Embed Size (px)
DESCRIPTION
no description
Citation preview

This journal is c the Owner Societies 2010 Phys. Chem. Chem. Phys., 2010, 12, 11015–11020 11015
Redox behavior of small metal clusters with respect to hydrogen.
The effect of the cluster charge from density functional resultsw
Galina P. Petrova,a Georgi N. Vayssilov*a and Notker Rosch*b
Received 16th March 2010, Accepted 10th May 2010
DOI: 10.1039/c004377j
Tetrahedral model iridium species [Ir4Hn]q+ of different charge and hydrogen loading
were described at the density functional level. The energy of dissociative adsorption of hydrogen
was calculated to vary in the small interval from �63 kJ mol�1 to �77 kJ mol�1 (per H atom).
Adsorption of hydrogen on Ir4 and Ir4+ induces an oxidation of the metal moiety, whereas the
highly charged cluster Ir43+ is reduced upon hydrogen adsorption. The ligand shell acts as charge
buffer as the metal moieties of the complexes [Ir4H12]q+ with maximum hydrogen loading carry
very similar effective charges, irrespective of the total charge q. Similar effects were confirmed
to occur on small clusters of other 4d and 5d transition metals.
1. Introduction
Small transition metal clusters in the gas phase or supported
on a metal-oxide surface have been thoroughly studied by
both experimental1–6 and computational techniques.1,7–9 Such
systems have been found useful in various catalytic or sorption
processes as supported species1–3 or directly in the gas phase as
neutral and charged clusters.1,4,10 The interaction of such
clusters with molecular hydrogen is particularly important,
for characterizing the properties of the clusters and their
application as catalysts.5,6 Therefore, we previously explored
computationally the dissociative adsorption of H2 from the
gas phase on isolated and zeolite-supported tetrairidium
clusters.11–13 These model studies showed both supported metal
clusters and metal clusters in the gas phase to adsorb up to
12 hydride ligands, i.e. 3 H ligands per Ir atom. Hydrogenated
Ir4 clusters adsorbed on a dehydroxylated zeolite support were
calculated to be the most stable. The formal charge of such
Ir4Hn moieties is 3 e as a result of reverse proton spillover from
bridging hydroxyl groups of the zeolite fragment. Yet, calcu-
lated charges of the Ir4Hn moiety were estimated to be notably
smaller, 1.07–1.64 e.13 From a comparison of the results for
supported clusters with those for neutral model clusters in the
gas phase, one expects the (effective) charge of the cluster to
have a notable influence on the properties of the hydrogenated
metal clusters.
To explore this effect of the cluster charge on the hydrogen
loading, we extended our studies on tetrairidium species to a
series of cationic species [Ir4Hn]q+ with total charges q= 1–3 e.
The highest value of q corresponds to the formal charge of the
zeolite-supported Ir4Hn moiety, while the value at the low end,
1 e, is close to the estimated charges of the supported species, as
just mentioned.13 In the present work, we show that this
variation by only of a few electrons on the whole cluster
changes the direction of the oxidation: the metal moiety is
oxidized for neutral systems or clusters with q = 1 e, but the
hydrogen ligands are oxidized when the cluster carries a charge
q = 3 e. Thus, the charge of the system strongly affects the
direction of the oxidation or reduction during hydrogen loading
of the metal cluster. These effects, first observed for iridium
clusters, also have been confirmed for hydrogenated clusters of
other 4d (Ru, Rh, Pd) and 5d (Os, Pt) transition metals.
2. Method and models
The electronic structure calculations were carried out with the
linear combination of Gaussian-type orbitals fitting-functions
density functional method (LCGTO-FF-DF)14,15 as imple-
mented in the program PARAGAUSS.16,17 We employed the
gradient-corrected exchange-correlation functional suggested
by Becke (exchange) and Perdew (correlation) (BP).18 We
applied a scalar relativistic variant of the LCGTO-FF-DF
method that affords an explicit description of relativistic
effects by treating all electrons with the Douglas–Kroll–Hess
approach of second order.15,19,20 The Kohn–Sham (KS)
orbitals were represented by flexible Gaussian-type basis
sets, contracted in generalized form: (6s1p) - [4s1p] for
H,21 (18s13p9d) - [7s6p4d] for Ru, Rh and Pd,22,23 and
(21s17p12d7f) - [9s8p6d4f] for Ir, Os, and Pt.22,23 The
auxiliary basis set, used in the LCGTO-FF-DF method to
represent the Hartree part of the electron–electron interaction,
was derived from the orbital basis set in a standard fashion.14
On each center (except H), this set was augmented by five
p- and five d-type polarization exponents, constructed as geometric
series with a factor 2.5, starting from 0.1 au (p) or 0.2 au (d). Only
the p-type series was added at hydrogen centers.
