Embed Size (px)
Citation preview

ISSN 1063-7745, Crystallography Reports, 2007, Vol. 52, No. 6, pp. 1025–1029. © Pleiades Publishing, Inc., 2007.Original Russian Text © A.N. Rudenko, V.G. Mazurenko, 2007, published in Kristallografiya, 2007, Vol. 52, No. 6, pp. 1062–1066.
1025
INTRODUCTION
Currently, information about the vibrational spectraof quasicrystals is very poor and mainly based on theresults of experimental investigations. There have beenonly two theoretical studies devoted to the analysis ofthe vibrational spectra of quasicrystals. In one of them,the general properties of the vibrational behavior ofquasicrystalline objects were discussed for the exampleof a 1D quasi-periodic chain [1]. In the other study, theresults of calculations of the vibrational spectra andsome thermodynamic properties of a number of 2Dquasicrystals of octagonal and decagonal types werereported [2]. Currently, theoretical investigations of thevibrational properties of icosahedral quasicrystals areabsent. The information about the vibrational spectra ofquasicrystals is necessary for understanding the pro-cesses occurring in quasicrystals, conditions of theirformation, and stability as well as for predicting otherproperties.
The purpose of this study is to calculate the vibra-tional spectra of an icosahedral quasicrystal
Al
0.62
Cu
0.255
Fe
0.125
and compare them with the experi-mental neutron inelastic scattering data [3].
We can separate the next stages in calculation of thevibrational spectra of quasicrystals:
(i) simulation of the atomic structure of a quasicrys-tal;
(ii) obtainment of the parameters of interatomicinteraction; and
(iii) calculation of the vibrational spectra.
STRUCTURAL MODEL OF AN i-AlCuFe QUASICRYSTAL
Simulation of the atomic structure of a quasicrystalcan be divided into two stages:
(i) construction of the geometric structure of aquasi-periodic lattice and
(ii) decoration of the obtained lattice with atomsaccording to the chemical composition of the sampleunder consideration.
In construction of the atomic structure of ani-AlCuFe quasicrystal, we used the model of a crystal-line 1/1 approximant proposed in [4]. Within thismodel, a quasicrystal is a periodic crystal structure witha unit cell containing 128 atoms with the lattice param-eter
a
= 12.3
Å. The unit cell of such an approximant isshown in Fig. 1.
Within this model, the geometric structure was con-structed on the basis of the method of projection of a 6Dfcc lattice with the parameter
a
= 4.465
Å on the realspace [5] and the lattice obtained was decorated withatoms on the basis of the experimental X-ray diffractiondata on the i-AlCuFe quasicrystal.
MODEL OF INTERATOMIC INTERACTION
To describe the interatomic interaction in a quasi-crystal, we used the EAM semiempirical model [6].Within this model, the lattice energy is represented as asum of two terms:
(1)
where the first term corresponds to pair interatomicinteractions and the second is the lattice energy,
E12--- ϕ rij( )
i j≠∑ F ρi( ),
i
∑+=
QUASICRYSTALS
Calculation of Vibrational Spectra of an Icosahedral Quasicrystal AlCuFe
A. N. Rudenko and V. G. Mazurenko
Ural State University, pr. Lenina 51, Yekaterinburg, 620083 Russia
e-mail: [email protected]
Received September 14, 2006
Abstract
—Vibrational spectra of an icosahedral quasicrystal AlCuFe have been calculated on the basis of acrystalline 1/1 approximant by the recurrence method. To describe the interaction of atoms in a quasicrystal,the semiempirical EAM model was used. It is shown that the calculated spectra are in satisfactory agreementwith the experimental neutron inelastic scattering data.
PACS numbers: 61.44.Br, 63.20.-e, 07.05.Tp
DOI:
10.1134/S1063774507060156
1026
CRYSTALLOGRAPHY REPORTS
Vol. 52
No. 6
2007
RUDENKO, MAZURENKO
which is related to the interaction between electrondensities.
