-os.
5. Finalmente, pratique adicionando meias gotas no frasco. Calcule ovalor médio de várias meias gotas e compare seus resultados comaqueles que você obteve com as gotas inteiras. Quando for realizar ti-tulações, você deve estar atento para determinar o ponto final empre-gando meias gotas para obter uma boa precisão.
37A-7 Amostragem1
Discussão
Em muitos métodos analíticos somente uma pequena fração de umapopulação inteira é analisada. Os resultados obtidos a partir da determi-nação de um analito em uma amostra de laboratório são admitidos comosimilares às concentrações do analito em toda a população. Conseqüen-temente, uma amostra de laboratório obtida a partir de um grupo deveser representativa da população.
Nesse experimento, você vai investigar como as dimensões daamostra influenciam na incerteza associada com a etapa de amostragem.Geralmente, a dimensão de determinada amostra deve aumentar con-forme a heterogeneidade da amostra se eleva, a fração do analitodiminui ou se reduz a incerteza desejada. O sistema modelo usado nesseexperimento consiste em uma porção de esferas de plástico que sãoidênticas no tamanho, forma e densidade, mas de cores diferentes. Se p
representa a fração de partículas do analito (esferas de uma cor), então 1 � p é a fração de um segundo tipode partículas (esferas de uma segunda cor). Se uma amostra contendo n partículas é retirada de uma popu-lação, o número de partículas do analito na amostra será np. Pode-se demonstrar que o desvio padrão donúmero de partículas do analito np, obtido a partir de uma amostra contendo uma mistura de dois compo-nentes, é . O desvio padrão relativo (sr) é portanto,
sr � �C1 � p
np
2np(1 � p)np
2np(1 � p)
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1005
Uma secção de bureta construída apartir de uma bureta quebrada. Asburetas quebradas são cuidadosamentelimpas e cortadas em pedaços de cerca de 10 cm de comprimento. A extremidade superior de cada secçãoé cuidadosamente selada fundindo-se o vidro, e a extremidade oposta éalongada para formar uma ponta. A extremidade alongada é entãocortada para permitir uma abertura de aproximadamente 1 mm naextremidade da secção de bureta. Umaseringa hipodérmica equipada comuma agulha é utilizada para adicionarágua destilada em cada secção até queela esteja preenchida pela metade. A extremidade alongada de cadasecção é então selada e as secções sãoguardadas com a parte superior parabaixo em um suporte para tubos deensaio ou em um bloco de madeiracom espaços apropriados paraacomodar as secções. Cada secção de bureta deve ser numerada.
1J. E. Vitt e R. C. Engstrom, J. Chem. Educ., v. 76, p. 99, 1999.

Essa equação sugere que, conforme o número de partículas amostradas aumenta, a incerteza relativadiminui. Usando uma mistura de esferas de duas cores, você vai determinar a incerteza da amostragem emfunção da dimensão da amostra.
PROCEDIMENTO
1. Mexa bem o recipiente contendo as esferas e retire uma amostra usando um béquer pequeno. Assegure-se de que o béquer esteja totalmente cheio, mas não transbordando.
2. Transfira as esferas para uma bandeja e conte o número de esferas de cada cor.3. Repita a etapa número 1 utilizando um béquer de tamanho intermediário e depois um béquer grande.
Anote o número total de esferas na sua amostra e a porcentagem de esferas de uma das cores indicadaspelo seu professor. Cada estudante de sua sala deverá coletar e contar três amostras similares e colocaros resultados em um formulário que será fornecido pelo seu professor. Depois que todos os dados foremcoletados, o formulário será copiado e distribuído para todos os estudantes de sua sala.
CÁLCULOS
1. Usando os dados coletados no formulário, calcule a porcentagem média de esferas de uma cor especí-fica e o desvio padrão relativo da porcentagem de cada amostra de diferentes dimensões.
2. Utilizando a equação mostrada previamente, baseada na teoria da amostragem, calcule o desvio padrãorelativo teórico usando os valores de p e o número médio de partículas para cada uma das três amostrasde dimensões diferentes.
3. Compare os dados obtidos pela sua sala com os resultados teóricos. O desvio padrão relativo diminuiconforme a dimensão da amostra aumenta, de acordo com a teoria da amostragem?
4. Utilize a equação do desvio padrão relativo para calcular o número de esferas que deveriam seramostradas para atingir um desvio padrão relativo de 0,002.
5. Sugira duas razões pelas quais essa teoria não seria adequada para descrever a amostragem de muitosmateriais para análises químicas.
37A-8 Determinando Erros de Amostragem por meio de Análise por Injeção em Fluxo2
Discussão
A variância total ao se analisar uma amostra de laboratório pode ser considerada a soma da variância dométodo e da amostragem (ver a Seção 8B-2). Além disso, pode-se decompor a variância do métodona soma das variâncias que surgem na preparação da amostra e na etapa de medição final .
Pode-se estimar a variância de medição final por meio da determinação em replicata de uma mesmaamostra. A variância da preparação da amostra pode ser estimada pela propagação das incertezas asso-ciadas a essa etapa. Se for obtida a variância total a partir de medidas em replicata de amostras diferentes,a variância da amostragem é facilmente conseguida pela diferença.
A determinação de fosfato pelo procedimento colorimétrico por injeção em fluxo é usada para se obteros dados necessários. A reação é
H3PO4 � 12Mo � 24H� [H3PMo12O40] � 12H2O8O2�4
s2a
s2o
s2p
s2f
s2o � s2
a � s2p � s2
f
s2fs2
p
s2as2
m
s2o
1006 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON
2R. D. Guy, L. Ramaley, e P. D. Wentzell, J. Chem. Educ., v. 75, p. 1.028–1.033, 1998.

O ácido 12-molibdofosfórico [H3PMo12O40], normalmente abreviado como 12-MPA, é então reduzido paraazul de fosfomolibdênio, PMB, por um agente redutor apropriado como o ácido ascórbico.
12-MPA � ácido ascórbico PMB � ácido deidroascórbico
A absorbância do PMB é então medida em 650 nm no colorímetro por injeção em fluxo.
PREPARO DAS SOLUÇÕES
1. Solução de ácido nítrico 0,4 mol L�1. Adicione 26 mL de HNO3 concentrado em um balão volumétricode 1 L e dilua até a marca com água destilada.
2. Reagente molibdato 0,005 mol L�1, (NH4)6Mo7O24�4H2O. Em um balão volumétrico de 100 mL, dis-solva 0,618 g de heptamolibdato de amônio em uma solução 0,40 mol L�1 de HNO3. Dilua até a marcacom HNO3 0,40 mol L–1.
3. Reagente ácido ascórbico 0,7% em glicerina a 1%. Adicione 0,7 g de ácido ascórbico e cerca de 0,8 mLde glicerina em um balão volumétrico de 100 mL e dilua até a marca com água destilada (Nota).
4. Solução estoque de fosfato, 100 ppm de fosfato. Adicione 0,0143 g de KH2PO4 em um balão volu-métrico de 100 mL e dilua até a marca com água destilada.
5. Soluções de trabalho de fosfato, 10, 20, 30, 40 e 60 ppm de fosfato. Cada estudante deve preparar essassoluções em balões volumétricos de 25 mL.
NotaA glicerina é usada como tensoativo no sistema de análise por injeção em fluxo.
PROCEDIMENTO
Os estudantes devem trabalhar em pares durante esse experimento. Se você for o Estudante 1, prepare umamistura sólida de concentração desconhecida (Nota 1). Misture e triture a amostra com o auxílio de umalmofariz e um pilão por pelo menos dez minutos. Em seguida, transfira a mistura para uma folha limpade papel branco para formar um monte circular. Utilizando uma espátula, divida o monte em seis partesiguais. Para cada parte, retire uma porção de 0,10 g e determine com exatidão a sua massa. Transfira cadaporção para balões volumétricos individuais de 10 mL e dilua com água destilada. Devolva a misturarestante para o almofariz e mexa rapidamente. Transfira novamente a mistura para uma folha de papel bran-co e forme uma nova pilha circular. Divida novamente em seis partes. Desta vez, retire uma porção de 0,25g e pese-a com exatidão. Repita para as outras cinco porções. Transfira-as para balões volumétricos de25mL e dilua à marca com água destilada. Repita esse processo para massas de 0,50 g, diluindo para 50mL; 1,0 g, diluindo para 100 mL; e 2,50 g, diluindo para 250 mL. No final, o Estudante 1 deve ter cincoconjuntos de amostras com seis soluções em cada conjunto. Cada conjunto deve possuir a mesma concen-tração nominal, porém com diferentes massas da mistura desconhecida.
Enquanto o Estudante 1 está preparando as amostras, o Estudante 2 deve obter os dados necessáriospara a confecção de uma curva analítica usando os padrões de fosfato. Se você for o Estudante 2, utilize osistema de análise por injeção em fluxo, como é mostrado na Figura 37-1. O produto é determinado apósa reação em 650 nm com uma célula de detecção em fluxo. Injete cada padrão de fosfato três vezes e meçao pico de absorbância para cada padrão. Determine os valores médios do pico de absorbância para cadapadrão em função da concentração. Neste momento, o Estudante 1 deve ter preparado as amostras de con-centração desconhecida.
A seguir, injete as amostras desconhecidas em triplicata. Dezoito injeções devem ser realizadas paracada conjunto de amostras. Para a última amostra do último conjunto de amostras, faça dez injeções pa-ra se obter uma boa estimativa da variância final de medição, .s2
f
S
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1007

Análise dos DadosDigite os dados da curva analítica realizada pelo Estudante 2 em uma planilha e utilize a regressão linearpor quadrados mínimos para obter uma equação da curva. Digite os dados para os cinco conjuntos deamostras desconhecidas e use a equação para calcular a concentração de fosfato em cada uma das 30amostras. Expresse a concentração de fosfato em termos da porcentagem de KH2PO4 na mistura original.Sua planilha deve se parecer com aquela exibida na Figura 37-2. Faça um gráfico da porcentagem deKH2PO4 em função da massa da amostra. Observe a importância da dimensão da amostra na dispersãodos dados.
1008 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON
Molibdato
Amostrade fosfato
Bombaperistáltica
Ácido ascórbico
0,5
0,550 cm 50 cm
Descarte
Válvulade injeção Detector
50 µLµ
Figura 37-1 Configuração do sistema de análise por injeção em fluxo para adeterminação de fosfato. As vazões são dadas em mL/min. O tubo de Tygonpossui 0,8 mm de diâmetro interno.
,
,
,
,
,
,,,,,,,,,,,,
,,,,,,
,,
,
,,,,,,,,,
Massa nominal, g Massa verdadeira, g Volume da solução, mL Absorbância do pico Conc. %KH2PO4Amostra No
Figura 37-2 Planilha para registro dos dados das amostras de fosfato.
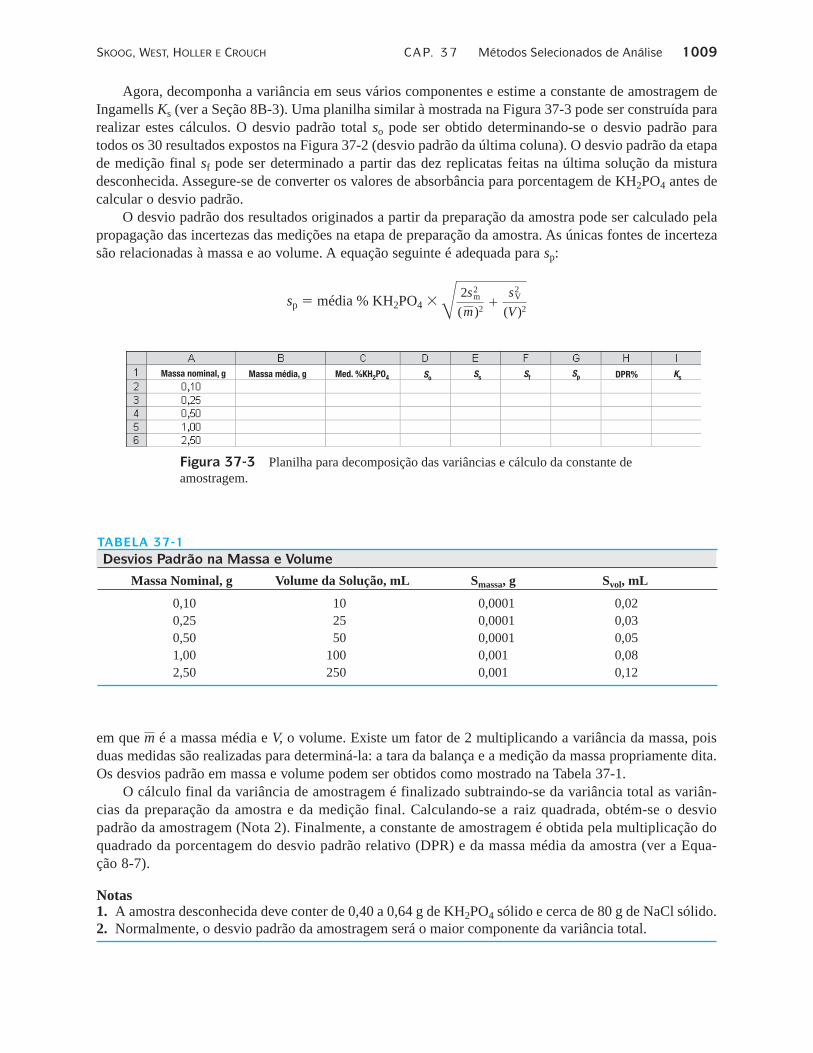
Agora, decomponha a variância em seus vários componentes e estime a constante de amostragem deIngamells Ks (ver a Seção 8B-3). Uma planilha similar à mostrada na Figura 37-3 pode ser construída pararealizar estes cálculos. O desvio padrão total so pode ser obtido determinando-se o desvio padrão paratodos os 30 resultados expostos na Figura 37-2 (desvio padrão da última coluna). O desvio padrão da etapade medição final sf pode ser determinado a partir das dez replicatas feitas na última solução da misturadesconhecida. Assegure-se de converter os valores de absorbância para porcentagem de KH2PO4 antes decalcular o desvio padrão.
O desvio padrão dos resultados originados a partir da preparação da amostra pode ser calculado pelapropagação das incertezas das medições na etapa de preparação da amostra. As únicas fontes de incertezasão relacionadas à massa e ao volume. A equação seguinte é adequada para sp:
sp � média % KH2PO4 �C 2s2m
( m )2�
s2V
(V )2
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1009
TABELA 37-1Desvios Padrão na Massa e Volume
Massa Nominal, g Volume da Solução, mL Smassa, g Svol, mL
0,10 10 0,0001 0,020,25 25 0,0001 0,030,50 50 0,0001 0,051,00 100 0,001 0,082,50 250 0,001 0,12
Massa nominal, g Massa média, g Med. %KH2PO4 So Ss Sf Sp DPR% Ks
Figura 37-3 Planilha para decomposição das variâncias e cálculo da constante deamostragem.
em que é a massa média e V, o volume. Existe um fator de 2 multiplicando a variância da massa, poisduas medidas são realizadas para determiná-la: a tara da balança e a medição da massa propriamente dita.Os desvios padrão em massa e volume podem ser obtidos como mostrado na Tabela 37-1.
O cálculo final da variância de amostragem é finalizado subtraindo-se da variância total as variân-cias da preparação da amostra e da medição final. Calculando-se a raiz quadrada, obtém-se o desviopadrão da amostragem (Nota 2). Finalmente, a constante de amostragem é obtida pela multiplicação doquadrado da porcentagem do desvio padrão relativo (DPR) e da massa média da amostra (ver a Equa-ção 8-7).
Notas1. A amostra desconhecida deve conter de 0,40 a 0,64 g de KH2PO4 sólido e cerca de 80 g de NaCl sólido.2. Normalmente, o desvio padrão da amostragem será o maior componente da variância total.
m

37B MÉTODOS GRAVIMÉTRICOS DE ANÁLISE
Aspectos gerais, cálculos e aplicações típicas das análises gravimétricas são discutidas no Capítulo 12.
37B-1 Determinação Gravimétrica de Cloreto em Amostra Solúvel
Discussão
A quantidade de cloreto em um sal solúvel pode ser determinada pela precipitação do cloreto de prata.
Ag� � Cl� AgCl(s)
O precipitado é coletado em um cadinho de filtração previamente pesado e, em seguida, é lavado. Depoisde o precipitado ter sido seco a 110 °C até massa constante, determina-se a sua massa.
A solução contendo a amostra permanece em meio levemente ácido durante a precipitação para seeliminar a possível interferência de ânions de ácidos fracos (como o CO ) que forma sais solúveis de prataem quantidades irrisórias em ambiente neutro. É necessário um leve excesso de íons prata para diminuir asolubilidade do cloreto de prata, porém deve-se evitar um grande excesso para minimizar a co-precipitaçãode nitrato de prata.
Primeiramente, o cloreto de prata forma colóides que são, em seguida, coagulados por aquecimento.O ácido nítrico e o pequeno excesso de nitrato de prata provocam a coagulação a partir de uma concen-tração eletrolítica relativamente alta. O ácido nítrico presente na solução de lavagem mantém a concentra-ção eletrolítica e elimina a possibilidade de peptização durante a etapa de lavagem; subseqüentemente oácido se decompõe a produtos voláteis quando o precipitado é seco. Ver a Seção 12A-2 para informaçõesadicionais relativas às propriedades e o tratamento de precipitados coloidais.
Assim como outros haletos de prata, o cloreto de prata, finamente dividido, pode sofrer fotodecom-posição:
2AgCl(s) 2Ag(s) � Cl2(g)
A prata elementar produzida nessa reação é responsável pelo desenvolvimento de uma coloração violetano precipitado. Teoricamente, essa reação provoca resultados baixos para o íon cloreto. Na prática, entre-tanto, seu efeito torna-se desprezível evitando-se a exposição direta e prolongada do precipitado à luz.
Se a fotodecomposição do cloreto de prata ocorre antes da filtração, a seguinte reação
3Cl2(aq) � 3H2O � 5Ag� 5AgCl(s) � ClO � 6H�
tende a provocar resultados elevados.No procedimento normal, uma pequena fotodecomposição do cloreto de prata é inevitável. É impor-
tante minimizar ao máximo possível a exposição do sólido a fontes intensas de luz.Em razão do custo elevado do nitrato de prata, qualquer reagente que não for usado deve ser armazena-
do em um recipiente apropriado; da mesma maneira, os precipitados de cloreto de prata devem ser guarda-dos depois do término da análise.3
PROCEDIMENTO
Limpe três cadinhos com fundo de vidro sinterizado de porosidade média ou três cadinhos de filtração de
�3S
¡hv
2�3
S
1010 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON
3A prata pode ser removida do cloreto de prata e do excesso de reagente pela redução com ácido ascórbico; ver J. W. Hill e L. Bellows. J. Chem.Educ., v. 63, n. 4, p. 357, 1986; ver também J. P. Rawat e S. Iqbal M. Kamoonpuri. J. Chem. Educ., v. 63, n. 4, p. 537, 1986; para recuperação(como AgNO3) baseada na troca iônica. Para consultar os possíveis riscos envolvidos na recuperação do nitrato de prata, ver D. D. Perrin, W. L. F.Armarego, e D. R. Perrin. Chem. Int., v. 9, n. 1, p. 3, 1987.

porcelana colocando em cada um deles, durante cerca de cinco minutos, aproximadamente 5 mL de HNO3
concentrado. Utilize vácuo (ver Seção 2-16) para escoar o ácido pelo cadinho. Enxágüe cada cadinho comtrês porções de água de torneira e interrompa o vácuo. Em seguida, adicione cerca de 5 mL de NH3 6 molL�1 e espere aproximadamente cinco minutos antes de escoar a solução pelo filtro. Finalmente, lave cadacadinho com seis a oito porções de água destilada ou desionizada. Identifique cada cadinho com umamarca. Seque os cadinhos a 110 °C até massa constante enquanto as outras etapas da análise estão sendorealizadas. A primeira secagem deve durar pelo menos uma hora; depois, os períodos de secagem podemser mais curtos (30 a 40 min). Este processo de aquecimento e secagem deve ser repetido até que a massa
se torne constante com uma precisão de 0,2 a 0,3 mg.Transfira a amostra desconhecida para um recipiente de pesagem e
seque-a a 110 °C (ver a Figura 2-9) por uma a duas horas; deixe o reci-piente e seu conteúdo resfriarem à temperatura ambiente em umdessecador. Pese por diferença (com uma precisão de 0,1 mg) amostrasindividuais em béqueres de 400 mL (Nota 1). Dissolva cada amostra comcerca de 100 mL de água destilada e 2 a 3 mL de HNO3 6 mol L�1.
Adicione lentamente, sob agitação, uma solução de AgNO3 0,2mol L�1 em cada uma das amostras, previamente resfriadas, até queseja observada a coagulação de AgCl (Notas 2 e 3) e, então, adicionemais 3 a 5 mL. Aqueça até quase fervura e faça a digestão dos sólidos
por cerca de 10 min. Adicione algumas gotas de AgNO3 para confirmar o fim da precipitação. Se ocor-rer a formação de mais precipitado, adicione cerca de 3 mL de AgNO3, faça a digestão, e teste nova-mente até a completa precipitação. Despeje a solução de AgNO3 que não foi usada em um recipiente dedescarte (não no frasco original do reagente). Cubra cada béquer e deixe-os em um lugar escuro por,pelo menos, duas horas (preferencialmente até o próximo período no laboratório).
Leia as instruções da Seção 2F para a filtração. Retire o líquido sobrenadante passando-o através doscadinhos de filtração de porcelana. Lave os precipitados várias vezes (enquanto eles ainda estão no béquer)com uma solução preparada adicionando-se 2 a 5 mL de HNO3 6 mol L�1 por litro de água destilada;remova essas soluções de lavagem pelo filtro. Transfira quantitativamente o AgCl dos béqueres para ca-dinhos individuais com pequenas porções da solução de lavagem; utilize um bastão de borracha para soltarqualquer partícula aderida às paredes dos béqueres. Continue a lavagem até que os filtrados estejam livresdo íon Ag+ (Nota 4).
Seque o precipitado a 110 °C por, pelo menos, uma hora. Guarde os cadinhos em um dessecadorenquanto eles resfriam. Determine a massa dos cadinhos contendo o precipitado. Repita esse ciclo de aque-cimento, resfriamento e pesagem até que a massa apresente uma concordância de 0,2 mg após pesagensconsecutivas. Calcule a porcentagem de Cl� na amostra.
Depois do término da análise, remova os precipitados batendo levemente os cadinhos sobre um pedaçode papel liso. Transfira o precipitado de AgCl para um recipiente de descarte de prata. Remova o excessode AgCl das cadinhos preenchendo-os com uma solução de NH3 6 mol L�1.
Notas1. Consulte seu instrutor com relação à quantidade apropriada da amostra.2. Determine, aproximadamente, a quantidade necessária de AgNO3 calculando-se o volume que seria
preciso se a amostra desconhecida fosse constituída por NaCl puro.3. Utilize um bastão de agitação para cada amostra e deixe-os dentro dos seus béqueres durante as deter-
minações.4. Para testar as lavagens para a remoção de Ag�, colete um volume pequeno em um tubo de ensaio e adi-
cione algumas gotas de HCl. Considera-se que a lavagem esteja completa quando nenhuma ou umafraca turbidez surge na solução.
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1011
� Assegure-se de identificar claramente seus béqueres e cadinhos.
Digerir significa aquecer semagitação um precipitado na soluçãomãe, isto é, na solução da qual elefoi formado.