The current study comprises four series of hydrogenated
clusters in the gas phase with net charges q = 0–3 e (e denotes
a Faculty of Chemistry, University of Sofia, 1126 Sofia,Bulgaria. E-mail: [email protected]
bDepartment Chemie and Catalysis Research Center,Technische Universitat Munchen, 85747 Garching, Germany.E-mail: [email protected]
w Electronic supplementary information (ESI) available: Table withcalculated energy characteristics and interatomic distances of theoptimized structures [Ir4Hn]
q+ (q = 0–3; n = 0, 3, 6, 9, 12); tablewith various characteristics of the electronic structure of the clustersmodeled; figure showing density of state plots of the Ir 2p core levels asa function of the cluster charge and the hydrogen loading; figure withthe optimized structures of [M4Hn]
q+ species (M = Ru, Rh, Pd, Os,Pt); figure with variations of the average shifts of the M 2p levels in theclusters [M4H12]
q+ for various charges q. See DOI: 10.1039/c004377j
PAPER www.rsc.org/pccp | Physical Chemistry Chemical Physics
Publ
ishe
d on
29
July
201
0. D
ownl
oade
d by
UN
IVE
RSI
TY
OF
NE
BR
ASK
A o
n 15
/04/
2015
02:
34:1
3.
View Article Online / Journal Homepage / Table of Contents for this issue

11016 Phys. Chem. Chem. Phys., 2010, 12, 11015–11020 This journal is c the Owner Societies 2010
the elementary charge) of the complexes: Ir4Hn, [Ir4Hn]+,
[Ir4Hn]2+, and [Ir4Hn]
3+. Unlike in our previous study,13 we
did not apply any symmetry constraints during the structure
optimization. We considered the consecutive dissociative
adsorption of (formally) 3/2 H2 to the species Ir4Hn and in
this way generated structures that contain n = 0, 3, 6, 9, or
12 hydrogen ligands.13,24 The initial positions of the ligands
were chosen similar to those reported earlier for the zeolite-
supported hydrogenated moieties.13 Each stationary point was
checked with a normal mode analysis that incorporated all
degrees of freedom to ensure that the reported structures
correspond to local minima. In the spirit of this model study,
we did not search for isomeric structures.
We quantified the stability of a cluster [Ir4Hn]q+ via the
energy change DE of the formal reaction [Ir4]q+ + n/2H2 -
[Ir4Hn]q+, i.e. via its relative energies with respect to the
corresponding bare cluster [Ir4]q+:
DE([Ir4Hn]q+)=Etot([Ir4Hn]
q+)� Etot([Ir4]q+)� n/2Etot(H2)
(1)
Etot is the total energy of a system. A negative value of
DE reflects the favorable formation of [Ir4Hn]q+ from the bare
cluster after dissociative adsorption of the proper amount of
hydrogen. The basis set superposition error was estimated via
the counterpoise method to 3–6% of DE. The adiabatic ioni-
zation potentials of the bare, [Ir4]q+, and the hydrogenated
clusters, [Ir4Hn]q+ (q = 0–2), were calculated as differences of
appropriate total energies Etot. The spin contamination of the
KS determinant of open-shell systems never exceeded 2%.
Experimental core level energies cannot directly be com-
pared with energies of KS orbitals, but changes of KS energies
with respect to a reference provide adequately approximate
core level shifts.25 We estimated average energy shifts of the
Ir 4f shell of [Ir4Hn]q+ clusters, using the Ir 4f KS energies of
the bare tetrahedral clusters [Ir4]q+ as reference. A positive
value of the shift corresponds to a stabilization of the core
levels relative to the reference. We determined effective charges
q of metal moiety Ir4 by fitting the electrostatic potential.26
The quoted charges should be taken with due caution as
Kohn–Sham methods with common exchange-correlation
functionals tend to overestimate electron delocalization.27
To probe the general validity of our analysis, we also
modeled [M4Hn]q+ clusters, q = 0–3, of other 4d and 5d late
transition metals in the same way, but only for the loadings
n = 0, 6, and 12.
3. Results and discussion
3.1 Structures and stability
The optimized structures of the clusters [Ir4Hn]q+ are provided
in Fig. 1 The average values hIr–Iri and the spread DR of the
nearest-neighbor distances in the metal moiety are given in
Table 1. As in the case of zeolite-supported species,13 the
average Ir–Ir distance increases with the number of H ligands
coordinated at the cluster, from 247� 1 pm in the bare clusters
to 268 � 2 pm in the clusters with 12 H ligands. These averages
include clusters of all charges studied. The nearest-neighbor
Ir–Ir distances are not uniform within a cluster, as illustrated
by rather large values DR, up to B40 pm (Table 1). The
clusters [Ir4H9]3+ and [Ir4H12]
3+ exhibit a ‘‘butterfly’’ struc-
ture with one of the Ir–Ir distances elongated up to 308 pm.
The terminal H–Ir bonds, 158–163 pm, vary by a few
picometres only as the charge of the cluster changes. In
clusters with a large hydrogen loading, n = 9, 12, and a high
charge, q = 2 e or 3 e, activated H2 molecules, with inter-
atomic distances less than 94 pm,28 form at one of the Ir centers.
Such paired hydrogen ligands feature longer H–Ir bonds,
169–171 pm, than common terminal H ligands.