As the potential of pair interatomic interaction andelectron density, we used the functions proposed in [7,8]. In particular, the pair potential was chosen in theform of a sixth-order polynomial:
(2)
The single-electron density is taken in the form of rap-idly damping oscillating function:
ϕ r( ) r rm–( )4d0 d1r d2r
2d3r
3+ + +[=
+ d4r4
d5r5
d6r6
+ + ].
Al
Cu
Fe
0.25
0.20
0.15
0.10
0.05
0
–0.052018141210 1686420
f
,
THz
g
,
arb. units
Fig. 1.
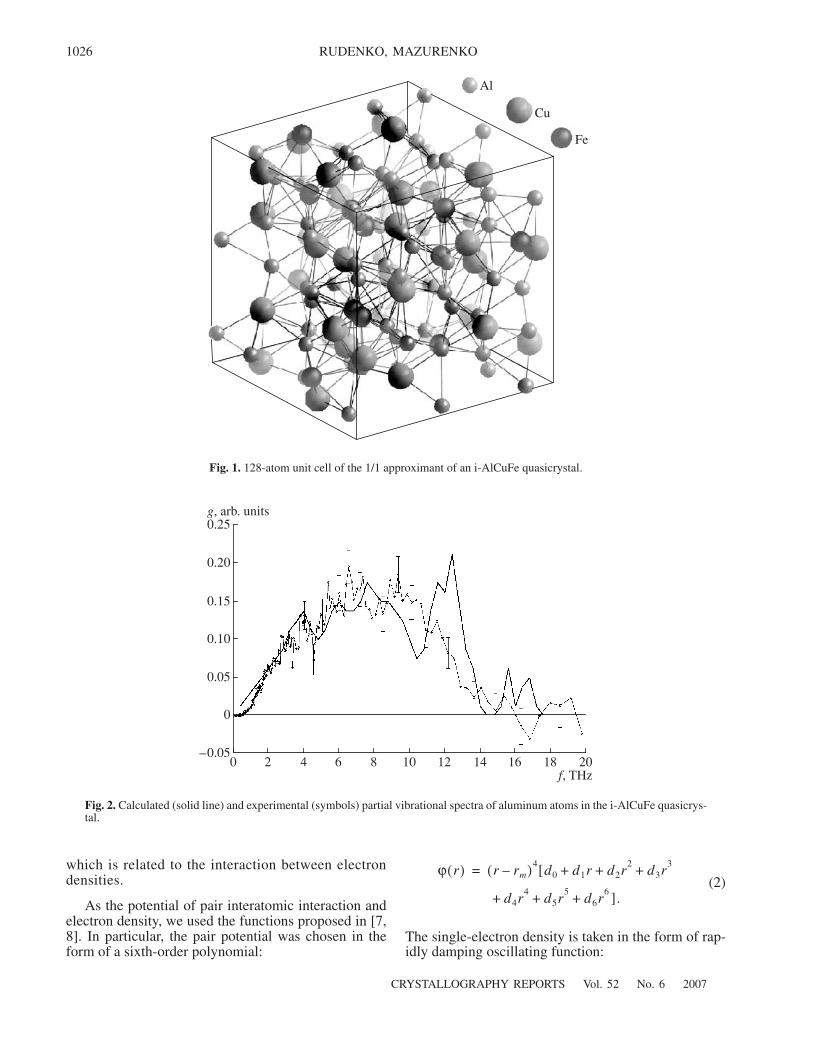
128-atom unit cell of the 1/1 approximant of an i-AlCuFe quasicrystal.
Fig. 2.
Calculated (solid line) and experimental (symbols) partial vibrational spectra of aluminum atoms in the i-AlCuFe quasicrys-tal.

CRYSTALLOGRAPHY REPORTS
Vol. 52
No. 6
2007
CALCULATION OF VIBRATIONAL SPECTRA 1027
(3)
The electron-density functional
F
(
ρ
)
was found fromthe universal state equation [9].
Fitting of the parameters and determination of theexact form of the functions were performed on the basisof empirical data, such as the moduli of elasticity of thesecond and third orders and the vacancy-formationenergies for pure fcc, fcc, and bcc crystals of alumi-num, copper, and iron, respectively.