37B-2 Determinação Gravimétrica de Estanho em Latão
Discussão
O latão é uma liga importante. Geralmente, o cobre é o principal componente com pequenas quantidadesde chumbo, zinco, estanho e outros elementos. O tratamento do latão com ácido nítrico provoca a formaçãode “ácido metaestânico” H2SnO3�xH2O que é pouco solúvel; todos os outros componentes são dissolvidos.O sólido é filtrado, lavado e calcinado para formar SnO2.
A determinação gravimétrica de estanho permite praticar o emprego de filtros de papel isentos de cin-zas e freqüentemente é conduzida em conjunto com uma análise mais abrangente de uma amostra de latão.
PROCEDIMENTO
Rotule três cadinhos de porcelana com tampa. Durante os intervalos do experimento, leve cada conjuntode cadinhos com as tampas para ignição a 900 °C em uma mufla até peso constante.
Não seque a amostra desconhecida. Se for instruído, lave-a com acetona para remover resíduos de óleoou graxa. Pese (com precisão de 0,1 mg) alíquotas de 1 g da amostra desconhecida em béqueres de 250mL. Cubra os béqueres com vidros de relógio. Coloque os béqueres na capela, e adicione, cuidadosamente,uma solução contendo cerca de 15 mL de HNO3 concentrado e 10 mL de H2O. Proceda à digestão dasamostras durante 30 min; adicione mais HNO3 se necessário. Lave os vidros de relógio e evapore assoluções até a redução de volume a 5 mL, mas não até a secura (Nota 1)
Adicione cerca de 5 mL de HNO3 3 mol L�1, 25 mL de água destilada e um quarto de tablete de polpade papel de filtro em cada amostra; aqueça durante 45 min sem levar à ebulição. Separe o precipitadoH2SnO3�xH2O em um filtro de papel isento de cinzas com baixa porosidade (Ver a Seção 2F-3 e as Notas2 e 3). Utilize pequenas quantidades de HNO3 0,3 mol L�1 aquecidas para lavar os últimos traços de cobredo precipitado. Faça um teste para verificar a eficiência da lavagem adicionando uma gota de NH3(aq) nasuperfície do precipitado; lave mais vezes se o precipitado ficar azul.
Remova o filtro de papel e seu conteúdo do funil, dobre-o e coloque-o nos cadinhos (com suas tam-pas) que foram levados a peso constante (Ver a Figura 2-14). Queime o filtro de papel à temperatura maisbaixa possível. Deve haver acesso livre ao ar durante a carbonização (ver a Seção 2F-3 e a Figura 2-15).Aumente a temperatura aos poucos até que todo o carbono seja removido. Em seguida, leve os cadinhoscobertos e seus conteúdos a massa constante em uma mufla a 900 °C (Nota 4).
Calcule a porcentagem de estanho na amostra desconhecida.
Notas1. Muitas vezes, redissolver os componentes solúveis dos resíduos após uma amostra ter sido evaporada
até a secura é difícil e consome tempo.2. A etapa de filtração pode consumir muito tempo e, uma vez iniciada, não pode ser interrompida.3. Se a amostra desconhecida for analisada eletroliticamente com relação ao seu conteúdo de chumbo e
cobre (ver Seção 37K-1), colete os filtrados em béqueres longos. O volume final deve ser de cerca de125 mL; evapore até este volume se for necessário. Se a análise for apenas do estanho, o volume daslavagens não é importante.
4. A redução parcial do SnO2 pode fazer que o precipitado incinerado fique acinzentado. Nesse caso, adi-cione uma gota de ácido nítrico, evapore cuidadosamente e incinere novamente.
37B-3 Determinação Gravimétrica de Níquel em Aço
Discussão
O níquel presente em uma amostra de aço pode ser precipitado a partir de um meio levemente alcalino comuma solução alcoólica de dimetilglioxima (ver Seção 12D-3). A interferência do ferro(III) é eliminada pelomascaramento com ácido tartárico. O produto é seco a 110 °C.
1012 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON

A característica volumosa do precipitado de níquel-dimetilglioxima limita a massa de níquel que podeser convenientemente manipulada e, dessa forma, a massa da amostra. Deve-se tomar cuidado para con-trolar o excesso de dimetilglioxima alcoólica utilizada. Se for adicionada uma quantidade muito grande, aconcentração de álcool poderá ser suficiente para dissolver quantidades apreciáveis do precipitado, provo-cando resultados negativos. Se a concentração de álcool for muito baixa, entretanto, o reagente pode pre-cipitar e provocar erros positivos.
PREPARO DAS SOLUÇÕES
1. Dimetilglioxima, 1% (m/v). Dissolva 10 g de dimetilglioxima em 1 L de etanol. (Essa solução é sufi-ciente para cerca de 50 precipitações.)
2. Ácido tartárico 15% (m/v). Dissolva 225 g de ácido tartárico em água suficiente para resultar em 1.500mL de solução. Se a solução não estiver transparente filtre-a antes de utilizá-la. (Essa solução é sufi-ciente para cerca de 50 precipitações.)
PROCEDIMENTO
Limpe e marque três cadinhos de vidro sinterizado com porosidade média (Nota 1); leve-os à massa cons-tate pela secagem a 110 °C durante pelo menos uma hora. Pese (com precisão de 0,1 g) amostras contendode 30 a 35 mg de níquel em béqueres individuais de 400 mL (Nota 2). Na capela, dissolva cada amostraem cerca de 50 mL de HCl 6 mol L�1 sob aquecimento brando. Adicione aproximadamente 15 mL deHNO3 6 mol L�1, e ferva moderadamente para eliminar quaisquer óxidos de nitrogênio que possam ter sidoproduzidos. Dilua até cerca de 200 mL e aqueça até fervura. Adicione cerca de 30 mL de ácido tartárico15% e NH3(aq) concentrado suficiente para produzir um odor fraco de amônia nos vapores sobre assoluções (Nota 3); então adicione mais 1 a 2 mL de NH3(aq). Se a solução não estiver transparente nestaetapa, siga os procedimentos da Nota 4. Acidifique as soluções com HCl (sem odor de NH3), aqueça até60 °C a 80 °C, e adicione cerca de 20 mL da solução de dimetilglioxima 1%. Sob agitação vigorosa, adi-cione NH3 6 mol L�1 até que um leve excesso apareça (odor fraco de NH3) e mais 1 a 2 mL adicionais.Digira os precipitados por 30 a 60 min, resfrie por pelo menos uma hora, e filtre.
Lave os sólidos com água até a ausência de Cl� na água de lavagem (Nota 5). Leve os cadinhos e seusconteúdos a massa constante a 110 °C. Anote a porcentagem de níquel na amostra. O precipitado seco tema composição Ni(C4H7O2N2)2 (288,92 g mol�1).
Notas1. Cadinhos de filtração de porcelana com porosidade média ou cadinhos de Gooch com fundo de vidro
podem substituir os cadinhos de vidro sinterizado nessa determinação.2. Utilize uma barra magnética de agitação diferente para cada amostra e deixe-a no béquer durante o pro-
cedimento.3. A presença ou ausência de excesso de NH3 é prontamente reconhecida pelo seu odor: faça movimentos
com as mãos para levar os vapores até seu nariz.4. Se houver a formação de Fe2O3 xH2O pela adição de NH3, acidifique a solução com HCl, adicione
mais ácido tartárico e neutralize novamente. Alternativamente, remova o sólido por filtração. É neces-sário finalizar a lavagem com uma solução aquecida de NH3/NH4Cl; as lavagens são combinadas com asolução contendo a maior parte da amostra.
5. Teste a presença de Cl� coletando uma pequena porção da água de lavagem em um tubo de ensaio, aci-dificando com HNO3 e adicionando uma ou duas gotas de AgNO3 0,1 mol L�1. As lavagens poderão serfinalizadas quando pouca ou uma fraca turbidez for observada.
#
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1013

37C TITULAÇÕES DE NEUTRALIZAÇÃO
Discussão
Titulações de neutralização são realizadas com soluções padrão de ácidos ou bases fortes. Enquanto umasolução simples (de ácido ou base) é suficiente para a titulação de determinado tipo de analito, é conve-niente ter soluções padrão disponíveis para ambos, ácido e base, no caso de ser necessária a retrotitulaçãopara localizar o ponto final com mais exatidão. A concentração de uma das soluções é estabelecida pela ti-tulação contra um padrão primário; a concentração da outra é então determinada a partir da razãoácido/base (isto é, o volume de ácido necessário para neutralizar 1,000 mL da base).
37C-1 O Efeito do Dióxido de Carbono Atmosférico nas Titulações de Neutralização
A água em equilíbrio com a atmosfera contém cerca de 1 � 10�5 mol L�1 de ácido carbônico em decor-rência do equilíbrio
CO2(g) � H2O H2CO3(aq)
Nesse nível de concentração, a quantidade de base 0,1 mol L�1 consumida pelo ácido carbônico em umatitulação típica é desprezível. Com reagentes mais diluídos (60,05 mol L�1), entretanto, o ácido carbôni-co presente na água, utilizada como solvente do analito e no preparo dos reagentes, pode ser liberado porfervura durante um período curto.
A água que foi purificada por destilação em vez da desionização está, muitas vezes, supersaturada comdióxido de carbono e pode, portanto, conter ácido suficiente para afetar os resultados de uma análise.4 Asinstruções a seguir assumem que a quantidade de dióxido de carbono na água utilizada pode ser negligen-ciada sem causar erros graves. Para mais discussões sobre os efeitos do dióxido de carbono nas titulaçõesde neutralização, ver a Seção 16A-3.
37C-2 Preparo de Soluções Indicadoras para Titulações de Neutralização
Discussão
A teoria dos indicadores ácido/base é discutida na Seção 14A-2. Existe um indicador para, virtualmente,qualquer faixa de pH variando entre 1 e 13.5 As instruções a seguir referem-se à preparação de soluções deindicadores adequados para a maioria das titulações de neutralização.
PROCEDIMENTO
As soluções estoque contêm, geralmente, 0,5 a 1,0 g de indicador por litro. (Um litro de indicador ésuficiente para centenas de titulações.)
1. Verde de bromocresol. Dissolva o sal de sódio diretamente em água destilada.2. Fenolftaleína, timolftaleína. Dissolva o indicador sólido em uma solução preparada com 800 mL de
etanol e 200 mL de água destilada ou desionizada
8
1014 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON
4A água que é utilizada para titulações de neutralização pode ser testada adicionando-se cinco gotas de fenolftaleína em uma porção de 500 mL.Menos que 0,2 a 0,3 mL de OH� 0,1 mol L�1 deve ser suficiente para produzir um leve início de coloração rosa do indicador. Se um volume maiorfor necessário, a água deve ser fervida e resfriada antes de sua utilização para o preparo de soluções padrão ou para a dissolução de amostras.
5Ver, por exemplo, J. A. Dean. Analitical Chemistry Handbook, p. 3.31-3.33. Nova York: McGraw-Hill, 1995.

37C-3 Preparo de Soluções Diluídas de Ácido Clorídrico
Discussão
O preparo e a padronização de ácidos são considerados nas Seções 16A-1 e 16A-2.
PROCEDIMENTO
Para uma solução 0,1 mol L�1, adicione cerca de 8 mL de HCl concentrado em aproximadamente 1 L deágua destilada (Nota). Misture bem e armazene em um frasco de vidro com rolha.
NotaÉ conveniente eliminar o CO2 da água fervendo-a previamente se for preparada uma solução muito diluída(60,05 mol L�1).
37C-4 Preparo de Hidróxido de Sódio Isento de Carbonato
Discussão
Ver as Seções 16A-3 e 16A-4 para informações envolvendo o preparo e a padronização de bases. Soluções padrão de base são razoavelmente estáveis enquanto elas forem protegidas do contato com
a atmosfera. A Figura 37-4 mostra um arranjo para prevenir a absorção de dióxido de carbono atmosféricodurante o armazenamento e quando o reagente é dispensado. O ar que entra no frasco passa por um sólidoabsorvente de CO2 como soda cáustica ou Ascarita II.6 A contaminação que ocorre conforme a solução étransferida do frasco de armazenamento para a bureta é, geralmente, desprezível.
Como alternativa ao sistema de armazenamento mostrado na Figura 37-4, um frasco de polietileno debaixa densidade bem fechado fortemente pode, freqüentemente, fornecer proteção suficiente a curto prazocontra a absorção de dióxido de carbono atmosférico. Antes de ser fechado, o frasco flexível deve ser com-primido para minimizar o espaço de ar no seu interior. Também deve-se tomar cuidado para manter o fras-co fechado, com exceção do período no qual seu conteúdo estiver sendo transferido para uma bureta.Soluções de hidróxido de sódio poderão, eventualmente, tornar o frasco de polietileno quebradiço.
A concentração das soluções de hidróxido de sódio diminui lentamente (0,1 a 0,3% por semana)quando a base é armazenada em frascos de vidro. A perda de concentração é causada pela reação da base
com o vidro para formar silicatos de sódio. Poresse motivo, soluções padrão de base nãodevem ser armazenadas por períodos longos(mais que uma ou duas semanas) em reci-pientes de vidro. Além disso, as bases nuncadevem ser mantidas em recipientes com tampade vidro, pois a reação entre a base e a tampapode prendê-la rapidamente no frasco de for-ma irreversível. Finalmente, para evitar essemesmo tipo de problema, as buretas comtorneiras de vidro devem ser imediatamenteesvaziadas e lavadas abundantemente comágua após terem sido utilizadas com soluçõespadrão de base. Esse problema pode ser evita-do com o uso de buretas equipadas comtorneiras de Teflon.
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1015
Algodão
Rolha de borrachacom dois furos
Algodão
Rolha comfenda
Frasco de plástico ourecoberto com parafina Pinça
de Mohr
Absorventede CO2
Figura 37-4 Arranjo para o armazenamento de soluçõespadrão de base.
6Thomas Scientific, Swedesboro, NJ. A Ascarita II consiste em hidróxido de sódio depositado sobre uma estrutura não-fibrosa de silicato.

PROCEDIMENTO
Se o professor pedir, prepare um frasco protegido para armazenamento (ver a Figura 37-4). Transfira 1 L deágua destilada para o frasco de armazenamento (ver a Nota na Seção 37C-3). Retire cuidadosamente 4 a 5mL de NaOH a 50% e transfira para um recipiente pequeno (Nota 2), adicione-o na água e misturevigorosamente. Seja extremamente cuidadoso ao manusear a solução de NaOH a 50%, que é altamentecorrosiva. Se os reagentes entrarem em contato com a pele, lave imediatamente a área com quantidadesabundantes de água.
Proteja a solução do contato desnecessário com a atmosfera.
Notas1. Uma solução de base que deverá ser utilizada por até duas semanas pode ser armazenada em um frasco
de polietileno bem fechado. Após cada retirada da base, comprima o frasco ao fechar a tampa para mini-mizar o espaço de ar acima do reagente. O frasco se tornará quebradiço após sua utilização por muitotempo como recipiente de soluções de bases.
2. Assegure-se de que qualquer Na2CO3 sólido na solução de NaOH a 50% foi decantado no fundo dofrasco e que o líquido sobrenadante está absolutamente transparente. Se necessário, filtre a base em umcadinho de Gooch; colete o filtrado em um tubo de ensaio colocado dentro do frasco de filtração.
37C-5 Determinação da Razão Ácido/Base
Discussão
Se ambas as soluções de ácido e base forem preparadas, é importante determinar suas razões volumétricas.O conhecimento dessa razão e da concentração de uma das soluções permite o cálculo da concentraçãomolar da outra.
PROCEDIMENTO
As instruções para preparação prévia de uma bureta são dadas nas Seções 2G-4 e 2G-6; consulte essasinstruções se necessário. Coloque um tubo de ensaio ou um béquer pequeno sobre a parte de cima da bure-ta para minimizar o contato da solução de NaOH com a atmosfera.
Anote os volumes iniciais do ácido e da base com uma precisão de 0,01 mL. Não tente ajustar o vo-lume inicial em zero. Transfira entre 35 e 40 mL da solução de ácido para um elenmeyer de 250 mL. Toquea ponta da bureta na parede interna do frasco e lave com um pouco de água destilada. Adicione duas gotasde fenolftaleína (Nota 1) e depois uma quantidade suficiente de base para fazer com que a solução fiquepermanentemente rosa. Adicione o ácido gota a gota até a eliminação da coloração e, novamente, lave asparedes internas do frasco. Adicione cuidadosamente a base até que a solução adquira uma tonalidade leve-mente rosa que se mantém por pelo menos 30 s (Notas 2 e 3). Anote o volume final na bureta (novamentecom uma precisão de 0,01 mL). Repita a titulação. Calcule a razão volumétrica ácido/base. As razões paraas titulações em duplicata devem concordar em até 1 a 2 partes por mil. Se necessário, realize mais titu-lações para atingir esse nível de precisão.
Notas1. A razão volumétrica também pode ser determinada com um indi-
cador que possui uma faixa de transição ácida, tal como o verde debromocresol. Se o NaOH estiver contaminado com carbonato, arazão obtida com esse indicador vai diferir significativamente do va-lor obtido com a fenolftaleína. Geralmente, a razão ácido/base deveser avaliada com o indicador que deverá ser usado nas titulações sub-seqüentes.
1016 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON
� Soluções de bases devem serarmazenadas em frascos de polietileno em vez de frascos devidro em decorrência da reaçãoentre as bases e o vidro. Essassoluções nunca devem serarmazenadas em frascos comtampa de vidro; depois de algumtempo a remoção da tampa torna-se, muitas vezes, impossível.

2. As meias gotas podem ser formadas na ponta da bureta, transferidas para a parede do frasco, tocando-ana ponta da bureta e lavando-as com uma pequena quantidade de água com uma pisseta.
3. O ponto final da fenolftaleína varia conforme o CO2 é absorvido da atmosfera.
37C-6 Padronização de Ácido Clorídrico com Carbonato de Sódio
Discussão
Ver Seção 16A-2
PROCEDIMENTO
Seque uma quantidade de padrão primário de Na2CO3 por cerca de duas horas a 110 °C (ver Figura 2-9) eresfrie em um dessecador. Pese massas de 0,20 a 0,25 g (com uma precisão de 0,1 mg) em erlenmeyers de250 mL e dissolva cada uma com cerca de 50 mL de água destilada. Adicione três gotas de verde de bro-mocresol e titule com HCl até que a solução comece a passar de azul para verde. Ferva a solução por doisa três minutos, resfrie à temperatura ambiente (Nota 1) e finalize a titulação (Nota 2 ).
Determine a correção do indicador titulando aproximadamente 100 mL de uma solução de NaCl 0,05mol L�1 com três gotas do indicador. Ferva rapidamente, resfrie e finalize a titulação. Anote qualquer volu-me que for necessário ao branco e subtraia dos volumes da titulação. Calcule a concentração das soluçõesde HCl.
Notas1. O indicador deve mudar de verde para azul conforme o CO2 é removido durante o aquecimento. Se ne-
nhuma mudança de coloração ocorrer, um excesso de ácido foi originalmente adicionado. Esse excessopode ser retrotitulado com base, considerando-se que a razão ácido/base é conhecida; caso contrário, asolução deve ser descartada.
2. É permitido retrotitular com base para estabelecer o ponto final com mais certeza.
37C-7 Padronização de Hidróxido de Sódio com Biftalato de Potássio
Discussão
Ver Seção 16A-4.
PROCEDIMENTO
Seque uma quantidade do padrão primário de hidrogenoftalato de potássio (KHP) por cerca de duas horasa 110 °C (ver a Figura 2-9) e deixe resfriar em dessecador. Pese amostras individuais entre 0,7 e 0,8 g (comprecisão de 0,1 mg) em erlenmeyers de 250 mL e dissolva em 50 a 75 mL de água destilada. Adicione duasgotas de fenolftaleína; titule com a base até que a cor rosa do indicador persista por 30 s (Nota). Calcule aconcentração da solução de NaOH.
NotaÉ permitido realizar a retrotitulação com ácido para estabelecer o ponto final mais precisamente. Registreo volume gasto na retrotitulação. Empregue a razão ácido/base para calcular o volume real de base uti-lizado na padronização.
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1017