The relative energies DE of the hydrogenated species are
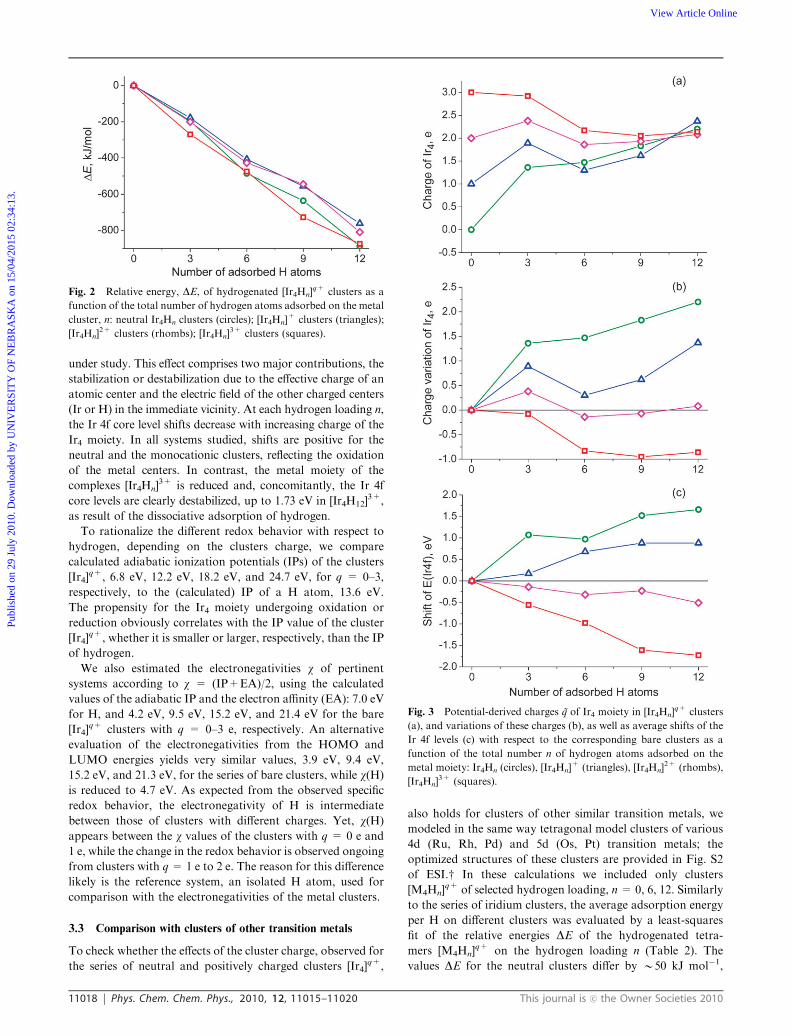
provided in Table 1 and compared in Fig. 2 as a function of
the number n of hydrogen ligands of the clusters. Similarly to
the data for the neutral tetrairidium clusters in the gas phase,13
the relative energies of the charged clusters [Ir4Hn]q+ increase
(by absolute value) almost linearly with the hydrogen loading
n along each series of clusters with a fixed charge q (Fig. 2).
The dissociative adsorption of H2 releases, on average,
D2E = 70 � 20 kJ mol�1 per H atom, but the variations
within each series at fixed q clusters are notably smaller, at
most �8 kJ mol�1. Hydrogen adsorption is calculated to be
most favorable on complexes with q = 3 e and on neutral
clusters, where D2E = �77 kJ mol�1 and �73 kJ mol�1,
respectively. The value for the neutral clusters is close to the
adsorption energy, �70 kJ mol�1, calculated for models with
C3 symmetry restrictions,13 but smaller than the corresponding
energies calculated for the neutral clusters Ir4H and Ir4H2 of
similar structure,�75 kJ mol�1 and�90 kJ mol�1, respectively.11
The corresponding adsorption energies for the clusters
[Ir4Hn]+ and [Ir4Hn]
2+ areB10 kJ mol�1 smaller,�63 kJ mol�1
and �66 kJ mol�1, respectively. This may be related to the
stronger redox interactions in complexes with charges q=0, 3 e,
compared to those with q = 1, 2 e (section 3.2). In summary, H
adsorption is quite favorable in all systems studied.11,13
3.2 Electron density distribution
Now, we turn to the central part of this work, the analysis of
the electron density distribution and of the (first) ionization
potentials of hydrogenated metal clusters of different charge.