According to the EAM model data, we calculatedthe dynamic matrix
D
αβ
(
ij
)
for a spherical cluster withthe radius
R
= 41.21
Å, composed of 20196 atoms.
f r( )1 a1 αr( )cos a2 αr( )sin+ +
rβ------------------------------------------------------------------.=
CALCULATIONOF THE VIBRATIONAL SPECTRUM
To determine the vibrational eigenfrequencies, it isnecessary to solve the equation
(4)
However, in direct space, solution of such an equationrequires a very long computation time in view of thehigh order of the dynamic matrix. Therefore, to deter-mine the vibrational spectra, we used the recurrencemethod, whose main advantage is that it does notrequire complete diagonalization of the dynamic matrixfor determining the vibrational frequencies.
det Dαβ ij( ) δαβδijωs–( ) 0.=
0.16
0.14
0.12
0.10
0.08
0.06
0.04
0.02
0
–0.0220181614121086420
f
,
THz
g
,
arb. units
0.06
0.05
0.04
0.03
0.02
0.01
0
–0.01
g
,
arb. units
20181614121086420
f
,
THz
Fig. 3.
Calculated (solid line) and experimental (symbols) partial vibrational spectra of copper atoms in the i-AlCuFe quasicrystal.
Fig. 4.
Calculated (solid line) and experimental (symbols) partial vibrational spectra of iron atoms in the i-AlCuFe quasicrystal.
1028
CRYSTALLOGRAPHY REPORTS
Vol. 52
No. 6
2007
RUDENKO, MAZURENKO
In the recurrence method, the total density of vibra-tional states is the sum of local densities of vibrationalstates over all atoms and directions in the cluster underconsideration:
(5)
The sum of local densities of vibrational states isexpressed in terms of the Fourier transform of Green’sfunction:
(6)
which, in turn, can be represented as a continuous frac-tion with the recurrence coefficients {
a
n
,
b
n
}:
(7)
The recurrence coefficients are calculated by construct-ing the so-called Lanczos basis of orthogonal vectors
u
n
from the recurrence relation
(8)
where the recurrence coefficients are expressed in theform
(9)
From these relations, 16 exact pairs of recurrence coef-ficients {
a
n
,
b
n
} were calculated; then, using the extrap-olation procedure, we determined 50 more pairs ofcoefficients and their asymptotic values. The number of
g ω( ) 1n--- giα ω( ).
iα
n
∑=
giα ω( ) 2ωπ
-------Im Guiαω( ),–=
Gu ω( ) 1
ω2a1–
b1
ω2a2–
b2
bn 1–
ω2an– bn–
------------------------------------------------------------------------------------–
----------------------------------------------------–---------------------------------------------------------------------------.=
…
un 1+ D an–( )un bn 1– un 1– ,–=
an
unTDun
unTun
----------------, bn 1–
unTun
un 1–T
un 1–
----------------------.= =
recurrence coefficients was determined according tothe sizes of the dynamic matrix and the specified fre-quency calculation step. From the found recurrencecoefficients, we calculated the local densities of vibra-tional states for each atom and direction from a chosencluster and then the partial densities and the total den-sity of vibrational states of the cluster. The calculationstep for the local densities and partial densities and thetotal density of vibrational states was chosen to be0.4 THz.