37C-8 Determinação de Hidrogenoftalato de Potássio em uma Amostra Impura
Discussão
A amostra desconhecida é uma mistura de KHP e um sal neutro. Essa análise é convencionalmente reali-zada de maneira concomitante à padronização da base.
PROCEDIMENTO
Consulte o professor a respeito da quantidade apropriada de amostra. Então, siga as instruções contidas naSeção 37C-7.
37C-9 Determinação do Teor de Ácido em Vinagre e Vinho
Discussão
O teor total de ácido de vinagre ou vinho é prontamente determinado pela titulação com uma base padrão.Costuma-se descrever o teor de ácido em vinagre em termos de ácido acético, seu principal constituinteácido, ainda que outros ácidos estejam presentes. De maneira similar, o teor de ácido de vinho é expressoem porcentagem de ácido tartárico, embora existam outros ácidos na amostra. A maioria dos vinagres con-tém cerca de 5% de ácido (m/v) expresso como ácido acético; geralmente, vinhos contêm menos de 1% deácido (m/v) expresso como ácido tartárico.
PROCEDIMENTO
1. Se a amostra desconhecida for vinagre (Nota 1), pipete 25,00 mL em um frasco volumétrico de 250 mLe dilua até a marca com água destilada. Misture vigorosamente e pipete alíquotas contendo 50,00 mL em erlenmeyers de 250 mL. Adicione cerca de 50 mL de água e duas gotas de fenolftaleína (Nota 2)em cada frasco e titule com NaOH 0,1 mol L�1 padrão até quando surgir uma cor rosa que se mantenhapor � 30 s.
Descreva a acidez do vinagre como porcentagem (m/v) de CH3COOH (60,053 g/mol).2. Se a amostra desconhecida for vinho, pipete alíquotas de 50,00 mL em erlenmeyers de 250 mL, adi-
cione cerca de 50 mL de água destilada e duas gotas de fenolftaleína a cada frasco (Nota 2) e titule atéquando surgir uma cor rosa que se mantenha por � 30 s.
Expresse a acidez da amostra como porcentagem (m/v) de ácido tartárico, C2H4O2(COOH)2 (150,09g/mol) (Nota 3).
Notas1. A acidez do vinagre engarrafado tende a diminuir com sua exposição ao ar. Recomenda-se que a
amostra seja armazenada em frascos individuais com tampas bem fechadas.2. A quantidade de indicador empregada deve ser aumentada, se necessário, para tornar a mudança de cor
visível em amostras coloridas.3. O ácido tartárico tem dois hidrogênios ácidos, ambos os quais são titulados com fenolftaleína.
37C-10 Determinação de Carbonato de Sódio em uma Amostra Impura
Discussão
A titulação do carbonato de sódio foi discutida na Seção 16A-2, em conjunto com o seu emprego comopadrão primário; as mesmas considerações se aplicam à determinação de carbonato em uma amostradesconhecida que não tenha interferentes.
1018 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON

PROCEDIMENTO
Seque a amostra a 110 °C por duas horas e então deixe resfriar em um dessecador. Consulte o professorsobre a quantidade apropriada de amostra. Em seguida, siga as instruções da Seção 37C-6.
Descreva a porcentagem de Na2CO3 na amostra.
37C-11 Determinação de Nitrogênio Amoniacal pelo Método de Kjeldahl
Discussão
Essas instruções são adequadas para a determinação de Kjeldahl em proteínas e materiais, como carnes,farinha de trigo, massas, cereais secos e rações para animais domésticos. Uma modificação simples per-mite a análise de amostras desconhecidas que contenham formas de nitrogênio em estados de oxidaçãomais altos.7
No método de Kjeldahl (ver a Seção 16B-1), a amostra orgânica é digerida em ácido sulfúrico con-centrado a quente, que converte o nitrogênio na forma de amina presente na amostra em sulfato de amônio.Após o resfriamento, o ácido sulfúrico é neutralizado pela adição de hidróxido de sódio concentrado.Então, a amônia liberada por esse tratamento é destilada e recolhida em um volume conhecido e em exces-so de uma solução padrão de um ácido; o excesso é determinado por uma retrotitulação com uma basepadronizada.
A Figura 37-5 ilustra um equipamento típico para a destilação de Kjeldahl. O frasco de gargalo longo,empregado tanto na digestão quanto na destilação, é chamado frasco de Kjeldahl. No sistema mostrado naFigura 37-5a, a base é adicionada lentamente abrindo-se parcialmente a torneira do frasco de estocagemde NaOH; assim, a amônia liberada é arrastada para o frasco coletor por destilação com arraste por vapor.
Em um método alternativo (Figura 37-5b), uma solução densa e concentrada de hidróxido de sódio écuidadosamente introduzida pela parede lateral do frasco de Kjeldahl para formar uma segunda camada naparte inferior. Dessa forma o frasco é rapidamente conectado a um retentor de spray e um condensadorcomum antes que ocorra qualquer perda de amônia. Só então as duas camadas são misturadas por movi-mentos suaves do frasco.
A coleta quantitativa da amônia requer que a ponta do condensador se estenda até o líquido contidono frasco coletor durante a etapa de destilação. No entanto, a ponta precisa ser removida antes do términodo aquecimento. Caso contrário, o líquido será succionado de volta ao sistema.
Dois métodos são comumente empregados para coletar e determinar a amônia liberada da amostra. Emum deles, a amônia é destilada em um volume conhecido de ácido padrão. Após a destilação se completar,o excesso de ácido é retrotitulado com uma base padrão. Um indicador com uma faixa de transição ácidaé requerido por causa da acidez dos íons amônio presentes no ponto de equivalência. Uma alternativa con-veniente, que requer apenas uma solução padrão, envolve a coleta da amônia em um excesso não medidode ácido bórico, o qual retém a amônia pela reação
H3BO3 � NH3 NH � H2BO
O íon diidrogenoborato produzido é uma base razoavelmente forte que pode ser titulada com uma soluçãopadrão de ácido clorídrico.
H2BO � H3O� H3BO3 � H2O
No ponto de equivalência, a solução contém ácido bórico e íons amônio; um indicador que tenha um inter-valo de transição ácido (como, o verde de bromocresol) é, novamente, requerido.
S�3
�3
�4S
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1019
7Ver Official Methods of Analysis. 14. ed., p. 16. Washington, DC: Association of Official Analytical Chemists, 1984.

PROCEDIMENTO
Preparo de AmostrasConsulte seu professor sobre a quantidade de amostra. Se a amostra for úmida (como carne), pese-a empapéis-filtro de 9 cm (Nota 1). Dobre o papel em torno da amostra e introduza-a no frasco de Kjeldahl. (Opapel não deixa a amostra aderir ao gargalo do frasco.) Se a amostra desconhecida não for úmida (comocereais ou massa), ela pode ser pesada por diferença diretamente no frasco de Kjeldahl.
Adicione 25 mL de H2SO4 concentrado, 10 g de K2SO4 em pó e o catalisador (Nota 2) em cada frasco.
DigestãoPrenda os frascos em uma posição inclinada em uma capela ou sistema de digestão ventilado. Aqueçacuidadosamente até a ebulição. Interrompa o aquecimento brevemente se o sistema formar quantidadesexcessivas de espuma; nunca deixe que a espuma alcance o gargalo do frasco. Uma vez que a espuma desa-pareça e o ácido esteja fervendo vigorosamente, as amostras podem permanecer sem acompanhamento;prepare o sistema de destilação durante esse período. Continue a digestão até que a solução se torne inco-lor ou amarelo-pálida; duas a três horas podem ser necessárias para alguns materiais. Se necessário, repo-nha o ácido perdido por evaporação.
Quando a digestão estiver completa, desligue o aquecimento e deixe os frascos resfriarem até a tem-peratura ambiente; agite-os, se o conteúdo mostrar sinais de solidificação. Cuidadosamente, adicione 250mL de água em cada e deixe novamente a solução resfriar até a temperatura ambiente.
Destilação da AmôniaMonte um sistema de destilação similar ao mostrado na Figura 37-5. Pipete 50,00 mL do padrão de HCl0,1 mol L�1 no frasco coletor (Nota 3). Prenda o frasco de maneira que a ponta do adaptador se estendaabaixo da superfície do ácido. Circule água pelo sistema de refrigeração do condensador.
Mantenha o frasco de Kjeldahl sob certo ângulo e introduza cuidadosamente cerca de 60 mL de umasolução de NaOH a 50% (m/v), tomando cuidado para minimizar a mistura com a solução contida no fras-co. A solução de hidróxido de sódio concentrada é altamente corrosiva e deve ser manipulada com grandecuidado (Nota 4). Adicione vários pedaços de zinco granulado (Nota 5) e um pequeno pedaço de papel delitmus. Conecte imediatamente o frasco de Kjeldahl ao retentor de spray. Misture cuidadosamente o con-teúdo através de agitação suave. O papel de litmus deve se tornar azul após a mistura estar completa, indi-cando que a solução está alcalina.
1020 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON
(a)
Geradorde vapor
Frasco deKjeldahl
NaOHconcentrado
Frasco deKjeldahl
Retentorde spray
Frascocoletor
Frascocoletor
(b)
Figura 37-5 Sistemas de destilação de Kjeldahl.

Leve a solução à fervura e destile a uma velocidade constante até que o volume remanescente sejaigual à metade ou um terço do volume original. Controle a velocidade de aquecimento para evitar queo líquido do frasco coletor seja sugado de volta para o frasco de Kjeldahl. Após a destilação ser con-siderada completa, abaixe o frasco coletor para remover o adaptador do líquido. Interrompa o aqueci-mento, desconecte o sistema e enxágüe a parte interna do condensador com pequenas porções de águadestilada, coletando a água de lavagem no frasco coletor. Adicione duas gotas de verde de bromocre-sol ao frasco coletor e titule o HCl residual com um padrão de NaOH 0,1 mol L�1 até obter a mudançade cor do indicador.
Relate a porcentagem de nitrogênio e a porcentagem de proteína (Nota 6) na amostra.
Notas1. Se for empregado papel-filtro para envolver a amostra, repita o experimento utilizando apenas um
pedaço de papel similar como controle (branco). O papel-filtro lavado com ácido geralmente contémquantidades consideráveis de íons amônio e, portanto, deve ser evitado, quando possível.
2. Qualquer um dos seguintes produtos pode catalisar a digestão: um cristal de CuSO4, 0,1 g de selênio,0,2 g de CuSeO3. O catalisador pode ser omitido, se desejado.
3. Uma modificação desse procedimento emprega 50 mL de uma solução de ácido bórico a 4% em vez doHCl padrão no frasco coletor. Após a destilação ter se completado, o borato de amônio produzido é titu-lado com HCl 0,1 mol L�1 padrão, com duas ou três gotas de verde de bromocresol como indicador.
4. Se a solução de hidróxido de sódio tiver contato com sua pele, lave a área afetada imediatamente comgrandes quantidades de água.
5. O zinco granulado (10 a 20 mesh) é adicionado para minimizar a projeção por superebulição da soluçãodurante a destilação; o zinco reage vagarosamente com a base para produzir pequenas bolhas dehidrogênio, que previnem o superaquecimento do líquido.
6. A porcentagem de proteína na amostra é calculada multiplicando-se a porcentagem de N por um fatorapropriado: 5,70 para cereais, 6,25 para carnes e 6,38 para laticínios.
37D TITULAÇÕES DE PRECIPITAÇÃO
Conforme observado na Seção 13F, a maioria das titulações de precipitação usa uma solução padrão denitrato de prata como titulante. As instruções que se seguem referem-se à titulação volumétrica de cloretoempregando um indicador de adsorção.
37D-1 Preparação da Solução Padrão de Nitrato de Prata
PROCEDIMENTO
Utilize uma balança de topo para transferir a massa aproximada de AgNO3 para um pesafiltro (Nota 1).Seque a 110 °C por aproximadamente uma hora, mas não por mais tempo (Nota 2), e então deixe resfriaraté a temperatura ambiente em um dessecador. Pese o frasco e o conteúdo (com precisão de 0,1 mg).Transfira quantitativamente o AgNO3 para um frasco volumétrico empregando um funil. Tampe o pesafil-tro e pese novamente. Enxágüe o funil extensivamente. Dissolva o AgNO3, dilua até a marca com água emisture bem (Nota 3). Calcule a concentração dessa solução.
Notas1. Consulte o professor sobre o volume e concentração da solução de AgNO3 a ser preparada. A massa de
AgNO3 a ser tomada deve ser a seguinte:
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1021

Massa aproximada de AgNO3Necessária para Preparar
Concentração doíon parata, mol L�1 1.000 mL 500 mL 250 mL
0,10 16,9 8,5 4,20,05 8,5 4,2 2,10,02 3,4 1,8 1,0
2. O aquecimento prolongado provoca a decomposição parcial do AgNO3. Alguma coloração pode ocorrermesmo após uma hora de aquecimento a 110 °C; o efeito dessa decomposição na pureza do reagente énormalmente imperceptível.
3. Soluções de nitrato de prata devem ser mantidas no escuro quando não estão em uso.
37D-2 A Determinação de Cloreto pela Titulação com Indicador de Adsorção
Discussão
Nessa titulação, o indicador de adsorção aniônico diclorofluoresceína é empregado para localizar o pontofinal. Com o primeiro excesso de titulante, o indicador é incorporado ao contra-íon da camada vizinha aocloreto de prata dando cor ao sólido (página 342). Para obter uma mudança de cor satisfatória, é desejávelmanter as partículas de cloreto de prata no estado coloidal. A dextrina é adicionada à solução para estabi-lizar o colóide prevenindo sua coagulação.
PREPARO DAS SOLUÇÕES
Indicador diclorofluoresceína (suficiente para várias centenas de titulações). Dissolva 0,2 g de diclorofluo-resceína em uma solução preparada pela mistura de 75 mL de etanol e 25 mL de água.
PROCEDIMENTO
Seque a amostra a 110 °C por cerca de uma hora; deixe retornar à temperatura ambiente em um dessecador.Pese amostras individuais (com precisão de 0,1 mg) em erlenmeyers e dissolva-as em volumes apropria-dos de água destilada (Nota 1). Adicione cerca de 0,1 g de dextrina e cinco gotas do indicador a cada umdos frascos. Titule (Nota 2) com AgNO3 até o aparecimento da primeira cor rosa permanente de dicloro-fluoresceinato de prata. Expresse o resultado na forma da porcentagem de Cl� na amostra.
Notas1. Empregue 0,25 g de amostras para AgNO3 0,1 mol L�1 e cerca de metade dessa massa para AgNO3 0,05
mol L�1. Dissolva o primeiro em aproximadamente 200 mL de água destilada e o último em aproxi-madamente 100 mL. Se for feito uso de AgNO3 0,02 mol L�1, pese uma porção de 0,4 g da amostra emum balão volumétrico de 500 mL de capacidade e tome alíquotas de 50 mL para a titulação.
2. O AgCl coloidal é sensível à fotodecomposição, particularmente na presença do indicador; tentativasde realizar a titulação à luz do sol não darão certo. Se a fotodecomposição parece ser um problema,estabeleça um ponto final aproximado com uma titulação preliminar e empregue essa informação pa-ra estimar os volumes de AgNO3 necessários para as outras amostras. Para cada amostra subseqüente,adicione o indicador e a dextrina após a adição da maior parte do AgNO3 e então complete a titulaçãoimediatamente.
1022 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON

37D-3 Determinação de Cloreto via Titulação Gravimétrica
Discussão
O método de Mohr emprega íons CrO como indicador na titulação do íon cloreto com o nitrato de prata.O primeiro excesso de titulante resulta na formação de um precipitado vermelho de cromato de prata, quesinaliza o ponto final da titulação.
Em vez da bureta, uma balança é empregada nesse procedimento para determinar a massa da soluçãode nitrato de prata necessária para alcançar o ponto final. A concentração de nitrato de prata é mais con-venientemente determinada pela padronização contra um padrão primário de cloreto de sódio, embora apreparação direta pela massa também seja viável. A concentração do reagente é expressa como a molari-dade em peso (massa), mmol de AgNO3/g de solução. Ver a Seção 13D-1 para mais detalhes.
PREPARO DAS SOLUÇÕES
(a) Nitrato de prata, aproximadamente 0,1 mmol/g de solução (suficiente para cerca de dez titulações).Dissolva aproximadamente 4,5 g de AgNO3 em cerca de 500 mL de água destilada. Padronize asolução contra quantidades pesadas de NaCl grau reagente, como descrito na Nota 1 do procedimento.Expresse a concentração como molaridade em peso (massa), isto é, mmol AgNO3/g solução. Quandonão estiver em uso, guarde a solução em um local escuro.
(b) Cromato de potássio, 5% (suficiente para cerca de dez titulações). Dissolva aproximadamente 1,0 g deK2Cr2O4 em cerca de 20 mL de água destilada.
NotaAlternativamente, o AgNO3 pode ser preparado diretamente por pesagem. Para tanto, siga as instruçõesfornecidas na Seção 37D-1 para a pesagem de uma quantidade conhecida do padrão primário de AgNO3.Empregue um funil para transferir a massa de AgNO3 pesada para um frasco de polietileno de 500 mL quetenha sido previamente pesado com precisão de 10 mg. Adicione cerca de 500 mL de água e pese nova-mente. Calcule a molaridade em peso.
INSTRUÇÕES PARA A REALIZAÇÃO DE UMA TITULAÇÃO GRAVIMÉTRICA
Prepare um dispensador de reagente a partir de um frasco de polietileno munido com uma tampa de roscae equipado com uma ponta dispensadora fina. Esta pode ser preparada reduzindo-se a ponta de um tubo depolietileno na chama. Com um furador de rolhas, faça um furo na tampa que seja ligeiramente menor queo diâmetro externo da ponta do tubo. Cuidadosamente, force a ponta através do furo, aplique uma camadade cola epóxi para vedar a ponta à tampa. Rotule o frasco.
Encha o dispensador de reagente com uma quantidade do titulante e rosqueie a tampa firmemente. Peseo frasco e seu conteúdo com precisão de um miligrama. Introduza um indicador adequado na solução con-tendo o analito. Segure o dispensador de maneira que sua ponta fique abaixo da boca do frasco. Adicionevários incrementos do reagente apertando o dispensador e agitando o frasco de titulação com a outra mão.Quando considerar que apenas mais algumas gotas do reagente são necessárias, diminua a pressão no fras-co interrompendo o fluxo; toque a ponta na parte interna do frasco e reduza ainda mais a pressão no dis-pensador para que o líquido da ponta do tubo seja sugado de volta para o frasco à medida que o tubo vaisendo retirado do frasco de titulação. Coloque o dispensador sobre um pedaço de papel laminado limpo eseco e enxágüe as paredes internas do frasco de titulação com um jato de água destilada ou desionizada.Adicione o reagente gota a gota até alcançar o ponto final (Nota). Pese o dispensador e registre os dados.
NotaIncrementos menores podem ser adicionados deixando formar parcialmente uma gota na ponta do tubo etocando o mesmo contra a parede do frasco. Enxágüe as paredes do frasco com água para misturar a meiagota com a solução contida no frasco.
2�4
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1023

PROCEDIMENTO
Seque a amostra a 110 °C por pelo menos uma hora (Nota). Resfrie em um dessecador. Consulte o pro-fessor em relação à quantidade adequada de amostra. Pese (com precisão de 0,1 mg) amostras individuaisem erlenmeyers de 250 mL de capacidade e dissolva com aproximadamente 100 mL de água destilada.Adicione pequenas quantidades de NaHCO3 até cessar a efervescência. Introduza cerca de 2 mL da soluçãode K2CrO4 e titule até o aparecimento da primeira cor permanente de Ag2CrO4.
Determine um “branco” para o indicador pela suspensão de uma pequena quantidade de CaCO3 livrede cloreto em 100 mL de água destilada contendo 2 mL de K2CrO4.
Corrija a massa do reagente gasta na titulação subtraindo o volume do branco. Expresse o resultado como porcentagem de Cl� na amostra. Descarte o AgCl e os reagentes de acordo com as instruções do professor.
Nota 1O AgNO3 é convenientemente padronizado de forma simultânea à análise. Seque o NaCl grau reagente porcerca de uma hora. Resfrie e então pese (com precisão de 0,1 mg) uma porção de 0,25 g em erlenmeyers etitule conforme descrito anteriormente.
TITULAÇÕES DE FORMAÇÃO DE37E COMPLEXOS COM EDTA
Ver a Seção 17D para uma discussão sobre o emprego analítico do EDTA como um agente quelante. Asinstruções que seguem são para a titulação direta de magnésio e determinação da dureza de águas naturais.
37E-1 Preparo das Soluções
PROCEDIMENTO
Um tampão pH 10 e uma solução de um indicador são necessários para essas titulações.
1. Solução tampão, pH 10 (suficiente para 80 a 100 titulações). Dilua 57 mL de NH3 concentrado e 7 g deNH4Cl em quantidade de água destilada suficiente para completar 100 mL de solução.
2. Indicador Negro de Eriocromo T (suficiente para cerca de 100 titulações). Dissolva 100 mg do sólidoem uma solução contendo 15 mL de etanolamina e 5 mL de etanol absoluto. Essa solução deve serpreparada a cada duas semanas; a refrigeração diminui sua deterioração.
37E-2 Preparação da Solução Padrão de EDTA 0,01 mol L–1
Discussão
Ver a Seção 17D-1 para uma descrição sobre as propriedades do Na2H2Y�2H2O grau reagente e seuemprego na preparação direta das soluções padrão de EDTA.
PROCEDIMENTO
Seque cerca de 4 g do Na2H2Y�2H2O purificado (Nota 1) a 80 °C para remover a umidade superficial.Resfrie até a temperatura ambiente em dessecador. Pese (com precisão de 1 mg) aproximadamente 3,8 gem um balão volumétrico de 1 L (Nota 2). Utilize um funil para assegurar que a transferência seja quanti-tativa; enxágüe bem o funil com água antes de removê-lo do balão. Adicione entre 600 mL e 800 mL deágua (Nota 3) e agite freqüentemente. A dissolução pode demorar 15 min ou mais. Quando todo o sólido
1024 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON

tiver dissolvido, dilua até a marca com água e misture bem (Nota 4). No cálculo da concentração molar dasolução, corrija a massa do sal em relação ao teor de 0,3% de umidade que normalmente fica retido apósa secagem a 80 °C.
Notas1. Instruções para a purificação do sal dissódico são descritas por W. J. Blaedel e H. T. Knight., Anal.
Chem., v. 26, n. 4, p. 741, 1954.2. A solução pode ser preparada a partir do sal dissódico anidro, se preferir. A massa tomada, nesse caso,
deve ser de aproximadamente 3,6 g.3. A água empregada na preparação de soluções padrão de EDTA deve ser totalmente isenta de cátions
polivalentes. Se existir qualquer dúvida em relação à sua qualidade, passe a água por uma resina detroca catiônica antes do seu emprego.
4. Como alternativa, uma solução de EDTA que seja aproximadamente 0,01 mol L�1 pode ser preparada epadronizada pela titulação direta contra uma solução de Mg2� de concentração conhecida (empregandoas instruções contidas na Seção 37E-3).
37E-3 Determinação de Magnésio por Titulação Direta
Discussão
Ver a Seção 17D-7.
PROCEDIMENTO
Empregue um balão volumétrico limpo de 500 mL para preparar a solução da amostra; dilua até a marcacom água e misture vigorosamente. Transfira alíquotas de 50,00 mL para frascos erlenmeyers de 250 mL;adicione 1 a 2 mL do tampão pH 10 e três a quatro gotas do indicador negro de eriocromo T em cada fras-co. Titule com EDTA 0,01 mol L�1 até que a cor do indicador mude de vermelho para azul (Notas 1 e 2).
Expresse os resultados em partes por milhão de Mg2� na amostra
Notas1. A mudança de cor tende a ser lenta nas proximidades do ponto final. É necessário tomar cuidado para
não excedê-lo.2. Outros elementos alcalinos, se presentes, são titulados juntamente com o Mg2�; a remoção de Ca2� e
Ba2� pode ser realizada com (NH4)2CO3. A maioria dos cátions polivalentes também é titulada. A pre-cipitação na forma de hidróxidos ou o emprego de agentes mascarantes pode ser necessária para elimi-nar essa fonte de interferência.
37E-4 Determinação de Cálcio por Titulação de Substituição
Discussão
Uma solução do complexo magnésio/EDTA é útil para a titulação de cátions que formam complexos maisestáveis que o de magnésio, mas para os quais não há indicador disponível. Os íons magnésio presentes nocomplexo são substituídos por uma quantidade quimicamente equivalente dos cátions do analito.
O analito não complexado remanescente e os íons magnésio liberados são, então, titulados comnegro de eriocromo T como indicador. Observe que a concentração de magnésio na solução não é impor-tante; tudo o que é necessário é que a razão molar entre Mg2� e EDTA no reagente seja exatamente iguala unidade.
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1025