Metal moieties of zeolite-supported tetrairidium clusters and
of neutral clusters in the gas phase are oxidized after adsorp-
tion of hydrogen ligands and the ligands carry a partial
negative charge.11,13 In the following we will discuss the
changes in the electron density distribution as the hydrogen
loading and the cluster charge increase. We will diagnose
these changes via potential-derived charges as well as average
shifts of selected Ir core levels relative to the energies of
the corresponding bare cluster (Table 1, Fig. 3, and Fig. S1
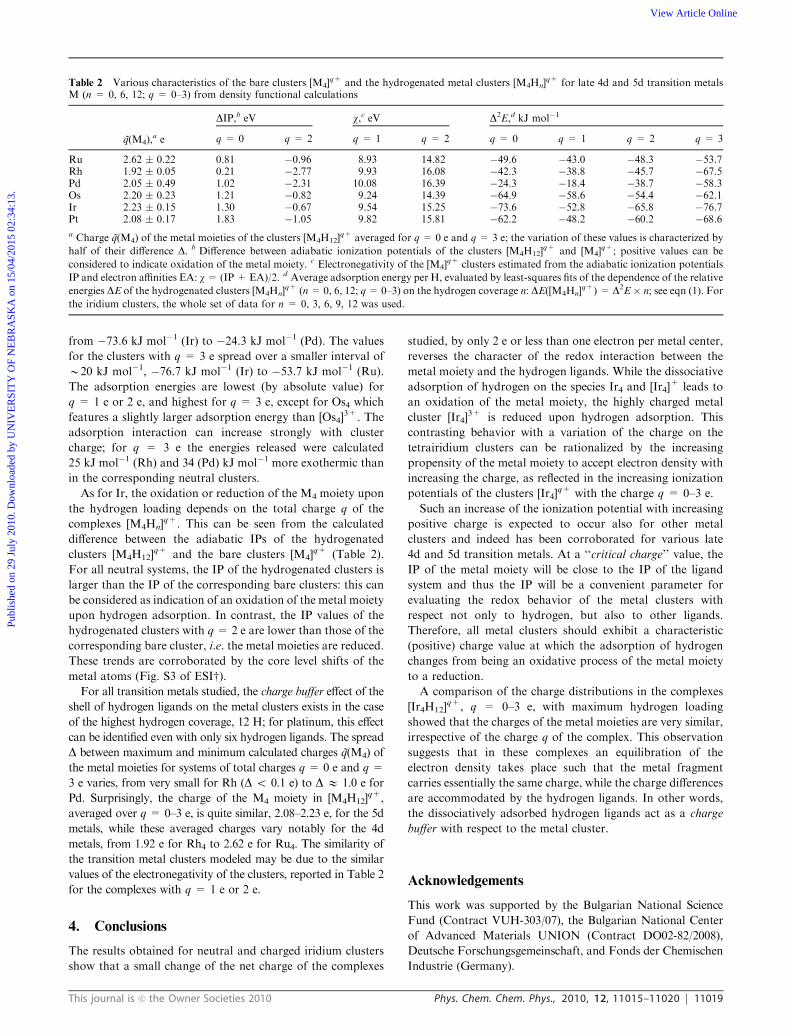
of ESIw).In the hydrogenated clusters Ir4Hn and [Ir4Hn]
+ (n 4 0) the
charge of the metal moiety is above the total charge q of the
system (Fig. 3b). Thus the metal moieties are oxidized through
the adsorption of hydrogen, similar to our earlier results for
zeolite-supported Ir4 clusters.13 The effect increases with the
hydrogen loading. At maximum hydrogen loading, n = 12,
the charge of the Ir4 moiety is 2.20 e in the neutral complex
and 2.37 e in the corresponding monocation. In the corres-
ponding dicationic hydrogenated complexes the charge
Publ
ishe
d on
29
July
201
0. D
ownl
oade
d by
UN
IVE
RSI
TY
OF
NE
BR
ASK
A o
n 15
/04/
2015
02:
34:1
3.
View Article Online

This journal is c the Owner Societies 2010 Phys. Chem. Chem. Phys., 2010, 12, 11015–11020 11017
alterations of the Ir4 moiety are quite small, from only �0.14 eto 0.38 e. On the other hand, the dissociative adsorption of
hydrogen on iridium tetramers of charge q = 3 e results in a
reduction of the metal moiety, as the positive charge of the
iridium moiety in the hydrogenated clusters is always below
the total charge q of the system. In [Ir4H12]3+ the charge of the
metal moiety is by 0.86 e lower than the charge of the bare
cluster, 3 e.
Interestingly, with increasing hydrogen loading, the
charges of the metal moiety approach one and the same value,
2.23 � 0.15 e (n = 12), independently of the net charge
q = 0–3 e of the complex (Fig. 3a). A Mulliken analysis
(see ESIw) reveals the same trend: here, the charge of the metal
moiety gradually decreases to a common value, 0.67 � 0.23 e,
in the clusters with the highest hydrogen loading. Thus, a shell
of adsorbed hydrogen ligands acts as a ‘‘charge buffer’’ with
respect to the metal cluster.
These charge effects are reflected by the shifts of the metal
core levels as a comparison of Fig. 3b and c reveals for the
example of the Ir 4f levels.29 Although core level shifts in
general are the compound result of several factors,30 the
charge of the metal moiety clearly dominates for the systems
Fig. 1 Optimized structures of [Ir4Hn]q+ species for q = 0, 1, 2, and 3 and energy changes for the subsequent adsorption of 3/2 H2 (in kJ mol�1).