RESULTS OF THE CALCULATIONS
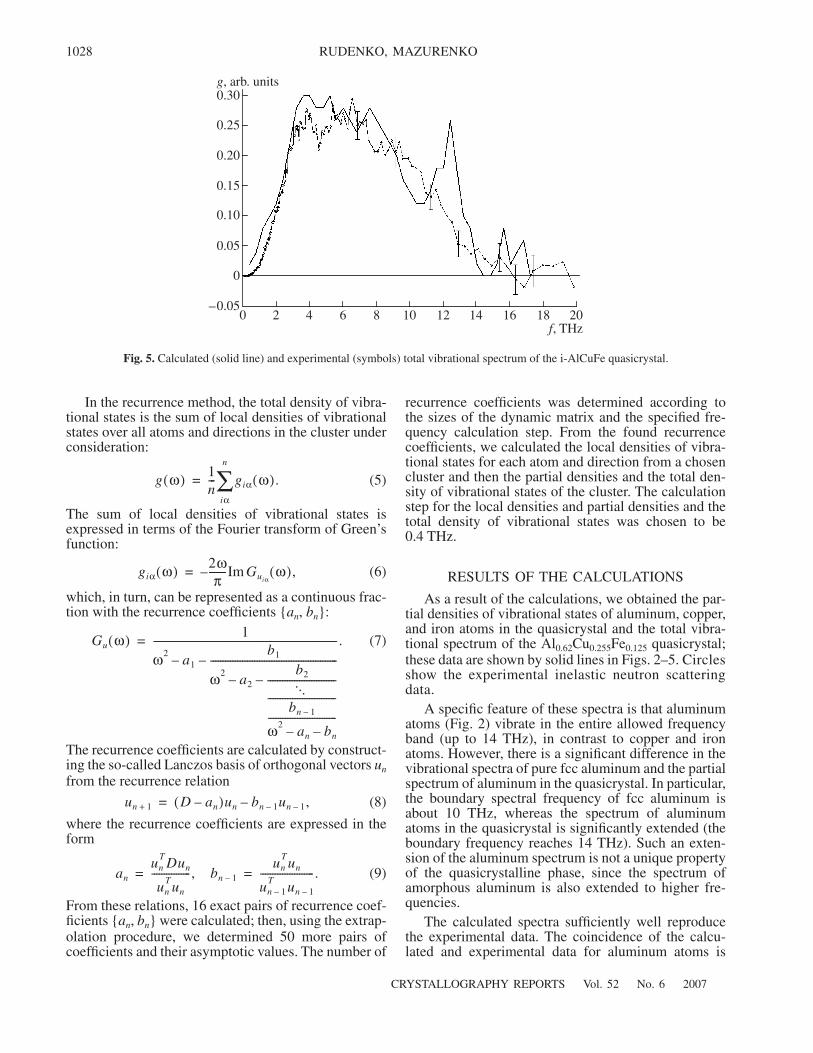
As a result of the calculations, we obtained the par-tial densities of vibrational states of aluminum, copper,and iron atoms in the quasicrystal and the total vibra-tional spectrum of the Al0.62Cu0.255Fe0.125 quasicrystal;these data are shown by solid lines in Figs. 2–5. Circlesshow the experimental inelastic neutron scatteringdata.
A specific feature of these spectra is that aluminumatoms (Fig. 2) vibrate in the entire allowed frequencyband (up to 14 THz), in contrast to copper and ironatoms. However, there is a significant difference in thevibrational spectra of pure fcc aluminum and the partialspectrum of aluminum in the quasicrystal. In particular,the boundary spectral frequency of fcc aluminum isabout 10 THz, whereas the spectrum of aluminumatoms in the quasicrystal is significantly extended (theboundary frequency reaches 14 THz). Such an exten-sion of the aluminum spectrum is not a unique propertyof the quasicrystalline phase, since the spectrum ofamorphous aluminum is also extended to higher fre-quencies.
The calculated spectra sufficiently well reproducethe experimental data. The coincidence of the calcu-lated and experimental data for aluminum atoms is
0.30
0.25
0.20
0.15
0.10
0.05
0
–0.05
g, arb. units
20181614121086420f, THz
Fig. 5. Calculated (solid line) and experimental (symbols) total vibrational spectrum of the i-AlCuFe quasicrystal.

CRYSTALLOGRAPHY REPORTS Vol. 52 No. 6 2007
CALCULATION OF VIBRATIONAL SPECTRA 1029
especially pronounced below 9 THz. Above 9 THz,some deviations of the calculated data from the experi-mental results are observed. However, the behavior ofthe calculated and experimental data correlates well inthis part of the spectrum. For copper (Fig. 3) and iron(Fig. 4) atoms, an expansion of the vibrational spec-trum of the quasicrystal in comparison with ideal crys-tals of pure elements is observed also. The strongestdeviation of the total spectrum of the quasicrystal(Fig. 5) from the experimental one is observed in therange from 12 to 13 THz in the form of an additionalpeak. Its appearance is likely due to the contribution ofvibrations of aluminum atoms.