PROCEDIMENTO
Preparação do Complexo Magnésio/EDTA, 0,1 mol L�1 (suficiente para 90 a 100 titulações)Para um volume de solução de 50 mL contendo 3,72 g de Na2H2Y2�2H2O em água destilada, adicione umaquantidade equivalente (2,46 g) de MgSO4�7H2O. Adicione algumas gotas de fenolftaleína, seguida por uma quantidade suficiente de NaOH 0,1 mol L�1 para tornar a solução levemente rósea. Dilua para cerca de100 mL com água. A adição de algumas gotas de negro de eriocromo T a uma porção dessa solução tam-ponada a pH 10 deve provocar o aparecimento de uma cor violeta. Além disso, uma única gota de Na2H2Y0,01 mol L�1 adicionada à solução violeta deve provocar a mudança de cor para azul e uma quantidade igual de uma solução de Mg2� 0,01 mol L�1 deve causar uma mudança de cor para vermelho. A composiçãoda solução original deve ser ajustada com Mg2� ou H2Y2� até que esses critérios sejam atingidos.
TitulaçãoPese uma quantidade da amostra (com precisão de 0,1 mg) em um béquer de 500 mL (Nota 1). Cubra comum vidro de relógio e adicione cuidadosamente 5 a 10 mL de HCl 6 mol L�1. Após a dissolução daamostra, remova o CO2 pela adição de cerca de 50 mL de água desionizada, fervendo vagarosamente poralguns minutos. Resfrie, adicione uma gota ou duas de vermelho de metila e neutralize com NaOH 6 molL�1 até o desaparecimento da cor vermelha. Transfira quantitativamente a solução para um balãovolumétrico de 500 mL e dilua até a marca. Tome alíquotas de 50,00 mL da solução diluída para titulação,tratando cada uma delas da seguinte forma. Adicione 2 mL do tampão pH 10,1 mL da solução Mg/EDTA,adicione três a quatro gotas de negro de eriocromo T ou indicador Calmagita. Titule (Nota 2) com oNa2H2Y 0,01 mol L�1 padrão até que a cor da solução mude de vermelho para azul.
O resultado deve ser expresso em termos de miligramas de CaO presentes na amostra.
Notas1. A amostra tomada deve conter entre 150 e 160 mg de Ca2�.2. Interferências nessas titulações são basicamente as mesmas daquelas encontradas na titulação direta de
Mg2� e são eliminadas da mesma forma.
37E-5 Determinação da Dureza na Água
Discussão
Ver a Seção 17D-9.
PROCEDIMENTO
Acidifique alíquotas contendo 100,0 mL da amostra com algumas gotas de HCl e ferva gentilmente poralguns minutos para eliminar o CO2. Resfrie, adicione três a quatro gotas de vermelho de metila e neu-tralize com NaOH 0,1 mol L�1. Introduza 2 mL do tampão pH 10 e adicione três a quatro gotas de negrode eriocromo T e titule com Na2H2Y 0,01 mol L�1 padrão até a mudança do indicador de vermelho paraazul (Nota).
Expresse o resultado em termos de miligramas de CaCO3 por litro de água.
NotaA mudança de cor é gradual na ausência de Mg2�. Nesse caso, adicione 1 a 2 mL de MgY2� 0,1 mol L�1
antes de iniciar a titulação. Esse reagente é preparado pela adição de 2,645 g de MgSO4�7H2O a 3,722 g deNa2H2Y�2H2O em 50 mL de água destilada. Essa solução é levemente alcalina na presença de fenolftaleínae é diluída a 100 mL. Uma pequena porção, misturada com o tampão pH 10 e algumas gotas do indicadornegro de eriocromo T deve ter uma cor violeta intensa. Uma única gota de uma solução de EDTA 0,01 molL�1 deve provocar a alteração da cor para o azul, enquanto um volume igual de Mg2� 0,01 mol L�1 deve
1026 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON

provocar a modificação para o vermelho. Se necessário, ajuste a composição com EDTA ou com Mg2� atéque esses critérios sejam atingidos.
TITULAÇÕES COM37F PERMANGANATO DE POTÁSSIO
As propriedades e a utilização do permanganato de potássio estão descritas na Seção 20C-1. As instruçõesa seguir relacionam-se à determinação de ferro em minério e cálcio em calcário.
37F-1 Preparação do Permanganato de Potássio 0,02 mol L–1
Discussão
Ver a página 533 para uma discussão sobre as precauções necessárias na preparação e estocagem desoluções de permanganato.
PROCEDIMENTO
Dissolva aproximadamente 3,2 g de KMnO4 em 1 L de água destilada. Mantenha a solução sob agitaçãobranda por cerca de uma hora. Cubra e deixe em repouso por uma noite. Remova o MnO2 por filtração(Nota 1) em um cadinho de placa porosa (Nota 2) ou em um cadinho de Gooch com fundo de vidrosinterizado. Transfira a solução para um frasco com tampa de vidro; guarde no escuro enquanto ela nãoestiver sendo utilizada.
Notas1. O aquecimento e a filtração podem ser dispensados se a solução de permanganato for padronizada e uti-
lizada no mesmo dia.2. Remova o MnO2 que for coletado no filtro com H2SO4 1 mol L�1 contendo algumas gotas de H2O2 a
3%, seguido pelo enxágüe com grandes quantidades de água.
37F-2 Padronização de Soluções de Permanganato de Potássio
Discussão
Ver a Seção 20C-1 para uma discussão sobre padrões primários para soluções de permanganato. Asinstruções que seguem são relacionadas à padronização com oxalato de sódio.
PROCEDIMENTO
Seque cerca de 1,5 g do padrão primário de Na2C2O4 a 110 °C por pelo menos uma hora. Resfrie emdessecador; pese (com precisão de 0,1 mg) amostras individuais de 0,2 a 0,3 g em béqueres de 400 mL.Dissolva cada amostra em cerca de 250 mL de H2SO4 1 mol L–1. Aqueça cada solução entre 80 °C e 90 °Ce titule com KMnO4, agitando com um termômetro. Deve-se esperar o desaparecimento da coloração róseagerada pela primeira adição do titulante para que as próximas adições sejam feitas (Notas 1 e 2). Reaqueçaa solução se a temperatura cair abaixo de 60 °C. Considere o ponto final quando a cor rosa permanecer por30 s (Notas 3 e 4). Determine o volume gasto para uma titulação em branco empregando um volumeidêntico de H2SO4 1 mol L–1.
Corrija os dados da titulação em função do branco e calcule a concentração da solução de perman-ganato (Nota 5).
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1027

Notas1. Lave prontamente com água eventuais gotas de KMnO4 que porventura foram espirradas para as pare-
des do béquer.2. Haverá formação de MnO2 juntamente com o Mn2� se o KMnO4 for adicionado muito rapidamente,
causando a formação de uma coloração castanho-pálida. A formação do precipitado não é um problemasério desde que haja oxalato suficiente para reduzir o MnO2 formado a Mn2�; a titulação deve ser inter-rompida até o desaparecimento da cor castanha. A solução precisa estar livre de MnO2 no ponto final.
3. Em vez da parte de baixo do menisco, a superfície superior da solução de permanganato deve ser uti-lizada para medir os volumes de titulante gastos. Alternativamente, pode-se iluminar a parte detrás dabureta com uma lanterna ou fósforo para melhor leitura do menisco da maneira convencional.
4. Uma solução de permanganato não deve ficar na bureta por mais tempo que o necessário porque podeocorrer a decomposição parcial do reagente para formar MnO2. A remoção do MnO2 formado recente-mente pode ser feita com uma solução de H2SO4 1 mol L�1 contendo uma pequena quantidade deH2O2 a 3%.
5. Conforme observado na página 536, esse procedimento fornece concentrações molares que podem seralguns décimos de porcentagem menores que o esperado. Para resultados mais exatos, adicione a partirda bureta, permanganato suficiente para reagir com 90% a 95% do oxalato presente (cerca de 40 mL deKMnO4 0,02 mol L�1 para 0,3 g de amostra). Deixe a solução em repouso até o desaparecimento da corvioleta do permanganato. Então, aqueça a solução a 60 °C e complete a titulação, tomando como pontofinal o volume obtido quando a cor rosa permanecer por 30 s. Determine o volume gasto para titular umbranco contendo igual volume de H2SO4 1 mol L�1.
37F-3 Determinação de Cálcio em Calcário
Discussão
Com outros cátions, o cálcio é determinado, convencionalmente, pela precipitação com íons oxalato. Ooxalato de cálcio sólido é filtrado, lavado até ficar livre do excesso do reagente precipitante e dissolvidoem ácido diluído. Então, o ácido oxálico liberado nessa etapa é titulado com uma solução padrão de per-manganato ou algum outro agente oxidante. Esse método pode ser aplicado a amostras que contenhammagnésio e outros metais alcalinos. A maioria dos outros cátions precisa estar ausente uma vez que eles seprecipitam ou co-precipitam na forma de oxalato e provocam erros positivos na análise.
Fatores que Afetam a Composição de Precipitados de Oxalato de Cálcio É essencial que arazão molar entre o cálcio e o oxalato seja exatamente unitária no precipitado e, portanto, na solução nomomento da titulação. Diversos cuidados são necessários para garantir essa condição. Por exemplo, ooxalato de cálcio formado em soluções neutras ou amoniacais pode estar contaminado com hidróxido decálcio ou com um oxalato de cálcio básico, que podem gerar resultados mais baixos que os esperados. Aformação desses compostos é prevenida adicionando-se o oxalato a soluções ácidas contendo a amostra eformando-se vagarosamente o precipitado desejado pela adição, gota a gota, de amônia. O oxalato de cál-cio cristalino formado sob essas condições pode ser filtrado prontamente. Perdas resultantes da solubiliza-ção do oxalato de cálcio são negligenciáveis acima de pH 4, desde que a lavagem seja limitada à liberaçãodo excesso de oxalato presente.
A co-precipitação do oxalato de sódio torna-se uma fonte de erros positivos na determinação de cál-cio quando a concentração de sódio na amostra excede a de cálcio. Esse tipo de erro pode ser eliminadopor reprecipitação.
O magnésio, se presente em concentrações elevadas, também pode ser uma fonte de contaminação.Um excesso de íons oxalato ajuda a prevenir a interferência por meio da formação de complexos solúveisde oxalato com magnésio. A rápida filtração do oxalato de cálcio também pode prevenir interferências emrazão da tendência pronunciada do oxalato de magnésio de formar soluções supersaturadas a partir dasquais a formação do precipitado só ocorre após uma ou mais horas. Essas medidas não são suficientes para
1028 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON

amostras que contenham mais magnésio que cálcio. Nesse caso, areprecipitação do oxalato de cálcio torna-se necessária.
A Composição do Calcário Os calcários são compostos principal-mente de carbonato de cálcio; calcários dolomíticos também contêmgrandes quantidades de carbonato de magnésio. Silicatos de cálcio e
magnésio também estão presentes em pequenas quantidades, juntamente com os carbonatos e silicatos deferro, alumínio, manganês, titânio, sódio e de outros metais.
O ácido clorídrico é um solvente eficiente para a maioria dos calcários. Apenas a sílica, que não inter-fere na análise, permanece insolúvel. Alguns calcários são mais facilmente decompostos após serem cal-cinados, outros necessitam de tratamento por fusão com carbonato.
O método que segue é bastante eficiente na determinação de cálcio na maioria dos calcários. Ferro ealumínio, quando presentes em quantidades equivalentes às de cálcio, não interferem. Pequenas quanti-dades de manganês e titânio também podem ser toleradas.
PROCEDIMENTO
Preparo da AmostraSeque a amostra por uma a duas horas a 110 °C e resfrie em um dessecador. Se o material for facilmentedecomposto em ácido, pese entre 0,25 e 0,30 g (com precisão de 0,1 mg) em béqueres de 250 mL. Adicione10 mL de água a cada amostra e cubra com um vidro de relógio. Acrescente 10 mL de HCl concentrado,gota a gota, tomando cuidado para evitar perdas por projeção à medida que o ácido é adicionado.
Precipitação do Oxalato de CálcioAdicione cinco gotas de água saturada com bromo para oxidar qualquer ferro presente na amostra e fervacom cuidado (utilize a capela de exaustão) por cinco minutos para remover o excesso de Br2. Dilua cadasolução contendo a amostra a cerca de 50 mL, aqueça até a fervura e acrescente 100 mL de uma soluçãode (NH4)2C2O4 a 6% (m/v) a quente. Adicione três a quatro gotas de vermelho de metila e precipite oCaC2O4 pela adição lenta de NH3 6 mol L�1. Quando o indicador começar a mudar de cor, adicione o NH3
a uma velocidade de três a quatro gotas por segundo. Continue até que as soluções tomem a coloraçãointermediária amarelo-laranja do indicador (pH 4,5 a 5,5). Deixe a solução em repouso por 30 min (Nota)e filtre; cadinhos de filtração de porosidade média ou cadinhos de Gooch são satisfatórios. Lave os pre-cipitados com várias porções de 10 mL de água gelada. Enxágüe a parte externa dos cadinhos para removero (NH4)2C2O4 residual e retorne-os para os béqueres nos quais o CaC2O4 foi formado.
TitulaçãoAdicione 100 mL de água e 50 mL de H2SO4 3 mol L�1 a cada um dos béqueres contendo o precipitadode oxalato de cálcio e o cadinho. Aqueça entre 80 °C e 90 °C e titule com permanganato 0,02 mol L�1. Atemperatura deve ser maior que 60 °C durante a titulação; reaqueça se necessário.
Apresente o resultado como porcentagem de CaO na amostra.
NotaO período em repouso pode ser mais longo se a amostra não contiver Mg2�.
37F-4 Determinação de Ferro em Minério
Discussão
Os minérios de ferro mais comuns são a hematita (Fe2O3), magnetita (Fe3O4) e limonita (2Fe2O3�3H2O).As etapas de análise desses minérios são (1) dissolução da amostra, (2) redução do ferro ao estado biva-lente e (3) titulação do ferro(II) com um oxidante padrão.
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1029
A reprecipitação é um método de minimização de erros devido à co-precipitação por meio de dissolução do precipitado obtido inicialmente e formação novamentedo sólido.

Decomposição de Minérios de Ferro Normalmente, os minérios de ferro são completamentedecompostos em ácido clorídrico concentrado. A velocidade do ataque por esse reagente é aumentada pelapresença de uma pequena quantidade de cloreto de estanho(II). A tendência do ferro(II) e ferro(III) de for-mar cloro complexos é o aspecto responsável pela maior eficiência do ácido clorídrico sobre o ácido nítricoou ácido sulfúrico como solvente para minérios de ferro.
Muitos minérios de ferro contêm silicatos que podem não ser inteiramente decompostos pelo trata-mento com ácido clorídrico. A decomposição incompleta é indicada pela presença de um resíduo de corescura que permanece após o tratamento prolongado com o ácido. Um resíduo branco de sílica hidratada,que não interfere de forma alguma na análise, é indicativo da completa decomposição.
A Pré-redução do Ferro Uma vez que uma parte ou todo o ferro está presente no estado trivalente apósa decomposição da amostra, a pré-redução a ferro(II) precisa ocorrer antes da titulação com o oxidante.Qualquer um dos métodos descritos na Seção 20A-1 pode ser utilizado. Talvez, o pré-redutor mais eficientepara o ferro seja o cloreto de estanho(II).
2Fe3� � Sn2� 2Fe2� � Sn4�
As únicas outras espécies reduzidas por esse reagente são arsênio, cobre, mercúrio, molibdênio, tungstênioe vanádio em seus altos estados de oxidação.
O excesso de agente redutor é eliminado pela adição de cloreto de mercúrio(II):
Sn2� � 2HgCl2 Hg2Cl2(s) � Sn4� � 2Cl�
O cloreto de mercúrio(I) (Hg2Cl2) pouco solúvel não reduz o permanganato, tampouco o excesso de clore-to de mercúrio(II) (HgCl2) reoxida o ferro(II). É preciso ter cuidado, entretanto, para prevenir a ocorrênciada reação alternativa
Sn2� � HgCl2 Hg(l) � Sn4� � 2Cl�
O mercúrio elementar reage com permanganato e provoca um erro positivo na análise. A formação do mer-cúrio, que é favorecida por um apreciável excesso de estanho(II), é prevenida pelo controle cuidadoso desseexcesso e pela rápida adição do excesso de cloreto de mercúrio(II). Uma redução apropriada é indicadapelo aparecimento de uma pequena quantidade de um precipitado branco sedoso após a adição de mer-cúrio(II). A formação de um precipitado cinza nesse momento indica a presença de mercúrio metálico; atotal ausência de um precipitado revela que uma quantidade insuficiente de cloreto de estanho(II) foiempregada. Em qualquer um dos casos, a amostra deve ser descartada.
A Titulação do Ferro(II) A reação do ferro(II) com permanganato é rápida. A presença do ferro(II) namistura de reação, entretanto, induz a oxidação do cloreto pelo íon permanganato, uma reação que não seprocessa normalmente de forma rápida o suficiente para provocar erros sérios. Erros positivos ocorrem seessa reação paralela não for controlada. Seus efeitos podem ser eliminados por meio da remoção do ácidoclorídrico pela evaporação com ácido sulfúrico ou pela introdução do reagente de Zimmermann-Reinhardt,que contém manganês(II) e uma mistura levemente concentrada dos ácidos sulfúrico e fosfórico.
Acredita-se que a oxidação do íon cloreto durante a titulação envolva uma reação direta entre essaespécie e os íons manganês(III) que se formam como intermediários na redução do íon permanganato peloferro(II). Postula-se que a presença de manganês(II) no reagente de Zimmermann-Reinhardt iniba a for-mação de cloro em virtude da diminuição do potencial do par manganês(III)/manganês(II). Os íons fosfa-to podem exercer um efeito similar pela formação de complexos estáveis de manganês(III). Além disso, osíons fosfato reagem com ferro(III) para formar complexos quase incolores de maneira que a cor amarelado complexo de ferro(II)-cloro não interfira no ponto final.8
S
S
S
1030 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON
8O mecanismo pelo qual o reagente de Zimmermann-Reinhardt age tem sido sujeito a vários estudos. Para uma discussão desse trabalho, ver H. A.Laitinen. Chemical Analysis. Nova York: McGraw-Hill, p. 369-372, 1960.