Table 1 Various characteristics of the bare clusters [Ir4]q+ and the hydrogenated clusters [Ir4Hn]
q+ (n = 0, 3, 6, 9, 12; q = 0–3) from densityfunctional calculations
Nsa DE,b kJ mol�1 hIr–Iri,c pm DR,d pm q(Ir4),e e DE(Ir 4f),f eV
Ir4 0 — 248 1 0 —[Ir4]
+ 1 — 246 9 1 —[Ir4]
2+ 2 — 247 14 2 —[Ir4]
3+ 3 — 247 17 3 —Ir4H3 3 �196 252 17 1.36 1.07[Ir4H3]
+ 2 �179 252 14 1.89 0.17[Ir4H3]
2+ 1 �201 251 12 2.38 �0.14[Ir4H3]
3+ 0 �270 251 3 2.47 �0.56Ir4H6 0 �487 256 24 1.47 0.97[Ir4H6]
+ 1 �408 256 28 1.30 0.68[Ir4H6]
2+ 0 �427 255 26 1.86 �0.32[Ir4H6]
3+ 1 �475 258 38 2.17 �0.97Ir4H9 1 �637 262 32 1.83 1.52[Ir4H9]
+ 0 �555 262 38 1.62 0.89[Ir4H9]
2+ 1 �544 261 50 1.93 �0.23[Ir4H9]
3+ 0 �727 257g 60 2.05 �1.61Ir4H12 0 �888 267 7 2.20 1.67[Ir4H12]
+ 1 �761 270 14 2.37 0.88[Ir4H12]
2+ 0 �809 268 28 2.08 �0.50[Ir4H12]
3+ 1 �874 266g 42 2.14 �1.72a Number of unpaired electrons in the complex. b Relative stability of the hydrogenated cluster, see eqn (1). c Average nearest-neighbor distance of
the metal moiety. d Difference between the largest and the smallest nearest-neighbor distances of the optimized structure. e Potential-derived
charge of the metal moiety q(Ir4).f Average shift of the Ir 4f core levels with respect to the value of the corresponding bare cluster; positive values
can be considered to indicate oxidation of the metal moiety. g Distances exceeding 290 pm are not included when calculating the average.
Publ
ishe
d on
29
July
201
0. D
ownl
oade
d by
UN
IVE
RSI
TY
OF
NE
BR
ASK
A o
n 15
/04/
2015
02:
34:1
3.
View Article Online
11018 Phys. Chem. Chem. Phys., 2010, 12, 11015–11020 This journal is c the Owner Societies 2010
under study. This effect comprises two major contributions, the
stabilization or destabilization due to the effective charge of an
atomic center and the electric field of the other charged centers
(Ir or H) in the immediate vicinity. At each hydrogen loading n,
the Ir 4f core level shifts decrease with increasing charge of the
Ir4 moiety. In all systems studied, shifts are positive for the
neutral and the monocationic clusters, reflecting the oxidation
of the metal centers. In contrast, the metal moiety of the
complexes [Ir4Hn]3+ is reduced and, concomitantly, the Ir 4f
core levels are clearly destabilized, up to 1.73 eV in [Ir4H12]3+,
as result of the dissociative adsorption of hydrogen.
To rationalize the different redox behavior with respect to
hydrogen, depending on the clusters charge, we compare
calculated adiabatic ionization potentials (IPs) of the clusters
[Ir4]q+, 6.8 eV, 12.2 eV, 18.2 eV, and 24.7 eV, for q = 0–3,
respectively, to the (calculated) IP of a H atom, 13.6 eV.
The propensity for the Ir4 moiety undergoing oxidation or
reduction obviously correlates with the IP value of the cluster
[Ir4]q+, whether it is smaller or larger, respectively, than the IP
of hydrogen.
We also estimated the electronegativities w of pertinent
systems according to w = (IP+EA)/2, using the calculated
values of the adiabatic IP and the electron affinity (EA): 7.0 eV
for H, and 4.2 eV, 9.5 eV, 15.2 eV, and 21.4 eV for the bare
[Ir4]q+ clusters with q = 0–3 e, respectively. An alternative
evaluation of the electronegativities from the HOMO and
LUMO energies yields very similar values, 3.9 eV, 9.4 eV,
15.2 eV, and 21.3 eV, for the series of bare clusters, while w(H)
is reduced to 4.7 eV. As expected from the observed specific
redox behavior, the electronegativity of H is intermediate
between those of clusters with different charges. Yet, w(H)
appears between the w values of the clusters with q = 0 e and
1 e, while the change in the redox behavior is observed ongoing
from clusters with q= 1 e to 2 e. The reason for this difference
likely is the reference system, an isolated H atom, used for
comparison with the electronegativities of the metal clusters.