On the whole, the obtained calculated spectra are insatisfactory agreement with the experimental data. Thedifference between them is mainly due to the approxi-mations chosen in the simulation of the atomic struc-ture of a quasicrystal and description of the interatomicinteraction.
CONCLUSIONSThe partial vibrational spectra of aluminum, copper,
and iron atoms in an Al0.62Cu0.255Fe0.125 icosahedralquasicrystal and the total vibrational spectrum of thequasicrystal have been calculated for the first time onthe basis of realistic model representations about theatomic structure and interatomic interaction. The spec-tra obtained are in satisfactory agreement with theexperimental vibrational spectrum of the quasicrystal.Hence, we can conclude that the models used are appli-cable in the first-order approximation.
We believe that the deviations between the calcu-lated and experimental data are primarily due to theapproximations used in the simulation of the atomicstructure of the quasicrystal (the lowest possible degreeof quasicrystal approximation and the semiempiricalcharacter of the interatomic interaction model) as wellas to the error of the recurrence method used to calcu-late the vibrational spectra.
The most promising calculation methods for obtain-ing more exact vibrational spectra of quasicrystals arethe following ones:
The most modern ab initio method, based on theperturbation theory of density functional (the methodof linear response [10]) with description of the inter-atomic interaction on the basis of pseudopotentials.
This method makes it possible to deal with crystallineapproximants in the reciprocal space, which simplifiesthe problem of calculation of the vibration frequencies;however, application of this method meets difficultiesin the case of high-order approximants.
The molecular-dynamics method [11], in which thevibrational spectrum is the Fourier transform of theautocorrelation function of the velocity of the systemunder study. This method makes it possible to considerapproximants of arbitrarily high orders. Its advantage isthat it allows consideration of anharmonic effects andphason dynamics of quasicrystals and makes it possibleto consider a system at temperatures different fromabsolute zero. A drawback of this method is that it actu-ally does not allow study of large systems by ab initiomethods.
REFERENCES
1. T. Janssen, in Quasicrystals: An Introduction to Structure,Physical Properties, and Applications, Ed. by J.-B. Suck,M. Schreiber, and P. Haussler (Springer Ser. Mater. Sci.,2002), Vol. 55, p. 423.
2. H. Elhor, Dissertation (Tech. Univ. Chemnitz, 2003).3. P. P. Parshin, M. G. Zemlyanov, A. V. Mashkov, et al.,
Fiz. Tverd. Tela (St. Petersburg) 46 (3), 510 (2004)[Phys. Solid State 46 (3), 526 (2004)].
4. E. Cockayne, R. Phillips, X. B. Kan, et al., J. Non-Cryst.Solids 153–154, 140 (1993).
5. M. Cornier-Quiquandon, A. Quivy, S. Lefebvre, et al.,Phys. Rev. B: Condens. Matter Mater. Phys. 44, 2071(1991).
6. M. S. Daw and M. I. Baskes, Phys. Rev. B: Condens.Matter Mater. Phys. 29, 6443 (1984).
7. S. Chantasiriwan and F. Milstein, Phys. Rev. B: Con-dens. Matter Mater. Phys. 53, 14082 (1996).
8. S. Chantasiriwan and F. Milstein, Phys. Rev. B: Con-dens. Matter Mater. Phys. 58, 5996 (1998).
9. J. H. Rose, J. R. Smith, F. Guinea, et al., Phys. Rev. B:Condens. Matter Mater. Phys. 29, 2963 (1984).
10. S. Baroni, S. de Gironcoli, and A. Dal Corso, Rev. Mod.Phys. 73, 516 (2001).
11. F. Ercolessi, A Molecular Dynamics Primer (Spring Col-lege Computat. Phys., ICTP, Trieste, 1997), p. 49.
Translated by A. Madonov



























![A complicated quasicrystal approximant [epsilon]16 predicted by](https://img.dokumen.tips/doc/110x75/613d3a34736caf36b75ad4c8/a-complicated-quasicrystal-approximant-epsilon16-predicted-by.jpg)