PREPARO DOS REAGENTES
As seguintes soluções são suficientes para cerca de 100 titulações.
1. Cloreto de estanho(II) 0,25 mol L�1. Dissolva 60 g de SnCl2�2H2O isento de ferro em 100 mL de HClconcentrado; aqueça se necessário. Após a dissolução do sólido, dilua para 1 L com água destilada eguarde em um frasco bem fechado. Adicione alguns pedaços de estanho finamente pulverizado parapreservar a solução.
2. Cloreto de mercúrio(II), a 5% (m/v). Dissolva 50 g de HgCl2 em 1 L de água destilada.3. Reagente de Zimmermann-Reinhardt. Dissolva 300 g de MnSO4�4H2O em 1 L de água. Cuidadosa-
mente, adicione 400 mL de H2SO4 e 400 mL de H3PO4 a 85% e dilua para 3 L.
PROCEDIMENTO
Preparo da AmostraSeque o minério a 110 °C por pelo menos três horas e então deixe resfriar até a temperatura ambiente emum dessecador. Consulte o professor sobre a quantidade de amostra que deverá requerer entre 25 e 40 mLdo padrão de KMnO4 0,02 mol L–1. Pese amostras em erlenmeyers de 500 mL.
Em cada um, adicione 10 mL de HCl concentrado e aproximadamente 3 mL de SnCl2 0,25 mol L–1
(Nota 1). Cubra cada frasco com um pequeno vidro de relógio. Aqueça os frascos em uma capela à tem-peratura próxima ao ponto de ebulição até que as amostras sejam decompostas e o sólido não-dissolvido,se houver, seja branco (Nota 2). Utilize outros 1 a 2 mL de SnCl2 para eliminar qualquer cor amarela quese desenvolva à medida que a solução é aquecida. Aqueça uma solução em branco consistindo em 10 mLde HCl e 3 mL de SnCl2 pelo mesmo intervalo de tempo.
Após a decomposição do minério, remova o excesso de Sn(II) pela adição lenta de KMnO4 0,02 molL�1 até que a solução se torne suavemente amarela. Dilua a cerca de 15 mL. Adicione KMnO4 emquantidade suficiente para gerar uma cor rosa-clara no branco, então adicione uma gota de SnCl2 paradescolorir a solução.
Processe as amostras e brancos individuais em etapas subseqüentes para minimizar a oxidação doferro(II) pelo ar.
Redução do FerroAqueça a solução contendo a amostra até próximo à ebulição e faça adições gota a gota de SnCl2 0,25 molL�1 até o desaparecimento da cor amarela, depois adicione mais duas gotas (Nota 3). Resfrie até a tempera-tura ambiente e acrescente rapidamente 10 mL da solução de HgCl2 a 5%. Uma pequena quantidade de umprecipitado sedoso de Hg2Cl2 deve se formar (Nota 4). O branco deve ser tratado com a solução de HgCl2.
TitulaçãoApós a adição de HgCl2, espere entre dois e três minutos. Então, adicione 25 mL do reagente deZimmermann-Reinhardt e 300 mL de água. Titule imediatamente com o KMnO4 0,02 mol L�1 padrão atéo aparecimento da cor rósea que persista por 15 a 20 s. Não adicione o KMnO4 rapidamente durante a titu-lação. Corrija o volume do titulante para o branco. Expresse os resultados como porcentagem de Fe2O3
na amostra.
Notas1. O SnCl2 acelera a decomposição do minério pela redução dos óxidos de ferro(III) para ferro(II). Quan-
tidades de SnCl2 insuficientes são indicadas pelo aparecimento de uma cor amarela em razão do com-plexo ferro(III)-cloreto.
2. Se partículas escuras persistirem após a amostra ter sido aquecida com ácido por várias horas, filtre asolução em um papel filtro isento de cinzas, lave o resíduo com 5 a 10 mL de HCl 6 mol L�1 e guarde ofiltrado e as águas de lavagem. Calcine o papel e seu conteúdo em um cadinho de platina pequeno. Mis-
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1031

ture 0,5 a 0,7 g de Na2CO3 ao resíduo e aqueça até que um fundido claro seja obtido. Resfrie, adicione5 mL de água e então, cuidadosamente, adicione uns poucos mililitros de HCl 6 mol L�1. Aqueça o ca-dinho até a dissolução do fundido e junte ao filtrado inicial. Evapore a solução até um volume de 15 mLe continue a análise.
3. A solução pode não se tornar inteiramente incolor, mas, ao contrário, pode adquirir uma coloraçãoamarelo-esverdeada pálida. Quantidades adicionais de SnCl2 não alterarão essa cor. Se um excesso deSnCl2 for adicionado, esse excesso pode ser removido pela adição de KMnO4 0,2 mol L�1, repetindo-sea redução.
4. A ausência de precipitado indica que o SnCl2 foi utilizado em quantidades insuficientes e que a reduçãodo ferro(III) foi incompleta. Um resíduo cinza indica a presença de mercúrio elementar, que reage comKMnO4. A amostra precisa ser descartada em qualquer uma dessas circunstâncias.
5. Essas instruções podem ser empregadas para padronizar uma solução de permanganato contra o ferrograu padrão primário. Pese (com precisão de 0,1 mg) 0,2 g de pedaços de ferro eletrolítico em erlen-meyers de 250 mL e dissolva em cerca de 10 mL de HCl concentrado. Dilua cada amostra até apro-ximadamente 75 mL. Então, proceda, para cada alíquota, as etapas de redução e titulação.
37G TITULAÇÕES COM IODO
As propriedades de oxidação do iodo, a composição e a estabilidade de soluções de triiodeto e as aplicaçõesdesse reagente na análise volumétrica são discutidas na Seção 20C-3. O amido é normalmente empregadocomo um indicador para as titulações iodométricas.
37G-1 Preparo dos Reagentes
PROCEDIMENTO
(a) Iodo aproximadamente 0,05 mol L�1. Pese cerca de 40 g de KI em um béquer. Adicione 12,7 g de I2 e10 mL de água. Agite por vários minutos (Nota 1). Introduza mais 20 mL de água e agite novamentepor vários minutos. Decante e transfira cuidadosamente o sobrenadante para um frasco contendo 1 L deágua destilada. É essencial que qualquer quantidade de iodo não-dissolvido remanescente permaneçano béquer (Nota 2).
(b) Indicador de amido (suficiente para 100 titulações). Misture 1 g de amido solúvel e 15 mL de água paraformar uma pasta. Dilua a cerca de 500 mL com água fervente e aqueça até que a mistura fique translú-cida. Resfrie, armazene em um frasco firmemente tampado. Utiliza-se entre 3 e 5 mL para a maioriadas titulações.
O indicador é facilmente atacado por microrganismos presentes no ar e deve ser preparado em intervalosde poucos dias.
Notas1. O iodo dissolve-se lentamente na solução de KI. A agitação enérgica é necessária para acelerar o
processo.2. Qualquer I2 sólido transferido de maneira descuidada para o frasco de armazenamento, vai fazer que a
concentração da solução aumente de forma gradual. A filtração com uso de um cadinho de vidro sinte-rizado elimina essa fonte de problema.
37G-2 Padronização de Soluções de Iodo
Discussão
O óxido de arsênio, desde há muito tempo um padrão primário preferido para soluções de iodo, é raramenteutilizado atualmente em virtude do rigor das leis ambientais responsáveis por controlar até mesmo a uti-
1032 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON

lização de quantidades pequenas de compostos contendo arsênio. O tiossulfato de bário mono-hidratado eo tiossulfato de sódio anidro têm sido sugeridos como padrões alternativos.9 Provavelmente, o método maisconveniente para a determinação da concentração de uma solução de iodo é a titulação de alíquotas comuma solução de tiossulfato de sódio que tenha sido padronizada com iodeto de potássio puro. Instruçõespara esse método encontram-se descritas a seguir.
PREPARO DOS REAGENTES
1. Tiossulfato de sódio, 0,1 mol L�1. Siga as instruções nas Seções 37H-1 e 37H-2 para o preparo epadronização dessa solução.
2. Indicador de amido. Ver a Seção 37G-1(b).
PROCEDIMENTO
Transfira alíquotas de 25,00 mL da solução de iodo para um erlenmeyer de 250 mL e dilua para cerca de50 mL. Processe cada alíquota individualmente em etapas subseqüentes. Adicione aproximadamente 1 mLde H2SO4 3 mol L�1 e titule imediatamente com tiossulfato de sódio padrão até que a solução apresenteuma leve coloração amarelo-palha. Adicione cerca de 5 mL do indicador de amido e continue a titulaçãoconsiderando como ponto final a mudança da cor de azul para incolor (Nota).
NotaA coloração azul do complexo amido/iodo pode reaparecer depois do final da titulação em decorrência daoxidação do íon iodeto.
37G-3 Determinação de Antimônio em Estibinita
Discussão
A análise da estibinita, um mineral comum de antimônio, é uma aplicação típica da iodometria e baseia-sena oxidação do Sb(III) para Sb(V):
SbO � I2 � H2O SbO � 2I� � 2H�
A posição desse equilíbrio é fortemente dependente da concentração de íons hidrogênio. Para forçar areação para a direita, é comum conduzir a titulação na presença de um excesso de bicarbonato de sódio, queconsome os íons hidrogênio quando eles são produzidos.
A estibinita é um mineral de sulfeto de antimônio contendo sílica e outros contaminantes. Desde queesse material esteja livre de ferro e arsênio, a análise da estibinita visando à determinação de seu conteú-do de antimônio é adequada. As amostras são decompostas em ácido clorídrico concentrado a quente paraeliminar o sulfeto na forma de sulfeto de hidrogênio. Para prevenir a perda de cloreto antimônio(III) volátildurante essa etapa, alguns cuidados são necessários. A adição de cloreto de potássio favorece a formaçãode complexos de cloro não-voláteis tais como SbCl e SbCl .
Sais básicos pouco solúveis de antimônio, como o SbOCl, são formados com freqüência quando oexcesso de ácido clorídrico é neutralizado; estes reagem incompletamente com iodo provocando resulta-dos negativos. A dificuldade é superada pela adição de ácido tartárico que forma um complexo solúvel(SbOC4H4O ) do qual o antimônio é rapidamente oxidado pelo reagente.�
6
3�6
�4
3�483�
3
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1033
9W. M. McNevin e O. H. Kriege. Anal. Chem., v. 25, n. 5, p. 767, 1953.

PROCEDIMENTO
Seque a amostra desconhecida a 110 °C por uma hora e deixe resfriar em um dessecador. Pese amostrasindividuais (Nota 1) em um erlenmeyer de 500 mL. Adicione cerca de 0,3 g de KCl e 10 mL de HClconcentrado em cada frasco. Aqueça as misturas (utilize a capela) sem deixar ferver até que permaneçamapenas resíduos brancos ou levemente acinzentados de SiO2.
Adicione 3 g de ácido tartárico em cada amostra e aqueça por mais dez a 25 minutos. Depois, sob agi-tação intensa, adicione água (Nota 2) com uma pipeta ou bureta até que o volume seja de cerca de 100 mL.Se o Sb2S3 avermelhado se formar, interrompa a diluição e aqueça mais um pouco até eliminar H2S; adi-cione HCl se necessário.
Adicione três gotas de fenolftaleína e neutralize com NaOH 6 mol L�1 até que uma leve coloração rosado indicador apareça. Remova a cor pela adição de gotas de HCl 6 mol L�1 e então adicione 1 mL emexcesso. Introduza de 4 a 5 g de NaHCO3 tomando cuidado para evitar perdas da solução por projeçãodurante a adição. Adicione 5 mL do indicador de amido, lave a parte interna do frasco e titule com o padrãode I2 0,05 mol L�1 até que a primeira coloração azul persista por 30 s.
Informe a porcentagem de Sb2S3 na amostra desconhecida.
Notas1. As amostras devem conter de 1,5 a 2 mmol de antimônio; consulte seu professor sobre a massa apro-
priada da amostra. Pesagens com precisão de miligramas são adequadas para amostras maiores que 1 g.2. A adição lenta de água, com uma agitação eficiente, é essencial para prevenir a formação de SbOCl.
37H TITULAÇÕES COM TIOSSULFATO DE SÓDIO
Vários métodos baseiam-se em propriedades redutoras do íon iodeto:
2I� I2 � 2e�
O iodo, produto da reação, é normalmente titulado com uma solução padrão de tiossulfato de sódio comamido servindo como indicador.
I2 � 2S2O 2I� � S4O
Uma discussão sobre os métodos empregando tiossulfato é encontrada na Seção 20B-2.
37H-1 Preparo de Tiossulfato de Sódio 0,1 mol L–1
PROCEDIMENTO
Ferva cerca de 1 L de água destilada por dez a 15 minutos. Deixe a água resfriar à temperatura ambiente;adicione então cerca de 25 g de Na2S2O3�5H2O e 0,1 g de Na2CO3. Agite até que o sólido tenha sidodissolvido. Transfira a solução para um frasco limpo de vidro ou plástico e guarde no escuro.
37H-2 Padronização de Tiossulfato de Sódio com Iodato de Potássio
Discussão
Soluções de tiossulfato de sódio são convenientemente padronizadas pela titulação do iodo produzidoquando um excesso não medido de iodeto de potássio é adicionado a um volume conhecido de uma soluçãopadrão de iodato de potássio acidificada. A reação é:
IO � 5I� � 6H� 3I2 � 3H2OS�3
2�6S2�
3
S
1034 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON

Note que cada mol de iodato resulta na produção de três mols de iodo. O procedimento a seguir baseia-senessa reação.
PREPARO DAS SOLUÇÕES
1. Iodato de potássio, 0,0100 mol L�1. Seque cerca de 1,2 g do padrão primário KIO3 a 110 °C por pelomenos uma hora e resfrie em um dessecador. Pese (com precisão de 0,1 mg) cerca de 1,1 g em um balãovolumétrico de 500 mL; utilize um funil para garantir a transferência quantitativa do sólido. Lave bem ofunil, dissolva o KIO3 em cerca de 200 mL de água destilada, dilua até a marca e agite bem.
2. Indicador de amido. Ver Seção 37G-1(b).
PROCEDIMENTO
Pipete alíquotas de 50,00 mL da solução padrão de iodato em erlenmeyers de 250 mL. Processe cadaamostra individualmente a partir desse ponto para minimizar erros provenientes da oxidação do íon iodetopelo ar. Introduza 2 g de KI isento de iodato e movimente o frasco em círculos para acelerar a dissolução.Adicione 2 mL de HCl 6 mol L�1 e titule imediatamente com tiossulfato até que a solução torne-seamarelo-pálida. Acrescente 5 mL do indicador de amido e titule, sob agitação constante, até o desapareci-mento da coloração azul. Calcule a concentração molar da solução de iodo.
37H-3 Padronização de Tiossulfato de Sódio com Cobre
Discussão
Soluções de tiossulfato também podem ser padronizadas com fio ou folha de cobre metálico puro. Esseprocedimento é vantajoso quando a solução for utilizada para a determinação de cobre, pois qualquer errosistemático no método tende a ser cancelado.
O cobre(II) é reduzido quantitativamente a cobre(I) pelo íon iodeto:
2Cu2� � 4I� S 2CuI(s) � I2
A importância da formação do CuI para favorecer essa reação pode ser vista a partir dos seguintes poten-ciais padrão de eletrodo:
Cu2� � e� 8 Cu� E0 � 0,15 V
I2 � 2e� 8 2I� E0 � 0,54 V
Cu2� � I� � e� 8 CuI(s) E0 � 0,86 V
Os dois primeiros potenciais sugerem que o iodeto não deve reduzir o cobre(II); a formação do CuI, entre-tanto, favorece a redução. A solução deve conter pelo menos 4% de iodeto em excesso para tornar a reaçãocompleta. Além disso, o pH deve ser menor que 4 para prevenir a formação de espécies básicas de cobreque reagem lentamente e de forma incompleta com o íon iodeto. Entretanto, a acidez da solução não podeser maior que cerca de 0,3 mol L�1, em virtude da tendência do íon iodeto a ser oxidado pelo ar em umprocesso catalisado por sais de cobre. Óxidos de nitrogênio também catalisam a oxidação do íon iodetopelo ar. Uma fonte comum desses óxidos é o ácido nítrico normalmente utilizado para dissolver o cobremetálico e outros sólidos contendo cobre. A uréia é usada para retirar óxidos de nitrogênio dessas soluções:
(NH2)2CO � 2HNO2 S 2N2(g) � CO2(g) � 3H2O
A titulação do iodo com tiossulfato tende a provocar resultados ligeiramente negativos por causa daadsorção de quantidades pequenas, porém mensuráveis, de iodo no CuI sólido. O iodo adsorvido é liberado
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1035

lentamente, até mesmo quando o tiossulfato estiver em excesso, resultando em pontos finais transientes eprematuros. Essa dificuldade é grandemente superada pela adição de íon tiocianato. O tiocianato decobre(I), que é pouco solúvel, substitui parte do iodeto de cobre na superfície do sólido:
CuI(s) � SCN� S CuSCN(s) � I�
Essa reação é acompanhada pela liberação do iodo, tornando-o, portanto, disponível para a titulação. Aadição de tiocianato deve ser retardada até que a maior parte do iodo tenha sido titulada, como forma deprevenir a interferência a partir de uma reação lenta entre as duas espécies, possivelmente:
2SCN� � I2 S 2I� � (SCN)2
PREPARO DAS SOLUÇÕES
1. Uréia, 5% (m/v). Dissolva cerca de 5 g de uréia em água o suficiente para resultar em 100 mL desolução. Serão necessários aproximadamente 10 mL para cada titulação.
2. Indicador de amido. Ver a Seção 37G-1(b).
PROCEDIMENTO
Use uma tesoura para cortar o fio ou folha de cobre em porções de 0,20 a 0,25 g. Limpe o metal com umpapel-filtro para deixá-lo isento de poeira e graxas; não o seque por aquecimento. Os pedaços de cobredevem ser manuseados com tiras de papel, luvas de tecido de algodão ou pinças para prevenir a conta-minação pelo contato com a pele.
Use um vidro de relógio previamente pesado ou um frasco de pesagem para obter a massa individualdas amostras de cobre (com precisão de 0,1 mg) por diferença. Transfira cada amostra para um erlenmey-er de 250 mL. Adicione 5 mL de HNO3 6 mol L�1, cubra com um vidro de relógio pequeno e aqueça bran-damente (utilize a capela) até que o metal tenha sido dissolvido. Dilua com cerca de 25 mL de água des-tilada, adicione 10 mL de uréia a 5% (m/v) e ferva rapidamente para eliminar óxidos de nitrogênio. Laveo vidro de relógio coletando a água de lavagem no frasco. Resfrie.
Adicione NH3 concentrada gota a gota sob agitação para produzir o Cu(NH3) de cor azul intensa; asolução deve ter um leve odor de amônia (Nota). Faça adições, gota a gota, de H2SO4 3 mol L�1 até que acor do complexo desapareça e então adicione 2,0 mL de H3PO4 a 85%. Resfrie à temperatura ambiente.
Processe cada amostra individualmente a partir desse ponto para minimizar a oxidação do íon iodetopelo ar. Adicione 4,0 g de KI na amostra e titule imediatamente com Na2S2O3 até que a solução se torneamarelo-pálida. Acrescente 5 mL do indicador de amido e continue a titulação até que a cor azul fique maisfraca. Adicione 2 g de KSCN; agite vigorosamente por 30 s. Complete a titulação utilizando o desapare-cimento da cor azul do amido/I2 como ponto final.
Calcule a concentração molar da solução de Na2S2O3
NotaNão aspire os vapores diretamente do frasco; em vez disso, leve-os ao seu nariz através de movimentos coma sua mão.
37H-4 Determinação de Cobre em Latão
Discussão
O procedimento de padronização descrito na Seção 37H-3 é facilmente adaptado para a determinação decobre em latão, uma liga que também contém quantidades apreciáveis de estanho, chumbo e zinco (eprovavelmente quantidades menores de níquel e ferro). O método é relativamente simples e aplicável para
2�4
1036 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON

latões com menos de 2% de ferro. Uma amostra previamente pesada é tratada com ácido nítrico que provo-ca a precipitação do estanho como óxido hidratado de composição desconhecida. A evaporação com oácido sulfúrico até o aparecimento de trióxido de enxofre elimina o excesso de nitrato, redissolve os com-postos de estanho e, possivelmente, provoca a formação de sulfato de chumbo. O pH é ajustado pela adiçãode amônia, seguido pela acidificação com uma quantidade conhecida de ácido fosfórico. Um excesso deiodeto de potássio é adicionado e o iodo liberado é titulado com padrão de tiossulfato. Ver a Seção 37H-3para discussões adicionais.
PROCEDIMENTO
Se você for orientado, limpe o óleo do metal por meio do tratamento com um solvente orgânico; aqueçarapidamente em uma estufa para eliminar o solvente. Pese (com precisão de 0,1 mg) amostras de 0,3 g emerlenmeyers de 250 mL e acrescente 5 mL de HNO3 6 mol L�1 em cada frasco; aqueça (utilize a capela)até obter uma solução. Adicione 10 mL de H2SO4 concentrado e evapore (utilize a capela) até que osvapores brancos do SO3 sejam totalmente eliminados. Deixe a mistura resfriar. Adicione cuidadosamente30 mL de água destilada, ferva por um a dois minutos e resfrie novamente.
Siga as instruções no terceiro e quarto parágrafos do procedimento descrito na Seção 37H-3.Informe a porcentagem de Cu na amostra.
37I TITULAÇÕES COM BROMATO DE POTÁSSIO
As aplicações de soluções padrão de bromato para a determinação de grupos funcionais orgânicos sãodescritas na Seção 20C-4. As instruções a seguir referem-se à determinação de ácido ascórbico em pasti-lhas de vitamina C.
37I-1 Preparo das Soluções
PROCEDIMENTO
1. Bromato de potássio, 0,015 mol L�1. Transfira cerca de 1,5 g de bromato de potássio de grau analíticoem um frasco de pesagem e seque a 110 °C por pelo menos uma hora. Resfrie em um dessecador. Pese,aproximadamente, 1,3 g (com precisão de 0,1 mg) e transfira para um balão volumétrico de 500 mL; useum funil para garantir a transferência quantitativa do sólido. Lave bem o funil e dissolva o KBrO4 emcerca de 200 mL de água destilada. Dilua até a marca e agite vigorosamente.
O bromato de potássio pode provocar chamas se ficar em contato com materiais orgânicos umedeci-dos (como toalhas de papel em um recipiente de descarte). Consulte seu professor com relação aodescarte de qualquer excesso.
2. Tiossulfato de sódio, 0,05 mol L�1. Siga as instruções da Seção 37G-1; utilize cerca de 12,5 g deNa2S2O3�5H2O por litro de solução.
3. Indicador de amido. Ver a Seção 37G-1(b).
37I-2 Padronização de Tiossulfato de Sódio com Bromato de Potássio
Discussão
O iodo é gerado pela reação entre um volume conhecido do padrão de bromato de potássio e um excessonão medido de iodeto de potássio:
BrO � 6I� � 6H� S Br� � 3I2 � 3H2O
O iodo produzido é titulado com a solução padrão de tiossulfato de sódio.
�3
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1037

PROCEDIMENTO
Pese (com precisão de 1 mg) três a cinco pastilhas de vitamina C (Nota 1). Pulverize as pastilhascompletamente em um almofariz e transfira o pó para um frasco de pesagem seco. Pese amostrasindividuais de 0,40 a 0,50 g (com precisão de 0,1 mg) em um erlenmeyer seco. Processe cada amostraindividualmente a partir desse ponto. Dissolva a amostra (Nota 2) em 50 mL de H2SO4 1,5 mol L�1; entãoadicione cerca de 5 g de KBr. Titule imediatamente com padrão de KBrO3 até o primeiro sinal de coramarela decorrente do excesso de Br2. Anote o volume de KBrO3 utilizado. Adicione 3 g de KI e 5 mL doindicador de amido; titule por retorno (Nota 3) com padrão de Na2S2O3 0,05 mol L�1.
Calcule a massa média (em miligramas) de ácido ascórbico (176,12 g mol�1) em cada pastilha.
Notas1. Esse método não é aplicável para pastilhas mastigáveis de vitamina C.2. O excipiente em muitas pastilhas de vitamina C permanece em suspensão durante a análise. Se o exci-
piente for amido, a coloração característica com complexo com iodo aparece após a adição de KI.3. O volume de tiossulfato necessário para a titulação de retorno quase nunca excede alguns mililitros.
PROCEDIMENTO
Pipete alíquotas de 25,00 mL da solução de KBrO3 em erlenmeyers de 250 mL e lave a parte interior comágua destilada. Processe cada amostra individualmente a partir desse ponto. Acrescente 2 a 3 g de KI ecerca de 5 mL de H2SO4 3 mol L�1. Titule imediatamente com Na2S2O3 até que a cor da solução fiqueamarelo-pálida. Adicione 5 mL do indicador de amido e titule até que a cor azul desapareça.
Calcule a concentração da solução de tiossulfato.
37I-3 Determinação de Ácido Ascórbico em Pastilhas de Vitamina C com Bromato de Potássio
Discussão
O ácido ascórbico, C6H8O6, é oxidado a ácido deidroascórbico pelo bromo.Um excesso não medido de brometo de potássio é adicionado a uma solução acidificada da amostra.
A solução é titulada com padrão de bromato de potássio até o primeiro sinal permanente do excesso debromo; esse excesso é então determinado iodometricamente com padrão de tiossulfato. A titulação com-pleta deve ser realizada sem demoras para prevenir a oxidação do ácido ascórbico pelo ar.
1038 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON
OC⁄C⁄C⁄H � Br2
C£C
O£C
O
O
O
C⁄C⁄C⁄H � 2Br� � 2H�
C⁄C
O£C
H
O
H
HH H
O
H
O
HO
H
O
H
HH H
O
Oxidação do ácido ascórbicopelo bromo.