3.3 Comparison with clusters of other transition metals
To check whether the effects of the cluster charge, observed for
the series of neutral and positively charged clusters [Ir4]q+,
also holds for clusters of other similar transition metals, we
modeled in the same way tetragonal model clusters of various
4d (Ru, Rh, Pd) and 5d (Os, Pt) transition metals; the
optimized structures of these clusters are provided in Fig. S2
of ESI.w In these calculations we included only clusters
[M4Hn]q+ of selected hydrogen loading, n= 0, 6, 12. Similarly
to the series of iridium clusters, the average adsorption energy
per H on different clusters was evaluated by a least-squares
fit of the relative energies DE of the hydrogenated tetra-
mers [M4Hn]q+ on the hydrogen loading n (Table 2). The
values DE for the neutral clusters differ by B50 kJ mol�1,
Fig. 2 Relative energy, DE, of hydrogenated [Ir4Hn]q+ clusters as a
function of the total number of hydrogen atoms adsorbed on the metal
cluster, n: neutral Ir4Hn clusters (circles); [Ir4Hn]+ clusters (triangles);
[Ir4Hn]2+ clusters (rhombs); [Ir4Hn]
3+ clusters (squares).
Fig. 3 Potential-derived charges q of Ir4 moiety in [Ir4Hn]q+ clusters
(a), and variations of these charges (b), as well as average shifts of the
Ir 4f levels (c) with respect to the corresponding bare clusters as a
function of the total number n of hydrogen atoms adsorbed on the
metal moiety: Ir4Hn (circles), [Ir4Hn]+ (triangles), [Ir4Hn]
2+ (rhombs),
[Ir4Hn]3+ (squares).
Publ
ishe
d on
29
July
201
0. D
ownl
oade
d by
UN
IVE
RSI
TY
OF
NE
BR
ASK
A o
n 15
/04/
2015
02:
34:1
3.
View Article Online
This journal is c the Owner Societies 2010 Phys. Chem. Chem. Phys., 2010, 12, 11015–11020 11019
from �73.6 kJ mol�1 (Ir) to �24.3 kJ mol�1 (Pd). The values
for the clusters with q = 3 e spread over a smaller interval of
B20 kJ mol�1, �76.7 kJ mol�1 (Ir) to �53.7 kJ mol�1 (Ru).
The adsorption energies are lowest (by absolute value) for
q = 1 e or 2 e, and highest for q = 3 e, except for Os4 which
features a slightly larger adsorption energy than [Os4]3+. The
adsorption interaction can increase strongly with cluster
charge; for q = 3 e the energies released were calculated
25 kJ mol�1 (Rh) and 34 (Pd) kJ mol�1 more exothermic than
in the corresponding neutral clusters.
As for Ir, the oxidation or reduction of the M4 moiety upon
the hydrogen loading depends on the total charge q of the
complexes [M4Hn]q+. This can be seen from the calculated
difference between the adiabatic IPs of the hydrogenated
clusters [M4H12]q+ and the bare clusters [M4]
q+ (Table 2).
For all neutral systems, the IP of the hydrogenated clusters is
larger than the IP of the corresponding bare clusters: this can
be considered as indication of an oxidation of the metal moiety
upon hydrogen adsorption. In contrast, the IP values of the
hydrogenated clusters with q = 2 e are lower than those of the
corresponding bare cluster, i.e. the metal moieties are reduced.
These trends are corroborated by the core level shifts of the
metal atoms (Fig. S3 of ESIw).For all transition metals studied, the charge buffer effect of the
shell of hydrogen ligands on the metal clusters exists in the case
of the highest hydrogen coverage, 12 H; for platinum, this effect
can be identified even with only six hydrogen ligands. The spread
D between maximum and minimum calculated charges q(M4) of
the metal moieties for systems of total charges q = 0 e and q =
3 e varies, from very small for Rh (D o 0.1 e) to D E 1.0 e for
Pd. Surprisingly, the charge of the M4 moiety in [M4H12]q+,
averaged over q = 0–3 e, is quite similar, 2.08–2.23 e, for the 5d
metals, while these averaged charges vary notably for the 4d
metals, from 1.92 e for Rh4 to 2.62 e for Ru4. The similarity of
the transition metal clusters modeled may be due to the similar
values of the electronegativity of the clusters, reported in Table 2
for the complexes with q = 1 e or 2 e.
4. Conclusions
The results obtained for neutral and charged iridium clusters
show that a small change of the net charge of the complexes
studied, by only 2 e or less than one electron per metal center,
reverses the character of the redox interaction between the
metal moiety and the hydrogen ligands. While the dissociative
adsorption of hydrogen on the species Ir4 and [Ir4]+ leads to
an oxidation of the metal moiety, the highly charged metal
cluster [Ir4]3+ is reduced upon hydrogen adsorption. This
contrasting behavior with a variation of the charge on the
tetrairidium clusters can be rationalized by the increasing
propensity of the metal moiety to accept electron density with
increasing the charge, as reflected in the increasing ionization
potentials of the clusters [Ir4]q+ with the charge q = 0–3 e.
Such an increase of the ionization potential with increasing
positive charge is expected to occur also for other metal
clusters and indeed has been corroborated for various late
4d and 5d transition metals. At a ‘‘critical charge’’ value, the
IP of the metal moiety will be close to the IP of the ligand
system and thus the IP will be a convenient parameter for
evaluating the redox behavior of the metal clusters with
respect not only to hydrogen, but also to other ligands.
Therefore, all metal clusters should exhibit a characteristic
(positive) charge value at which the adsorption of hydrogen
changes from being an oxidative process of the metal moiety
to a reduction.