37J MÉTODOS POTENCIOMÉTRICOS
As medidas potenciométricas fornecem um método altamente seletivo para a determinação quantitativade vários cátions e ânions. Uma discussão sobre os princípios e aplicações das medidas potenciométri-cas é encontrada no Capítulo 21. Instruções detalhadas são dadas nesta seção para a utilização de medi-das potenciométricas na localização dos pontos finais em titulações volumétricas. Além disso, é descritoum procedimento para a determinação potenciométrica direta do íon fluoreto em água potável e em pas-tas de dente.
37J-1 Instruções Gerais para a Realização de uma Titulação Potenciométrica
O procedimento a seguir é aplicável para os métodos de titulação descritos nesta seção. Com a escolhaapropriada do eletrodo, também pode ser aplicado para a maioria dos métodos volumétricos descritos nasSeções 37B até 37H.
PROCEDIMENTO
1. Dissolva a amostra em 50 a 250 mL de água. Lave o par de eletrodos apropriado com água desionizadae mergulhe-os na solução da amostra. Providencie agitação magnética (ou mecânica). Posicione abureta de forma que o reagente possa ser dispensado sem respingar
2. Conecte os eletrodos no medidor, inicie a agitação e anote o volume inicial da bureta e o potencial (oupH) inicial.
3. Anote a leitura do medidor e o volume da bureta após cada adição do titulante. Adicione volumes eleva-dos (cerca de 5 mL) no início. Evite adicionar o titulante antes que a leitura do medidor se mantenhaconstante entre 1 e 2 mV (ou 0,05 unidades de pH) por pelo menos 30 s (Nota). Considere o volume doreagente a ser adicionado pela estimativa do valor de ¢E/¢V após cada adição. Nas proximidades doponto de equivalência adicione o reagente em intervalos de 0,1 mL. Continue a titulação com adições de 2 a 3 mL depois do ponto de equivalência aumentando o intervalo de volume adicionado conforme ovalor de ¢E/¢V for se tornando novamente menor.
NotaOs motores dos agitadores podem, ocasionalmente, provocar erros nas leituras do medidor; é aconselháveldesligar o motor enquanto as leituras estão sendo feitas.
37J-2 Titulação Potenciométrica de Cloreto e Iodeto em uma Mistura
Discussão
A Figura 13-6, página 340, mostra uma curva de titulação argentométrica teórica para uma mistura de íonscloreto e iodeto. Adições iniciais de nitrato de prata resultam exclusivamente na formação de iodeto deprata, pois a solubilidade deste sal é cerca de 5 � 10�7 menor que a do cloreto de prata. Pode ser mostra-do que essa diferença de solubilidade é suficiente para adiar a formação do cloreto de prata até que prati-camente todo o iodeto, com exceção de 7 � 10�5%, tenha sido precipitado. Dessa forma, um pouco antesdo ponto de equivalência a curva é praticamente igual àquela obtida quando somente iodeto é titulado (vera Figura 13-6). Logo após o ponto de equivalência do iodeto, a concentração do íon prata é determinadapela concentração do íon cloreto em solução e a curva de titulação torna-se essencialmente igual àquelaobtida quando somente íons cloreto são titulados.
Curvas similares à da Figura 13-6 podem ser obtidas experimentalmente medindo-se o potencial deum eletrodo de prata imerso na solução contendo o analito. Portanto, uma mistura de cloreto/iodeto podeser analisada para cada um de seus componentes. Entretanto, essa técnica não é aplicável para a análise demisturas iodeto/brometo ou brometo/cloreto, pois as diferenças de solubilidade entre os seus sais de prata
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1039

não são grandes o suficiente. Assim, o sal mais solúvel começa a se formar em quantidades significativasantes que a precipitação do sal menos solúvel tenha acabado. O eletrodo indicador de prata pode ser sim-plesmente um fio do metal polido. Um eletrodo de calomelano pode ser utilizado como referência, embo-ra a difusão do íon cloreto da ponte salina possa fazer que os resultados da titulação sejam mais elevados.Essa fonte de erro pode ser eliminada ao colocar o eletrodo calomelano em uma solução de nitrato de potás-sio que estará em contato com a solução contendo o analito por meio de uma ponte salina de KNO3.Alternativamente, a solução contendo o analito pode ser levemente acidificada com algumas gotas de ácidonítrico; um eletrodo de vidro pode então servir como eletrodo de referência, pois o pH da solução e, con-seqüentemente, seu potencial vão permanecer essencialmente constantes durante a titulação.
A titulação de misturas demonstra como uma titulação potenciométrica pode apresentar múltiplos pon-tos finais. O potencial do eletrodo de prata é proporcional ao pAg. Assim um gráfico de EAg contra o vo-lume do titulante renderá uma curva experimental com a mesma forma da curva teórica mostrada na Figura13-6. (As unidades da ordenada serão diferentes, certamente.)
Curvas experimentais para a titulação de misturas contendo I�/Cl� não apresentam a descontinuidadeque ocorre no primeiro ponto de equivalência da curva teórica (ver a Figura 13-6). Mais importante, o vo-lume de nitrato de prata necessário para atingir o ponto final do I� é, geralmente, um pouco maior que oobtido teoricamente. Esse efeito é resultado da co-precipitação do AgCl, mais solúvel, durante a formaçãodo AgI, menos solúvel. Dessa forma, ocorre um consumo excessivo do reagente na primeira parte da titu-lação. O volume total aproxima-se muito do valor correto.
Apesar desse erro de co-precipitação, o método potenciométrico é útil na análise de misturas de hale-tos. Com quantidades aproximadamente iguais de iodeto e cloreto, os erros relativos podem ser mantidosdentro da faixa de 2%.
PREPARO DOS REAGENTES
1. Nitrato de prata, 0,05 mol L�1. Siga as instruções na Seção 37C-1.2. Ponte salina de nitrato de potássio. Dobre um tubo de vidro de 8 mm de diâmetro na forma de U com
extremidades longas o suficiente para ficarem próximas do fundo de dois béqueres de 100 mL. Aqueça50 mL de água até a fervura e adicione, sob agitação, 1,8 g de agar em pó; continue o aquecimento e aagitação até a formação de uma suspensão uniforme. Dissolva 12 g de KNO3 na suspensão ainda quente.Permita que a suspensão resfrie um pouco. Prenda o tubo em U com as aberturas para cima e utilize umconta-gotas para preenchê-lo com a suspensão quente de agar. Resfrie o tubo em uma torneira de águacorrente para formar um gel. Quando a ponte não estiver sendo utilizada insira as extremidades em umasolução KNO3 2,5 mol L�1.
PROCEDIMENTO
Obtenha uma amostra desconhecida em um balão volumétrico de 250 mL limpo; dilua até a marca comágua e misture bem.
Transfira 50,00 mL da amostra para um béquer de 100 mL limpo e adicione uma ou duas gotas de HNO3
concentrado. Acrescente cerca de 25 mL de KNO3 2,5 mol L�1 em um segundo béquer de 100 mL e coloqueas duas soluções em contato com a ponte salina de agar. Mergulhe o eletrodo de prata na solução contendoo analito e o eletrodo de referência calomelano dentro do segundo béquer. Titule com AgNO3 como descritona Seção 37J-1. Adicione pequenos volumes do titulante nas proximidades dos dois pontos finais.
Elabore um gráfico dos dados e identifique os pontos finais para os dois íons. Faça um gráfico de umacurva de titulação teórica considerando que as medidas das concentrações dos dois constituintes estejamcorretas.
Anote a massa, em miligramas, do I� e Cl� na amostra ou proceda de outra maneira conforme orientação.
1040 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON

37J-3 Determinação Potenciométrica de Espécies de Solutos em uma MisturaContendo Carbonato
Discussão
Um sistema de eletrodos de vidro/calomelano pode ser usado para localizar pontos finais em titulações deneutralização e para estimar constantes de dissociação. Como uma etapa preliminar à titulação, o sistemade eletrodos é padronizado contra um tampão de pH conhecido.
A amostra desconhecida é considerada como uma solução aquosa preparada a partir de uma ou talvezduas substâncias adjacentes presentes na série a seguir: NaHCO3, Na2CO3 ou NaOH (ver a Seção 16B-2).O objetivo é determinar quais desses componentes foram utilizados para preparar a solução desconhecida,assim como a porcentagem de massa de cada soluto.
A maioria das amostras desconhecidas requer uma titulação com um ácido ou base padrão. Algumaspoucas requerem titulações separadas, uma com ácido e outra com base. O pH inicial da solução desco-nhecida serve como guia para definição do(s) titulante(s) apropriado(s); a avaliação da Figura 16-3 e daTabela 16-2 pode ser útil na interpretação dos dados.
PREPARO DAS SOLUÇÕES
Solução padrão de HCl 0,1 mol L�1 e/ou NaOH 0,1 mol L�1. Siga as instruções das Seções 37C-3 até 37C-7.
PROCEDIMENTO
Obtenha a amostra em um balão volumétrico de 250 mL limpo. Dilua até a marca e misture bem. Transfirauma pequena quantidade da amostra diluída para um béquer e determine o pH. Titule uma alíquota de50,00 mL com solução padrão de ácido ou base (ou talvez ambas). Utilize as curvas de titulação resultantespara selecionar o(s) indicador(es) apropriado(s) para a detecção do ponto final e realize titulações emduplicata com esse(s) indicador(es).
Identifique as espécies de soluto presentes na amostra desconhecida e anote a porcentagem de massa/volume para cada uma delas. Calcule a constante de dissociação aproximada que pode ser obtida para cadaespécie contendo carbonato a partir dos dados da titulação. Estime a força iônica da solução e corrija aconstante calculada para uma constante aproximada de dissociação termodinâmica.
37J-4 Determinação Potenciométrica Direta do Íon Fluoreto
Discussão
O eletrodo de estado sólido sensível a fluoreto (ver a Seção 21D-6) tem sido empregado extensivamente nadeterminação de fluoreto em uma variedade de materiais. As instruções seguintes referem-se à determinaçãodesse íon em água potável e em pasta de dente. Uma solução tampão de ajuste da força iônica total (TISAB)é utilizada para ajustar à mesma força iônica todas as amostras desconhecidas e as soluções padrão; quan-do esse reagente é usado, determina-se a concentração de fluoreto em vez de sua atividade. O pH do tam-pão é de aproximadamente 5, um nível no qual F– é predominante entre as espécies contendo fluoreto. Otampão também contém ácido ciclo-hexilaminadinitrilotetra-acético que forma quelatos estáveis comferro(III) e alumínio(II) e, dessa forma, libera os íons fluoreto de seus complexos com esses cátions.
Ver novamente as Seções 21D e 21F antes de realizar esses experimentos.
PREPARO DAS SOLUÇÕES
1. Solução tampão de ajuste da força iônica total (TISAB). Essa solução é comercializada sob o nome deTISSAB.10 Uma quantidade de tampão suficiente para 15 a 20 determinações pode ser preparada mistu-
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1041
10Thermo Electron Corp., Beverly, MA

rando-se (sob agitação) 57 mL de ácido acético glacial, 58 g de NaCl, 4 g de ácido ciclo-hexilamina-dinitrilotetra-acético e 500 mL de água destilada em um béquer de 1 L. Resfrie a solução em um banhode água ou gelo e adicione cuidadosamente NaOH 6 mol L�1 até um pH de 5,0 a 5,5. Dilua para 1 L comágua e armazene em um frasco de plástico.
2. Solução padrão de fluoreto, 100 ppm. Seque uma quantidade de NaF a 110 °C por duas horas. Resfrieem um dessecador; pese (com precisão de 0,1 mg) 0,22 g em um balão volumétrico de 1 L. (Cuidado! ONaF é altamente tóxico. Lave imediatamente qualquer parte da pele que entre em contato com essecomposto, com quantidades abundantes de água.) Dissolva em água, dilua até a marca, misture bem earmazene em um frasco de plástico. Calcule a concentração exata em partes por milhão.
Uma solução padrão de F� pode ser obtida também, a partir de fornecedores comerciais.
PROCEDIMENTO
Os instrumentos para esse experimento consistem em um eletrodo de fluoreto de membrana sólida, umeletrodo saturado de calomelano e um medidor de pH. Um eletrodo de calomelano com junção de vidroesmerilhado é necessário para a determinação em pasta de dente, pois a medida é feita em uma suspensãoque tende a obstruir a junção líquida. A junção de vidro deve ser deslocada de tempos em tempos pararenovar a interface após cada série de medições.
Determinando Fluoreto em Água PotávelTransfira porções de 50,00 mL de água para balões volumétricos de 100 mL e dilua até a marca com asolução TISAB.
Prepare uma solução de F� 5 ppm, diluindo 25,0 mL da solução padrão de 100 ppm em um balãovolumétrico de 500 mL. Transfira alíquotas de 5,00; 10,0; 25,0; e 50,0 mL da solução TISAB e dilua até amarca. (Essas soluções correspondem a concentrações de F� de 0,5; 1,0; 2,5; e 5,0 ppm, respectivamente,nas amostras.)
Depois de lavar abundantemente e secar com um lenço de papel, mergulhe os eletrodos na soluçãopadrão de 0,5 ppm. Agite mecanicamente por três minutos; então anote o potencial. Repita esse procedi-mento com as soluções padrão restantes e as amostras.
Em um gráfico relacione o potencial medido em função do log da concentração das soluções padrão.Utilize esse gráfico para determinar a concentração de fluoreto, em partes por milhão, na amostra des-conhecida.
Determinando Fluoreto em Pasta de Dente11
Pese (com uma precisão de 0,1 mg) 0,2 g de pasta de dente em um béquer de 250 mL. Adicione 50 mL dasolução tisab e ferva por dois minutos sob agitação vigorosa. Resfrie e então transfira a suspensãoquantitativamente para um balão volumétrico de 100 mL, dilua até a marca com água destilada e misturebem. Siga as instruções acima para a análise da água potável começando do segundo parágrafo.
Informe a concentração de F� na amostra em partes por milhão.
37K MÉTODOS ELETROGRAVIMÉTRICOS
Um exemplo conveniente de um método eletrogravimétrico de análise é a determinação simultânea decobre e chumbo em uma amostra de latão. Informações adicionais relativas aos métodos eletrogravimé-tricos são encontradas na Seção 22C.
1042 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON
11De T. S. Light e C. C. Cappuccino. J. Chem. Educ., v. 52, p. 247, 1975.

37K-1 Determinação Eletrogravimétrica de Cobre e Chumbo em Latão
Discussão
Esse procedimento é baseado na deposição de cobre metálico em um cátodo e do chumbo como PbO2 emum ânodo. Como primeiro passo, os óxidos hidratados de estanho (SnO2 xH2O), que se formam quandouma amostra é tratada com ácido nítrico, devem ser removidos por filtração. O dióxido de chumbo édepositado quantitativamente no ânodo a partir de uma solução com uma elevada concentração de íonnitrato; o cobre é apenas parcialmente depositado no cátodo sob essas condições. Portanto, é necessárioeliminar o excesso de nitrato depois que a deposição do PbO2 estiver completa. A remoção é realizada pelaadição de uréia:
6NO � 6H� � 5(NH2)2CO S 8N2(g) � 5CO2(g) � 13H2O
O cobre então se deposita quantitativamente a partir da solução depois que a concentração do íon nitrato forreduzida.
PROCEDIMENTO
Preparo dos EletrodosMergulhe os eletrodos de platina em uma solução a quente de HNO3 6 mol L�1 por cerca de cinco minu-tos (Nota 1), lave-os abundantemente com água destilada, enxágüe com várias porções de etanol e seque-os em uma estufa a 110 °C por dois a três minutos. Resfrie e pese ambos, os ânodos e os cátodos, com umaprecisão de 0,1 mg.
Preparo das AmostrasNão é necessário secar as amostras desconhecidas. Pese (com precisão de 0,1 mg) amostras de 1 g embéqueres de 250 mL. Cubra os béqueres com vidros de relógio. Adicione cuidadosamente cerca de 35 mLde HNO3 6 mol L�1 (use a capela). Deixe reagir por pelo menos 30 min; adicione mais ácido se necessário.Evapore até cerca de 5 mL, mas nunca até a secagem (Nota 2).
Para cada amostra, adicione 5 mL de HNO3 3 mol L�1, 25 mL de água e um pedaço de um tablete depolpa de papel-filtro; deixe reagir sem ferver por cerca de 45 min. Remova o SnO2 xH2O por filtração uti-lizando um papel-filtro de baixa porosidade (Nota 3); colete os filtrados em béqueres alongados. Façavárias lavagens com pequenas porções de HNO3 0,3 mol L�1 a quente para remover os últimos traços decobre; verifique a eficiência das lavagens com algumas gotas de NH3(aq). O volume final do filtrado comas lavagens deve estar entre 100 e 125 mL; adicione água ou evapore a solução para atingir esse volume.
EletróliseCom a corrente desligada, prenda o cátodo no terminal negativo e o ânodo no terminal positivo do apare-lho de eletrólise. Ligue rapidamente o motor de agitação para ter certeza de que os eletrodos não o toquem.Cubra os béqueres com vidros de relógio e inicie a eletrólise. Mantenha uma corrente de 1,3 A por 35 min.
Lave os vidros de relógio e adicione 10 mL de H2SO4 3 mol L�1 seguido de 5 g de uréia em cadabéquer. Mantenha a corrente em 2 A até que as soluções percam suas colorações. Para testar a eficiênciada eletrólise, retire uma gota da solução com um conta-gotas e misture com algumas gotas de NH3(aq) emum tubo de ensaio pequeno. Se a mistura ficar azul, transfira o conteúdo do tubo de volta para o frasco deeletrólise e continue a eletrólise por mais dez minutos. Repita este teste até que nenhuma coloração azuldo Cu(NH3) seja produzida.
Quando a eletrólise estiver completa, desligue a agitação, porém deixe a corrente ligada. Lave os eletro-dos abundantemente com água enquanto eles são removidos da solução. Depois da lavagem, desligue oaparelho de eletrólise (Nota 4), desconecte os eletrodos e mergulhe-os em acetona. Seque os cátodos porcerca de três minutos e os ânodos por 15 min a 110 °C. Deixe os eletrodos resfriarem ao ar e então pese-os.
Informe a porcentagem de chumbo (Nota 5) e de cobre no latão.
2�4
#
�3
#
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1043

Notas1. Alternativamente, graxa e outros materiais orgânicos podem ser removidos pelo aquecimento dos
eletrodos de platina até a incandescência em uma chama. Não toque na superfície dos eletrodos comseus dedos depois de limpá-los, pois graxas e óleos levam à formação de depósitos não-aderentes quepodem se soltar durante a lavagem e a pesagem.
2. O íon cloreto deve ser totalmente eliminado nessa determinação, pois ele reage com o ânodo de platinadurante a eletrólise. Essa reação não é somente destrutiva, mas também provoca erros positivos naanálise pela co-deposição de platina com cobre no cátodo.
3. Se desejado, o conteúdo de estanho pode ser determinado gravimetricamente pela combustão doSnO2 xH2O para SnO2.
4. É importante manter o potencial entre os eletrodos até que eles sejam removidos da solução e lavados.Uma pequena quantidade de cobre pode redissolver se esse cuidado não for observado.
5. A experiência tem mostrado que uma pequena umidade é retida pelo PbO2 e que resultados melhoressão obtidos se o fator estequiométrico de 0,8643 for utilizado em vez de 0,8662.
37L TITULAÇÕES COULOMÉTRICAS
Em uma titulação coulométrica, o “reagente” é uma corrente contínua constante com grandeza exatamenteconhecida. O tempo requerido para essa corrente oxidar ou reduzir o analito quantitativamente (direta ouindiretamente) é medido. Ver a Seção 22D-5 para uma discussão desse método eletroanalítico.
37L-1 A Titulação Coulométrica do Ciclo-hexeno
Discussão12
Muitas olefinas reagem com bromo rapidamente, o bastante para permitir suas titulações. A reação é con-duzida em um ambiente altamente não-aquoso com mercúrio(II) como catalisador. Uma maneira conve-niente de realizar essa titulação é adicionar íon brometo em excesso em uma solução da amostra e gerar obromo em um ânodo que é conectado a uma fonte de corrente constante. Os processos do eletrodo são:
O hidrogênio produzido não reage com o bromo rápido o suficiente para interferir. O bromo reage com umaolefina, tal como o ciclo-hexeno, resultando no produto de adição:
O método amperométrico com eletrodos gêmeos polarizados (página 644) fornece uma maneira conve-niente para detectar o ponto final nessa titulação. Uma diferença de potencial de 0,2 a 0,3 V é mantida entredois pequenos eletrodos. Esse potencial não é suficiente para causar a geração de hidrogênio no cátodo.Dessa forma, próximo ao ponto final, o cátodo está polarizado e nenhuma corrente é observada. No pontofinal, o primeiro excesso de bromo despolariza o cátodo e produz uma corrente. As reações nos eletrodosindicadores gêmeos são:
A corrente é proporcional à concentração de bromo e é rapidamente medida com um microamperímetro.
cátodo Br2 � 2e� S 2Br�
ânodo 2Br� S 2 Br2 � 2e�
¡
Br
Br
Br2 �
cátodo 2H� � 2e� S H2(g)
ânodo 2 Br� S 2 Br2 � 2e�
#
1044 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON
12Esse procedimento foi descrito por D. H. Evans em J. Chem. Educ., v. 45, n. 1, p. 88, 1968.