A comparison of the charge distributions in the complexes
[Ir4H12]q+, q = 0–3 e, with maximum hydrogen loading
showed that the charges of the metal moieties are very similar,
irrespective of the charge q of the complex. This observation
suggests that in these complexes an equilibration of the
electron density takes place such that the metal fragment
carries essentially the same charge, while the charge differences
are accommodated by the hydrogen ligands. In other words,
the dissociatively adsorbed hydrogen ligands act as a charge
buffer with respect to the metal cluster.
Acknowledgements
This work was supported by the Bulgarian National Science
Fund (Contract VUH-303/07), the Bulgarian National Center
of Advanced Materials UNION (Contract DO02-82/2008),
Deutsche Forschungsgemeinschaft, and Fonds der Chemischen
Industrie (Germany).
Table 2 Various characteristics of the bare clusters [M4]q+ and the hydrogenated metal clusters [M4Hn]
q+ for late 4d and 5d transition metalsM (n = 0, 6, 12; q = 0–3) from density functional calculations
q(M4),a e
DIP,b eV w,c eV D2E,d kJ mol�1
q = 0 q = 2 q = 1 q = 2 q = 0 q = 1 q = 2 q = 3
Ru 2.62 � 0.22 0.81 �0.96 8.93 14.82 �49.6 �43.0 �48.3 �53.7Rh 1.92 � 0.05 0.21 �2.77 9.93 16.08 �42.3 �38.8 �45.7 �67.5Pd 2.05 � 0.49 1.02 �2.31 10.08 16.39 �24.3 �18.4 �38.7 �58.3Os 2.20 � 0.23 1.21 �0.82 9.24 14.39 �64.9 �58.6 �54.4 �62.1Ir 2.23 � 0.15 1.30 �0.67 9.54 15.25 �73.6 �52.8 �65.8 �76.7Pt 2.08 � 0.17 1.83 �1.05 9.82 15.81 �62.2 �48.2 �60.2 �68.6a Charge q(M4) of the metal moieties of the clusters [M4H12]
q+ averaged for q = 0 e and q = 3 e; the variation of these values is characterized by
half of their difference D. b Difference between adiabatic ionization potentials of the clusters [M4H12]q+ and [M4]
q+; positive values can be
considered to indicate oxidation of the metal moiety. c Electronegativity of the [M4]q+ clusters estimated from the adiabatic ionization potentials
IP and electron affinities EA: w= (IP+EA)/2. d Average adsorption energy per H, evaluated by least-squares fits of the dependence of the relative
energies DE of the hydrogenated clusters [M4Hn]q+ (n=0, 6, 12; q= 0–3) on the hydrogen coverage n: DE([M4Hn]
q+)= D2E� n; see eqn (1). For
the iridium clusters, the whole set of data for n = 0, 3, 6, 9, 12 was used.
Publ
ishe
d on
29
July
201
0. D
ownl
oade
d by
UN
IVE
RSI
TY
OF
NE
BR
ASK
A o
n 15
/04/
2015
02:
34:1
3.
View Article Online

11020 Phys. Chem. Chem. Phys., 2010, 12, 11015–11020 This journal is c the Owner Societies 2010
References
1 Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knozingerand J. Weitkamp, Wiley-VCH, Weinheim, 1997; A. W. Castleman,Jr. and P. Jena, Proc. Natl. Acad. Sci. U. S. A., 2006, 103,10560–10569.
2 A. M. Argo, J. F. Odzak, J. F. Goellner, F. S. Lai, F.-S. Xiao andB. C. Gates, J. Phys. Chem. B, 2006, 110, 1775–1786; F. Li andB. C. Gates, J. Phys. Chem. C, 2007, 111, 262–267.
3 J. Wei and E. Iglesia, Angew. Chem., Int. Ed., 2004, 43, 3685–3688.4 S. Liu, H. J. Zhai and L. S. Wang, J. Chem. Phys., 2002, 117,9758–9765; C. Adlhart and E. Uggerud, Chem.–Eur. J., 2007, 13,6883–6890.
5 S. T. Homeyer, Z. Karpinski and W. M. H. Sachtler, J. Catal.,1990, 123, 60–73; W. M. H. Sachtler and Z. Zhang, Adv. Catal.,1993, 39, 129–220; T. J. McCarthy, G.-D. Lei and W. M. H.Sachtler, J. Catal., 1996, 159, 90–98.
6 B. J. Kip, F. B. M. Duivenvoorden, D. C. Koningsberger andR. Prins, J. Catal., 1987, 105, 26–38; F. W. H. Kampers andD. C. Koningsberger, Faraday Discuss. Chem. Soc., 1990, 89, 137–141.
7 A. Genest, S. Kruger and N. Rosch, J. Phys. Chem. A, 2008, 112,7739–7744; K. M. Neyman, C. Inntam, L. V. Moskaleva andN. Rosch, Chem.–Eur. J., 2007, 13, 277–286.