Uma maneira conveniente de realizar várias análises é gerar inicialmente bromo o suficiente no sol-vente para produzir uma rápida medida de corrente, digamos 20 A. Uma alíquota da amostra é entãointroduzida e a corrente imediatamente diminui e aproxima-se de zero. A geração de bromo inicia-se nova-mente e o tempo necessário para recuperar uma corrente de 20 A é medido. Uma segunda alíquota daamostra é adicionada à mesma solução e o processo é repetido. Várias amostras podem então ser analisadassem a troca do solvente.
O procedimento a seguir refere-se à determinação de ciclo-hexeno em uma solução de metanol. Outrasolefinas também podem ser determinadas dessa forma.
PREPARO DO SOLVENTE
Dissolva cerca de 9 g de KBr e 0,5 g de acetado de mercúrio(II) (Nota 1) em uma mistura contendo 300 mLde ácido acético glacial, 130 mL de metanol e 65 mL de água (suficiente para cerca de 35 mmol de Br2)(Cuidado! Compostos de mercúrio são altamente tóxicos e o solvente é um irritante da pele. Se houvercontato, enxágüe a área afetada com quantidades abundantes de água.)
PROCEDIMENTO
Obtenha uma amostra desconhecida em um balão volumétrico de 100 mL; dilua até a marca com metanole misture bem. A temperatura do metanol deve estar entre 18 °C e 20 °C (Nota 2).
Adicione solvente ácido acético/metanol suficiente para cobrir os eletrodos indicador e gerador norecipiente de eletrólise. Aplique cerca de 0,2 V sobre os eletrodos indicadores. Acione o sistema de eletro-dos geradores e produza bromo até que uma corrente de, digamos, 20 A seja indicada no microam-perímetro. Pare a geração de bromo, anote a corrente no indicador com precisão de 0,1 A e ajuste ocronômetro para zero. Transfira 10,00 mL da amostra desconhecida para o solvente; a corrente no indi-cador deverá diminuir para quase zero. Recomece a geração de bromo. Gere o bromo em incrementos cadavez menores ativando o gerador por períodos cada vez mais curtos enquanto a corrente aumenta e aproxi-ma-se do valor registrado anteriormente. Observe e anote o tempo necessário para atingir o valor de cor-rente original. Zere o cronômetro, introduza uma segunda alíquota (use um volume maior se o temponecessário para a primeira titulação foi curto) e repita o processo. Titule várias alíquotas.
Informe a massa em miligramas de ciclo-hexeno na amostra.
Notas1. Íons mercúrio(II) catalisam a adição de bromo em duplas ligações olefínicas.2. O coeficiente de expansão do metanol é 0,11% °C�1; portanto, se a temperatura não for controlada,
podem ocorrer erros volumétricos.
37M VOLTAMETRIA
Vários aspectos dos métodos polarográficos e amperométricos são considerados no Capítulo 23. Dois exem-plos que ilustram esses métodos são descritos nesta seção. Existe uma grande diversidade de instrumentosdisponíveis para essas determinações. Dessa forma, será necessário que você consulte as instruções de opera-ção dos fabricantes para conhecer os detalhes de operação do equipamento em particular que será utilizado.
37M-1 Determinação Polarográfica de Cobre e Zinco em Latão
Discussão
A porcentagem de cobre e zinco em uma amostra de latão pode ser determinada a partir de medidas polaro-gráficas. O método é particularmente útil para análises rápidas e de rotina; apesar da rapidez, entretanto, aexatidão é consideravelmente mais baixa que aquela obtida com métodos volumétricos ou gravimétricos.
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1045

A amostra é dissolvida em uma quantidade mínima de ácido nítrico, Não é necessário remover oSnO2 xH2O produzido. A adição de um tampão amônia/cloreto de amônio causa a precipitação de ferrocomo um óxido básico. Um polarograma do líquido sobrenadante apresenta duas ondas do cobre. Aprimeira, em aproximadamente �0,2 V (versus ECS), corresponde à redução do cobre(II) ao cobre(I) e aoutra em aproximadamente �0,5 V representa a redução completa do metal. A análise é baseada na cor-rente de difusão total das duas ondas. A concentração de zinco é determinada a partir de sua onda em �1,3V. Em equipamentos que permitem a compensação de corrente, as ondas do cobre são medidas na maiorsensibilidade possível. Essas ondas são então suprimidas pelo controle de compensação do equipamento ea onda do zinco é obtida, novamente, com a sensibilidade mais elevada possível.
PREPARO DAS SOLUÇÕES
1. Solução de cobre(II), 2,5 � 10�2 mol L�1. Pese (com precisão de 0,1 mg) 0,4 g de fio de cobre. Dissolvaem 5 mL de HNO3 concentrado (utilize a capela). Ferva brevemente para remover óxidos de nitrogênio;então resfrie, dilua com água, transfira quantitativamente para um balão volumétrico de 250 mL, diluaaté a marca com água e misture vigorosamente.
2. Solução de zinco(II), 2,5 � 10�2 mol L�1. Seque o ZnO (grau analítico) por uma hora a 110 °C, resfrieem um dessecador e pese 0,5 g (com precisão de 0,1 mg) em um béquer pequeno. Dissolva uma misturade 25 mL de água e 5 mL de HNO3 concentrado. Transfira para um balão volumétrico de 250 mL e diluaaté a marca com água.
3. Gelatina, 0,1%. Adicione cerca de 0,1 g de gelatina em 100 mL de água fervente.4. Tampão amônia/cloreto de amônio (suficiente para cerca de 15 polarogramas). Misture 27 g de NH4Cl
e 35 mL de amônia concentrada em água destilada suficiente para produzir 500 mL. Essa solução con-tém aproximadamente 1 mol L–1 de NH3 e 1 mol L–1 de NH4
�.
PROCEDIMENTO
CalibraçãoUse uma bureta para transferir porções de 0,00; 1,00; 8,00; e 15,00 mL da solução padrão de Cu(II) parabalões volumétricos de 50 mL. Adicione 5 mL da solução de gelatina e 30 mL do tampão em cada balão.Dilua até a marca e misture bem. Prepare séries idênticas para as soluções de Zn(II).
Lave a célula polarográfica três vezes com pequenas porções de uma solução de cobre(II) e entãopreencha a célula. Borbulhe nitrogênio na solução por dez a 15 minutos para remover oxigênio. Apliqueum potencial de �1,6 V e ajuste a sensibilidade até que o detector forneça uma resposta equivalente àescala completa. Obtenha um polarograma, varrendo o potencial de 0 a �1,5 V (versus ECS). Meça a cor-rente limite em um potencial logo depois da segunda onda. Obtenha a corrente de difusão pela subtraçãoda corrente do branco (Nota) no mesmo potencial. Calcule id/c.
Preparo da amostraPese amostras de latão de 0,10 g a 0,15 g (com precisão de 0,5 mg) em béqueres de 50 mL e dissolva com2 mL de HNO3 concentrado (utilize a capela). Ferva brandamente para eliminar óxidos de nitrogênio.Resfrie, adicione 10 mL de água destilada e transfira quantitativamente cada amostra para balões volu-métricos de 50 mL; dilua até a marca com água e misture bem.
Transfira 10,00 mL da amostra diluída para outro balão volumétrico de 50 mL, adicione 5 mL degelatina e 30 mL do tampão, dilua até a marca com água e misture bem.
AnáliseSiga as instruções do segundo parágrafo do procedimento de calibração acima, avalie a corrente de difusãopara o cobre e o zinco.
Calcule a porcentagem de Cu e Zn na amostra de latão.
#
1046 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON

NotaO polarograma para o branco (isto é, 0 mL de padrão) deve ser obtido sob o mesmo ajuste de sensibilidadeutilizado para os padrões contendo a concentração mais baixa dos íons metálicos.
37M-2 Titulação Amperométrica de Chumbo
Discussão
As titulações amperométricas são discutidas na Seção 23B-4. No procedimento a seguir, a concentração de chumbo em uma solução aquosa é determinada pela titulação com uma solução padrão de dicromato depotássio. A reação é:
Cr2O � 2Pb2� � H2O S 2PbCrO4(s) � 2H�
A titulação pode ser realizada com um eletrodo gotejante de mercúrio mantido tanto em 0 quanto em �1,0 V(versus ECS). A 0 V, a corrente permanece próxima de zero pouco antes do ponto final, mas aumenta rapi-damente a partir daí em conseqüência da redução do dicromato, o qual está agora em excesso. Em �1,0 V,
ambos os íons chumbo e dicromato são reduzidos. A corrente entãodiminui na região mais próxima do ponto final (refletindo a diminuiçãoda concentração do íon chumbo conforme o titulante é adicionado),passa por um valor mínimo no ponto final e aumenta conforme o dicro-mato se torna disponível. O ponto final em �1,0 V deve ser, entre osdois, o mais fácil de ser localizado com exatidão. Entretanto, a remoçãodo oxigênio é desnecessária para a titulação em 0 V.
PREPARO DAS SOLUÇÕES
1. Eletrólito de suporte. Dissolva 10 g de KNO3 e 8,2 g de acetato de sódio em cerca de 500 mL de águadestilada. Adicione ácido acético glacial e leve o pH para 4,2 (use o pH-metro); cerca de 20 mL de ácidoserão necessários (suficiente para cerca de 20 titulações).
2. Gelatina, 0,1%. Adicione 0,1 g de gelatina em 100 mL de água fervente.3. Dicromato de potássio, 0,01 mol L�1. Utilize um funil para pesar cerca de 0,75 g (com precisão de
0,5 mg) do padrão primário K2Cr2O7 em um balão volumétrico de 250 mL. Dilua até a marca com águadestilada e misture bem.
4. Ponte salina de nitrato de potássio. Ver a Seção 37J-2.
PROCEDIMENTO
Obtenha uma amostra (Nota) em um balão volumétrico de 100 mL limpo; dilua até a marca com águadestilada e misture bem. Realize a titulação em um béquer de 100 mL. Coloque o eletrodo saturado decalomelano em um segundo béquer de 100 mL e promova o contato entre as soluções nos dois recipientescom a ponte salina de KNO3.
Transfira uma alíquota de 10,00 mL da amostra desconhecida para o béquer de titulação. Adicione 25mL do eletrólito de suporte e 5 mL de gelatina. Coloque um eletrodo gotejante de mercúrio na solução con-tendo a amostra. Conecte ambos os eletrodos ao polarógrafo. Meça a corrente no potencial aplicado de 0V. Adicione K2Cr2O7 0,01 mol L�1 em incrementos de 1 mL; anote a corrente e o volume após cada adição.Continue a titulação até cerca de 5 mL depois do ponto final. Corrija as correntes pelas variações do vo-lume do titulado e faça um gráfico com os dados obtidos. Calcule o volume no ponto final.
Repita as titulações em �1,0 V. Nesse caso, você deve borbulhar nitrogênio na solução por cerca dedez a 15 minutos antes de iniciar a titulação e depois de cada adição de reagente. O fluxo de nitrogêniodeve, é claro, ser interrompido durante as medições da corrente. Novamente, corrija as correntes pela va-riação do volume, faça um gráfico e determine o volume no ponto final.
2�7
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1047
� A corrente corrigida é dada por
(id)corr � id
em que V é o volume original, v éo volume do reagente adicionado e id é a corrente não-corrigida.
aV � v
Vb

Relate a massa de Pb, em miligramas, na amostra desconhecida.
NotaUma solução estoque 0,0400 mol L�1 pode ser preparada dissolvendo-se 13,5 g de Pb(NO3)2 em 10 mL deuma solução HNO3 6 mol L�1 com água. As amostras devem conter de 15 a 25 mL dessa solução.
MÉTODOS BASEADOS NA37N ABSORÇÃO DE RADIAÇÃO
Os métodos de absorção molecular são discutidos no Capítulo 26. As instruções a seguir envolvem (1) autilização de uma curva analítica para a determinação de ferro em água, (2) o emprego do procedimentode adição de padrão para a determinação de manganês em aço e (3) a determinação espectrofotométrica dopH em uma solução tampão.
37N-1 Lavagem e o Manuseio das Células Espectrofotométricas
A exatidão das medições espectrofotométricas depende criticamente da disponibilidade de células idênti-cas e de boa qualidade. Elas devem ser calibradas entre si a intervalos regulares para detectar diferençasresultantes de arranhões, corrosão e desgaste. É igualmente importante a limpeza apropriada das facesexternas (as janelas) exatamente antes de as células serem inseridas em um fotômetro ou espectro-fotômetro. O método preferido consiste em limpar as janelas com um lenço de papel embebido commetanol; o metanol é então facilmente evaporado, deixando as janelas livres de contaminantes. Tem sidomostrado que esse método é muito superior ao procedimento usual de limpeza das janelas com lenços depapel secos, que tende a deixar resíduos de papel e um filme sobre a janela.13
37N-2 Determinação de Ferro em Água Natural
Discussão
O complexo vermelho-alaranjado que se forma entre ferro(II) e 1,10-fenantrolina (ortofenantrolina) é útilna determinação de ferro em águas de abastecimento. O reagente é uma base fraca que reage para formaro íon fenantrolínio, fenH�, em meio ácido. Assim, a formação do complexo com o ferro é mais bem descri-ta pela equação
Fe2� � 3phenH� 8 Fe(phen) � 3H�
A estrutura do complexo é mostrada na Seção 19E-1. A constante de formação para o equilíbrio é 2,5 � 106
a 25 °C. O ferro(II) é quantitativamente complexado em uma faixa de pH variando entre 3 e 9. Um pH decerca de 3,5 é geralmente recomendado para prevenir a precipitação de sais de ferro, tais como os fosfatos.
Um excesso de um agente redutor, como hidroxilamina ou hidroquinona, é necessário para manter oferro no estado de oxidação �2. O complexo, uma vez formado, é muito estável. Essa determinação podeser realizada com um espectrofotômetro ajustado em 508 nm ou com um fotômetro equipado com um fil-tro verde.
PREPARO DAS SOLUÇÕES
1. Solução padrão de ferro, 0,01 mg mL�1. Pese (com precisão de 0,2 mg) 0,0702 g de Fe(NH4)2(SO4)2
6H2O em um balão volumétrico de 1 L. Dissolva em 50 mL de água contendo de 1 a 2 mL de ácidosulfúrico concentrado; dilua a marca e misture bem.
#
2�3
1048 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON
13Para mais informações, ver a J. O. Erickson e T. Surles. Amer.Lab., v. 8, n. 6, p. 50, 1976.

2. Cloreto de hidroxilamina (suficiente para 80 a 90 medidas). Dissolva 10 g de H2NOH � HCl em cerca de100 mL de água destilada.
3. Solução de ortofenantrolina (suficiente para 80 a 90 medidas). Dissolva 1,0 g de ortofenantrolinamonoidratada em cerca de 1 L de água. Aqueça suavemente se necessário. Cada mililitro é suficientepara até 0,09 mg de Fe. Prepare apenas a quantidade de reagente necessária; ele escurece com o passardo tempo e então precisa ser descartado.
4. Acetato de sódio, 1,2 mol L�1 (suficiente para 80 a 90 medidas). Dissolva 166 g de NaOAc � 3H2O em1 L de água destilada.
PROCEDIMENTO
Preparo da Curva AnalíticaTransfira 25,00 mL da solução padrão de ferro para um balão volumétrico de 100 mL e 25 mL de água emum segundo balão volumétrico de 100 mL. Adicione 1 mL de hidroxilamina, 10 mL de acetato de sódio e10 mL de ortofenantrolina em cada balão. Deixe as misturas em repouso por cinco minutos; dilua até amarca e misture bem.
Limpe um par de células espectrofotométricas apropriadas para o equipamento. Lave cada uma delaspelo menos três vezes com a solução a ser analisada. Determine a absorbância do padrão com relação aobranco.
Repita esse procedimento com pelo menos outros três volumes da solução padrão de ferro; assegure-se de trabalhar com uma faixa de absorbância de 0,1 a 1,0. Faça um gráfico da curva analítica.
Determinação de FerroTransfira 10,00 mL da amostra desconhecida para um balão volumétrico de 100 mL; proceda exatamenteda mesma maneira empregada para as soluções padrão, medindo a absorbância com relação ao branco.Altere o volume da amostra desconhecida para obter medidas de absorbância em replicata que se encon-trem dentro da faixa de trabalho da curva de calibração.
Informe a concentração de ferro na amostra desconhecida em partes por milhão.
37N-3 Determinação de Manganês em Aço
Discussão
Quantidades pequenas de manganês são facilmente determinadas fotometricamente pela oxidação deMn(II) ao íon permanganato de coloração intensa. O periodato de potássio é um agente oxidante efetivopara esse propósito. A reação é
5IO � 2Mn2� � 3H2O S 5IO � 2MnO � 6H�
Soluções de permanganato contendo periodato em excesso são muito estáveis.Existem poucos interferentes nesse método. A presença de íons coloridos pode ser compensada com
um branco. Cério(III) e crômio(III) são exceções; esses íons geram produtos de oxidação com o periodatoque absorvem, com alguma intensidade, no mesmo comprimento de onda usado para a medida do per-manganato.
O método fornecido aqui é aplicável para aços que não contêm quantidades elevadas de crômio. Aamostra é dissolvida em ácido nítrico. Qualquer quantidade de carbono presente no aço é oxidada com pe-roxodissulfato. O ferro(III), uma fonte de interferência, é eliminado pela complexação com o ácido fos-fórico. O método da adição de padrões (ver a Seção 8C-3) é utilizado para estabelecer a relação entre aabsorbância e a quantidade de manganês na amostra.
Um espectrofotômetro ajustado em 525 nm ou um fotômetro com um filtro verde de radiação podemser empregados para as medidas de absorbância.
�4
�3
�4
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1049

PREPARO DAS SOLUÇÕES
Solução padrão de manganês(II) (suficiente para centenas de análises). Pese 0,1 g (com precisão de 0,1mg) de manganês em um béquer de 50 mL e dissolva em cerca de 10 mL de HNO3 6 mol L�1 (utilize acapela). Ferva cuidadosamente para eliminar óxidos de nitrogênio. Resfrie; então, transfira a soluçãoquantitativamente para um balão volumétrico de 1 L. Dilua até a marca com água e misture muito bem.Depois de ser convertido para permanganato, o manganês presente em 1 mL da solução padrão provocaaumento da absorbância de cerca de 0,09, quando diluído a um volume de 50 mL.
PROCEDIMENTO
A amostra não precisa ser seca. Se existirem evidências da presença de graxa, lave a amostra com acetonae seque rapidamente. Pese (com precisão de 0,1 mg) amostras em duplicata (Nota 1) em béqueres de 150mL. Adicione cerca de 50 mL de HNO3 6 mol L�1 e ferva cuidadosamente (utilize a capela); o aqueci-mento por cinco a dez minutos deve ser suficiente. Adicione com cautela cerca de 1 g de peroxidissulfatode amônio e ferva cuidadosamente por mais dez a 15 minutos. Se a solução ficar rosa ou se apresentar umdepósito de MnO2, adicione 1 mL de NH4HSO3 (ou 0,1 g de NaHSO3) e aqueça por cinco minutos. Resfrie;transfira quantitativamente (Nota 2) para balões volumétricos de 250 mL. Dilua até a marca com água emisture bem. Utilize uma pipeta de 20,00 mL para transferir três alíquotas de cada amostra para béqueresindividuais. Proceda da seguinte maneira:
Alíquota Volume de H3PO4, a 85%, mL Volume do Padrão de Mn, mL Massa de KIO4, g
1 5 0,00 0,42 5 5,00 (Nota 3) 0,43 5 0,00 0,0
Ferva cuidadosamente cada solução por cinco minutos, resfrie e transfira quantitativamente para um frascovolumétrico de 50 mL. Misture bem. Meça a absorbância das alíquotas 1 e 2 utilizando a alíquota 3 como obranco (Nota 4).
Informe a porcentagem de manganês na amostra desconhecida
Notas1. A massa da amostra depende do seu conteúdo de manganês; consulte o professor.2. Se existir evidência de turbidez; filtre as soluções enquanto elas são transferidas para os balões
volumétricos.3. O volume de cada adição de padrão deve ser definido pela absorbância da amostra. É importante obter
uma estimativa grosseira pela geração de permanganato em cerca de 20 mL da amostra. Dilua para 50mL e meça a absorbância.
4. Um único branco pode ser utilizado para todas as medidas, desde que as massas das amostras estejamdentro de 50 mg umas das outras.
37N-4 Determinação Espectrofotométrica do pH
Discussão
O pH de um tampão desconhecido é determinado pela adição de um indicador ácido/base e medidas espec-trofotométricas da absorbância das soluções resultantes. Em virtude da existência de sobreposição entre osespectros para as formas ácidas e básicas do indicador, é necessário avaliar as absortividades molares indi-viduais para cada forma em dois comprimentos de onda. Ver a página 754 para mais discussões.
1050 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON

A relação entre as duas formas do verde de bromocresol em uma solução aquosa é descrita pelo equi-líbrio
HIn � H2O 8 H3O� � In�
para o qual
Ka �
A avaliação espectrofotométrica do [In�] e do [HIn] permite o cálculo de [H3O�] e, portanto, do pH.
PREPARO DAS SOLUÇÕES
1. Verde de bromocresol, 1,0 � 10�4 mol L�1 (suficiente para cerca de cinco determinações). Dissolva40,0 mg (com precisão de 0,1 mg) do sal de sódio do verde de bromocresol (720 g mol�1) em água edilua para 500 mL em um frasco volumétrico.
2. HCl, 0,5 mol L�1. Dilua cerca de 4 mL de HCl concentrado para aproximadamente 100 mL com água.3. NaOH, 0,4 mol L�1. Dilua cerca de 7 mL de NaOH 6 mol L�1 para cerca de 100 mL com água.
PROCEDIMENTO
Determinação dos Espectros de Absorção IndividuaisTransfira alíquotas de 25,00 ml da solução indicadora de verde de bromocresol para dois balões volumétri-cos de 100 mL. Em um deles, adicione 25 mL de HCl 0,5 mol L�1; no outro, acrescente 25 mL de NaOH0,4 mol L�1. Dilua até a marca e misture bem.
Obtenha o espectro de absorção do indicador entre 400 e 600 nm, para suas formas ácida e base con-jugadas, utilizando água como um branco. Registre os valores de absorbância em intervalos de 10 nm, aprincípio, e a intervalos mais próximos, conforme necessário, para definir os valores dos máximos e míni-mos. Determine a absortividade molar para HIn e In� nos comprimentos de onda correspondentes às suasabsorções máximas.
Determinação do pH de um Tampão DesconhecidoTransfira 25,00 mL da solução estoque do indicador verde de bromocresol para um balão volumétrico de100 mL. Adicione 50,00 mL do tampão desconhecido, dilua até a marca e misture bem. Meça a absorbân-cia da solução diluída nos comprimentos de onda para os quais as absortividades molares foram calculadas.
Relate o pH do tampão.
37O FLUORESCÊNCIA MOLECULAR
O fenômeno de fluorescência e suas aplicações analíticas são discutidos no Capítulo 27. As instruções aseguir referem-se à determinação de quinino em bebidas nas quais a concentração de quinino é tipicamentede 25 a 60 ppm.
37O-1 Determinação de Quinino em Bebidas14
Discussão
Soluções de quinino fluorescem fortemente quando excitadas por radiação de 350 nm. A intensidade rela-tiva do pico de fluorescência em 450 nm fornece um método sensível para a determinação de quinino em
[H3O� ] [In�]
[HIn]� 1,6 � 10�5
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1051
14Essas intruções foram adaptadas a partir de J. E. O’Reilly. J. Chem. Educ., v. 52, n. 9, p. 610, 1975.

bebidas. Medições preliminares são necessárias para definir a faixa de concentração na qual a intensidadede fluorescência é linear ou quase linear. A amostra desconhecida é então diluída conforme for necessáriopara permitir leituras dentro dessa faixa.
PREPARO DOS REAGENTES
1. Ácido sulfúrico, 0,05 mol L�1. Adicione cerca de 17 mL de H2SO4 6 mol L�1 em 2 L de água destilada.2. Padrão de sulfato de quinino, 1 ppm. Pese (com precisão de 0,5 mg) 0,100 g de sulfato de quinino em
um balão volumétrico de 1 L e dilua até a marca com H2SO4 0,05 mol L�1 (suficiente para 60 a 70análises). Transfira 10,00 mL dessa solução para outro balão volumétrico de 1 L e, novamente, dilua atéa marca com H2SO4 0,05 mol L�1. Essa última solução contém 1 ppm de quinino; deve ser preparada nodia e mantida no escuro quando não estiver sendo utilizada.
PROCEDIMENTO
Determinação da Faixa de Concentração ApropriadaAjuste o fluorímetro para um comprimento de onda de excitação de 350 nm. Para encontrar uma faixa detrabalho apropriada, meça a fluorescência relativa do padrão de 1 ppm em um comprimento de onda deemissão de 450 nm (ou com um filtro apropriado que transmita nessa faixa). Use uma proveta graduadapara diluir 10 mL da solução 1 ppm com 10 mL de H2SO4 0,05 mol L�1; novamente, meça a fluorescên-cia relativa. Repita esse processo de diluição e medição até que a intensidade relativa se aproxime daque-la obtida com um branco constituído por H2SO4 0,05 mol L�1. Faça um gráfico com os dados e selecionea faixa para a análise (isto é, a região dentro da qual o gráfico é linear).
Preparo da Curva AnalíticaUtilize uma vidraria volumétrica para preparar três ou quatro padrões que se distribuam pela faixa linear;meça a intensidade de fluorescência para cada um deles. Faça um gráfico com os dados.
AnáliseObtenha uma amostra. Faça diluições apropriadas com H2SO4 0,5 mol L�1 para fazer que sua intensidadede fluorescência fique dentro da curva de calibração.
Determine a concentração de quinino na amostra desconhecida em partes por milhão.
37P ESPECTROSCOPIA ATÔMICA
Vários métodos de análise baseados na espectroscopia atômica são discutidos no Capítulo 28. Uma dessasaplicações é a absorção atômica, demonstrada no experimento a seguir.
37P-1 Determinação de Chumbo em Latão por Espectroscopia de Absorção Atômica
Discussão
O latão e outras ligas metálicas de cobre contêm de 0% a cerca de 10% de chumbo, assim como estanho ezinco. A espectroscopia de absorção atômica permite a determinação quantitativa desses elementos. Aexatidão desse procedimento não é tão elevada como aquela obtida por medições gravimétricas ouvolumétricas, mas o tempo necessário para obter a informação analítica é consideravelmente menor.Uma amostra de massa conhecida é dissolvida em uma mistura de ácidos nítrico e clorídrico, o segundo énecessário para prevenir a precipitação de estanho como ácido metaestânico, SnO2 � xH2O. Depois de umadiluição adequada, a amostra é aspirada para a chama e a absorção da radiação gerada a partir de uma lâm-pada de cátodo oco é medida.
1052 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON

PREPARO DAS SOLUÇÕES
Solução padrão de chumbo, 100 mg L�1. Seque uma quantidade de Pb(NO3)2 de grau analítico por cerca deuma hora à 110 °C. Resfrie. Pese (com precisão de 0,1 mg) 0,17 g e transfira para um balão volumétrico de1 L. Dissolva em uma solução contendo 5 mL de água e 1 a 3 mL de HNO3 concentrado. Dilua até a marcacom água destilada e misture bem.
PROCEDIMENTO
Pese, em duplicata, porções da amostra (Nota 1) em béqueres de 150 mL. Cubra os béqueres com vidros derelógio e então dissolva (use a capela) em uma mistura contendo cerca de 4 mL de HNO3 concentrado e 4mL de HCl concentrado (Nota 2). Ferva cuidadosamente para remover óxidos de nitrogênio. Resfrie,transfira as soluções quantitativamente para um balão volumétrico de 250 mL, dilua até a marca com águae misture bem.
Use uma bureta para dispensar porções de 0, 5, 10, 15 e 20 mL da solução padrão de chumbo parabalões volumétricos individuais de 50 mL. Adicione 4 mL de HNO3 concentrado e 4 mL de HCl concen-trado em cada balão volumétrico e dilua até a marca com água.
Transfira alíquotas de 10,00 mL de cada amostra para balões volumétricos de 50 mL, adicione 4 mLde HCl concentrado e 4 mL de HNO3 concentrado e dilua até a marca com água.
Ajuste o monocromador para 283,3 nm e meça a absorbância para cada padrão e para a amostra nessecomprimento de onda. Faça pelo menos três – e preferencialmente mais – leituras para cada medição.
Construa com os dados um gráfico da curva analítica. Reporte a porcentagem de chumbo no latão.
Notas1. A massa da amostra depende do conteúdo de chumbo no latão e da sensibilidade do equipamento uti-
lizado para as medições da absorção. Uma amostra contendo de 6 a 10 mg de chumbo é aceitável. Con-sulte seu professor.
2. Latões que contenham uma porcentagem elevada de estanho requerem mais HCl para prevenir a for-mação de ácido metaestânico. As amostras diluídas podem apresentar turbidez quando guardadas porperíodos prolongados, uma turbidez leve não afeta a determinação do chumbo.
37P-2 Determinação de Sódio, Potássio e Cálcio em Águas Minerais porEspectroscopia de Emissão Atômica
Discussão
Um método conveniente para a determinação de metais alcalinos ealcalinos-terrosos em água e em plasma sangüíneo é baseado nos espec-tros característicos que esses elementos emitem quando são aspiradosem uma chama de ar/gás natural. As instruções seguintes são apropria-das para a determinação desses três elementos em amostras de água.Tampões de radiação (página 823) são utilizados para minimizar oefeito de cada elemento na intensidade de emissão dos outros.
PREPARO DAS SOLUÇÕES
1. Solução padrão de cálcio, aproximadamente 500 ppm. Seque uma quantidade de CaCO3 por cerca deuma hora a 110 °C. Resfrie em um dessecador; pese (com precisão de 1 mg) 1,25 g em um béquer de 600 mL. Adicione cerca de 200 mL de água destilada e cerca de 10 mL de HCl concentrado; cubra o
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1053
Um tampão de radiação é umasubstância que é adicionada emgrande excesso nas amostras e nospadrões para se sobrepor ao efeitode matriz das espécies e assimminimizar interferências (ver aSeção 8C-3)

béquer com um vidro de relógio durante a adição de ácido para evitar a perda por respingamento.Depois de a reação ter sido completada, transfira a solução quantitativamente para um balão volumétricode 1 L, dilua até a marca e misture bem.
2. Solução padrão de potássio, aproximadamente 500 ppm. Seque uma quantidade de KCl por cerca deuma hora a 110 °C. Resfrie em um dessecador; pese (com precisão de 1 mg) cerca de 0,95 g em umbalão volumétrico de 1 L. Dissolva em água destilada e dilua até a marca.
3. Solução padrão de sódio, aproximadamente 500 ppm. Proceda como no item 2 utilizando 1,25 g (comprecisão de 1 mg) de NaCl seco.
4. Tampão de radiação para a determinação de cálcio. Prepare cerca de 100 mL de uma solução que tenhasido saturada com NaCl, KCl e MgCl2, nessa ordem.
5. Tampão de radiação para a determinação de potássio. Prepare cerca de 100 mL de uma solução quetenha sido saturada com NaCl, CaCl2 e MgCl2, nessa ordem.
6. Tampão de radiação para a determinação de sódio. Prepare cerca de 100 mL de uma solução que tenhasido saturada com CaCl2, KCl e MgCl2, nessa ordem.
PROCEDIMENTO
Preparação das curvas de trabalhoAdicione 5,00 mL do tampão de radiação apropriado para cada um dos balões volumétricos de 100 mL deuma seqüência. Adicione volumes conhecidos do padrão de forma a produzir soluções com concentraçõesna faixa de 0 a 10 ppm do cátion a ser determinado. Dilua até a marca e misture bem.
Meça a intensidade de emissão para cada solução, fazendo pelo menos três leituras para cada uma.Aspire água destilada entre cada conjunto de medidas. Corrija os valores médios para a radiação de fundoe prepare uma curva analítica com os dados.
Repita esse procedimento para os outros dois cátions.
Análise de uma amostra de águaPrepare alíquotas em duplicata para a amostra como indicado para a preparação da curva analítica. Se fornecessário, utilize um padrão para calibrar a resposta do equipamento para a curva de trabalho; então meçaa intensidade de emissão da amostra desconhecida. Corrija os dados para a radiação de fundo. Determine aconcentração do cátion na amostra desconhecida pela comparação com a curva analítica.
37Q APLICAÇÃO DE RESINAS DE TROCA IÔNICA
37Q-1 A Separação de Cátions
A aplicação de resinas de troca iônica para a separação de espécies iônicas de cargas opostas é discutidana Seção 30D. As instruções a seguir referem-se à separação por troca iônica de níquel(II) do zinco(II)baseada na conversão dos íons de zinco para cloro complexos carregados negativamente. Depois da sepa-ração, cada um dos cátions é determinado por meio de titulação com EDTA.
Discussão
A separação dos dois cátions baseia-se nas diferenças em suas tendências a formar complexos aniônicos.Complexos estáveis de clorozincatos(II) (tais como ZnCl e ZnCl ) são formados em uma solução 2 molL�1 de ácido clorídrico e retidos em uma resina de troca aniônica. Ao contrário, o níquel(II) não é com-plexado apreciavelmente nesse meio e passa rapidamente por esse tipo de coluna. Depois da separaçãocompleta, a eluição com água decompõe efetivamente os complexos de cloro e permite a remoção do zinco.
O níquel e o zinco são determinados pela titulação com padrão de EDTA em pH 10. O negro de erio-cromo T é o indicador utilizado para a titulação do zinco. O vermelho de bromopirogalol ou murexida éusado para a titulação do níquel.
2�4
�3
1054 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON

PREPARO DAS SOLUÇÕES
1. Padrão de EDTA. Ver a Seção 37E-2.2. Tampão pH 10. Ver a Seção 37E-1.3. Indicador negro de Ério T. Ver a Seção 37E-1.4. Indicador vermelho de bromopirogalol (suficiente para 100 titulações). Dissolva 0,5 g do indicador
sólido em 100 mL de uma solução de etanol 50% (v/v).5. Indicador murexida. O reagente sólido consiste em cerca de 0,2% em massa do indicador em NaCl.
Aproximadamente 0,2 g são necessários para cada titulação. A preparação sólida é utilizada, pois assoluções do indicador são muito instáveis.
PREPARO DAS COLUNAS DE TROCA IÔNICA
Uma coluna de troca iônica típica é composta por um cilindro de 25 a 40 cm de comprimento e 1 a 1,5 cmde diâmetro. Uma torneira na extremidade inferior permite o ajuste da vazão do líquido que elui da coluna.Uma bureta pode ser usada convenientemente como uma coluna. É recomendado que duas colunas sejampreparadas para permitir o tratamento simultâneo de amostras em duplicata.
Insira uma porção de lã de vidro para reter as partículas da resina. Então, introduza a resina de trocaaniônica básica forte (Nota) suficiente para preencher de 10 a 15 cm da coluna. Lave a coluna com cercade 50 mL de uma solução de NH3 6 mol L�1, seguida por 100 mL de água e 100 mL de HCl 2 mol L�1.Ao final desse ciclo, a vazão deve ser interrompida para que o nível de líquido permaneça cerca de 1 cmacima da resina na coluna. Em nenhum momento permita que o nível de líquido desça abaixo do topoda resina.
NotaAmberlite CG 400 ou uma sua equivalente podem ser utilizadas.
PROCEDIMENTO
Obtenha uma amostra desconhecida que deve conter de 2 a 4 mmol de Ni2� e Zn2� em um balãovolumétrico de 100 mL limpo. Adicione 16 mL de HCl 12 mol L�1, dilua até a marca com água destiladae misture bem. A solução resultante é aproximadamente 2 mol L�1 em ácido. Transfira 10,00 mL daamostra desconhecida diluída para a coluna. Coloque um frasco de erlenmeyer de 250 mL debaixo da co-luna e esgote lentamente até que o nível de líquido esteja um pouco acima da resina. Lave o interior dacoluna com várias porções de 2 a 3 mL de HCl 2 mol L�1; diminua o nível de líquido para perto da super-fície da resina depois de cada lavagem. Faça a eluição do níquel com cerca de 50 mL de HCl 2 mol L�1
em uma vazão de 2 a 3 mL min�1.Faça a eluição do Zn(II) passando cerca de 100 mL de água pela coluna, utilizando a mesma vazão;
colete o líquido em um erlenmeyer de 500 mL.
Titulação do NíquelEvapore a solução contendo o zinco até a secura para eliminar o excesso de HCl. Evite aquecer demais;não permita que o NiCl2 residual se decomponha a NiO. Dissolva o resíduo em 100 mL de água destiladae adicione 10 a 20 mL do tampão pH 10. Adicione 15 gotas do indicador vermelho de bromopirogalol ou0,2 g de murexida. Titule até a mudança da cor (azul para roxo com o vermelho de bromopirogalol, amare-lo para roxo com murexida).
Calcule o número de miligramas de níquel na amostra desconhecida.
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1055

Titulação do ZincoAdicione 10 a 20 mL do tampão pH 10 e uma a duas gotas de negro de Ério T no eluato. Titule com asolução padrão de EDTA até a mudança de coloração de vermelho para azul.
Calcule a massa de zinco na amostra desconhecida em miligramas.
37Q-2 Determinação de Magnésio por Cromatografia de Troca Iônica
Discussão
O hidróxido de magnésio em pastilhas de leite de magnésia pode ser determinado pela dissolução daamostra em uma quantidade mínima de ácido e diluição para um volume conhecido. O excesso de ácido éestabelecido pela titulação de várias alíquotas da amostra diluída com uma base padrão. Outras alíquotassão passadas por uma coluna de troca de cátions, na qual o íon magnésio é retido e trocado na solução poruma quantidade quimicamente equivalente de íons hidrogênio:
Mg2� � 2H S Mg � 2H�
O ácido no eluato é então titulado; a diferença entre o volume de base necessária para as duas séries de ti-tulações é proporcional ao íon magnésio na amostra. Portanto,
mmol H� eluato = 2 � mmol Mg2�
PROCEDIMENTO
Etapa no 1. Prepare aproximadamente 1 L de NaOH 0,05 mol L�1. Padronize essa solução contra porçõespreviamente pesadas do padrão primário de biftalato de potássio seco; utilize amostras comcerca de 0,4 g (com precisão de 0,1 mg) de biftalato de potássio para cada padronização.
Etapa no 2. Anote o número de pastilhas em sua amostra. Transfira as pastilhas para um balão volumétricode 500 mL limpo (Nota 1). Dissolva-as em um volume pequeno de HCl 3 mol L�1 (4 a 5 mLpor pastilha devem ser suficientes). Então dilua até a marca com água destilada.
Etapa no 3. Titule o ácido livre em várias alíquotas de 15,00 mL da amostra dissolvida; a fenolftaleína éum indicador satisfatório.
Etapa no 4. Condicione a coluna com cerca de 15 mL de HCl 3 mol L�1 (Nota 2), seguido por três porçõesde 15 mL de água destilada. Nunca permita que o nível de líquido desça abaixo do topo dorecheio da coluna.
Etapa no 5. Carregue cada coluna com uma alíquota de 15,00 mL da amostra. Faça a eluição a uma vazão decerca de 2 a 3 mL min�1. Lave as colunas com três porções de 15 mL de água. Colete o eluato eas águas de lavagem em um erlenmeyer. Repita essa etapa com outras alíquotas da amostra(Nota 3).
Etapa no 6. Titule as amostras eluídas (e lavadas) com um padrão alcalino. Corrija o volume total paraaquele necessário para titular o ácido livre e calcule a massa de Mg(OH)2 em miligramas emcada pastilha.
Notas1. A amostra será dissolvida mais rapidamente se as pastilhas forem trituradas previamente em um almo-
fariz. Se optar por essa alternativa, você deve saber (a) a massa total de suas pastilhas e (b) a massa daamostra triturada que você vai transferir para o balão volumétrico.
2. O volume das alíquotas da amostra e o volume de base utilizado nas várias titulações devem ser medi-dos cuidadosamente (com precisão de 0,01 mL). Todos os outros volumes podem e devem ser apenasaproximados.
2�res
�res
1056 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON

3. Uma bureta de 25 mL preenchida até uma profundidade de cerca de 15 cm com a resina de trocacatiônica Dowex-50 constitui uma coluna adequada. É recomendado o recondicionamento após quatroou cinco eluições.
37R CROMATOGRAFIA GÁS-LÍQUIDO
Como mostrado no Capítulo 31, a cromatografia gás-líquido permite ao analista separar os componentesde uma mistura complexa. As instruções a seguir referem-se à determinação de etanol em bebidas.
37R-1 Determinação de Etanol em Bebidas por Cromatografia Gasosa15
Discussão
O etanol é convenientemente determinado em soluções aquosas por meio da cromatografia gasosa. O méto-do é facilmente aplicável para medir o teor alcoólico de bebidas.
As instruções de operação referem-se a uma coluna Poropack de 0,64 cm de diâmetro � 0,5 m,preenchida com partículas de 80 a 100 mesh. É necessário um detector de condutividade térmica. (A ioni-zação em chama não é satisfatória em virtude de a sua falta de sensibilidade para a água.)
A determinação baseia-se em uma curva analítica na qual a razão entre a área do pico de etanol e aárea do pico de etanol mais água é colocada em um gráfico em função do volume percentual de etanol:
vol % EtOH � � 100%
Essa relação não é exatamente linear. Pelo menos duas razões podem ser consideradas para explicar a cur-vatura. Na primeira, o detector por condutividade térmica responde linearmente para razões em massa em vez de razões em volume. Na segunda, para concentrações elevadas, os volumes de etanol e água nãosão exatamente complementares como seria necessário para a linearidade. Ou seja,
vol EtOH � vol H2O vol soln
PREPARO DAS SOLUÇÕES
Use uma bureta para medir 10,00; 20,00; 30,00; e 40,00 mL de etanol absoluto em erlenmeyers de 50 mL(Nota). Dilua com água destilada e misture bem.
NotaO coeficiente de expansão térmica para o etanol é aproximadamente cinco vezes maior que o da água. Énecessário, portanto, manter a temperatura das soluções utilizadas nesse experimento a �1 °C durante asmedições do volume.
PROCEDIMENTO
As seguintes condições experimentais têm resultado em cromatogramas satisfatórios para essa determi-nação:
vol EtOH
vol soln
SKOOG, WEST, HOLLER E CROUCH CAP. 37 Métodos Selecionados de Análise 1057
15Adaptado a partir de J. J. Leary. J. Chem. Educ., v. 60, p. 675, 1983.

Temperatura da coluna 100 °CTemperatura do detector 130 °CTemperatura de injeção 120 °CCorrente da ponte 100 mAVazão 60 mL/min�1
Injete uma amostra de 1 L do padrão 20% (v/v) e registre o cromatograma. Obtenha mais cromatogra-mas ajustando a velocidade de registro até que o pico da água apresente uma largura de cerca de 2 mm à meiaaltura. Varie, então, o volume da amostra injetada e a atenuação até que sejam produzidos picos com uma al-tura de pelo menos 40 mm. Obtenha cromatogramas para o restante dos padrões (incluindo água pura e etanolpuro) da mesma maneira. Meça a área de cada pico e faça um gráfico com áreaEtOH/(áreaEtOH � área )em função do volume percentual de etanol.
Obtenha cromatogramas para a amostra desconhecida. Relate o volume percentual de etanol.
H2O
1058 FUNDAMENTOS DE QUÍMICA ANALÍTICA – EDITORA THOMSON
Fichas de Dados de Segurança (MSDS) constituem-se em uma fonte impor-tante para qualquer pessoa que utilize substâncias químicas. Esses docu-mentos fornecem informações essenciais sobre as propriedades e toxici-dade das substâncias químicas que são utilizadas no laboratório. Acessehttp://chemistry.brookscole.com/skoogfac/. A partir do menu ChapterResources, escolha Web Works. Localize as seções do Capítulo 37 e cliqueno link para uma listagem abrangente da maioria das páginas na Internetque oferecem MSDS. Vá para uma dessas páginas e procure o MSDS parao ácido oxálico. Pesquise por todo o MSDS, veja os dados de reatividade,observe suas propriedades químicas e encontre os tratamentos de pri-meiros-socorros para a ingestão do ácido oxálico. Os fabricantes de subs-tâncias químicas são obrigados a fornecer um MSDS para todas as substân-cias que eles vendem, e muitas delas podem ser encontradas na Internet. Éuma boa idéia examinar o MSDS para qualquer substância que você utilizano laboratório.
TRABALHOS NA WEB





