8 S. Sicolo, C. Di Valentin and G. Pacchioni, J. Phys. Chem. C, 2007,111, 5154–5161.
9 Y. Dong and M. Springborg, Eur. Phys. J. D, 2007, 43, 15–18;R. Grybos, L. Benco, T. Bucko and J. Hafner, J. Chem. Phys.,2009, 130, 104503.
10 I. Swart, F. M. F. de Groot, B. M. Weckhuysen, P. Grune,G. Meijer and A. Fielicke, J. Phys. Chem. A, 2008, 112,1139–1149; D. J. Harding, T. R. Walsh, S. M. Hamilton,W. S. Hopkins, S. R. Mackenzie, P. Grune, M. Hartelt, G. Meijerand A. Fielicke, J. Chem. Phys., 2010, 132, 011101; M. Citir, F. Liuand P. B. Armentrout, J. Chem. Phys., 2009, 130, 054309.
11 C. Bussai, S. Kruger, G. N. Vayssilov and N. Rosch, Phys. Chem.Chem. Phys., 2005, 7, 2656–2663; S. Kruger, C. Bussai, A. Genestand N. Rosch, Phys. Chem. Chem. Phys., 2006, 8, 3391–3398.
12 G. P. Petrova, G. N. Vayssilov and N. Rosch, Chem. Phys. Lett.,2007, 444, 215–219.
13 G. P. Petrova, G. N. Vayssilov and N. Rosch, J. Phys. Chem. C,2007, 111, 14484–14492.
14 B. I. Dunlap and N. Rosch, Adv. Quantum Chem., 1990, 21,317–339.
15 N. Rosch, S. Kruger, M. Mayer and V. A. Nasluzov, in: RecentDevelopment and Applications of Modern Density Functional Theory.
Theoretical and Computational Chemistry, ed. J. M. Seminario,Elsevier, Amsterdam, 1996, vol. 4, pp. 497–566.
16 T. Belling, T. Grauschopf, S. Kruger, M. Mayer, F. Nortemann,M. Staufer, C. Zenger and N. Rosch, inHigh Performance Scientificand Engineering Computing, Lecture Notes in Computational Scienceand Engineering, ed. H.-J. Bungartz, F. Durst and C. Zenger,Springer, Heidelberg, 1999, vol. 8, pp. 439–453.
17 T. Belling, T. Grauschopf, S. Kruger, F. Nortemann, M. Staufer,M. Mayer, V. A. Nasluzov, U. Birkenheuer, A. Hu,A. V. Matveev, A. M. Shor, M. S. K. Fuchs-Rohr,K. M. Neyman, D. I. Ganyushin, T. Kerdcharoen, A. Woiterski,A. B. Gordienko, S. Majumder and N. Rosch, PARAGAUSS
version 3.0, Technische Universitat Munchen, Munchen,2004.
18 A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38,3098–3100; J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986,33, 8822–8824; J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986,34, 7406.
19 O. D. Haberlen and N. Rosch, Chem. Phys. Lett., 1992, 199,491–496.
20 N. Rosch, A. Matveev, V. A. Nasluzov, K. M. Neyman,L. Moskaleva and S. Kruger, in Relativistic Electronic StructureTheory. Part II: Applications, Theoretical and ComputationalChemistry Series, ed. P. Schwerdtfeger, Elsevier, Amsterdam,2004, vol. 14, pp. 656–722.
21 F. B. van Duijneveldt, IBM Res. Report RJ 945, 1971.22 O. Gropen, J. Comput. Chem., 1987, 8, 982–1003.23 K. M. Neyman, C. Inntam, V. A. Nasluzov, R. Kosarev and
N. Rosch, Appl. Phys. A: Mater. Sci. Process., 2004, 78, 823–828;K. M. Neyman, C. Inntam, A. V. Matveev, V. A. Nasluzov andN. Rosch, J. Am. Chem. Soc., 2005, 127, 11652–11660.
24 G. P. Petrova, G. N. Vayssilov and N. Rosch, J. Phys. Chem. C,2008, 112, 18572–18577.
25 G. N. Vayssilov and N. Rosch, J. Catal., 1999, 186, 423–432.26 B. H. Besler, K. M. Merz and P. A. Kollman, J. Comput. Chem.,
1990, 11, 431–439.27 A. J. Cohen, P. Mori-Sanchez and W. Yang, Science, 2008, 321,
792–794.28 G. J. Kubas, Chem. Rev., 2007, 107, 4152–4205.29 Other core levels shift by similar amounts. For instance, the
average shifts of the Ir 2p and Ir 5s levels change synchronouslywith the energies of the Ir 4f levels, with deviations of at most 0.03eV; see Table S2 of ESI.
30 P. S. Bagus, F. Illas, G. Pacchioni and F. Parmigiani, J. ElectronSpectrosc. Relat. Phenom., 1999, 100, 215–236.
Publ
ishe
d on
29
July
201
0. D
ownl
oade
d by
UN
IVE
RSI
TY
OF
NE
BR
ASK
A o
n 15
/04/
2015
02:
34:1
3.
View Article Online





























